
Abstract
Bioluminescence resonance energy transfer (BRET) is a prominent biophysical technology for monitoring molecular interactions, and has been widely used to study protein-protein interactions (PPI) in live cells. This technology requires proteins of interest to be associated with an energy donor (i.e., luciferase) and an acceptor (e.g., fluorescent protein) molecule. Upon interaction of the proteins of interest, the donor and acceptor will be brought into close proximity and energy transfer of chemical reaction-induced luminescence to its corresponding acceptor will result in an increased emission at an acceptor-defined wavelength, generating the BRET signal. We leverage the advantages of the superior optical properties of the NanoLuc® luciferase (NLuc) as a BRET donor coupled with Venus, a yellow fluorescent protein, as acceptor. We term this NLuc-based BRET platform “BRETn”. BRETn has been demonstrated to have significantly improved assay performance, compared to previous BRET technologies, in terms of sensitivity and scalability. This chapter describes a step-by-step practical protocol for developing a BRETn assay in a multi-well plate format to detect PPIs in live mammalian cells.
Access provided by CONRICYT – Journals CONACYT. Download protocol PDF
Similar content being viewed by others


Key words
- Bioluminescence resonance energy transfer (BRET)
- Nanoluc luciferase (NLuc)
- Hippo signaling pathway
- YAP-TEAD interaction
- Ultra high-throughput screening (uHTS)
1 Introduction
Bioluminescence resonance energy transfer (BRET) is a naturally occurring phenomenon existing in some marine species such as Aequorea, where radiation-less transfer of energy is observed from an activated bioluminescent donor (e.g., photoprotein aequorin ) to its associated green fluorescent protein (GFP) . This natural physical process has been engineered to monitor direct molecular interactions, due to its stringent distance requirement (≤10 nm) to allow efficient energy transfer [1–3]. In principle, the photons from the light-emitting donor can excite an acceptor fluorophore by resonance energy transfer if the donor and acceptor fluorophore are in close proximity with proper orientation and have appropriate overlap between the donor emission and acceptor excitation spectra. Several versions of BRET have been reported by combining various donor and acceptor pairs [2–4]. Previously described BRET systems include BRET1 and BRET2, which use Renilla luciferase (RLuc) as BRET donors [4]. These systems have revolutionized the way we study PPIs , especially in live cells. Here we describe a working protocol of a Nanoluc luciferase (NLuc )-based BRET platform.
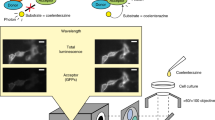
Nanoluc luciferase is a luminescent protein engineered from the luciferase of a luminous deep-sea shrimp, Oplophorus gracilirostris [5]. It has been shown that NLuc is the smallest (19 kDa) and brightest luciferase to date, with superior stability, glow-type luminescence , and narrow emission spectrum [5]. These improved properties of NLuc allowed us to generate a new BRET platform for PPI detection, termed BRETn to stand for NanoLuc-based BRET. For the BRETn PPI biosensor design, NLuc was used instead of the conventional RLuc as a BRETn donor. Venus [6], a yellow fluorescent protein variant, serves as a BRETn acceptor . The interaction of NLuc -fused protein X and Venus-fused protein Y brings the donor and acceptor into close proximity, leading to energy transfer of the luminescence signal from NLuc to Venus upon substrate , furimazine, and oxidation (Fig. 1). The emission signal of Venus can then be used as a measurement of BRET signal for detection of PPIs (Fig. 1).

Schematic illustration of BRETn design. NLuc and Venus are genetically fused to the N-terminal end of each protein of interest, X and Y, respectively. BRET signal can be detected when NLuc and Venus are brought into close proximity upon interaction of protein X and Y
The performance of BRETn has been demonstrated as a PPI biosensor for pathway profiling and PPI modulator screening [7]. The improved properties of BRETn enable miniaturization of the assay to a 1536-well plate uHTS format for large-scale screening . The following protocol describes a general step-by-step procedure for developing a BRETn assay to monitor PPIs in live cells. The results of BRETn assay development for monitoring the YAP-TEAD interaction [8–10], a key PPI involved in the Hippo signaling pathway , are presented as a case study. The Hippo signaling pathway plays a critical role in normal physiology, such as the control of organ size during development. Its dysregulation has been associated with tumorigenesis in a range of cancer types [8–10]. Targeting the YAP-TEAD interaction has been suggested to be a promising strategy for treatment of YAP-addicted cancers [11, 12]. However, no specific YAP-TEAD small molecule inhibitors are currently available [11, 12]. In order to better understand the function of the YAP-TEAD interaction, a highly sensitive BRETn assay for discovery of interaction disruptors has been designed and optimized. Small molecule chemical probes that disrupt the YAP-TEAD interaction will enable physiological studies and have potential for anti-cancer therapeutic development. Using the YAP-TEAD interaction as a model system, a detailed protocol for a BRETn assay for monitoring PPIs in live cells is described. While this chapter uses YAP-TEAD as an example, the procedure described in this chapter can be readily adapted and applied to other molecular interactions.
2 Materials
2.1 Plasmids for NLuc-tagged TEAD2 and Venus-tagged YAP1 Construction (see Note 1)
-
1.
Entry vector with TEAD2 in pDONR221.
-
2.
pENTR/D-TOPO vector.
-
3.
TOPO® Cloning kit.
-
4.
Gateway pDEST26 Vector.
-
5.
pFUW vector.
2.2 Other Reagents
-
1.
Cell culture media : 1× Dulbecco’s Modified Eagle Medium (DMEM) without phenol red indicator, supplemented with 4 mM l-glutamine, 4.5 g/L glucose, 1 mM sodium pyruvate, 100 U/mL penicillin, 100 mg/mL streptomycin, and 10 % fetal bovine serum.
-
2.
Human Embryonic Kidney 293T (HEK293T) cells (see Note 2 ).
-
3.
Polyethylenimine (PEI) transfection reagent stock solution (see Note 2 ): dissolve PEI (1 mg/mL) in deionized water at 80 °C with stirring. Adjust solution to pH 7.2 using 0.1 M HCl at room temperature. Filter-sterilize, aliquot, and store at −20 °C.
-
4.
Nano-Glo® luciferase assay system (Promega, Madison, WI).
-
5.
Plate (see Note 3 ): 1536-well solid bottom white plate and clear bottom black plate and 384-well polypropylene plate.
-
6.
Plate reader for BRET signal measurements (see Note 4 ): for YAP-TEAD BRETn assay development , BRETn signal measurement was performed using 460 and 535 nm emission filters.
3 Methods
3.1 Plasmid Construction
NLuc-tagged TEAD2 and Venus-tagged YAP1 were constructed using Gateway® cloning system as described [7] (see Note 1 ). Entry vector with TEAD2 in pDONR221 was purchased. Entry vector with YAP1 was cloned into pENTR/D-TOPO vector using PCR and TOPO® Cloning kit. The NLuc coding sequence (Promega, Madison, WI), along with a linker (see Note 1 ), was inserted into the pDEST26 (Invitrogen) to generate Gateway®-based NLuc destination expression vector. The Venus coding sequence, along with a linker (see Note 1 ), was inserted into the pFUW vector (Emory University) to generate the Gateway®-based Venus destination expression vector. NLuc -TEAD2 and Venus-YAP1 expression plasmids were constructed by performing LR reactions between corresponding entry and destination vectors using LR Clonase™ II enzyme mix.
3.2 Plating Cells
HEK293T cells (2000 cells in 4 μl per well (see Note 3 )) were dispensed into a 1536-well plate. Cells were plated side by side sequentially in both a solid bottom white plate and a clear bottom black plate (see Note 3 ). Plates were sealed with a gas permeable sealing membrane.
3.3 Transient Transfection
Below we describe a typical experiment where the donor and acceptor expression levels are varied by serial dilution of the amount of DNA to be transfected. This experimental setup will allow the acceptor to saturate the donor and provide a maximal BRET signal. A template design for a 384-well DNA plate format for orthogonal co-transfection is shown in Fig. 2 (see Note 5 ).

An example 384-well plate format for DNA transfection. Various amounts of NLuc and Venus plasmids were mixed orthogonally using multichannel pipette. The final DNA amount transfected into a single well of 1536-well cell plate is indicated
-
1.
Serially dilute NLuc-TEAD2 and NLuc-empty plasmid to 0.2 and 0.1 ng/mL in dH2O in sterile Eppendorf tube (see Note 6 ).
-
2.
Dilute Venus-YAP1 and Venus-empty plasmid to 40 ng/μl with dH2O in sterile Eppendorf tube. Then serially dilute Venus-YAP1 to 20 and 10 ng/μl, and Venus-empty to 20, 10, and 5 ng/mL using pcDNA (40 ng/μl) (see Note 6 ).
-
3.
Orthogonally mix 5 μl NLuc plasmids (0.1 and 0.2 ng/μl for both NLuc-TEAD2 and NLuc-empty) and 5 μl Venus plasmids (10, 20, and 40 ng/μl for Venus-YAP1 and 5, 10, and 20 ng/μl for Venus-empty) according to the format as shown in Fig. 2 in a 384-well plate.
-
4.
Dilute PEI to 60 ng/μl in cell culture media (see Note 2 ).
-
5.
Dispense 10 μl/well of the 60 ng/μl PEI dilution into the 384-well DNA plate to make the final ratio of PEI:DNA = 3:1.
-
6.
Incubate the DNA:PEI mix at 25 °C for 20 min.
-
7.
Perform forward in-well transfection using an automated liquid handler . Transfer 1 μl of DNA:PEI mix from 384-well plate into 1536-well plate in four replicates (Fig. 3). The final amount of DNA transfected into each 1536-well is indicated in Fig. 2. The 1536-well solid bottom white plate and clear bottom black plate are transfected side by side.
Fig. 3 
Schematic illustration of forward in-well transfection from 384-well DNA plate into 1536-well cell plate in four replicates
-
8.
Incubate cells in CO2 incubator at 37 °C for 2 days.

3.4 BRETn Measurement
-
1.
Load 1536-well clear bottom black plate into plate reader and detect fluorescence signal (FI) (excitation : 480 nm, emission : 535 nm) using microplate reader software.
-
2.
Immediately prior to the BRET measurement, prepare furimazine (i.e., Nano-Glo® Luciferase Assay Substrate) by diluting the stock solution 1:100 in cell culture media (see Note 7 ).
-
3.
Dispense 1 μl of 1:100 diluted substrate directly into a 1536-well solid bottom white plate (cell culture media does not need to be changed) (see Note 7 ).
-
4.
Load 1536-well solid bottom white plate into plate reader and take BRETn reading by measuring luminescence at 460 ± 15 nm (I460) and 535 ± 15 nm (I535).
3.5 Data Analysis
-
1.
The BRETn signal can be determined by I535/I460.
-
2.
The net BRETn signal is calculated by subtracting out background BRETn signal from cells transfected with donor plasmid only.
-
3.
Calculate the net fluorescence intensity (FI) by subtracting out background fluorescence from cells transfected with pcDNA only.
-
4.
Calculate the net I460 by subtracting out background luminescence from cells transfected only with pcDNA.
-
5.
Calculate the acceptor /donor ratio by dividing the net FI by net I460.
-
6.
Using graphing software, plot the acceptor/donor ratio against the net BRETn as in Fig. 4 (see Note 8 ). The data can then be fit with a nonlinear regression model.
Fig. 4 
BRETn saturation curve of NLuc-TEAD2 and Venus-YAP1 interaction. The error bar represents the s.d. from four replicates. AUC values are presented as mean ± s.d. from three independent experiments

4 Notes
-
1.
BRETn follows classical rules of energy transfer in that the energy transfer efficiency is inversely proportional to the sixth power of distance between two fluorophores , and is also correlated with the orientation of two fluorophores. Therefore, it is advantageous to use peptide linkers, such as GGGGSGGGGS used in this study, between the NLuc /Venus tag and the proteins of interest to allow sufficient movement of the tags with less steric hindrance. Moreover, there are eight possible fusion combinations for each pair of proteins considering different tag placements at N- or C-terminus of the gene of interest. It is highly recommended to try all eight pairing combinations in order to achieve a maximized BRETn dynamic window.
-
2.
Other cell lines and transfection reagents can be used. We choose to use HEK293T and PEI as they yield high transfection efficiency and reproducibility at affordable cost. For cell lines other than HEK293T cells, the amount of DNA transfected and DNA/PEI ratio need to be optimized empirically.
-
3.
Other types and formats of standard multiple well plates can be used with the method described above and the reagent amount suggested in Table 1. We use plates from Corning (Corning, NY). Other plate manufacturers may be acceptable but should be thoroughly tested prior to use.
Table 1 Recommended cell culture condition and DNA transfection range for performing BRETn assay in other multiple well plates -
4.
We use the EnVision® Multilabel Reader (PerkinElmer) for our BRETn assay. Other plate readers equipped with desired emission filters can be used. To collect BRETn data, we use the Wallac EnVision Manager software (PerkinElmer). Wallac is able to automatically calculate the BRETn signal from I460 and I535 channel, and export the data in a format that is compatible with Microsoft Excel.
-
5.
Controls are essential to assess the success of the BRETn assay. Negative controls, such as NLuc-TEAD and Venus, and NLuc and Venus-YAP1, are necessary to rule out false-positive signals due to nonspecific energy transfer. Positive controls, such as the NLuc-Venus fusion protein, allow the researcher to define positive BRET signals.
-
6.
The amount of NLuc and Venus plasmid used for transfection needs to be determined empirically based on the PPI pairs.
-
7.
BRETn assay can also be performed with cell lysates, instead of live cell detection described in this study with appropriate modifications of protocol.
-
8.
Conventionally, BRET data was quantified solely by calculating maximal BRET signal (BRETmax) based on nonlinear regression analysis, where BRET50 was often neglected. In this scenario, in order to get precise BRETmax , the BRET saturation curve needs to reach plateau. We employed an area under the curve (AUC) analysis method to quantitatively compare the BRETn signal between PPI and controls, as AUC reflects both BRETmax and BRET50 for various PPIs [7].
References
Xu Y, Kanauchi A, von Arnim AG, Piston DW, Johnson CH (2003) Bioluminescence resonance energy transfer: monitoring protein-protein interactions in living cells. Methods Enzymol 360:289–301
Pfleger KDG, Eidne KA (2006) Illuminating insights into protein-protein interactions using bioluminescence resonance energy transfer (BRET). Nat Methods 3(3):165–174. doi:10.1038/Nmeth841
Xu Y, Piston DW, Johnson CH (1999) A bioluminescence resonance energy transfer (BRET) system: application to interacting circadian clock proteins. Proc Natl Acad Sci U S A 96(1):151–156
Couturier C, Deprez B (2012) Setting up a bioluminescence resonance energy transfer high throughput screening assay to search for protein/protein interaction inhibitors in mammalian cells. Front Endocrinol 3:1–13. doi:10.3389/fendo.2012.00100
Hall MP, Unch J, Binkowski BF, Valley MP, Butler BL, Wood MG, Otto P, Zimmerman K, Vidugiris G, Machleidt T, Robers MB, Benink HA, Eggers CT, Slater MR, Meisenheimer PL, Klaubert DH, Fan F, Encell LP, Wood KV (2012) Engineered luciferase reporter from a deep sea shrimp utilizing a novel imidazopyrazinone substrate. ACS Chem Biol 7(11):1848–1857. doi:10.1021/Cb3002478
Nagai T, Ibata K, Park ES, Kubota M, Mikoshiba K, Miyawaki A (2002) A variant of yellow fluorescent protein with fast and efficient maturation for cell-biological applications. Nat Biotechnol 20(1):87–90. doi:10.1038/nbt0102-87
Mo X-L, Luo Y, Ivanov AA, Su R, Havel JJ, Li Z, Khuri F, Du Y, Fu H (2015) Enabling systematic interrogation of protein-protein interactions in live cells with a versatile ultra-high throughput biosensor platform. J Mol Cell Biol. doi: 10.1093/jmcb/mjv064
Johnson R, Halder G (2014) The two faces of Hippo: targeting the Hippo pathway for regenerative medicine and cancer treatment. Nat Rev Drug Discov 13(1):63–79. doi:10.1038/nrd4161
Harvey KF, Zhang X, Thomas DM (2013) The Hippo pathway and human cancer. Nat Rev Cancer 13(4):246–257. doi:10.1038/nrc3458
Huang J, Wu S, Barrera J, Matthews K, Pan D (2005) The Hippo signaling pathway coordinately regulates cell proliferation and apoptosis by inactivating Yorkie, the Drosophila homolog of YAP. Cell 122(3):421–434. doi:10.1016/j.cell.2005.06.007
Zhang Z, Lin Z, Zhou Z, Shen HC, Yan SF, Mayweg AV, Xu Z, Qin N, Wong JC, Zhang Z, Rong Y, Fry DC, Hu T (2014) Structure-based design and synthesis of potent cyclic peptides inhibiting the YAP-TEAD protein-protein interaction. ACS Med Chem Lett 5(9):993–998. doi:10.1021/ml500160m
Liu-Chittenden Y, Huang B, Shim JS, Chen Q, Lee SJ, Anders RA, Liu JO, Pan D (2012) Genetic and pharmacological disruption of the TEAD-YAP complex suppresses the oncogenic activity of YAP. Genes Dev 26(12):1300–1305. doi:10.1101/gad.192856.112
Acknowledgments
We thank Dr. Kun-Liang Guan for providing YAP1, and Dr. Atsushi Miyawaki for providing Venus cDNA plasmid as cloning template. We would like to thank Drs. Jonathan Havel and Zenggang Li for their contributions in generating NLuc destination vector and Venus-YAP1 construct. We also thank Dr. Yuhong Du for her constructive inputs to make this assay work, and Dr. Margaret Johns for editing the text. This study is supported in part by National Cancer Institute to H.F. (NIH U01CA168449) and to the Winship Cancer Institute of Emory University (NIH 5P30CA138292).
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2016 Springer Science+Business Media New York
About this protocol
Cite this protocol
Mo, XL., Fu, H. (2016). BRET: NanoLuc-Based Bioluminescence Resonance Energy Transfer Platform to Monitor Protein-Protein Interactions in Live Cells. In: Janzen, W. (eds) High Throughput Screening. Methods in Molecular Biology, vol 1439. Humana Press, New York, NY. https://doi.org/10.1007/978-1-4939-3673-1_17
Download citation
DOI: https://doi.org/10.1007/978-1-4939-3673-1_17
Published:
Publisher Name: Humana Press, New York, NY
Print ISBN: 978-1-4939-3671-7
Online ISBN: 978-1-4939-3673-1
eBook Packages: Springer Protocols