
Abstract
Coculture assays allow the investigation of the role of endothelial cell and mural cell interactions in small vessel development and function. Different setups for coculture can be used to assay questions of interest. We include here methods for direct coculture, indirect coculture, and coculture in a three-dimensional extracellular matrix scaffold for studies of either a simple and direct association between the two cell types, the exchange of soluble molecules, or the interaction within a biomimetic tissue microenvironment.
Access provided by CONRICYT – Journals CONACYT. Download protocol PDF
Similar content being viewed by others



Key words
- Angiogenesis
- Endothelial cells
- Pericytes
- Coculture
- Matrigel
- Small intestinal submucosa
- Intercellular interactions
- Paracrine
- Juxtacrine
- Microvessels
1 Introduction
Cocultures have been used successfully over the years to study the interactions between endothelial cells and cells collectively known as mural cells (smooth muscle cells and pericytes ). Two-dimensional (2D) direct and indirect coculture methods in cell culture plates are useful to examine interactions that require direct contact (juxtacrine) or those that are made through diffusion of soluble factors (paracrine). Three-dimensional (3D) techniques involve the use of agarose, matrigel , or a fibrous decellularized extracellular matrix (ECM) scaffold to investigate interactions within a 3D-biomimetic environment. We have investigated through the use of cocultures the effect of heterotypic cell interactions on cell proliferation [1–3], migration [4, 5], apoptosis [6], activation of growth factors [2], cord formation [6, 7], and Notch signaling [8]. Coculture set-ups have allowed us to examine signaling pathways that dictate the behavior of endothelial cells [2, 4–7] during vessel formation, and the maturation and stabilization of neovessels through communication with pericytes [1–3, 6, 8].
We describe here three different methods of coculture including direct coculture, indirect coculture using transwells, and coculture in a 3D matrix using an ECM scaffold [9]. The methodologies include detailed culture conditions for primary human endothelial cells and an updated protocol for isolating and culturing bovine retinal pericytes [2, 10].
2 Materials
All materials should be sterilized by autoclaving, filtering, or ethylene oxide sterilization.
2.1 Materials for the Culture of Human Retinal Microvascular Endothelial Cells (HRECs)
-
1.
Primary human retinal microvascular endothelial cells (catalog number ACBRI 181) (Cell Systems).
-
2.
0.2 % gelatin in PBS (must be tissue culture grade) autoclaved.
-
3.
HREC culture medium: EBM-2 supplemented with 2 % fetal bovine serum (FBS) (catalog number S11150) (Atlanta Biologicals), EBM-2, 100 U/mL penicillin and 100 mg/mL streptomycin (1 % P/S), 2 mM l-glutamine (1 % l-glutamine), Single Quot (catalog number CC-4176) (Lonza). FBS must be refiltered and complete media must be filtered after preparation .
-
4.
1× phosphate buffer saline (PBS) without calcium and magnesium, must be autoclaved.
-
5.
Trypsin-versene (catalog number 17-161E) (Lonza).
-
6.
HREC freezing media: 90 % FBS and 10 % DMSO solution.
-
7.
Six-well or 24-well transwell supports.
2.2 Materials for Isolation and Culture of Bovine Retinal Pericytes (BRPs)
-
1.
Calf eyes shipped on ice (see Note 1 ).
-
2.
4 L of sterile PBS.
-
3.
Two No. 15 surgical blades.
-
4.
Poly-lined sterile field drapes, 18″ × 26″, one per eye.
-
5.
Surgical spears, one per eye (catalog number Q604230) (Fabco).
-
6.
Cell strainers, 100 μm pore size mesh (catalog number 352360) (Corning).
-
7.
Cell strainers, 70 μm pore size mesh (catalog number 352350) (Corning).
-
8.
Two sets of sterile scissors and forceps.
-
9.
Two 150 × 25 mm culture dishes per eye.
-
10.
Two 50 mL conical tubes per eye.
-
11.
Two sterile 1 L beakers.
-
12.
One 2 L beaker.
-
13.
One L of 0.4 % betadine solution in PBS (see Note 2 ).
-
14.
15 mL of collagenase (digest enzyme) solution per eye (prepare the day prior to isolation): 51,051 units collagenase and 0.25 % of FBS in PBS; filter sterilize.
-
15.
PBS-EDTA solution: 2.5 mL of 0.5 M (pH 8.0) in 500 mL of PBS; filter sterilize.
-
16.
Trypsin-EDTA (0.25 %) (catalog number 25200-056) (Life Technologies).
-
17.
BRP washing medium (prepare the day prior to isolation): Dulbecco’s modified Eagle’s medium (DMEM) (catalog number DMEM 12-708F) (Lonza) supplemented with 10 % calf serum (catalog number SH30072.03) (HyClone), 1 % P/S, 1 % l-glutamine, 0.025 mg/mL nystatin (catalog number N3503-5MU) (Sigma); filter sterilize.
-
18.
BRP culture medium (prepare the day prior to isolation): DMEM (catalog number DMEM 12-708F) (Lonza) supplemented with 10 % calf serum (catalog number SH30072.03) (HyClone), 1 % P/S and 1 % l-glutamine; filter sterilize.
-
19.
BRP growth medium: DMEM (catalog number DMEM 12-708F) (Lonza), 20 % bovine calf serum (catalog number 12133C) (Sigma), 1 % P/S and 1 % l-glutamine; filter sterilize (see Note 3 ).
-
20.
BRP freezing medium: 85 % FBS, 10 % DMSO and 5 % DMEM (catalog number DMEM 12-708F) (Lonza).
2.3 Materials for the Preparation of an ECM Scaffold from Porcine Small Intestine and for 3D ECM Coculture
-
1.
Hydrogen peroxide solution (catalog number 216763) (Sigma).
-
2.
Sodium hypochlorite solution (catalog number 425044) (Sigma).
-
3.
1 L autoclaved deionized water (DI).
-
4.
500 mL autoclaved PBS.
-
5.
Four-well chamber slide system (catalog number 177399) (Thermo Scientific).
-
6.
Sterile forceps and scissors.
-
7.
ECM coculture medium: DMEM (catalog number DMEM 12-708F) (Lonza), 10 % FBS (catalog number S11150) (Atlanta Biologicals), 1 % P/S and 1 % l-glutamine; filter sterilize.
3 Methods
Work in a biosafety cabinet. Use a surgical mask to protect yourself from exposure to enzymes.
3.1 Culture of HRECs
3.1.1 Thawing of HRECs
-
1.
Coat T75 flasks with 5 mL of 0.2 % gelatin in PBS, by adding gelatin evenly to the culture surface of the flask, incubating at 37 °C (inside incubator) for at least 30 min, aspirating gelatin from flasks and washing twice with 10 mL of PBS.
-
2.
Warm to 37 °C the HREC culture medium.
-
3.
Transfer cells from a vial with 1 × 106 cells to a 15 mL tube.
-
4.
Add 5 mL of HREC culture medium to resuspend the cells and mix by pipetting.
-
5.
Spin at 423 × g for 4 min.
-
6.
Aspirate media without disturbing the pellet.
-
7.
Resuspend in 10–12 mL of media and mix by pipetting.
-
8.
Plate in one T75 flask.
3.1.2 Passaging of HRECs
-
1.
Passage at a 1:3 to 1:5 ratio (see Note 4 ).
-
2.
Warm to 37 °C the HREC culture medium, trypsin-versene and PBS without calcium and magnesium.
-
3.
Accordingly with the desired passage ratio, coat T75 flasks as indicated in Subheading 3.1.1.
-
4.
Wash cells to be passaged with 5 mL PBS.
-
5.
Add 3 mL of trypsin-versene and incubate at 37 °C for 3–5 min, until cells detach; gently tap the sides of the flask to assist detachment.
-
6.
Add 6 mL of complete media to quench the trypsin and mix by pipetting.
-
7.
Transfer cells to a 15 mL conical tube and spin at 423 × g for 4 min.
-
8.
Aspirate media without disturbing the pellet.
-
9.
Resuspend and plate in the chosen passage ratio in 10–12 mL of complete media.
-
10.
Change media every other day.
3.1.3 Freezing of HRECs
-
1.
Wash cells to be frozen with 5 mL PBS.
-
2.
Add 3 mL of trypsin-versene and place in 37 °C incubator for 3–5 min, until cells detach; gently tap the sides of the flask to assist detachment.
-
3.
Add 6 mL of complete media to quench the trypsin and mix by pipetting.
-
4.
Transfer cells to a 15 mL conical tube and spin at 423 × g for 4 min.
-
5.
Aspirate media without disturbing the pellet.
-
6.
Count cells using an automated cell counter or a hemocytometer and resuspend in freezing media as 1 × 106 cells/mL; freeze in 1 mL volumes.
3.2 Isolation of Bovine Retinal Pericytes (BRPs)
3.2.1 Isolation of BRPs
-
1.
Upon arrival place eyes on ice.
-
2.
Set up the following materials inside the biosafety cabinet: 1 L sterile beakers, sterile drape, culture dishes, surgical spears, surgical tools, surgical blades, 500 mL PBS, sterile filtered wash medium, 50 mL conical tubes and the collagenase solution.
-
3.
Outside the biosafety cabinet, immerse the eyes in betadine (enough to cover the eyes) inside a 2 L beaker for 15 min.
-
4.
Fill two sterile 1 L beakers with sterile PBS; remove the eyes from the betadine one at a time and submerge in one of the 1 L beakers with sterile PBS.
-
5.
Prepare 130 mL per eye of the stock BRP wash medium diluted 1/25 with sterile PBS. Arrange one separate conical tube per eye, each containing 25 mL of solution.
-
6.
Place the lid of one of the culture dishes upside down under the sterile drape and manipulate the eye on the drape (see Note 5 ). Place the bottom part of the dish diagonally behind the dissection area while on the sterile drape, and use for collecting waste tissue.
-
7.
Remove the orbital fat and muscle surrounding the eyeball using sterile scissors and forceps. This can be done for all eyes on the same sterile drape prior to beginning dissection. After trimming, place the eyes in the other 1 L beaker filled with PBS. Change the sterile drape.
-
8.
Place one eye on the clean sterile drape placed over the culture dish lid. While holding the eye with forceps by the optic nerve, make an incision with the surgical blade roughly 5 mm posterior from the iris. Using scissors, cut around the entire eyeball to remove the anterior segment and place in the waste dish (Fig. 1a). The vitreous body (which is also waste) should come out with the anterior segment. Pull the vitreous out using forceps if it did not come out with the anterior segment.
Fig. 1 
Dissection of a bovine retina. (a) Dissection of the anterior and posterior segments of the eye. (b) Removal of a circular section (dashed line) of the retina in the center of the posterior segment containing the larger retinal vasculature
-
9.
Applying very light pressure, use a scalpel blade to cut out a thin circular section in the center of the posterior segment (about 0.1 mm deep and 40 mm in diameter) (Fig. 1b). Use a moist surgical spear to gather up the sectioned tissue towards the center (see Note 6 ). Cut the optic nerve and place the circular section in the waste dish (see Note 7 ).
-
10.
Use a moist surgical spear to gather towards the center of the posterior segment the remainder of the retina (which will have a circular hole in the center after the removal of the circular section in the previous step) (see Note 8 ). Carefully lift the gathered retina with a pair of forceps. Cut the remaining attachments of the retina to the optic nerve and then remove the retina with the second pair of sterile forceps. Place in the conical tube containing the 25 mL diluted BRP wash medium solution.
-
11.
Repeat the dissection on the remaining eyes. Keep the retinas in the previously arranged separate conical tubes containing diluted BRP wash medium solution (see Note 9 ). Keep at room temperature for less than 1 h.
-
12.
Wash each retina by gently inverting the tube three to four times. Carefully pour out the wash solution into a waste container without pouring out the retina. Wash twice more with BRP wash medium solution for a total of three washes. Remove as much of wash solution as possible and then transfer the retina to a clean sterile culture dish by pouring (see Note 10 ).
-
13.
Aspirate 1 mL of collagenase solution into a p1000 pipette and force the tissue through the pipette four to seven times until it flows easily. After the first or second pipetting movements, check if there are any visible large vessels (such as those present in the circular section removed in step 9) and pipette them out in a separate area of the culture dish.
-
14.
Continue to inspect and pipette the retina in collagenase solution four to seven more times until fully digested. Remove any visible large vessels to the waste dish. Add 13 mL of collagenase solution to the dish and pipette up and down with a p1000 pipette four to seven times.
-
15.
Incubate at 37 °C and 5 % CO2 for 1 h.
-
16.
Pipette up and down the digested tissue 10–15 times with a 10 mL pipette, and check under the microscope for detached single cells.
-
17.
When a single cell suspension is obtained, pass the solution through a 100 μm mesh into a clean sterile 50 mL conical tube containing 25 mL of BRP wash medium solution.
-
18.
Spin at 751 × g for 4 min.
-
19.
Aspirate the supernatant and repeat washing twice with 10–15 mL of BRP wash medium solution.
-
20.
Resuspend cells in 25 mL of BRP culture medium and plate by passing through a 70 μm mesh into a T175 flask. Culture at 37 °C and 5 % CO2.
-
21.
The following day, aspirate the medium, wash twice with 10 mL of PBS and replenish with 25 mL of BRP culture medium. Repeat on the sixth day of culture. See Fig. 2a–c for images of cells on the second and fourth day of culture.
Fig. 2 
Isolation of bovine retinal pericytes. 2 (P0) (a, b), 4 (P0) (c), 8 (P0) (d), 10 (P0) (e), and 18 (P1) (f) days of culture. Pericytes begin spreading on the plate after 4 days and take around 10–12 days to achieve confluency. Contaminants in culture after 2 (b) and 10 days of culture (e, black solid line). Pericytes are shown in white dashed line circles in (e). Bar: 500 μm
-
22.
On the eighth day of culture (Fig. 2d), replace the media with PBS and observe each flask under a microscope to determine the presence of non-pericyte cells such as endothelial cells, astrocytes, and fibroblasts, considered here as “contaminants” (Fig. 2b, e). Mark the location of contaminant colonies on the bottom of the flask. Colonies are easily detached using a rapid EDTA treatment as follows: wash the cells with PBS and then add 10 mL of PBS-EDTA solution. After 2 min, contaminants will be fully lifted of the plate while pericytes will only start to round up. Tap firmly the sides of the flask to assist detachment of contaminants, aspirate and wash twice with PBS. Add 25 mL of BRP growth medium. Confirm that all contaminants are removed under the microscope using the marked location as guide. Make sure there are no more “contaminants” before passaging (see Notes 11 and 12 ).
-
23.
Passage cells at a 1:1.5 ratio when they reach 90 % confluency (between day 8 and 14) by using the standard passaging procedure described in Subheading 3.2.2. P1 cells can be maintained in culture for 1–2 weeks without changes in morphology (Fig. 2f).
-
24.
If “contaminant” cells are visible in the next 1–2 weeks, use trypsin-versene (5 mL for a T175 flask) to remove them. Generally, endothelial and smooth muscle cells will detach before pericytes . Wash cells with 13 mL PBS, aspirate, add trypsin-versene, swirl it around in the flask and use the microscope to observe detachment of “contaminants”. Add twice the volume of culture medium to quench the trypsin, aspirate and wash twice with 13 mL PBS. Then passage pericytes using trypsin-EDTA by the standard passaging procedure in Subheading 3.2.2. P2 and P3 cells can be frozen 2–3 days later, using the procedure described in Subheading 3.2.2.
-
25.
Use BRPS up to P6 and always passage before 90 % confluency (avoid 100 % confluence, since they start to pile up).


3.2.2 Passaging and Freezing of BRPs
-
1.
Wash cells with 13 mL PBS and aspirate.
-
2.
Add 5 mL of trypsin-EDTA and incubate at 37 °C for 3–5 min, until cells detach; gently tap the sides of the flask to assist detachment.
-
3.
Add 10 mL of BRP growth medium to quench the trypsin and mix by pipetting.
-
4.
Transfer cells to a 50 mL conical tube and spin at 423 × g for 4 min.
-
5.
Aspirate media without disturbing the pellet.
-
6.
To passage: resuspend in 6–9 mL of BRP growth medium and passage at a 1:3 ratio. Add media to complete a volume of 12 mL of BRP growth medium in T75 flasks or 25 mL BRP growth medium in T175 flasks.
-
7.
To freeze: count cells using an automated cell counter or a hemocytometer, resuspend in freezing medium as 1 × 106 cells/mL and freeze in 1.0 mL volumes.
3.3 Direct Coculture of Endothelial Cells and Pericytes
-
1.
Expand BRPs and HRECs in their respective culture media splitting at 1:3 ratios (Fig. 3a, b).
Fig. 3 
Direct endothelial cell-pericyte coculture. (a) Human retinal endothelial cells in monoculture. (b) Bovine retinal pericytes in monoculture. (c) Example of direct coculture of endothelial cells (circles) and pericytes (arrows) plated at a 1:1 ratio and allowed to grow for 2 days. Bar: 500 μm
-
2.
For assays of pericyte recruitment, pericyte differentiation or endothelial cell proliferation (determined through the final total number of endothelial cells plus pericytes in the plate), proliferation of pericytes can be arrested at 80 % confluence by incubating in mitomycin C (10 μg/mL) for 2 h.
-
3.
Wash BRPs with PBS and detach from flask with trypsin-EDTA.
-
4.
Plate in 24-well plates at a density of 2.0 × 104 cells per well in BRP culture media and allow to attach overnight.
-
5.
Next day, detach HRECs as described in Subheading 3.1.2 and add an equal number of HRECs in BRP culture medium.
-
6.
Incubate and assay as desired (Fig. 3c).
-
7.
Simultaneously and using identical conditions , establish individual cultures of HRECs and proliferation-arrested BRPs as controls.

3.4 Indirect Coculture of Endothelial Cells and Pericytes in Transwells
-
1.
Expand BRPs and HRECs in their respective culture media splitting at 1:3 ratios (see Note 13 ).
-
2.
Wash BRPs with PBS and detach from flask with trypsin-EDTA.
-
3.
Plate BRPs in 24-well plates at a density of 2.0 × 104 cells per well or in 6-well plates at a density of 1.5 × 105 cells per well in BRP culture media and allow to attach overnight.
-
4.
Next day, detach HRECs as described in Subheading 3.1.2 and plate an equal number of HRECs into the upper chamber of transwells and allow to attach for 90 min.
-
5.
Place transwells into the wells containing the BRPs (Fig. 4a).
Fig. 4 
(a) Indirect coculture of endothelial cells and pericytes in transwells. Pericytes are plated on 6- or 24-well plates overnight. Endothelial cells are plated at 1:1 ratio the next day on the upper chamber of correspondent 6- or 24-well transwells. (b) Control with direct contact through the transwell membranes. Pericytes are incubated overnight on the underside of the transwells, which are then flipped over and inserted in wells. Endothelial cells are plated on the upper chambers of the transwells
-
6.
Add 1 mL of BRP culture media.
-
7.
Incubate and assay as desired.
-
8.
Simultaneously and using identical conditions, establish individual cultures of HRECs and BRPs as controls, as well as a direct contact coculture control. For the latter, plate BRPs on the underside of the transwells and allow to attach overnight; flip over the transwells, insert into wells, and plate HRECs into the upper chamber of the transwells (Fig. 4b).

3.5 Coculture in a Three-Dimensional ECM Scaffold
3.5.1 Preparation of an ECM Scaffold from Porcine Small Intestine
-
1.
Obtain the jejunum portion of the small intestine of a market-weight porcine donor, section into 30 cm long segments and open each segment longitudinally to form a rectangle.
-
2.
Rinse well with tap water and scrape off the exterior (serosa and muscularis) and interior (mucosa) layers of the intestine with the edge of a scalpel handle. The remaining tissue is the submucosa layer (Fig. 5a).
Fig. 5 
Preparation of an extracellular matrix scaffold. (a) Cross-section of the porcine small intestine. (b) Rectangular scaffold after preparation
-
3.
Wash for 1 h in DI with rapid agitation on an orbital shaker.
-
4.
Decellularize and disinfect to obtain acellular ECM scaffolds, by washing for 15 min in a solution of oxidizing agents such as 2.0 % sodium hypochlorite and 2.0 % hydrogen peroxide in DI with fast agitation.
-
5.
Wash for 15 min once in sterile PBS and twice in sterile DI.
-
6.
Let dry for 1–2 h inside a biosafety cabinet on top of clean saran wrap cleaned with 70 % ethanol.
-
7.
Sterilize each ECM scaffold separately with ethylene oxide and use within 6 months.

3.5.2 Coculture
-
1.
Expand BRPs and HRECs in their respective culture media splitting at 1:3 ratios.
-
2.
Use sterile scissors and forceps to cut 27 × 21 mm rectangles from a sterile ECM scaffold (Fig. 5b).
-
3.
Use forceps to place a scaffold on the bottom of each well in a chamber slide (Fig. 6a).
Fig. 6 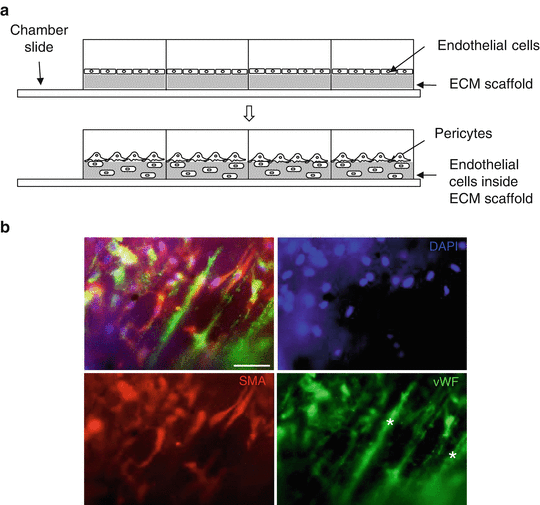
Coculture of endothelial cells and pericytes in an ECM scaffold (decellularized small intestinal submucosa ). (a) Endothelial cells are incubated in an ECM scaffold placed on the bottom of a chamber slide. Pericytes are added after 2 h. (b) Endothelial cells (stained with von Willebrand factor, vWF) and pericytes (stained with smooth muscle actin, SMA) locate inside the scaffold after 2 h of coculture. Scaffold fibers fluoresce in the green channel (stars). The out of focus location of multiple cells shows the 3D character of this coculture method. Bar: 50 μm
-
4.
Detach HRECs as described in Subheading 3.1.2, culture 5.0 × 104 HRECs per well on top of the ECM scaffold and incubate for 2 h in 1 mL of HREC culture media.
-
5.
Carefully aspirate the HREC media (without touching the ECM scaffold), add an equal number of BRPs in 500 μL of coculture media to each well and allow to attach for 2 h.
-
6.
Culture as desired; assay gene expression or image by immunofluorescent staining as desired (Fig. 6b) (see Notes 14 – 16 ).
-
7.
Simultaneously and using identical conditions, establish cocultures of HRECs and BRPs as controls on chamber slides without a scaffold.

4 Notes
-
1.
Calf eyes yield a higher number of pericytes with a higher proliferative potential than cells isolated from adult eyes.
-
2.
Use glass flasks, not plastic, to make dilution.
-
3.
Bovine calf serum 12133C from Sigma induces increased proliferation of pericytes.
-
4.
Passage at 90 % confluence. Confluence at these passage ratios is achieved in 3–6 days.
-
5.
The dish prevents the eye from rolling and maintains sterility.
-
6.
Keep the spear moist with PBS to avoid the retina from sticking to the spear.
-
7.
The larger vessels in the retina , where mural cells are mainly smooth muscle cells, will be removed with this section. These vessels are visible with the naked eye. The remaining retina in the posterior segment will contain the pericyte-covered microvasculature (which is not visible with the naked eye).
-
8.
Be careful not to remove any underlying pigmented tissue (i.e., the tapetum lucidum) or the retinal pigment epithelium.
-
9.
If any loose pigmented tissue is observed, carefully remove it by aspiration (do with care as it can result in the aspiration of the entire retina).
-
10.
Wash medium can be aspirated (instead of poured), but once again should be done with care to avoid the aspiration of the retina.
-
11.
If “contaminants” are still visible after the EDTA treatment, keep the cells in BRP culture medium for a few more days and then repeat this step.
-
12.
The most common “contaminants” are endothelial cells and fibroblasts. If left unattended, endothelial cells can become the 70 % of the total cell population. Fibroblasts hardly reach populations higher than the 10 % of the total cell population.
-
13.
Arrest of cell growth is only necessary for direct culture, where endothelial cell growth is determined by final cell number including both endothelial cell and pericyte total count. This is not needed in coculture since cells are physically separated.
-
14.
Interactions between HRECs and BRPs are observed after 2–4 days.
-
15.
Image scaffold without sectioning as cells tend to detach from thinner sections during the washes of staining procedures.
-
16.
Imaging of scaffolds works very well with upright fluorescence microscopy. Note in Fig. 4 the out of focus (i.e. out of plane) location of several cells, due to the 3D character of this coculture method. vWF antibody (catalog number A0082) (Dako) at a 1:100–200 dilution clearly stains endothelial cells (Fig. 6b). We have not obtained good results working with lectin for staining of endothelial cells on SIS membranes. For pericytes , SMA antibody (catalog number C6198) ( Sigma) works very well at a 1:400 dilution (Fig. 6b). Alternatively, live cells can be prelabeled before seeding on the chamber slide using a PKH26 cell membrane linker kit (catalog number PKH26GL) (Sigma) to label endothelial cells red and a PKH67 cell linker kit (catalog number PKH67GL) (Sigma) to label pericytes green. To label 1 × 106 live cells, detach cells from flask and wash twice with serum-free media. Add 0.5 μL of PKH dye to 50 μL of diluent-C and vortex. After second wash, resuspend cells in 50 μL diluent-C and mix with dye solution . Incubate for 3–5 min at room temperature. Add complete media (with serum) to quench reaction and wash two to three more times in complete media. Resuspend and culture as desired.
References
Orlidge J, D’Amore P (1987) Inhibition of capillary endothelial cell growth by pericytes and smooth muscle cells. J Cell Biol 105(3):1455–1462
Antonelli-Orlidge A, Saunders K, Smith S, D’Amore P (1989) An activated form of transforming growth factor beta is produced by cocultures of endothelial cells and pericytes. Proc Natl Acad Sci 86(12):4544–4548
Dodge A, Lu X, D’Amore P (1993) Density-dependent endothelial cell production of an inhibitor of smooth muscle cell growth. J Cell Biochem 53:21–31
Hirschi K, Rohovsky S, D’Amore P (1998) PDGF, TGFbeta and heterotypic cell-cell interactions mediate endothelial cell-induced recruitment of 10T1/2 cells and their differentiation to a smooth muscle fate. J Cell Biol 141(3):805–814
Hirschi K, Rohovsky S, Beck L, Smith S, D’Amore P (1999) Endothelial cells modulate the proliferation of mural cell precursors via platelet-derived growth factor-BB and heterotypic cell contact. Circ Res 84(3):298–305
Darland D, Massingham L, Smith S, Piek E, Saint-Geniez M, D’Amore P (2003) Pericyte production of cell-associated VEGF is differentiation-dependent and is associated with endothelial survival. Dev Biol 264(1):275–288
Darland D, D’Amore P (2001) TGFb is required for the formation of capillary-like structures in three-dimensional cocultures of 10T1/2 and endothelial cells. Angiogenesis 4:11–20
Arboleda-Velasquez JF, Primo V, Graham M, James A, Manent J, D’Amore P (2014) Notch signaling functions in retinal pericyte survival. Invest Ophthalmol Vis Sci 55(8):5191–5199
Sánchez-Palencia D, Rathan S, Ankeny C, Fogg R, Briceno J, Yoganathan A (2015) Mechanotransduction in small intestinal submucosa scaffolds: fabrication parameters potentially modulate the shear-induced expression of PECAM-1 and eNOS. J Tissue Eng Reg Med. doi:10.1002/term.2040
Bryan B, D’Amore P (2008) Pericyte isolation and use in endothelial/pericyte coculture models. In: Cheresh D (ed) Methods of enzymology, 1st edn. Elsevier, San Diego, CA
Acknowledgements
This work was supported by NIH grants R00EY021624 (JA) and R01EY005318 (PDA).
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2016 Springer Science+Business Media New York
About this protocol
Cite this protocol
Sánchez-Palencia, D.M., Bigger-Allen, A., Saint-Geniez, M., Arboleda-Velásquez, J.F., D’Amore, P.A. (2016). Coculture Assays for Endothelial Cells-Mural Cells Interactions. In: Ribatti, D. (eds) Tumor Angiogenesis Assays. Methods in Molecular Biology, vol 1464. Humana Press, New York, NY. https://doi.org/10.1007/978-1-4939-3999-2_4
Download citation
DOI: https://doi.org/10.1007/978-1-4939-3999-2_4
Published:
Publisher Name: Humana Press, New York, NY
Print ISBN: 978-1-4939-3997-8
Online ISBN: 978-1-4939-3999-2
eBook Packages: Springer Protocols