
Abstract
Nucleoli form around RNA polymerase I transcribed ribosomal RNA (rRNA) genes. The direct electron microscopy observation of rRNA genes after nucleolar chromatin spreading (Miller’s spreads) constitutes to date the only system to quantitatively assess transcription at a single molecule level. However, the spreading procedure is likely generating artifact and despite being informative, these spread rRNA genes are far from their in vivo situation. The integration of the structural characterization of spread rRNA genes in the three-dimensional (3D) organization of the nucleolus would represent an important scientific achievement. Here, we describe a correlative light and electron microscopy (CLEM) protocol allowing detection of tagged-Pol I by fluorescent microscopy and high-resolution imaging of the nucleolar ultrastructural context. This protocol can be implemented in laboratories equipped with conventional fluorescence and electron microscopes and does not require sophisticated “pipeline” for imaging.
Access provided by CONRICYT – Journals CONACYT. Download protocol PDF
Similar content being viewed by others


Key words
- Correlative light and electron microscopy (CLEM)
- Nucleolus
- RNA polymerase I (Pol I)
- Ribosomal RNA genes (rDNA)
- Pol I transcription
- Transmission electron microscopy (TEM)
- Fluorescence microscopy
- Yeast
- Saccharomyces cerevisiae
1 Introduction
Correlative light and electron microscopy (CLEM) combines the benefits of light microscopy (LM) and transmission electron microscopy (TEM ). On the one hand, LM can provide information about distribution of fluorescent tagged proteins and their dynamic properties when performing live-cell imaging ; rare events can also be preselected by LM before TEM analysis. On the other hand, TEM produces, at high resolution, images of the ultrastructural context of the proteins of interest. The main determinant of success in CLEM is the sample preparation. It should ensure the preservation of the cellular ultrastructure and the fluorescence of the tags simultaneously.
Here, we present a detailed protocol including sample preparation and an imaging workflow adapted for CLEM analysis of the transcription of ribosomal RNA (rRNA) genes by the RNA polymerase I (Pol I) in the nucleus of the budding yeast Saccharomyces cerevisiae. The advantages of this model stem from the repeated nature of the ribosomal DNA (rDNA) (~150 copies, half of them being transcribed) and the local very high transcriptional activity of the Pol I; it results in the formation of a subnuclear domain, the nucleolus, easy to observe and considered to be the “transcription factory” for rRNA production (Fig. 1a). Contrariwise, RNA polymerase II (Pol II) transcription, producing messenger RNA (mRNA), is a phenomenon intrinsically difficult to characterize at the single molecule level due to the thousands of different transcripts Pol II has to generate, scattered throughout the nucleoplasm. The ultrastructural characterization of Pol I transcribed genes is comparatively simple and was introduced in 1969 by O. Miller [1] by TEM analysis of extracted and spread nuclear chromatin (Fig. 1b, c). This direct electron microscopy observation constitutes to date the only system to quantitatively assess transcription at a single molecule level: each Pol I transcription unit adopts a characteristic “Christmas tree” conformation with each Pol I molecule loaded on the rDNA (tree’s trunk) associated with nascent rRNA appearing as growing “branches” decorated by “terminal knobs” (Fig. 1c). However, the spreading procedure is likely generating artifact and despite being informative, these spread rRNA genes are far from their in vivo situation! The integration of the structural characterization of spread rRNA genes in the three-dimensional (3D) organization of the nucleolus by CLEM would represent an important scientific achievement. S. cerevisiae provides an ideal model system for such studies, since rRNA genes represent about 10 % of the genome, rendering their detection relatively simple. In addition, thanks to a diversity of genetic tools, the Pol I transcription apparatus can be easily modified in baker’s yeast . Finally, while Pol I was for long time considered as a very specialized transcriptional machinery for the rDNA, recent structural studies provided evidences that molecular details of transcription by Pol I and Pol II are very related, consolidating the relevance of rDNA transcription as a model for highly transcribed genes [2].

Electron micrograph of the ribosomal RNA genes. (a) On a yeast section, the nucleus of S. cerevisiae displays a crescent-shaped dense nuclear subdomain, the nucleolus organized around the repeated rRNA genes transcribed by the Pol I machinery. Note that rod-like structures strongly counterstained, reminiscent of spread transcription units are visible on sections (red rectangles). (b) By Miller’s treatment, the head-to-tail repetitions of rRNA genes are spread and can be individually observed. (c) High magnification of one rDNA transcription unit adopting characteristic “Christmas tree” configuration
The CLEM method here described allows detection of Pol I and histones molecules tagged with fluorophores on yeast sections which are then imaged by TEM. This long procedure includes (1) the fixation step and resin embedding that preserve both ultrastructure of the cell nucleus and the fluorescence of tagged proteins, (2) the sectioning and harvesting of the sections in a compatible setup for fluorescent and TEM microscopy, and (3) the development of an “imaging workflow” to sequentially image sections in fluorescence and in TEM and align the pictures.
2 Materials
Prepare all solutions using deionized ultrapure water with a resistivity of 18 MΩ at 25 °C (milli-Q water) and analytical grade reagents. Carefully follow all waste disposal instructions when disposing waste materials; pay especially attention with reagents for TEM which are often hazardous.
2.1 Fixation and Resin Embedding
-
1.
Cryoprotective solution to resuspend the yeasts : dissolve 200 mg agarose Low Melting Point and 140 mg sucrose in 0.1 M sodium cacodylate buffer (EMS). 0.2 M cacodylate buffer (pH 7.2) in milli-Q water is stored at 4 °C for few months. (Take care, cacodylate contains sodium dimethyl arsenate!) Just before using, melt the cryoprotective solution few seconds in a microwave oven and keep it fluid on a water bath at 40–42 °C.
-
2.
Cryogenic fluid: liquid nitrogen (LN2) is extremely cold (−196 °C) and can cause severe burns to the skin and eyes. The experimenter has always to work with protective gloves and glasses. When LN2 evaporates, the air oxygen content decreases; therefore, LN2 is stored in Dewars stored in well ventilated area and the working place has to be kept in a well aerated room.
-
3.
The fixative: 0.1 % uranyl acetate, 2H2O (LauryLab) is dissolved in glass distilled acetone. The use of uranyl acetate is under strict regulation. To avoid any risk of contamination, manipulate the uranyl acetate exclusively under the fume hood. Dispose a small quantity of uranyl acetate crystals in a preweighed Eppendorf tube. Close the tube and weigh it to calculate the quantity of the uranyl acetate. Therefore, adapt the volume of acetone required to reach the correct dilution. A quantity of crystals equivalent to a matchstick tip weighs approximately 50 mg and has therefore to be dissolved in 50 ml of acetone.
-
4.
The resin: the lowicryl HM20 resin (EMS) is a highly cross-linked acrylate-based embedding resin, providing low viscosity at low temperature. It is stored at 4 °C for few months and ready, meaning that no addition of any catalyzer or accelerator is required. Before use, it is precooled in the freeze substitution system or in a −80 °C freezer. The resin is photo-polymerized by wavelength (360 nm) with a LED UV lamp adapted to the freeze substitution system. Pay attention with resin, it is armful if inhaled, and causes skin and eyes irritation.
-
5.
Freeze Substitution system: Leica EM AFS2 is a user-programmed freeze substitution and low temperature embedding system. It consists of a freeze substitution chamber with a stainless steel working platform equipped with an integrated LED UV lamp. The chamber is cooling down thanks to LN2 stored in a Dewar vessel mounted on a mobile trolley. The programming is made with a control unit and program steps are displayed on a color screen. The AFS2 is equipped with an automatic reagent handling system EM Freeze Substitution Processor (EM FSP, Leica) which dispenses reagents, dilute media and resins from 100 % stock solutions in containers.
2.2 Sectioning and Sections Harvesting
-
1.
Plastic support on TEM grids: collodion (Parlodion) 2 % in amyl acetate (EMS).
-
2.
Carbon rods 3 mm diameter (EMS).
-
3.
Ultra-microtome: Ultracut (Reichert).
-
4.
Diamond knife 35° (Diatome Inc).
-
5.
Finder grids with an alphanumerical code: Gilder Grids G200F2, in copper or nickel (EMS).
-
6.
Carbon Coater: the Q150T Turbo-Pumped Carbon Coater (Quorom Technologies) is a compact turbomolecular-pumped coating system that can evaporate carbon from carbon rods (EMS) with a precise thickness control (quartz crystal oscillator). The grids are installed on a stage that can rotate for a homogeneous covering.
2.3 Fluorescence and TEM Acquisitions
-
1.
Glass coverslips for fluorescence acquisition: long (24 × 60 mm) glass coverslips (Menzel-Gläser) are used. This size is required because these coverslips are used as slides and have to fit with the fluorescent microscope stage.
-
2.
Fluorescence imaging was performed with a Nikon TI-E/B inverted microscope featuring an EMCCD camera (Ixon Ultra DU897-ANDOR) and a HG intensilight illumination. Images were acquired using Nikon CFI Apo TIRF 100× (NA = 1.49) objective and Semrock filters sets for GFP (Ex: 482BP35; DM: 506; Em: 536BP40) and mCherry (Ex. 562BP40, DM593, Em.641BP75).
-
3.
Heavy metals for contrast:
Uranyl acetate: uranyl acetate (LauryLab) 5 % in water is stored at 4 °C, for months, protected from light.
Lead Citrate: lead citrate, trihydrate (EMS) 0.3 % is prepared in 0.1 M NaOH and stored at room temperature.
-
4.
Sodium hydroxide: ultra, pellets (Fluka).
-
5.
Transmission electron microscope: Jeol-JEM 1400.
3 Methods
In order to study the nuclear organization of the Pol I transcription , we generated a collection of yeast strains bearing genetically engineered fluorescent tags on proteins of the Pol I transcription machinery (Pol I subunits, transcription factors, etc.). In order to establish preparative conditions for CLEM, i.e., that preserve both ultrastructure of cells and the fluorescence of tagged proteins, we performed experiments with a strain containing a reduced number of rRNA genes copies (25 instead of 150) and expressing the Pol I subunit Rpa190 in fusion with the GFP and the H2B histone in fusion with m-Cherry (see Note 1 ). The advantages of using this strain stem from the abundance of H2B and Pol I correlated with very strong unambiguous fluorescent signals in living cells, therefore increasing the chance to keep fluorescence after the long procedure to prepare sections for TEM analysis. Moreover, in this strain all the rRNA genes are transcriptionally active; therefore, the Pol I is highly concentrated on the rDNA in the nucleolus, while the histones are concentrated in the nucleoplasm (actively transcribed rRNA genes are largely devoid of histones molecules, [3]); therefore the green and red fluorescent signals are complementary in the nucleus allowing the clear identification of each domain and reducing the probability of confusion with cellular auto-fluorescence emanating, for example, from vacuoles (see Note 2 ).
3.1 Fixation and Resin Embedding
The unique nucleolus of S. cerevisiae is a heterogeneous electron dense crescent-shaped nuclear subdomain attached to the nuclear envelope (Fig. 1a); it is organized into distinct morphological regions (related to functional regions) defined by their fibrillar or granular appearance [4]. The nucleolus of yeast is sensitive to fixation conditions and its structure is only well preserved using methods without chemical fixation of cells. To minimize artifactual structural reorganizations within the cell and to produce biological material embedded in resin blocks that can be sectioned and observed at room temperature, we developed cryo-fixation and cryo-substitution methods [5]. Numerous resins with various properties are available and should be tested since embedding is highly sample specific. For CLEM, resins should have besides basic features (low shrinkage and absence of specimen damage during cure, minimal compression of sections, relative absence of electron beam-induced section damage, etc.) the lowest auto-fluorescent properties without quenching fluorescence from genetically encoded fluorescence epitope (GFP and m-Cherry ).
-
1.
Harvest 1 ml of the cell culture in exponential phase growth (OD = 0.5 or 3.5 × 106 cells) (see Notes 3 and 4 ). Centrifuge 3 min at 800 rcf to pellet the cells and trash the supernatant. Resuspend the yeast pellet in 5–20 μl (according to the size of the pellet) prewarmed (42 °C) 1 % Agarose Low Melting Point and 7 % sucrose dissolved in 0.1 M cacodylate buffer (pH 7.2).
-
2.
Immediately freeze the cells embedded in Agarose by high-pressure cryo-fixation (EMPACT, Leica) (see Note 5 ).
-
3.
Transfer in liquid nitrogen the frozen pellets into glass distilled acetone containing 0.1 % uranyl acetate at −90 °C in a programmed Freeze Substitution System equipped with an automatic reagent handling system. This machine controls temperature and incubation times and dispenses reagents for freeze substitution, resin infiltration, and resin embedding steps according to the following program:
48 h
0.1 % uranyl acetate in acetone
−90 °C
10 h
acetone
increase gradually the temperature from −90 °C to −50 °C
24 h
HM20 30 % in acetone
−50 °C
24 h
HM20 70 % in acetone
−50 °C
12 h
HM20 100 %
−50 °C
8 h
HM20 100 %
−50 °C (see Note 6 )
48 h
HM20 100 %
−50 °C with UV for polymerization (see Note 7 )
20 h
HM20 100 %
increase the temperature from −50 °C to 20 °C (3.5 °C/h) with UV
24 h
HM20 100 %
20 °C with UV
Store the blocks, shielded from light at RT.
3.2 Sectioning and Sections Harvesting
-
1.
With an ultra-microtome, cut the blocks to produce sections of 100–200 nm thickness using an ultra 35° diamond knife.
-
2.
Deposit the sections on collodion-carbon coated Finder grids (EMS) (see Note 8 ).
3.3 Fluorescence and TEM Acquisitions
-
1.
For observation of the fluorescent signal (see Note 9 ), sink one grid in a water drop spread on a glass coverslip, paying attention that the sections are on the side in contact to the coverslip to allow correct focusing. To avoid any fluorescence background, the coverslips are treated (see Note 10 ).
Do the observations using an inverted fluorescent microscope (see Note 11 ).
-
2.
First, acquire the red fluorescence which corresponds to the strongest signal due to the abundance of the histones (see Note 12 ).
-
3.
Then acquire the green fluorescence.
-
4.
Finally, acquire the light transmitted image (conventional or interferential contrast for better contrast) at the same magnification and also at a lower magnification (or of a microscopic field a bit shifted) in order to locate the cells of interest using the alphanumerical code of the Finder grids (see Note 13 ).
-
5.
Carefully, remove the grids from the drop and let it dry protected from dust.
-
6.
Counterstain the sections with 12 min incubation in uranyl acetate 5 % in water, shielded from light. Rinse extensively in milli-Q water. Incubate 30 s in lead citrate in NaOH saturated atmosphere (see Note 14 ). Rinse extensively in milli-Q water.
-
7.
Evaporate 5 nm of carbon over the sections in order to spread the electron charges during TEM process (see Note 15 ).
-
8.
Sections are analyzed with a Jeol-JEM 1400 electron microscope . With the help of the alpha numerical code, locate the region of interest where fluorescent signals were detected (see Note 16 ). Do acquisition at low magnification in order to be able to recognize patterns possibly formed by the cells distribution in the section. Also do acquisitions at high magnification of cells that were positive in fluorescence (see Note 17 ).
3.4 Superimposition of the LM and TEM Images
-
1.
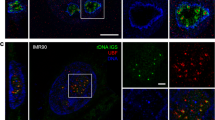
Align the pictures using particular areas visible both in LM and TEM (e.g., grids bars or collodion cracks, etc.) (see Note 18 ). In Fig. 2, two examples of microscopic fields imaged in TEM, fluorescence and Interferential contrast (DIC) are presented.
Fig. 2 
CLEM—correlative light-electron microscopy. Sections of yeast strains expressing H2B histone fused to mCherry (red) and Pol I fused to GFP (green) are imaged by interferential- and fluorescence microscopy and by transmission electron microscopy (TEM) . The green fluorescent signal corresponds to the GFP-Pol I which are concentrated in the nucleolus. The red fluorescent signal corresponds to mCherry-histones which are concentrated in the nucleoplasm. The same cells imaged by TEM providing the ultrastructural context of the fluorescent proteins. Scale bars: 1 μm

3.5 Conclusion
The CLEM protocol described here can be implemented in laboratories equipped with conventional fluorescence and electron microscopies facilities and does not require sophisticated “pipeline” for imaging . It is adapted to strong fluorescent signals. It requires manual and tedious exploration of the grids; semiautomation of samples screening and improvement of images alignment have still to be developed. Moreover, even if some patterns are clearly visible on fluorescent pictures of yeast sections, the conventional fluorescence microscopy cannot distinguish between objects separated by less than 200 nm (resolution inherently limited by the wavelength of the light); enhanced resolution is required to precisely relate fluorescent signal to nucleolar subcompartments. Therefore, the challenge is now to develop CLEM method making use of advanced high resolution fluorescent imaging. The group of A. S. Frangakis in Frankfurt (Germany) published CLEM of a tagged nucleolar protein using Direct Stochastic Optical Reconstruction Microscopy (STORM ) [6]. We are currently developing CLEM for analysis of nucleolar transcription with PALM. PALM (PhotoActivated Localization Microscopy) is based on the serial photoconversion and localization of hundreds to thousands of individual molecules to build an image at near-molecular resolution [7]. For this “high resolution CLEM procedure,” our group is establishing a collection of yeast strains producing proteins of the Pol I machinery in fusion with photoactivable tags (mEos2) adapted for detection of single molecules. By correlating the PALM images of Pol I machinery distribution with electron microscope images of the nucleolar structure, it will be possible to decipher if the observed rods on TEM micrographs (Fig. 1a) could correspond to individual rRNA genes. CLEM with PALM would provide new possibilities to understand how individual transcribed genes are organized in the volume of the nucleolus.
4 Notes
-
1.
The genotype of the strain we are using is as follows: ade2-1 ura3-1 his3-1,15, trp1-1 leu 2-3,112 can1-100 fob1D::KAN-MX4, rDNA copies 25, RPA190::GFP-URA3, HTB1::mcherry -HIS3. HTB1 is the gene coding the H2B histone. GFP (Green Fluorescent Protein) has a major excitation peak at a wavelength of 488 nm (blue) and its emission peak is at 509 nm (green). m-Cherry has a major excitation peak at a wavelength of 587 nm (green) and its emission peak is at 610 nm (red).
-
2.
The vacuole is in yeast sample a potential problem for fluorescence detection. Indeed, the vacuoles contain pigments that can be excited at certain wavelengths and therefore generate a fluorescent signal. The number, the size, and the composition of the yeast vacuole vary from one strain to another and depend on the physiological state of the cells (growth phase, cell cycle phase, nutrient starvation, carbon source, etc.). Note also, that we generally add an excess of adenine in the growth medium to inhibit formation of metabolic intermediates that are pigmented and can also be a source of auto-fluorescence.
-
3.
The nucleolus is the morphological counterpart of the ribosome biogenesis of the cells and its ultrastructure is therefore closely correlated to this activity. The harvesting of cells if they are not in exponential growth phase can give completely different results.
-
4.
Before any CLEM experiments, one should of course check the intensity of the fluorescence in living cells!
-
5.
To ensure a good morphological preservation, the decrease of temperature has to be around 10,000 °C/s and the pressure is 2000 bars. In such conditions, the water from the sample is transformed in amorphous (vitreous) ice because ice crystals that cause damage to the specimen’s ultrastructure are unable to form [8]. After cryofixation, cryomicroscopy of vitreous sections is possible but requires high skills and specific expensive microscope. We chose to substitute at low temperature the ice with chemical fixative in order to be able to work with samples at room temperature. Sectioning as well as imaging with “conventional” electron microscopy is much easier. Moreover, cryofixation/cryosubstitution provided very good quality of images of the yeast nucleolus [4, 5]. Finally, the correlation of the TEM picture with the fluorescent image is also much easier at room temperature.
-
6.
Long infiltration times guarantee a good quality of embedding.
-
7.
Other resins can be used for embedding samples dedicated to CLEM. Indeed, some fluorescent tags are quenched by long UV exposition. When using mEOS2 photo convertible tags (that are adapted to do high resolution fluorescent microscopy like PALM ), the GMA (EMS) resin that can polymerize at −20 °C without UV by adding N,N-dimethyl-p-toluidine is a good alternative [9].
-
8.
Without plastic support, the sections are not stable under the electron beam. Therefore, the grids are covered with a plastic film of collodion according to the following procedure. Fill a crystallizer of milli-Q water up to overflowing. Let fall a drop of collodion on the meniscus of water. When contacting the water, the collodion polymerizes. With a Pasteur pipette, remove the collodion film and trash it: this step will clear the water of any dust contaminant. Let fall another drop of collodion; when the collodion has polymerized, deposit carefully as many grids as possible on the plastic film. Thanks to parafilm or filter paper, recover all the collodion covered grids at the same time. Dry and store them shielded from dust. Handle collodion carefully because it forms toxic gases and can cause skin, eyes, respiratory, and central nervous system irritation. However, polymerization of the collodion under a fume hood is not advised because turbulences can cause wavy collodion films. When dried, the collodion can be covered with 5 nm of evaporated carbon/graphite (graphite is the softest stable form of carbon). Because of the low molecular weight and unique structure where atoms all lie in a plane, graphite films have excellent mechanical stability; moreover, carbon is able to disperse electron charges under the microscope beam. Therefore, carbon coating is increasing the resistance of the support.
-
9.
Observations in fluorescence have always to be done before observations in TEM because the electron beam bleaches fluorophores.
-
10.
To avoid fluorescence backgrounds, the coverslips are treated twice with sequential washes in methanol, acetone, and milli-Q water. The coverslips are then plunged in 1 M KOH and treated with ultra sounds for 1 h. Coverslips are finally rinsed and stored in milli-Q water at 4 °C.
-
11.
We never succeeded to perform CLEM with grids mounted between slide and coverslip (while that would have been adapted for observation with a straight microscope, more common in laboratories). The problem is to take apart the coverslip without damaging and cracking the collodion–carbon support and the sections. Contrariwise to published work, we were also not able to do CLEM with grids mounted in glycerol (even at low concentration); extensive washings did not remove fully the glycerol that impairs TEM observation.
-
12.
The acquisition time required to detect fluorescent signal from sections is drastically increased (×30) when compared to living cells. This decrease in signal intensity is not only due to the reduction of the quantity of the tagged protein but also to the degradation of the fluorescent signal during the procedure for preparation of the sample for TEM. Indeed, considering a 200 nm thick section of a nucleus with a radius of 0.87 μm (average radius of yeast nucleus measured in living cells) (R. Wang, LBME, unpublished results), the maximal volume of the sectioned nucleus is only three times smaller than the volume of the full nucleus . The protocol here described is probably not adapted for detection of low fluorescent signal.
-
13.
The acquisitions at the fluorescent microscope have to be performed as fast as possible (or one has to take care to regularly add water on the grid), because the drop of water is drying by heating under the fluorescent lamp.
-
14.
When staining with lead citrate, do so in a closed container with sodium hydroxide pellets in order to absorb atmospheric CO2. Indeed, the lead citrate forms water-insoluble precipitate (lead carbonate) if not used under CO2-free conditions.
-
15.
Because carbon is uniformly amorphous and highly transparent to electron beam, it can be used to protect the sample during the TEM observation. Coating the sections with 5 nm of carbon will stabilize them under the electron beam because, as explained in Note 8 , carbon can spread the electron charges.
-
16.
Unfortunately, it often happens that due to the repeated handling of the grids, the collodion film breaks; therefore some regions of interest can have “disappeared” between the acquisition of the fluorescent signal and the TEM acquisition. We multiply the number of acquired regions and the number of grids to bypass this problem.
-
17.
We often print the photo of a region of interest in light microscopy (DIC) and indicate on this print which cells are positive for fluorescence. This print is used when acquisitions are performed on the TEM to try to recognize the cells. The alphanumerical code is essential to spot the region of interest, but often, the way cells are distributed in the section finally enables to recognize them. The manual screening of the grids is long and tedious. We are presently developing a semiautomated protocol using ImageJ macro to improve this step of exploring regions of interest.
-
18.
In parallel with the implementation of high resolution fluorescence microscopy instead of conventional microscopy, the alignment step of the LM and TEM photos has to be improved. The deposit of fiducial markers that can be visualized with LM and TEM will increase the superimposition accuracy.
References
Miller O, Beatty B (1969) Visualization of nucleolar genes. Science 164:955–957
Engel C, Sainsbury S, Cheung AC, Kostrewa D, Cramer P (2013) RNA polymerase I structure and transcription regulation. Nature 502:650–655
Merz K, Hondele M, Goetze H, Gmelch K, Stoeckl U, Griesenbeck J (2008) Actively transcribed rRNA genes in S. cerevisiae are organized in a specialized chromatin associated with the high-mobility group protein Hmo1 and are largely devoid of histone molecules. Genes Dev 22:1190–1204
Léger-Silvestre I, Trumtel S, Noaillac-Depeyre J, Gas N (1999) Functional compartmentalization of the nucleus in the budding yeast Saccharomyces cerevisiae. Chromosoma 108:103–113
Léger-Silvestre I, Noaillac-Depeyre J, Faubladier M, Gas N (1997) Structural and functional analysis of the nucleolus of the fission yeast Schizosaccharomyces pombe. Eur J Cell Biol 72:13–23
Perkovic M, Kunz M, Endesfelder U, Bunse S, Wigge C, Yu Z, Hodirnau VV, Scheffer MP, Seybert A, Malkusch S, Schuman EM, Heilemann M, Frangakis AS (2014) Correlative Light- and Electron Microscopy with chemical tags. J Struct Biol 186:205–213
Betzig E, Patterson GH, Sougrat R, Lindwasser OW, Olenych S, Bonifacino JS, Davidson MW, Lippincott-Schwartz J, Hess HF (2006) Imaging intracellular fluorescent proteins at nanometer resolution. Science 313:1642–1645
Kanno H, Speedy RJ, Angell CA (1975) Supercooling of water to -92°C under pressure. Science 189:880–881
Paez-Segala M, Sun MG, Shtengel G, Viswanathan S, Baird MA, Macklin JJ, Patel R, Allen JR, Howe ES, Piszczek G, Hess HF, Davidson MW, Wang Y, Looger LL (2015) Fixation-resistant photoactivatable fluorescent proteins for CLEM. Nat Methods 12:215–218
Acknowledgement
This work is supported by ATS-Nudgene and Emergence-CLEMgene of the Toulouse-IDEX. O.G. and C.N. are supported by Agence Nationale de la Recherche (ANDY). TEM images were taken with the facilities on the METI platform, part of TRI-Genotoul.
Author information
Authors and Affiliations
Corresponding authors
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2016 Springer Science+Business Media New York
About this protocol
Cite this protocol
Normand, C., Berthaud, M., Gadal, O., Léger-Silvestre, I. (2016). Correlative Light and Electron Microscopy of Nucleolar Transcription in Saccharomyces cerevisiae . In: Németh, A. (eds) The Nucleolus. Methods in Molecular Biology, vol 1455. Humana Press, New York, NY. https://doi.org/10.1007/978-1-4939-3792-9_3
Download citation
DOI: https://doi.org/10.1007/978-1-4939-3792-9_3
Published:
Publisher Name: Humana Press, New York, NY
Print ISBN: 978-1-4939-3790-5
Online ISBN: 978-1-4939-3792-9
eBook Packages: Springer Protocols