
Abstract
Mitochondrial DNA (mtDNA) can help in the identification of biological evidence recovered from crime scenes and human remains. Typically the hypervariable regions are targeted for sequencing; however, more discriminating profiles are obtained if the whole genome is sequenced. Different approaches exist as to how best amplify and sequence whole mtDNA from forensic specimens. Here, we describe a method based on two-round PCR, combining multiplex and simplex PCRs. This method has been used in the analysis of mitochondrial genomes from archival saliva samples applied to FTA® cards after 10 years of transportation and preservation, without special protection. It is expected that this technique can be also used for the analysis of other old biological specimens directly or with modifications related to the level of DNA degradation.
Access provided by CONRICYT – Journals CONACYT. Download protocol PDF
Similar content being viewed by others


Key words
1 Introduction
Mitochondrial DNA (mtDNA) is a trace evidence of biological origin used for identifying perpetrators. It is also valuable for the identification of human remains based on linkage to maternal relatives. Since the first publications on isolation and sequencing of mtDNA from a variety of biological specimens [1, 2], it became clear that this molecular marker can be of crucial significance in forensic identifications, particularly when samples with low amounts of DNA and/or degraded DNA are available.
The first application of mtDNA in forensics was based on the sequencing of hypervariable sequence regions I and II (HVS-I and HVS-II) [3]. These segments are relatively small (about 400 bp each) and accumulate far more nucleotide polymorphisms than other regions of the mitochondrial genome due to a comparatively high mutation rate. However, power of discrimination using solely this region can be low, as many people share identical sequences, otherwise known as haplotypes, in HVS-I and HVS-II.
Additionally, it is sometimes difficult to correctly determine an individual’s haplogroup because similar polymorphisms in HVS-I and HVS-II may found in different haplogroups, or the nucleotide polymorphisms are noninformative. To overcome this limitation of partial DNA analysis, the whole mitochondrial genome sequences can be amplified and sequenced. In the majority of cases, this allows for the unambiguous placement of a given mtDNA genome into a haplogroup.
There are two approaches to whole human mtDNA sequencing. The first traditional approach is based on amplification of overlapping segments covering the entire genome, followed by Sanger sequencing. The second method was recently developed, based on a next-generation sequencing technology. Because the latter approach is currently under evaluation and may not yet meet forensic standards [4, 5], this chapter focuses on the multiplex PCR and Sanger sequencing technique, which is achievable in the majority of forensic laboratories.
Numerous PCR-based protocols have been developed to sequence the whole human mtDNA. These protocols differ in the number of PCR primers encompassing amplicons of varying size or in the amount of sequencing primers annealing to large PCR fragments (up to approximately 8.5 kb) [6–11]. Procedures are optimal for different applications depending on the quality and quantity of biological specimens and DNA isolated from them. In general, whole mtDNA sequencing protocols applicable for forensics should pursue the following criteria developed in [11] and supplemented in this chapter:
-
1.
Each nucleotide should be determined from at least two independent sequencing reactions using different primers [11].
-
2.
The number of PCR amplicons or sequencing primers required to cover the entire mitochondrial genome should be kept minimal [11].
-
3.
Primers should produce an optimal signal-to-noise ratio, allowing for heteroplasmy detection [11].
-
4.
When analyzing degraded samples primers should encompass relatively short PCR segments to permit stable amplification of mtDNA amplicons from degraded DNA and samples with low amounts of DNA.
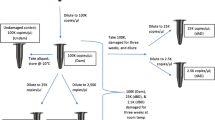
Old and partially degraded samples are frequently met in forensic investigations. However, no approach has been specifically designed for the reconstruction of whole mtDNA from such sampling. In this chapter, we demonstrate that the human whole mtDNA can be efficiently amplified and sequenced from saliva specimens collected in 1997 in Central Asia. These samples were placed in 1.5 ml tubes and transported at ambient temperature. Tubes were then stored in a freezer at −20 °C for 10 years before applying the saliva samples to FTA cards. Some recent protocols suggest carrying out amplification of the whole mitochondrial genome with only two overlapping amplicons of about 8.5 kb each [10, 11]. This technique is useful for relatively fresh blood and biopsy samples, but it was unable to produce results for old saliva samples with degraded DNA. Such difficult samples require an approach based on multiplex PCR followed by simplex PCRs with specific primer pairs and DNA sequencing of purified amplicons. We found that the primers published in [7] are better suited for successful reconstruction of whole mtDNA sequences from archival saliva specimens. Primers used for the first-round and second-round PCRs are listed in Table 1.
2 Materials
2.1 DNA Extraction
-
1.
FTA Purification Reagent (Whatman).
-
2.
Puncher for making 1.2 mm or 2.0 mm FTA disks (Whatman).
-
3.
Two 100 ml beakers.
-
4.
0.2 ml PCR tubes or 96-well PCR plates (Fisher Scientific, or any vendor).
-
5.
10 % bleach solution (Clorox).
2.2 PCR
-
1.
GoTaq DNA Polymerase (Promega).
-
2.
5× Colorless GoTaq Flexi Buffer (Promega).
-
3.
5× Green GoTaq Flexi Buffer (Promega).
-
4.
25 mM MgCl2 solution (Promega; lots of alternative are available).
-
5.
dNTPs.
-
6.
Assorted forward and reverse primers (Tables 1 and 2).
Table 2 Appropriate primer mixing for the first-round PCRs -
7.
Nuclease-free water.
-
8.
0.2 ml PCR tubes or 96-well PCR plates.
2.3 Agarose Gel
-
1.
Quick Dissolve Agarose (BioExpress) or identical agarose from any vendor.
-
2.
10× TAE buffer (Fisher Scientific, or any vendor).
-
3.
Ethidium bromide solution at concentration 10 mg/ml (Fisher Scientific or any vendor; alternative stains as GelRed and GelGreen from Biotium are available).
-
4.
100 bp PCR DNA ladder (Fisher Scientific).
-
5.
6× Blue/orange loading dye (Promega).
2.4 PCR Purification
-
1.
DNA Clean & Concentrator-5 capped columns with DNA Binding Buffer and DNA Wash Buffer (Zymo Research).
-
2.
ZR-96 DNA Clean-up Kit with DNA Binding Buffer, Wash Buffer, Silicon-A Plate, Collection Plate, and Elution Plate (Zymo Research).
-
3.
Nuclease-free water.
2.5 DNA Sequencing
-
1.
BigDye Terminator v.3.1 Cycle Sequencing Kit with 5× Dilution Buffer and the BigDye reaction mixture (Life Technologies).
-
2.
Sequencing primers corresponding to the forward and reverse PCR primers.
-
3.
Nuclease-free water.
2.6 DNA Sequencing Purification
-
1.
BigDye XTerminator Purification Kit with XTerminator solution and SAM solution (Life Technologies).
3 Methods
At all times during the extraction and amplification of human mtDNA, rigorous precautions must be followed in order to minimize the possibility of cross-transfer of mtDNA molecules between different samples. Negative extraction and amplification controls should be included to detect such cross-transfer and contamination. Pre-PCR and post-PCR procedures are to be divided between separate, isolated lab rooms and performed inside DNA workstations specially dedicated for each technological step such DNA isolation, PCR setup, and preparing DNA sequencing reactions after at least 2 h of irradiation under UV light in the hood to be used. After amplification, PCR tubes should be opened and agarose gel electrophoresis runs in a specialized electrophoresis room. Lab coats, reagents, and supplies must not be brought to DNA isolation and pre-PCR room from post-PCR/electrophoresis room.
3.1 Puncher and Cutting Square Cleaning Procedure
-
1.
Set out two 100 ml beakers. Place 15 ml of 10 % bleach solution into one of the beakers and 20 ml of deionized water into the other beaker.
-
2.
Briefly dunk the puncher into the bleach and remove. Follow quickly with dunking the puncher into the water. Do not let the puncher sit in the liquids.
-
3.
Click the end of the puncher onto a clean paper towel to rid the inside of any liquid.
-
4.
Let puncher stand to air dry.
-
5.
Pour a small amount of 10 % bleach solution onto the cutting square.
-
6.
Wipe bleach across the cutting square with a clean paper towel. Be sure to wipe entire surface of the cutting square. Let stand to air dry.
3.2 DNA Extraction from FTA Discs
-
1.
In a biosafety cabinet, remove FTA card from packaging. Unfold and lay on clean cutting square. Hold the card only on its edge, being sure to not touch the card directly.
-
2.
Using a clean puncher for making 1.2 mm or 2.0 mm disks, press firmly with puncher on area where the specimen is distributed on card.
-
3.
Place FTA disc in 0.2 ml PCR tube by clicking end of puncher to release the disc. Clean puncher and cutting square (see above) and repeat process (steps 1–3) until all specimens have been punched.
-
4.
Finally, make a final card punch with a cleaned puncher in the area outside of the specimen area. Place this disc into a PCR tube labeled as a negative extraction control.
-
5.
Add 200 μl of FTA purification reagent to each PCR tube.
-
6.
Incubate at room temperature for 5 min, then remove and discard spent FTA reagent.
-
7.
Repeat steps 5 and 6 twice for a total of three washes with FTA reagent.
-
8.
Add 200 μl nuclease-free water to each PCR tube.
-
9.
Incubate for 5 min at room temperature, then remove and discard water.
-
10.
Repeat steps 8 and 9 once for total of two washes with water.
-
11.
Dry samples at 56 °C in heat block or incubation chamber with caps open until the discs are dry (approximately 30 min).
-
12.
Setup PCR as shown below; reaction will take place in same tube and will include FTA disc.
3.3 Polymerase Chain Reactions (PCRs)
3.3.1 First-Round (multiplex) PCR
-
1.
In a PCR workstation, prepare the primer mixture by pipetting the pairs of forward and reverse primers (5 μM) together into one tube to obtain a primer solution in which each primer has a concentration of 1 μM. Primer pairs should be mixed in combinations according to Table 2 (see Note 1 ).
-
2.
Prepare the multiplex PCR master mix as indicated in Table 3. After each master mix has been made, pipette 50 μl of master mix into each PCR tube (or the specific wells of a 96-well plate) that contain FTA disks, one PCR tube/well with a blank extraction control, and one empty PCR tube/well for a negative PCR control.
Table 3 First-round (multiplex) PCR recipe -
3.
Place PCR tubes or plate in thermocycler using parameters specified for a particular thermostable DNA polymerase and PCR primers. Thermal cycling conditions optimal for GoTaq DNA Polymerase included an initial denaturation at 95 °C for 2 min, followed by 30 cycles of denaturation for 1 min at 95 °C, primer annealing for 1 min at 58 °C, and extension for 2 min at 72 °C. After the last cycle, the final extension for 5 min at 72 °C was added (see Note 2 ).
3.3.2 Second-Round (Simplex) PCR
-
1.
In a PCR workstation after UV treatment lasting two hours, separately prepare the master mixtures for each primer pair used in second-round PCR according to the recipe indicated in Table 4.
Table 4 Second-round (simplex) PCR recipe -
2.
After each master mix is made, pipette 20 μl of the appropriate master mix into PCR tubes or specific wells of a 96 well plate.
-
3.
Add 5 μl of the appropriate template from the first-round PCR into the appropriate tube/well.
-
4.
For every primer pair, pipette 20 μl of master mix into a new PCR tube or an empty well, adding 5 μl of nuclease-free water instead of DNA template. This will be your second-round PCR negative control.
-
5.
Place PCR tubes or plate in thermocycler using parameters specified for a particular thermostable DNA polymerase and PCR primers. Thermal cycling conditions optimal for GoTaq DNA Polymerase included an initial denaturation at 95 °C for 2 min, followed by 30 cycles of denaturation for 1 min at 95 °C, primer annealing for 1 min at 58 °C, and extension for 1 min at 72 °C. After the last cycle the final extension for 5 min at 72 °C was added (see Note 2 ).
3.4 Agarose Gel Electrophoresis
-
1.
In an electrophoresis room, prepare a 1 % agarose gel by adding 1 g agarose to 100 ml of 1× TAE buffer in a 250 ml Pyrex glass bottle. Melt the agarose in a microwave oven by heating the mixture for 1–2 min (depending on the oven’s power) until dissolved, with the bottle’s lid placed ajar on top to reduce buffer evaporation. Do NOT seal the lid on the bottle!
-
2.
Warning: the bottle will be hot! Carefully remove bottle and let cool on bench to approximately 65 °C. Add 1 μl of ethidium bromide with a concentration of 10 mg/ml. Swirl bottle 2–3 times to mix solution. Ethidium bromide is considered to be a hazardous chemical; practice caution in handling using protective wear, such as gloves and a lab coat. Properly dispose of ethidium bromide-containing gels and buffers according to your institution’s biosafety regulation.
-
3.
Pour into a gel tray with a comb corresponding to the number of your samples. Let the gel solidify for at least 30 min for complete polymerization.
-
4.
Add 1 μl of a 100-bp DNA Ladder solution to the first well. Starting with the second well, pipette 3–5 μl of each second-round simplex PCR reaction and each negative control (FTA extraction, first-round, and second-round negative controls) into individual wells (see Note 3 ). Be sure each sample is loaded into the appropriate well and not drifting into the well next to it.
-
5.
Run electrophoresis for approximately 30–60 min at 80–100 V, remove the gel from tray, and view with UV light using a UV transilluminator or gel documentation system (see Note 4 ). In the image, find DNA bands analogous to the expected size as determined from the positions of primers on the human mtDNA . Second-round PCR products should generate a single bright band. If the negative controls do not produce bright or faint PCR bands (Fig. 1), move on to purification of the simplex PCR mixture remaining in the corresponding PCR sample tubes or plate’s wells (Subheading 3.5).
Fig. 1 
Agarose gel electrophoresis showing PCR results. Five second-round PCR products obtained with the D1F and D1R primers are in lanes 2–6. The exACTGene ladder (Fisher BioReagents) is in lane 1. The negative extraction and PCR controls containing no PCR fragments are in lanes 7 and 8, respectively
-
6.
If DNA products of the expected length are present in the negative extraction and the first-round PCR negative controls, dispose of the tubes or wells with corresponding PCRs and repeat the first- and second-round PCRs for the particular primer pairs. If DNA bands of the expected size are present only in the second-round negative controls, discard the second-round PCR tubes or wells with the particular primer pair and setup new second-round PCRs with the same primer pair.

3.5 PCR Purification
We recommend using the Zymo-Spin Column Purification for PCR products in individual tubes and the Zymo Silicon-A Plate protocol when there are PCR samples in a plate to clean.
3.5.1 Zymo-Spin Column PCR Purification
-
1.
Label a 1.5 ml reaction tube and spin column with specimen code.
-
2.
Add 100 μl of DNA Binding Buffer to each PCR reaction tube.
-
3.
Pipette PCR reaction mixture into Zymo-Spin Column in a collection tube and centrifuge at 13,000 rpm for 30 s.
-
4.
Discard flow through and place column back into collection tube.
-
5.
Add 200 μl Wash Buffer to the column and centrifuge at 14,549 × g for 30 s.
-
6.
Discard flow through, place column back into collection tube and repeat step 5 for a total of two washes.
-
7.
Discard flow through and collection tube. Place column into appropriate 1.5 ml reaction tube from step 1.
-
8.
Add 40 μl of water and let column stand for 1 min.
-
9.
Centrifuge at 13,000 rpm for 30 s and discard spin column. Store products in a freezer.
3.5.2 Zymo Silicon-A Plate PCR Purification
-
1.
If you carried out PCRs in the 96-well PCR plate, add 100 μl of DNA Binding Buffer to each DNA sample that is ready for clean up.
-
2.
Transfer sample mixtures to wells of the Silicon-A Plate, which should be mounted on a collection plate.
-
3.
Place specimen plate on scale and measure. Place balance plate on scale and add or remove water to balance both plates.
-
4.
Centrifuge at 4,000 × g for 5 min until sample mixtures have been completely filtered. Discard flow through.
-
5.
Add 300 μl of Wash Buffer to each well of the Silicon-A Plate. Rebalance the plates using the scale. Centrifuge at 4,000 × g for 5 min. Discard flow through. Repeat this step again for a total of two washes.
-
6.
Add 40 μl of nuclease-free water directly to the column matrix in each well. Transfer the Silicon-A Plate onto an elution plate or a new 96-well plate, balance on the scale, and centrifuge at 4,000 × g for 5 min.
-
7.
Products in the 96-well plate are ready for sequencing reactions. Label the plate and place in a fridge until sequencing.
3.6 DNA Sequencing
-
1.
Using a NanoDrop Spectrophotometer, determine the concentration of PCR amplicons after purification of the PCRs. (The concentration of PCR amplicons can be also quantified based on comparison with a DNA ladder as 100 bp exACTGene DNA ladder, Fisher Scientific using an agarose gel.)
-
2.
From Table 5 and the actual concentration of the PCR amplicons, estimate the volume of PCR fragments that should be added to the sequencing reactions.
Table 5 Quantity of PCR amplicons of different size that are added to the sequencing reaction -
3.
Using the master mix protocol shown in Table 6, prepare the sequencing reactions with the correct amount of each reagent. Vortex and pipette the sequencing master mix into new 0.2 ml PCR tubes or in the appropriate wells of a 96-well plate.
Table 6 DNA sequencing reaction recipea -
4.
Pipette correct volume of DNA template (PCR purification product) into each tube or well, ensuring the master mix being added contains the appropriate primer.
-
5.
Place in thermal cycler and perform the sequencing reaction using an initial denaturation at 95 °C for 1 min, followed by 25 cycles of 10 s at 95 °C, 5 s at 53 °C, and 4 min at 60 °C. The temperature of primer annealing can be adjusted to the actual annealing temperature of a particular sequencing primer.
3.7 Sequencing Reaction Cleanup
-
1.
After cycle sequencing, centrifuge the PCR tubes or the 96-well plate briefly, then pipette 20 μl of SAM Solution into every tube/well with the DNA sequencing products.
-
2.
Vortex the XTerminator Solution until homogeneous. Using wide-bore pipette tips, pipette 5 μl of XTerminator Solution into each PCR tube/well.
-
3.
Vortex for 30 min.
-
4.
Centrifuge the PCR tubes or the 96-well plate briefly to separate out the XTerminator Solution.
-
5.
The products can be analyzed on any Genetic Analyzer (Life Technologies) for Sanger sequencing (see Note 5 ). The reaction tubes or plate can also be kept in a fridge at 4 °C.
3.8 Data Analysis
-
1.
Sequence each PCR fragment in both forward and reverse directions.
-
2.
Open resulting electropherogram files and perform visual inspection of the mtDNA sequences by two researchers independently with BioEdit [12] or 4Peaks (http://www.nucleobytes.com). Many commercial softwares are available, but these are freeware that can be downloaded from the internet.
-
3.
Align sample sequences to the revised Cambridge Reference Sequence [13] using MEGA5 [14] and identify variant sites in the sequences. MEGA5 can be replaced with any other software that can process DNA sequences including alignment, trimming, and assembling.
-
4.
Submit mtDNA HVS1 and HVS2 polymorphisms to HaploGrep [15], an online application that associates mtDNA haplotypes with specific haplogroups by comparison to a phylogenetic tree of global human mtDNA variation (mtDNA tree Build 16 on PhyloTree.org) [16], to make a preliminary estimation of haplogroup association.
-
5.
Verify the haplogroups determined through the HVS1 and HVS2 sequences by searching for the association of particular mutations found in your whole mtDNA sequence with the mtDNA tree Build 16 using the “Find” function in PhyloTree.org.
4 Notes
-
1.
Do not amplify overlapping fragments of mtDNA in the same PCR, as it can lead to accumulation of short amplicons flanked by the reverse primer of the upstream fragment and the forward primer of the downstream fragment.
-
2.
Each primer pair for the first-round and second-round PCRs was designed to anneal at 58 °C [7]. If different primers are used for the amplification of shorter amplicons, adjust annealing temperature according to the melting temperature for new primers using a T m (melting temperature) Calculator on the BioMath page of the Promega web site at www.promega.com/biomath. The annealing temperature of the primer pair equals the lowest T m of any individual primer in the mixture, minus 3 °C. The time of extension for GoTaq DNA Polymerase is 1 kb/min. If a different thermostable polymerase is used, modify the cycling parameters according to the manufacturer’s recommendations.
-
3.
The Green and Colorless GoTaq Flexi Buffers give approximately equivalent yields. For PCRs containing the 5× Green GoTaq Flexi Buffer, load the reaction onto the agarose gel directly after amplification. Reactions containing the 5× Colorless GoTaq Flexi Buffer may also be loaded directly onto the agarose gel, but a tracking dye (e.g., 6× blue/orange loading dye) must be added to monitor the progress of electrophoresis [17]. Add a tracking dye to the DNA ladder solution.
-
4.
UV transilluminator produces potentially harmful UV light. Protect eyes and skin from exposure to UV light. For safety, a gel documentation system has an automatic shut off for UV light when door is opened.
-
5.
Each sequencing run results in a electropherogram file in .ab1 format.
References
Higuchi R, von Beroldingen CH, Sensabaugh GF, Erlich HA (1988) DNA typing from single hairs. Nature 332:543–546
Sullivan KM, Hopgood R, Lang B, Gill P (1991) Automated amplification and sequencing of human mitochondrial DNA. Electrophoresis 12:17–21
Sullivan KM, Hopgood R, Gill P (1992) Identification of human remains by amplification and automated sequencing of mitochondrial DNA. Int J Legal Med 105:83–86
Bandelt HJ, Salas A (2012) Current next generation sequencing technology may not meet forensic standards. Forensic Sci Int Genet 6:143–145
Parson W, Strobl C, Huber G, Zimmermann B, Gomes SM, Souto L et al (2013) Evaluation of next generation mtGenome sequencing using the Ion Torrent personal genome machine (PGM). Forensic Sci Int Genet 7:543–549
Rieder MJ, Taylor SL, Tobe VO, Nickerson DA (1998) Automating the identification of DNA variations using quality-based fluorescence re-sequencing: analysis of the human mitochondrial genome. Nucleic Acids Res 26:967–973
Taylor RW, Taylor GA, Durham SE, Turnbull DM (2001) The determination of complete human mitochondrial DNA sequences in single cells: implications for the study of somatic mitochondrial DNA point mutations. Nucleic Acids Res 29:e74
Levin BC, Holland KA, Hancock DK, Coble M, Parsons TJ, Kienker LJ et al (2003) Comparison of the complete mtDNA genome sequences of human cell lines - HL-60 and GM 10742A - from individuals with pro-myelocytic leukemia and leber hereditary optic neuropathy, respectively, and the inclusion of HL-60 in the NIST human mitochondrial DNA standard reference material - SRM 2392-I. Mitochondrion 2:387–400
Coble MD, Just RS, O'Callaghan JE, Letmanyi IH, Peterson CT, Irwin JA et al (2004) Single nucleotide polymorphisms over the entire mtDNA genome that increase the power of forensic testing in Caucasians. Int J Legal Med 118:137–146
Gonder MK, Mortensen HM, Reed FA, de Sousa A, Tishkoff SA (2007) Whole-mtDNA genome sequence analysis of ancient African lineages. Mol Biol Evol 24:757–768
Fendt L, Zimmermann B, Daniaux M, Parson W (2009) Sequencing strategy for the whole mitochondrial genome resulting in high quality sequences. BMC Genomics 10:139
Hall T (1999) BioEdit: a user-friendly biological sequence alignment editor and analysis program for Windows 95/98/NT. Nucleic Acids Symp Ser 41:95–98
Andrews RM, Kubacka I, Chinnery PF, Lightowlers RN, Turnbull DM, Howell N (1999) Reanalysis and revision of the Cambridge reference sequence for human mitochondrial DNA. Nat Genet 23:147
Tamura K, Peterson D, Peterson N, Stecher G, Nei M, Kumar S (2011) MEGA5: molecular evolutionary genetics analysis using maximum likelihood, evolutionary distance, and maximum parsimony methods. Mol Biol Evol 28:2731–2739
Kloss-Brandstätter A, Pacher D, Schönherr S, Weissensteiner H, Binna R, Specht G et al (2011) HaploGrep: a fast and reliable algorithm for automatic classification of mitochondrial DNA haplogroups. Hum Mutat 32:25–32
van Oven M, Kayser M (2008) Updated comprehensive phylogenetic tree of global human mitochondrial DNA variation. Hum Mutat 30:e386–e394
Glebs A, Stencel E, Knoche K (2003) The “Go-to” choice: GoTaq DNA polymerase. Promega Notes 83:21–24
Acknowledgments
The study was supported by the North Dakota EPSCoR and UND faculty start-up awards to I.V.O. The research protocol was approved by the University of North Dakota’s Institutional Review Board (Protocol number IRB-201307-009) and the University of North Dakota’s Institutional Biosafety Committee (Registration number IBC-201102-001).
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2016 Springer Science+Business Media New York
About this protocol
Cite this protocol
Ovchinnikov, I.V., Malek, M.J., Kjelland, K., Drees, K. (2016). Whole Human Mitochondrial DNA Sequencing. In: Goodwin, W. (eds) Forensic DNA Typing Protocols. Methods in Molecular Biology, vol 1420. Humana Press, New York, NY. https://doi.org/10.1007/978-1-4939-3597-0_13
Download citation
DOI: https://doi.org/10.1007/978-1-4939-3597-0_13
Published:
Publisher Name: Humana Press, New York, NY
Print ISBN: 978-1-4939-3595-6
Online ISBN: 978-1-4939-3597-0
eBook Packages: Springer Protocols