
Abstract
Integrating viral gene transfer vectors are commonly used gene delivery tools in clinical gene therapy trials providing stable integration and continuous gene expression of the transgene in the treated host cell. However, integration of the reverse-transcribed vector DNA into the host genome is a potentially mutagenic event that may directly contribute to unwanted side effects. A comprehensive and accurate analysis of the integration site (IS) repertoire is indispensable to study clonality in transduced cells obtained from patients undergoing gene therapy and to identify potential in vivo selection of affected cell clones. To date, next-generation sequencing (NGS) of vector-genome junctions allows sophisticated studies on the integration repertoire in vitro and in vivo. We have explored the use of the Illumina MiSeq Personal Sequencer platform to sequence vector ISs amplified by non-restrictive linear amplification-mediated PCR (nrLAM-PCR) and LAM-PCR. MiSeq-based high-quality IS sequence retrieval is accomplished by the introduction of a double-barcode strategy that substantially minimizes the frequency of IS sequence collisions compared to the conventionally used single-barcode protocol. Here, we present an updated protocol of (nr)LAM-PCR for the analysis of lentiviral IS using a double-barcode system and followed by deep sequencing using the MiSeq device.
Access provided by CONRICYT – Journals CONACYT. Download protocol PDF
Similar content being viewed by others


Key words
- Gene therapy
- Lentiviral vector
- (nr)LAM-PCR
- Clonality
- Integration sites
- Safety
- Next-generation sequencing (NGS)
- Double-barcoding strategy
1 Introduction
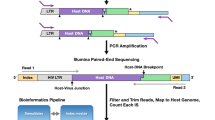
Gene therapy using integrating vector systems has been successfully applied for the treatment of monogenetic diseases in several clinical trials [1, 2]. The occurrence of severe adverse event in few clinical trials using gamma-retroviral vectors due to vector-induced overexpression of nearby cellular proto-oncogenes highlighted the necessity to comprehensively analyze the integration site (IS) repertoire of gene therapy-treated patients [3–6]. Concomitantly, investigators focused on the development of new vector systems supposed to offer advantageous biosafety features. These factors have led to the broad exchange of full long terminal repeat (LTR) -driven gamma-retroviral vectors by self-inactivating LTR lentiviral vectors in translational and clinical studies [7–9].
To identify IS in gene therapy-treated patients and/or to study integration profiles of different vector systems various PCR-based technologies are available. Among these, numerous variants of linker-mediated (LM)-PCR [10] and linear amplification-mediated (LAM)-PCR [11] are currently the most widely used methods. The resulting PCR amplicons can be sequenced to localize the vector ISs by aligning the individually trimmed sequence reads to the host genome. The development of next-generation sequencing technologies enabled researchers to sequence numerous thousands to millions sequences of individual PCR amplicons. To date, the MiSeq sequencing technology provides much higher sequence read numbers (up to 15 million) compared to Sanger sequencing (shotgun 96 reads) or 454 pyrosequencing technology (up to 1 million reads) [12], thus enabling a more profound representation of the IS repertoire. However, the huge increase in individual sequences bares an increased risk to detect false-positive ISs, i.e., contaminating IS which are shared between different samples.
Given the large size of the mammalian genome it is widely accepted that the likelihood to identify identical IS in individually transduced samples is close to zero. Thus, the same IS detected in multiple samples that are independent from the initially transduced target cell population (e.g., samples derived from different patients) have to be considered as collisions and to be removed from further analysis. To minimize the frequency of IS sequence collisions, the use of a double-barcoding strategy is indispensable. For (nr)LAM-PCR , we added the first barcode during ligation of the linker sequence (linker barcode) prior to any exponential amplification. The second barcode was introduced during preparation of LAM-PCR amplicons for sequencing by the MiSeq system (vector barcode). For downstream analyses only sequences that matched the unique combination of linker barcode and vector barcode are considered. Our data revealed that deep sequencing of LAM-PCR amplicons by this double-barcoding strategy designed for the MiSeq device is feasible and reaches high-quality accurate IS sequence retrieval.
2 Materials
2.1 DNA Extraction and Quantification
-
1.
DNA Isolation Kit for Cells and Tissues/Mammalian Blood (Roche Diagnostics, Germany).
-
2.
PCR grade water.
-
3.
Qubit 2.0 Fluorometer and Qubit dsDNA Assay Kit (Life Technologies, USA).
2.2 Linear PCR
-
1.
Taq DNA Polymerase (Genaxxon Bioscience, Germany).
-
2.
10× PCR buffer (Qiagen, Germany).
-
3.
dNTPs (Genaxxon Bioscience, Germany).
-
4.
PCR grade water.
-
5.
Human Genomic DNA (Negative Control, Roche Diagnostics, Germany).
-
6.
Linear PCR Primers (Eurofins MWG Biotech, Germany).
LV3LTR1bio: (B) 5′-AGCTTGCCTTGAGTGCTTCA-3′.
(B): This primer is biotinylated at the 5′-end.
2.3 Magnetic Capture
-
1.
Dynabeads M-280 Streptavidin (Life Technologies, USA).
-
2.
Magnetic separation units for 1.5 ml tubes and 96-well plate (Life Technologies, USA).
-
3.
Phosphate-buffered saline (PBS, pH 7.4).
-
4.
Bovine serum albumin.
-
5.
Lithium chloride.
-
6.
3 M/6 M Lithium chloride (LiCl) solution: Dissolve 6.36 g (3 M) or 12.72 g (6 M) of LiCl in 0.5 ml 1 M Tris–HCl (pH 7.5) and 0.1 ml 0.5 M EDTA (pH 8.0) and adjust the volume with PCR grade water to 50 ml. Filtrate the solution using a 0.45 μm filter. Solutions can be stored at room temperature for several months.
-
7.
PBS/0.1 % BSA: Dissolve 1 g of BSA in 1 L PBS , and aliquot into 1 ml sterile microfuge tube. Solution can be stored at −20 °C for up to 3 months.
2.4 Linker Cassette Construction
-
1.
250 mM Tris–HCl, pH 7.5.
-
2.
100 μM MaCl2.
-
3.
Barcoded oligonucleotides (Eurofins MWG Biotech, Germany).
Oligo1: 5′-GACCCGGGAGATCTGAATTCAGTGGCACAGCAGTTAGG(N)12bpCTA-3′.
Oligo2: 5′-TATAG(N)12bpCCTAACTGCTGTGCCACTGAATTCAGATC-3′.
(N)12bp: Barcode composed by 12 random nucleotides.
-
4.
Microcon-30 (Millipore, USA).
2.5 DNA Double-Strand Synthesis (Hexanucleotide Priming)
-
1.
Klenow Polymerase (Roche Diagnostics, Germany).
-
2.
Hexanucleotide mixture (Roche Diagnostics, Germany).
2.6 Restriction Digest
-
1.
Enzyme MluCI and MseI. (NEB, Germany).
2.7 Ligation of Linker Cassette
-
1.
Fast-link DNA ligation Kit (Epicentre Biotechnologies, USA).
2.8 Denaturation
-
1.
1 N NaOH.
2.9 Ligation of Single-Stranded Oligonucleotide
-
1.
10× Ligation buffer (Biozym, Germany).
-
2.
Single-stranded oligonucleotide (Eurofins MWG Biotech, Germany).
OligonrLAM: 5′-TAG(N)12bpCCTAACTGCTGTGCCACTGAATTCAGATCTCCCGGGT-3′.
(N)12bp: Barcode composed by 12 random nucleotides.
Modifications: Phosphate modification at the 5′-end and dideoxynucleotide modification at the 3′-end.
-
3.
Mangan chloride MnCl2.
-
4.
CircLigase ssDNA Ligase (Biozym, Germany).
2.10 Exponential PCRs
-
1.
Taq DNA polymerase.
-
2.
dNTPs.
-
3.
Primers (Eurofins MWG Biotech, Germany).
First exponential PCR:
LV3LTR2bio: (B) 5′-AGTAGTGTGTGCCCGTCTGT-3′.
LCI: 5′-GACCCGGGAGATCTGAATTC-3′.
(B): This primer is biotinylated at the 5′-end.
Second exponential PCR :
LV3LTR3: 5′-GTGTGACTCTGGTAACTAGAG-3′.
LCII: 5′-GATCTGAATTCAGTGGCACAG-3′.
2.11 Visualization of the (nr)LAM-PCR Product with Spreadex High-Resolution Gel- Electrophoresis
-
1.
Spreadex high-resolution agarose gel. Serva, Germany
-
2.
Gel electrophoresis apparatus.
-
3.
40× TAE buffer.
-
4.
5× Blue run loading buffer.
-
5.
Ethidium bromide.
-
6.
100 bp DNA ladder.
2.12 Sample Preparation for Miseq Platform
-
1.
Agencout AMPure XP Beads (Beckman Coulter, USA).
-
2.
Absolute ethanol.
-
3.
Taq DNA polymerase.
-
4.
dNTPs.
-
5.
Primers (Eurofins MWG Biotech , Germany).
MegaLinker: 5′-GCCTTGCCAGCCCGCTCAGAGTGGCACAGCAGTTAGG-3′.
BarcodedMega: 5′-GCCTCCCTCGCGCCATCAG(N)10bpACGAGTTTTAATGACTCCAAC-3′.
(N)10bp: Barcode composed by ten random nucleotides.
-
6.
Agilent Tapstation/Bioanalyzer 2100.
-
7.
Agilent High Sensitivity DNA kit.
3 Methods
3.1 DNA Extraction and Quantification
The procedure described here starts with the analysis of extracted DNA. A comparison of common DNA extraction procedures (organic, anion-exchange, or silica-based methods) did not reveal any differences on the qualitative outcome of the LAM-PCR technique. DNA samples extracted using commercial kits have consistently provided high-quality results, because the associated reagents have been subjected to quality control before use and are not likely to introduce problems. High-quality analytes ensure the best and most robust performance of following procedures and can provide reliable results. Perform DNA extraction and any following step of sample processing, e.g., dilution and quantification, in a dedicated hood for molecular biology in order to avoid any contamination.
3.2 Linear PCR
The first step of LAM-PCR is a linear amplification of the vector-genome junctions, accomplished with a 5′-biotinylated vector-specific primer(s) hybridizing to the U3- and/or U5 region of the vector long terminal repeat (LTR) . The primer sequences are given in Subheading 2.2.
-
1.
Mix the following components in a sterile nuclease-free PCR tube:
Input 100–500 ng genomic DNA.
1.67 nM 5′ Biotinylated primer.
10× PCR buffer.
200 μM dNTPs each.
0.5 μl (2.5 U) Taq polymerase.
Fill the reaction up with PCR grade water to a final volume of 50 μl.
-
2.
Mix by pipetting followed by a quick spin to collect all liquid from the sides of the tube.
-
3.
Place the PCR tube in a thermocycler , with the heated lid set to 105 °C, and run the following program:
$$ \begin{array}{l}2 \min \kern0.5em @\kern0.5em 95\ {}^{\circ}\mathrm{C}\kern0.28em \mathrm{Initial}\kern0.28em \mathrm{denaturation}\\ {}\begin{array}{l}45\mathrm{s}\kern0.5em @\kern0.5em 95\ {}^{\circ}\mathrm{C}\kern0.28em \mathrm{Denaturation}\\ {}45\mathrm{s}\kern0.5em @\kern0.5em 60\ {}^{\circ}\mathrm{C}\kern0.28em \mathrm{Annealing}\\ {}60\mathrm{s}\kern0.5em @\kern0.5em 72{}^{\circ}\mathrm{C}\kern0.28em \mathrm{Extension}\end{array}\Big\}\kern0.75em 50\kern0.28em \mathrm{cycles}\\ {}5 \min \kern0.5em @\kern0.5em 72{}^{\circ}\mathrm{C}\kern0.28em \mathrm{Final}\kern0.28em \mathrm{extension}\\ {}\mathrm{Hold}\kern0.5em @\kern0.5em 4{}^{\circ}\mathrm{C}\end{array} $$ -
4.
After completion of the PCR add another 0.5 μl (2.5 U) Taq polymerase to each PCR reaction and repeat the 50-cycle PCR.
3.3 Magnetic Capture (See Note 1 )
The magnetic capture allows enriching the amplified linear products through biotin-streptavidin binding.
-
1.
Expose 20 μl magnetic beads to a magnetic separation unit (MSU) . Incubate for 5 min at room temperature until the solution becomes clear.
-
2.
Carefully remove and discard the supernatant, remove the tube from the MSU, and resuspend the magnetic beads with 40 μl PBS/0.1 % BSA.
-
3.
Wash the magnetic beads again with 40 μl PBS/0.1 % BSA and carefully remove and discard the supernatant while the tube is on the MSU.
-
4.
Remove the tube from the MSU, resuspend the magnetic beads with 20 μl 3 M lithium chloride solution (see Note 2 ), and carefully remove and discard the supernatant while the tube is on the MSU.
-
5.
Remove the tube from the MSU, and resuspend the magnetic beads with 50 μl 6 M lithium chloride solution.
-
6.
Add 50 μl of prepared magnetic beads to the linear PCR product (1:1 ratio, see Note 3 ).
-
7.
Incubate the DNA/bead complexes on a horizontal shaker at 300 rpm overnight at room temperature (see Note 4 ).
3.4 Generation of a Linker Cassette
The oligonucleotide sequences are given in Subheading 2.4.
-
1.
Mix the following components in a sterile microfuge tube:
40 μl 100 pmol/μl Oligonucleotide oligo1.
40 μl 100 pmol/μl Oligonucleotide oligo2.
110 μl 250 mM Tris–HCl (pH 7.5).
10 μl 100 mM MgCl2.
-
2.
Mix by pipetting followed by a quick spin to collect all liquid from the sides of the tube.
-
3.
Incubate in a thermal heat block for 5 min at 95 °C.
-
4.
Switch the heat block off and let the sample cool down slowly overnight within the heat block.
-
5.
Add 300 μl of PCR grade water into the tube and transfer the sample on a Microcon-30 column.
-
6.
Centrifuge the sample for 10 min at room temperature and 14,000 × g.
-
7.
Place the column reversed onto a fresh tube and centrifuge the sample for 2 min at room temperature and 1000 × g.
-
8.
Fill the concentrated sample up with distilled water to a final volume of 80 μl.
-
9.
Aliquot the linker cassette and store it at −20 °C (see Note 5 ).
3.5 Hexanucleotide Priming
-
1.
Prepare hexanucleotide priming mixture in a sterile microfuge tube:
1× Concentrated hexanucleotide mixture.
200 μM dNTPs each.
1 U Klenow polymerase.
Fill the mixture up with PCR grade water to a final volume of 10 μl.
-
2.
Mix by pipetting followed by a quick spin to collect all liquid from the sides of the tube.
-
3.
Expose the DNA/bead complexes on the MSU for 60 s after overnight incubation.
-
4.
Remove and discard the supernatant while the tube is on the MSU. Be careful not to disturb the beads that contain the DNA targets.
-
5.
Wash the magnetic beads with 100 μl PCR grade water, and carefully remove and discard the supernatant while the tube is on the MSU.
-
6.
Remove the tube from the MSU, and resuspend the magnetic beads with 10 μl premade hexanucleotide mixture.
-
7.
Incubate in a thermal cycler for exactly 1 h at 37 °C.
-
8.
Add 90 μl of PCR grade water into the reaction, and expose the mixture on the MSU for 60 s.
-
9.
Remove and discard the supernatant while the tube is on the MSU, and wash the magnetic beads with 100 μl PCR grade water.
3.6 Restriction Digest (See Note 6 )
-
1.
Prepare restriction digest mixture in a sterile microfuge tube:
1 μl 10× Restriction buffer.
2 U MseI.
Fill the reaction mixture up with PCR grade water to a final volume of 10 μl.
-
2.
Mix by pipetting followed by a quick spin to collect all liquid from the sides of the tube.
-
3.
Carefully remove and discard the supernatant while the tube is on the MSU, remove the tube from the MSU, and resuspend the magnetic beads with 10 μl premade restriction digest mixture.
-
4.
Incubate in a thermal cycler for exactly 1 h at temperature recommended by the manufacturer (see Note 7 ).
-
5.
Add 90 μl of PCR grade water into the reaction, and expose the mixture on the MSU for 60 s.
-
6.
Remove and discard the supernatant while the tube is on the MSU, and wash the magnetic beads with 100 μl PCR grade water.
3.7 Linker Cassette Ligation
-
1.
Prepare ligation mixture in a sterile microfuge tube:
2 μl of restriction enzyme linker cassette.
1× Concentrated fast-link ligation buffer.
10 mM ATP.
2 U Fast-link ligase.
Fill the reaction mixture up with PCR grade water to a final volume of 10 μl.
-
2.
Mix by pipetting followed by a quick spin to collect all liquid from the sides of the tube.
-
3.
Carefully remove and discard the supernatant while the tube is on the MSU, remove the tube from the MSU, and resuspend the magnetic beads with 10 μl premade ligation mixture.
-
4.
Incubate for 5 min at room temperature.
-
5.
Add 90 μl of PCR grade water into the reaction, and expose the mixture on the MSU for 60 s.
-
6.
Remove and discard the supernatant while the tube is on the MSU, and wash the magnetic beads with 100 μl PCR grade water.
3.8 Alkaline Denaturation
-
1.
Prepare 1 ml of 0.1 N NaOH in a sterile microfuge tube:
PCR grade water (900 μl).
Stock 1.0 N NaOH (100 μl).
Invert the tube several times to mix.
-
2.
Carefully remove and discard the supernatant while the tube is on the MSU, remove the tube from the MSU, and resuspend the magnetic beads with 5 μl freshly prepared 0.1 N NaOH solution.
-
3.
Incubate the magnetic beads at room temperature on a shaker (300 rpm) for 10 min.
-
4.
Expose the denatured sample on the MSU for 60 s, and collect the supernatant into a fresh microfuge tube (see Note 8 ).
3.9 Ligation of a Single-Stranded Oligonucleotide (See Note 9 )
This step leads to the ligation of a single-stranded oligonucleotide to the unknown part of the DNA amplicons. As both ends after the ligation consist of known sequences, a subsequent exponential amplification of the PCR products is then possible. The sequence of the oligonucleotide and the modifications are shown in Subheading 2.9.
-
1.
Prepare ligation mixture in a sterile microfuge tube:
1 μl 10× Ligation buffer.
1 μl OligonrLAM (10 pmol/μl).
0.5 μl MnCl2 (10 mM).
0.5 μl ATP (10 mM).
0.5 μl Circligase ssDNA ligase.
Fill the reaction mixture up with PCR grade water to a final volume of 10 μl.
-
2.
Mix by pipetting followed by a quick spin to collect all liquid from the sides of the tube.
-
3.
Expose the DNA/bead complex (Subheading 3.3, step 7) to the MSU, and incubate for 5 min at room temperature until the solution becomes clear.
-
4.
Carefully remove and discard the supernatant , remove the tube from the MSU, and resuspend the magnetic beads with 10 μl of prepared ligation reaction.
-
5.
Incubate the reaction for at least 16 h and not longer than 24 h at 300 rpm on a horizontal shaker, at room temperature.
-
6.
Add 90 μl of PCR grade water into the reaction, and expose the mixture on the MSU for 60 s.
-
7.
Remove and discard the supernatant while the tube is on the MSU, and wash the magnetic beads with 100 μl PCR grade water.
-
8.
Carefully remove and discard the supernatant, remove the tube from the MSU, resuspend the magnetic beads with 10 μl PCR grade water, and transfer it to a fresh 0.5 ml microfuge tube.
3.10 Exponential PCRs and Magnetic Capture
Primer sequences for the first and second exponential PCR are listed in Subheading 2.10.
-
1.
Mix the following components in a sterile nuclease-free PCR tube:
1 μl of the denaturation product/2 μl ligation product of nrLAM-PCR as template.
8.3 μM of each primer.
10× PCR buffer.
200 μM dNTPs each.
0.25 μl (1.25 U) Taq polymerase.
Fill the reaction up with PCR grade water to a final volume of 25 μl.
-
2.
Place the PCR tube in a thermocycler , with the heated lid set to 105 °C, and run the following program:
$$ \begin{array}{l}2 \min\ @\ 95\ {}^{\circ}\mathrm{C}\kern0.28em \mathrm{Initial}\kern0.28em \mathrm{denaturation}\\ {}\begin{array}{l}45\mathrm{s}\kern0.5em @\kern0.5em 95\ {}^{\circ}\mathrm{C}\kern0.28em \mathrm{Denaturation}\\ {}45\mathrm{s}\kern0.5em @\kern0.5em 60\ {}^{\circ}\mathrm{C}\kern0.28em \mathrm{Annealing}\\ {}60\mathrm{s}\kern0.5em @\kern0.5em 72{}^{\circ}\mathrm{C}\kern0.28em \mathrm{Extension}\end{array}\Big\}\kern1em 36\kern0.28em \mathrm{cycles}\\ {}5 \min \kern0.5em @\kern0.5em 72{}^{\circ}\mathrm{C}\kern0.28em \mathrm{Final}\kern0.28em \mathrm{extension}\\ {}\mathrm{Hold}\kern0.5em @\kern0.5em 4{}^{\circ}\mathrm{C}\end{array} $$ -
3.
An additional magnetic capture step after the first exponential PCR is optional. For the detailed protocol please see Subheading 3.3 (see Note 10 ).
-
4.
Mix the following components for second exponential PCR in a sterile microfuge tube:
1 μl of the denaturation product of first exponential PCR as template.
8.3 μM of each primer.
10× PCR buffer.
200 μM dNTPs each.
0.5 μl (2.5 U) Taq polymerase.
Fill the reaction up with PCR grade water to a final volume of 50 μl.
-
5.
Carry out the PCR reaction using the same condition as the first exponential PCR.
3.11 Visualization of the (nr)LAM-PCR Product with Spreadex High-Resolution Gel Electrophoresis
-
1.
Fill the electrophoresis tank with 1.9 L of 1 × concentrated TAE buffer, fix a Spreadex gel within the electrophoresis tank using an appropriate catamaran.
-
2.
Load 10 μl of each (nr)LAM-PCR product with 2 μl of 5× concentrated blue run loading buffer.
-
3.
Add 1 kb plus DNA ladder for molecular weight reference.
-
4.
Let the gel run at 10 V/cm electrode gap.
-
5.
Switch the buffer pump 5 min later after the electrophoresis starts.
-
6.
After the electrophoresis, stain the gel for 20 min in ethidium bromide solution (~0.5 µg ethidium bromide/ml PCR grade water) on a shaker at 50 rpm and room temperature.
-
7.
Visualize the DNA on a gel documentation system.
3.12 Library Preparation of (nr)LAM-PCR Samples for High-Throughput Sequencing Using MiSeq Platform
The (nr)LAM-PCR samples are further prepared for high -throughput sequencing to identify the precise localization of the ISs in the host genome. In the following, we will give a guideline for how to proceed optimally with the (nr)LAM-PCR samples to allow subsequent high-throughput sequencing. In brief, 40 ng of purified (nr)LAM-PCR products are used to perform a third exponential PCR. This PCR step allows adding the Illumina-specific amplification and sequencing adaptors on both sides of the (nr)LAM-PCR amplicons. By incorporating a 10–12 bp barcode into customized sequencing adaptors and linker cassette, different samples can be finally pooled for multiplexing sequencing on MiSeq platform.
-
1.
Vortex AMPure XP beads to resuspend.
-
2.
Add 44 μl (1.1×) of resuspended AMPure XP Beads to the second exponential PCR product (~40 μl), mix well, and incubate for 5 min at room temperature.
-
3.
Place the tube on an appropriate MSU to separate beads from supernatant. After the solution is clear (about 5 min), carefully remove and discard the supernatant. Be careful not to disturb the beads that contain the DNA targets.
-
4.
Add 200 μl of 80 % freshly prepared ethanol to the sample while in the MSU. Incubate at room temperature for 30 s, and then carefully remove and discard the supernatant.
-
5.
Repeat step 4 once.
-
6.
Air-dry beads for 5 min while the tube is on the MSU with the lid open (see Note 11 ).
-
7.
Remove the tube from the MSU. Elute the DNA target by adding 24 μl of PCR grade water to the beads.
-
8.
Mix well on a vortex mixer or by pipetting up and down and incubate for 2 min at room temperature.
-
9.
Put the tube in the MSU until the solution is clear. Transfer 22 μl of supernatant (or desired volume) to a new tube, and proceed to third exponential PCR.
-
10.
Mix the following components for third exponential PCR in a sterile microfuge tube (for primer sequences for the third exponential PCR please see Subheading 2.12).
40 ng of the Ampure bead-purified second exponential PCR product.
0.5 μl of each primer (10 pmol/μl).
5 μl 10× PCR buffer.
1 μl dNTPs each (10 mM).
0.5 μl (2.5 U) Taq polymerase.
Fill the reaction up with PCR grade water to a final volume of 50 μl.
-
11.
Place the PCR tube in a thermocycler , with the heated lid set to 105 °C, and run the following program:
$$ \begin{array}{l}2 \min \kern0.5em @\kern0.5em 95\ {}^{\circ}\mathrm{C}\kern0.28em \mathrm{Initial}\kern0.28em \mathrm{denaturation}\\ {}\begin{array}{l}45\mathrm{s}\kern0.5em @\kern0.5em 95\ {}^{\circ}\mathrm{C}\kern0.28em \mathrm{Denaturation}\\ {}45\mathrm{s}\kern0.5em @\kern0.5em 60\ {}^{\circ}\mathrm{C}\kern0.28em \mathrm{Annealing}\\ {}60\mathrm{s}\kern0.5em @\kern0.5em 72{}^{\circ}\mathrm{C}\kern0.28em \mathrm{Extension}\end{array}\Big\}\kern1em 15\kern0.28em \mathrm{cycles}\\ {}5 \min \kern0.5em @\kern0.5em 72{}^{\circ}\mathrm{C}\kern0.28em \mathrm{Final}\kern0.28em \mathrm{extension}\\ {}\mathrm{Hold}\kern0.5em @\kern0.5em 4{}^{\circ}\mathrm{C}\end{array} $$ -
12.
Purify the third exponential PCR products with Ampure XP beads as steps 1–9.
-
13.
Measure the purified DNA concentration by using a Qubit fluorometer.
-
14.
Pool the desired DNA samples according to their multiplexes, and analyze the peak distribution using 1 μl pooled library by Agilent Bioanalyzer 2100/Tap station using DNA High Sensitivity Kit (see Note 12 ). The amount of DNA of each sample within the pool is proportional to the number of retrieved sequencing reads.
-
15.
Store the pooled library at −20 °C, or directly continue for MiSeq sequencing.
3.13 Bioinformatics/Sequence Analyses
Public available bioinformatics tools like Seqmap 2.0 [13], QuickMap [14], or our own developed HISAP pipelines, such as noted in Ref. 15, can process the retrieved sequences.
4 Notes
-
1.
If non-restrictive LAM-PCR is being performed, please go directly to Subheading 3.9 after this step.
-
2.
Alternatively binding solution provided by the manufacturer of the magnetic beads can also be used. LiCl solution in our hand performs in a comparable way and is cost effective.
-
3.
The ratio of PCR product and LiCl solution must always be 1:1.
-
4.
This capturing step needs to be carried out at least for 8 h.
-
5.
After thawing the aliquot of linker cassette, do not refreeze it.
-
6.
This protocol provides the detail procedure for restriction enzyme MseI; please adjust the components for this step if other restriction enzyme is used.
-
7.
Choose the restriction enzyme in a way that no restriction site is located within the known sequence of interest and the amplified part of the vector. Incubate the restriction digest mixture at the temperature recommended by the manufacturer to achieve maximum enzyme activity for 1 h in a thermocycler.
-
8.
Store the denatured LAM product at −20 °C.
-
9.
This step should be followed after subheading 3.3 if nrLAM-PCR is performed.
-
10.
This magnetic capture step should be performed to increase the sensitivity and specificity.
Minor changes to the magnetic capture protocol are described in Subheading 3.3.
-
Resuspend the magnetic beads in 25 μl 6 M LiCl instead of 50 μl.
-
After adding the magnetic beads to each first exponential PCR product (1:1 ratio), incubate the DNA/bead complexes for at least 1 h on a shaker at 300 rpm and room temperature.
-
Denature the DNA from DNA/bead complexes with 20 μl of freshly prepared 0.1 N NaOH solution.
-
-
11.
Do not overdry the beads. This may result in lower recovery of DNA target.
-
12.
To obtain the precise and reproducible results of NGS, high-quality analytes with Poisson distribution and precisely estimated DNA concentration are crucial.
References
Hacein-Bey-Abina S, Le Deist F, Carlier F et al (2002) Sustained correction of X-linked severe combined immunodeficiency by ex vivo gene therapy. N Engl J Med 346:1185–1193
Boztug K, Schmidt M, Schwarzer A et al (2010) Stem-cell gene therapy for the Wiskott-Aldrich syndrome. N Engl J Med 363:1918–1927. doi:10.1056/NEJMoa1003548
Hacein-Bey Abina S, von Kalle C, Schmidt M et al (2003) A serious adverse event after successful gene therapy for X-linked severe combined immunodeficiency. N Engl J Med 348:255–256
Braun CJ, Boztug K, Paruzynski A et al (2014) Gene therapy for Wiskott-Aldrich syndrome—long-term efficacy and genotoxicity. Sci Transl Med 6:227ra33. doi:10.1126/scitranslmed.3007280
Schwarzwaelder K, Howe SJ, Schmidt M et al (2007) Gammaretrovirus-mediated correction of SCID-X1 is associated with skewed vector integration site distribution in vivo. J Clin Invest 117:2241–2249. doi:10.1172/JCI31661
Deichmann A, Hacein-Bey-Abina S, Schmidt M et al (2007) Vector integration is nonrandom and clustered and influences the fate of lymphopoiesis in SCID-X1 gene therapy. J Clin Invest 117:2225–2232
Bartholomae CC, Arens A, Balaggan KS et al (2011) Lentiviral vector integration profiles differ in rodent postmitotic tissues. Mol Ther 19:703–710
Cartier N, Hacein-bey-abina S, Bartholomae CC et al (2009) Hematopoietic stem cell gene therapy with a lentiviral vector in X-linked adrenoleukodystrophy. Science 326:818–823
Aiuti A, Biasco L, Scaramuzza S et al (2013) Lentiviral hematopoietic stem cell gene therapy in patients with Wiskott-Aldrich syndrome. Science 341:1233151
Mueller PR, Wold B (1989) In vivo footprinting of a muscle specific enhancer by ligation mediated PCR. Science 246:780–786
Schmidt M, Schwarzwaelder K, Bartholomae C et al (2007) High-resolution insertion-site analysis by linear amplification-mediated PCR (LAM-PCR). Nat Methods 4:1051–1057
Paruzynski A, Arens A, Gabriel R et al (2010) Genome-wide high-throughput integrome analyses by nrLAM-PCR and next-generation sequencing. Nat Protoc 5:1379–1395
Hawkins TB, Dantzer J, Peters B et al (2011) Identifying viral integration sites using SeqMap 2.0. Bioinformatics 27:720–722
Appelt J-U, Giordano FA, Ecker M et al (2009) QuickMap: a public tool for large-scale gene therapy vector insertion site mapping and analysis. Gene Ther 16:885–893
Arens A, Appelt J-U, Bartholomae CC et al (2012) Bioinformatic clonality analysis of next-generation sequencing-derived viral vector integration sites. Hum Gene Ther Methods 23:111–118. doi:10.1089/hgtb.2011.219
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2016 Springer Science+Business Media New York
About this protocol
Cite this protocol
Wang, W., Bartholomae, C.C., Gabriel, R., Deichmann, A., Schmidt, M. (2016). The LAM-PCR Method to Sequence LV Integration Sites. In: Federico, M. (eds) Lentiviral Vectors and Exosomes as Gene and Protein Delivery Tools. Methods in Molecular Biology, vol 1448. Humana Press, New York, NY. https://doi.org/10.1007/978-1-4939-3753-0_9
Download citation
DOI: https://doi.org/10.1007/978-1-4939-3753-0_9
Published:
Publisher Name: Humana Press, New York, NY
Print ISBN: 978-1-4939-3751-6
Online ISBN: 978-1-4939-3753-0
eBook Packages: Springer Protocols