
Abstract
High-throughput sequencing technologies have made it possible for biologists to generate genome-wide profiles of chromatin features at the nucleotide resolution. Enzymes such as nucleases or transposes have been instrumental as a chromatin-probing agent due to their ability to target accessible chromatin for cleavage or insertion. On the scale of a few hundred base pairs, preferential action of the nuclear enzymes on accessible chromatin allows mapping of cell state-specific accessibility in vivo. Such accessible regions contain functionally important regulatory sites, including promoters and enhancers, which undergo active remodeling for cells adapting in a dynamic environment. DNase-seq and the more recent ATAC-seq are two assays that are gaining popularity. Deep sequencing of DNA libraries from these assays, termed genomic footprinting, has been proposed to enable the comprehensive construction of protein occupancy profiles over the genome at the nucleotide level. Recent studies have discovered limitations of genomic footprinting which reduce the scope of detectable proteins. In addition, the identification of putative factors that bind to the observed footprints remains challenging. Despite these caveats, the methodology still presents significant advantages over alternative techniques such as ChIP-seq or FAIRE-seq. Here we describe computational approaches and tools for analysis of chromatin accessibility and genomic footprinting. Proper experimental design and assay-specific data analysis ensure the detection sensitivity and maximize retrievable information. The enzyme-based chromatin profiling approaches represent a powerful and evolving methodology which facilitates our understanding of how the genome is regulated.
Access provided by CONRICYT – Journals CONACYT. Download protocol PDF
Similar content being viewed by others

Key words
- Chromatin remodeling
- DNase-seq
- ATAC-seq
- High-throughput sequencing
- Computational genomics
- Genomic footprinting
1 Introduction
Chromatin exerts significant regulation of the genome through tight packaging of DNA in the nucleus of a eukaryotic cell, preventing access of transcription factors and other proteins to their cognate sites [1, 2]. Accessibility at promoters, enhancers, or silencers is actively maintained and dynamically altered in a cell- and condition-specific manner [3–7]. Chromatin accessibility can be measured by the susceptibility of DNA either to cleavage by nucleases such as DNase I [8] or to transposition [9]. For example, DNase I hypersensitive sites (DHSs) are defined as the regions particularly prone to cutting by DNase I, and they represent regions with an “open chromatin” structure. DNase I hypersensitivity coupled with high-throughput sequencing (DNase-seq) has been used to provide genome-wide identification of functional regulatory elements [8, 10]. More recently, the assay for transposase-accessible chromatin using sequencing (ATAC-seq) was developed as a simpler method that can be performed on a small number of cells. Each assay generates a continuous high-resolution profile of chromatin accessibility along the genome in a given cell state [9]. DNase-seq and ATAC-seq have been shown to produce very similar signal profiles, in contrast to the poor concordance between DNase-seq and FAIRE (formaldehyde assisted isolation of regulatory elements)-seq [11]. FAIRE may not permit sensitive detection of regulatory regions due to high background signals. Here we focus on the enzyme-based chromatin assays DNase-seq and ATAC-seq and discuss computational analysis methods that extract epigenetic information from the data generated.
If a DNase-seq library is sequenced deeply to yield a large number (>300 million) of reads, the genomic loci which are highly occupied by transcription factors may be identified as narrow regions of protection against DNase I cleavage, termed “footprints” [12–14]. Although the cost of sequencing becomes an issue in practice, sufficient tag coverage allows pinpointing of specific binding sites at the nucleotide resolution. However, the detection of protein footprints and inferring the identity of factors are technically and computationally more challenging in comparison to the detection of accessible regions.
This chapter provides a description of the procedures that we have been employing to analyze DNase-seq and ATAC-seq data. Surveys of existing methods mostly cover analysis tools for ChIP-seq or RNA-seq [15], with fewer studies comparing different analysis methods for DNase-seq [3, 16–18]. The chapter is divided into two parts based on the resolution of analysis: First on analyses pertaining to chromatin accessibility on the scale of 100 bp to 1 kb, and the other on analyses of transcription factor footprints on the bp scale. Within each part, the algorithms are roughly categorized into different types of analyses: (1) generation of browser tracks for visual exploration; (2) detection of significant regions (hotspots or footprints) based on a background probability model and calculation of statistical measures; (3) artifact adjustment and filtering; (4) annotation of the identified regions with respect to other genomic features or related data; (5) downstream analyses and useful plotting strategies for delineating meaningful patterns from the combined set of regions across multiple conditions or time points.
2 Analysis of Chromatin Accessibility
2.1 Assay Protocols, Biases, and Data Reproducibility
It is worthwhile to note that distinct protocols exist under the same term “DNase-seq” (Table 1). Depending on the DNase-seq protocol, there are different data features and biases that one needs to take into consideration for the analysis and interpretation of the data. To distinguish between the protocols in this chapter, we denote the size selection-based methods as “DNase-seq I” and “DNase-seq II,” according to the sequencing type. We designate the biotin end-labeling method as “DNase-seq III.” With DNase-seq I and II, the reads (aka tags) come from the ends of the DNA fragments within accessible chromatin which are cleaved and released. The size selection for 100–500 bp range enriches for fragments that are doubly cut by DNase I. With DNase-seq III, individual DNA ends are labeled with biotin and captured for single-end sequencing. Interestingly, the sample processing protocols DNase-seq I/II and DNase-seq III produce different DNA sequence bias patterns [19]. Adjusting for the sequence-dependent cleavage bias becomes important for analysis of cut counts and TF footprint detection (Subheading 3.3).
ATAC-seq utilizes a completely different approach by inserting sequencing adaptors directly to accessible chromatin using a transposase. In contrast to DNase I whose DNA cleavage activity is used to mark open chromatin, this assay relies on transposition as the primary molecular reaction for targeting and sampling open chromatin. Therefore, reaction kinetics and targeting preferences are likely to be distinct from DNase-based methods. Despite the difference, the correlation, at least at the level of chromatin accessibility, between ATAC-seq and DNase-seq I was reported to be as high as that between DNase-seq I and III [9]. The correlation between ATAC-seq and DNase-seq III was slightly lower.
Current high throughput sequencing of a single or multiplexed sample routinely produces hundreds of millions of sequence reads of 35–100 bp in length from a lane. Quality-filtered sequence reads are then aligned to the reference genome. The regions densely populated with reads are putative DHSs or accessible chromatin regions. Even though accessibility data generated from proper experimental design are reproducible and visually convincing, there are systematic biases that should be corrected. For example, a proportion of the reads may not align to the reference genome simply because the genome of the cells used for the experiment is structurally different from the reference genome, containing aberrations such as polyploidy, translocations, or other mutations. Amplified regions would contribute more to the DNA sample and deleted regions would not produce any sequence reads.
Another source of sequencing data bias arises from the fact that the genomic locations of the sequenced fragments are inferred from finding the “best match” in the genome sequence. However, the accuracy of aligning a read back to the genome varies greatly depending on the sequence and read length. Hence it is necessary to consider the read “mappability” (Fig. 1). A given n-mer sequence read may occur at a unique location or at multiple genomic positions under a preset mismatch tolerance. Although reads with multiple genomic matches can, in principle, be probabilistically mapped, a common alignment approach allows only one genomic coordinate for each read and discards reads that cannot be uniquely mapped. The procedure creates “dark spots” across the genome and directly affects the background probability of observing reads at any given position in the genome (Fig. 1).

Mappability of sequence reads is far from uniform, across the scales from Mb to bps. The top plot shows the fluctuation of mappability along mm9 as the percentage of 35-mers in a 200 kb moving window which are uniquely mappable. The middle plot displays the mappability as the percentage of 35-mers in a 250 bp moving window which are uniquely mappable. The bottom plot shows the nucleotide-resolution mappability itself, i.e., the number of genome-wide occurrences of each 35-mer. The positions with the mappability count higher than 1 cannot have any reads mapped from commonly used parameter settings of an alignment tool
Identification of the genomic regions where reads are significantly enriched over the background must take into account these and other sources of bias and artifacts in the sequencing data. The objective of an algorithm for detecting accessible regions is to find all of the truly read-enriched sites while minimizing the false positive rate (Subheading 2.3).
2.2 Building a Profile for Data Visualization in a Genome Browser
Visualization of the data is important for assessing data quality and for confirming results from a global analysis. It is useful to note that there are a few different approaches even for this apparently simple practice. First, there are multiple ways of generating the data tracks depending on how the read distribution is summarized. A density profile or a coverage map can be generated by calculating (i) the number of reads overlapping each genomic bin of fixed size (ranging from 1 bp to 20 bp, for example), (ii) the number of reads whose starting nucleotides are in each bin, (iii) the number of reads whose fragments (extended from the starting nucleotide into the genomic sequence by a fixed length) overlap each genomic bin, or (iv) the number of paired-end reads whose spanned fragments overlap each genomic bin, etc. The differences resulting from methods (i)–(iv) are negligible when the tracks are browsed in a zoomed-out mode, but they become noticeable at a high resolution. We have used the approach (iii) for DNase-seq I data and (iv) for DNase-seq II data, both with a nucleotide resolution (no binning). For DNase-seq I, because the size-selected DNA fragments (100–1000 bp) are longer than the sequence reads (35–100 bp) from the 5′ ends, length adjustment is made to estimate the distribution of fragments in the DNase-treated sample. If the average fragment length is known from the sample QC, each sequence tag can be extended in the 3′ direction up to that length.
Another consideration for generating data track files stems from the occasional disconcordance between results from a statistical analysis and visual impressions from the browser tracks. For the purpose of assessing data quality, minimally processed data tracks are often used to display the “raw data” as well as the anomalies that are to be excluded from any systematic analysis, such as artifacts from repeat elements and PCR-duplicated reads. Such unadjusted data tracks are also used to convey the final analysis results in published data figures. However, the unadjusted data may not explain, for example, why some weakly accessible sites are detected as significant while other sites with similar read densities are not. These incidents arise often, because a detection algorithm adjusts for the systematic biases when assessing statistical significance (Subheading 2.3). Therefore, using adjusted data tracks might produce visualization more consistent with the results from statistical analyses, although this approach is not widely used.
There are several publicly accessible browser tools that accept users’ genomic data files and display them in the context of annotation tracks such as known genes, ncRNAs, repeat elements, and ENCODE data (Table 2). The University of California Santa Cruz Genome Browser has been popular and their website also provides the Table Browser from which one can download public data tracks for incorporation into further correlative analyses (http://genome.ucsc.edu/cgi-bin/hgTables). Integrated Genome Browser (IGB) is a genomic data browser which has undergone significant enhancements recently, supporting many file formats. The Integrative Genomics Viewer (IGV) and the Washington University Epigenome Browser have assay-specific capabilities for certain data types that other browsers do not provide. Hence, investigators who generate such data may benefit from the customized data exploration tools from these browsers.
2.3 Region Detection Algorithm
There are only a few algorithms specifically developed to identify accessible chromatin regions from DNase-seq (protocols I, II, and III) data, while numerous software packages exist now for calling peaks or enriched sites from ChIP-seq data. We have developed and described a one-pass algorithm for detecting “hotspots” in detail elsewhere, and refer the reader to [16] and the accompanying source code “DNase2Hotspots” and documentation at http://sourceforge.net/projects/dnase2hotspots.
Here we briefly outline the core components of the algorithm. DNase2Hotspots finds hotspots, or local enrichment of reads in a 250 bp target window relative to a local background (surrounding 200 kb window), based on the binomial distribution. The usage of a local background, rather than a genome-wide uniform background, adjusts for the local fluctuations in read distributions reflecting large-scale differences in chromatin accessibility or copy number variations. Significance of read enrichment within the target window over the local background is assessed using a binomial z score from counting expected read occurrences only at uniquely mappable genomic coordinates (Figs. 1 and 2). Hence the mappability is incorporated directly into the z score. An unthresholded hotspot is defined as a contiguous cluster of 250 bp windows whose z scores are nominally significant, i.e., greater than 2. The final z score threshold is imposed based on an empirically calculated false discovery rate (FDR). When the analysis calls for stringent region calling, hotspots are selected with 0 % FDR, i.e., the z score threshold is set by the minimum absolute value that does not allow any hotspots called from the randomized data. If it is desirable to include a larger number of accessible sites with a higher sensitivity of detection, then hotspots can be called with a higher FDR, such as 1 % or 5 %.

DNase2Hotspots assesses the enrichment of extended reads within a target window by computing the binomial z score with respect to the local background window. The top track shows a part of the larger 200 kb background window. The bottom track shows the distribution of individual reads (dark blue) and the estimated fragments (light blue) which are extended from the single end reads of DNase-seq I or III. For DNase-seq II and ATAC-seq, the ends of the fragments are defined by the paired end reads. The maximum read density or the average read density can be associated with each hotspot as a quantitative measure of chromatin accessibility
The ENCODE group at the University of Washington had developed the original hotspot detection algorithm which uses a two-pass procedure to capture weakly accessible sites that can be masked by nearby big DHSs. The ENCODE program is currently available at http://www.uwencode.org/proj/hotspot/.
F-seq was developed by the authors of DNase-seq III [20, 21] and an updated version is available at https://github.com/aboyle/F-seq. It is not unusual for the same data to produce significantly different sets of hotspots or peaks depending on the detection algorithm. To reconcile the different sets without relying on a single detection method, sometimes an ad hoc combination of the different sets is used to obtain the final set of accessible regions from the data [3].
2.4 Region Annotation and Integrative Analyses
Much of the biologically meaningful data analyses are performed during this stage of the analysis. When there are several accessibility profiles from different experimental conditions or cell states, it is very useful to have a “master set” of hotspots derived from reconciling the boundaries of overlapping hotspots. Essentially the same site may show up from multiple biological samples as hotspots with slightly different start and end coordinates. There can be different ways of defining the boundaries of hotspots to construct the master set for subsequent analyses. For example, each hotspot in the master set which represents overlapping hotspots detected in individual samples can be defined as their union, intersection, or union of the top three accessible sites, etc. The determination of the master hotspots is necessary for a comparative analysis which reveals chromatin accessibility changes across the samples or during a time course, based on a single convenient measure per hotspot. We have been using the maximum read density or the average read density as such a measure which reflects the extent of accessibility at each hotspot. Cluster analyses or supervised classification methods can then be applied to discern distinct patterns of chromatin behavior.
Once the hotspots are obtained, it is often desirable to annotate the sites with genomic information such as the closest genes, the distance to TSSs, or whether they overlap with regions found from other related data [16]. For instance, one can examine the proportions of accessible sites located at promoters, introns, or intergenic regions, or the extent of overlap with regions exhibiting other enhancer marks or repressive chromatin marks.
2.5 Motif Analysis on Hotspots
Motif analysis allows a higher resolution examination of the underlying genomic regions than any methods purely based on hotspots which can range up to a few kilobases. The presence of a TF binding motif element indicates a potential protein binding event within the accessible sites (see Subheading 3 for further discussions). There are two common types of sequence motif analyses that can be performed on the set of DNA sequences from a specific subset of the identified hotspots. One method is scanning the sequences for the presence of motifs for known TFs [22] (FIMO is available at http://meme.nbcr.net/meme/doc/fimo.html). It requires prior knowledge of TF binding motifs but the computation is straightforward.
Another analysis aims at discovery of novel motifs enriched in the target DNA sequences, which is computationally very intensive due to the large number of accessible sites that are often used as search input. A strategy to handle the computational demand is to reduce the total DNA content of the input set by narrowing down to the strongest signal regions, i.e., peaks or local summits. Deciding which regions to focus on critically affects the output motifs that are found to be enriched from the regions. One common caveat is selecting the top DHSs ranked by read density. Often the sites that produce the highest DNase-seq signal are constitutively open AT-rich regions whose accessibility may be governed more by their sequence characteristics than by dynamic chromatin regulation. By choosing the top 200 DHSs, for example, the investigator may only find simple repeat sequences that tend to avoid nucleosomes. Cell type-dependent and TF-specifically regulated sites are likely to reside in hotspots of modest read density. For this reason, we remove the top DHSs and include as many hotspots as possible for each genomic set of interest by limiting the searchable DNA sequences onto the narrow peaks within the hotspots [16]. For preparing input, the UCSC Table Browser can be used to extract the DNA sequences of specified genomic regions in the FASTA format.
The widely used de novo motif discovery tool MEME [23] uses an expectation maximization algorithm (http://meme.nbcr.net/meme/tools/meme). DREME is another discovery tool developed by the authors of MEME [24]. HOMER is a different tool that has gained popularity for ease of use [25]. These discovery tools seem to have different sensitivity for finding certain types of motifs. Therefore, it is recommended that users should try more than one method to discover a wider class of motifs. The enriched motifs found from the discovery step can be batch-queried against the known TF binding motifs available in motif databases such as JASPAR or UniPROBE, using the motif comparison tool TomTom (available at the same site for the MEME suite) [26].
3 Analysis of TF Footprints
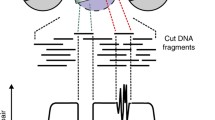
TF footprinting aims to detect sites bound by all protein factors from the same biological sample with a nucleotide precision [12–14]. To find TF footprints, one looks for narrow regions (8–30 bp) on which cleavage (or transposition in the case of ATAC-seq) is significantly reduced in comparison to the immediately surrounding regions (Fig. 3). The analysis requires ultra-deep sequencing to achieve reasonable coverage of cleavage events for all the hotspots in the genome.

Illustration of TF footprints which can be detected as protected regions from DNase cleavage and the corresponding narrow “valleys” in the cut count profile. DNase2TF begins with data-derived seeds and merges neighboring candidate regions until the significance of depletion no longer improves. Putative footprints are overlaid with known TF binding elements in the genome and assigned to best candidate TFs
3.1 Data Requirement
The same experimental protocols for DNase-seq or ATAC-seq are used to generate data for the purpose of genomic TF footprinting. However, additional data standards are imposed to determine the suitability of the data for higher resolution analyses. First, the depth of sequencing should be sufficient to provide at least 300 million uniquely mapped reads for a mammalian genome. Depending on the complexity of the sequencing library and the level of contaminating mitochondrial or other irrelevant DNA, the actual number of sequence reads needed may be much higher than the final target value. It is worth noting that, despite the decreasing cost and improved throughput of sequencing, currently feasible sequencing depths do not generally allow robust and reproducible detection of individual footprints for mammalian genomes.
Second, the data quality, as measured by the enrichment of nuclease activity within accessible chromatin, is useful to estimate the “signal-to-noise” ratio. We have used a quality score, similar to SPOT of the ENCODE team at the University of Washington, which is defined as proportion (ranging from 0 to 1) of reads overlapping FDR-unthresholded nominally defined hotspots. Datasets with low quality scores due to high background may be excluded or at least flagged for cautious data interpretation. Datasets with the quality score higher than 0.5 are generally considered to meet the suitability for TF footprinting analysis (Fig. 4).

Quality score is a genome-wide measure of read enrichment within the relevant regions versus the nonspecific background. Defined as the proportion of reads falling within nominally called hotspots, the Q score ranges from 0 to 1 and can be useful for deciding whether to advance to ultra-deep sequencing and footprinting analysis. Shown are read density tracks of example DNase-seq I samples with a range of quality scores
3.2 Cut Count Profiles
Although the cut count profile is generally thought to convey the raw data, there are data features which result in a few variant definitions that can potentially affect the visual representation of putative footprints. First, since the exact location of a DNase I cleavage event is between nucleotides, not at a nucleotide, cut counts are truly assigned to mid-points between bp coordinates. However, if the cut count data is to be uploaded to a browser, the obligatory assignment to integer coordinates necessitates a convention for the 1 bp-offset to be introduced to either the forward or the reverse direction reads [18]. While the choice is arbitrary, a consistent convention should be used throughout subsequent analyses.
ATAC-seq has an additional correction step to account for the distance between the sites of the sequencing adaptor insertion and the transposase binding [9]. The plus strand reads are shifted by +4 bps and the minus strand reads by −5 bps.
Analogous to the issue of raw versus adjusted data which was discussed in Subheading 2.2, the cut count profile may be generated to display the enzyme bias-corrected profile (see also Subheading 3.3). The choice depends on whether the resulting plot is intended to show technical features from the particular nuclease used to generate the data.
3.3 Artifacts from the Enzyme Bias on Sequence Patterns at Cut Sites
We and others have independently demonstrated that the sequence bias of DNase cleavage is quite pronounced [18, 27, 28], despite the previous assumption that DNase I cuts DNA in a sequence nonspecific manner. The cleavage bias generates distinct cut signatures when the cut count is averaged over TF binding motif elements. The cut signatures arise purely from the DNA sequence bias of DNase I, encoded in the tetramers or the hexamers surrounding the cleavage site, and are observed in deproteinized DNA [18, 27]. Analogously, distinct sequence biases have been observed for the transposase used in the ATAC-seq assay [9]. These findings raise doubts about the original interpretation of the cut signatures as reflecting the exact nucleotides bound and protected by sequence-specific proteins [14, 29, 30].
3.4 Cut Count Analysis When Matching ChIP-seq Data Are Available for TFs of Interest
To assess the true cut profiles at binding sites for a given TF, a reference dataset needs to be compiled: DNase-seq, ChIP-seq of the TF from a matching biological sample, and well-characterized PWM(s) for the TF. Then the TF binding motif elements called by FIMO can be separated based on whether they overlap ChIP peaks. The average cut count profiles can be computed over the two sets of motif elements (bound versus unbound) to delineate the effect of TF binding. The use of the bias-adjusted cut count profiles (Subheadings 3.2 and 3.3) may suppress the enzyme-specific artifacts and facilitate such comparisons.
3.5 Detection of Putative Footprints and Limitations in Inference of TF Occupancy In Vivo
The first footprint detection program, developed by Stamatoyannopoulos and coworkers, was used to identify footprints in DNase-seq data from S. cerevisiae [13]. The software does not scale well with large mammalian genomes. More recent detection programs have been developed based on completely different algorithms. The Wellington algorithm was designed to increase specificity of footprint calls by analyzing the plus and minus strands separately [31]. CENTIPEDE takes a different approach by making use of the a priori TF binding motifs, sequence conservation, and epigenetic marks [32]. However, the additional information available for making predictions about binding does not seem to result in higher accuracy [18]. We have developed an efficient computational algorithm that adjusts for the enzyme bias and read mappability [18]. The software package implementing the footprint detection algorithm is available as “DNase2TF” (http://sourceforge.net/projects/dnase2tfr) (Fig. 5).

Flow chart for DNase2TF and TF motif matching. Hotspots are precomputed as the set of regions within which the algorithm searches for footprints. For each hotspot, the reads are randomized to estimate the local FDR ten times. The FDR-thresholded footprints are called and matched with known TF binding motif elements
Despite the progress, it remains difficult to detect individual TF footprints with an acceptable accuracy and reproducibility. The high quality of the data necessary for TF footprinting and ultra-deep sequencing remain as nontrivial technical bottlenecks. One should also acknowledge the inherent limitation of TF footprinting arising from lack of footprint depths for TFs with short DNA binding residence times [18].
3.6 Sequence Motif Analysis
Even though a comprehensive TF discovery analysis is generally not possible with current tools, some novel TFs may still be found from significantly enriched footprints. For example, detected footprints which do not overlap any matches from known TF binding motifs can be analyzed separately for enrichment of de novo motifs. Since the genomic regions called as putative footprints are much narrower than accessible regions called as hotspots, the search for de novo motifs becomes more focused and specific.
References
Voss TC, Hager GL (2014) Dynamic regulation of transcriptional states by chromatin and transcription factors. Nat Rev Genet 15:69–81
Felsenfeld G, Groudine M (2003) Controlling the double helix. Nature 421:448–453
Hsiung CC, Morrissey CS, Udugama M, Frank CL, Keller CA, Baek S, Giardine B, Crawford GE, Sung MH, Hardison RC, Blobel GA (2015) Genome accessibility is widely preserved and locally modulated during mitosis. Genome Res 25:213–225
Morris SA, Baek S, Sung MH, John S, Wiench M, Johnson TA, Schiltz RL, Hager GL (2014) Overlapping chromatin-remodeling systems collaborate genome wide at dynamic chromatin transitions. Nat Struct Mol Biol 21:73–81
Thurman RE, Rynes E, Humbert R, Vierstra J, Maurano MT, Haugen E, Sheffield NC, Stergachis AB, Wang H, Vernot B, Garg K, John S, Sandstrom R, Bates D, Boatman L, Canfield TK, Diegel M, Dunn D, Ebersol AK, Frum T, Giste E, Johnson AK, Johnson EM, Kutyavin T, Lajoie B, Lee BK, Lee K, London D, Lotakis D, Neph S, Neri F, Nguyen ED, Qu H, Reynolds AP, Roach V, Safi A, Sanchez ME, Sanyal A, Shafer A, Simon JM, Song L, Vong S, Weaver M, Yan Y, Zhang Z, Zhang Z, Lenhard B, Tewari M, Dorschner MO, Hansen RS, Navas PA, Stamatoyannopoulos G, Iyer VR, Lieb JD, Sunyaev SR, Akey JM, Sabo PJ, Kaul R, Furey TS, Dekker J, Crawford GE, Stamatoyannopoulos JA (2012) The accessible chromatin landscape of the human genome. Nature 489:75–82
Siersbaek R, Nielsen R, John S, Sung MH, Baek S, Loft A, Hager GL, Mandrup S (2011) Extensive chromatin remodelling and establishment of transcription factor ‘hotspots’ during early adipogenesis. EMBO J 30:1459–1472
John S, Sabo PJ, Thurman RE, Sung MH, Biddie SC, Johnson TA, Hager GL, Stamatoyannopoulos JA (2011) Chromatin accessibility pre-determines glucocorticoid receptor binding patterns. Nat Genet 43:264–268
John S, Sabo PJ, Canfield TK, Lee K, Vong S, Weaver M, Wang H, Vierstra J, Reynolds AP, Thurman RE, Stamatoyannopoulos JA (2013) Genome-scale mapping of DNase I hypersensitivity. Curr Protoc Mol Biol Chapter 27:Unit 21.27
Buenrostro JD, Giresi PG, Zaba LC, Chang HY, Greenleaf WJ (2013) Transposition of native chromatin for fast and sensitive epigenomic profiling of open chromatin, DNA-binding proteins and nucleosome position. Nat Methods 10:1213–1218
Boyle AP, Davis S, Shulha HP, Meltzer P, Margulies EH, Weng Z, Furey TS, Crawford GE (2008) High-resolution mapping and characterization of open chromatin across the genome. Cell 132:311–322
Song L, Zhang Z, Grasfeder LL, Boyle AP, Giresi PG, Lee BK, Sheffield NC, Graf S, Huss M, Keefe D, Liu Z, London D, McDaniell RM, Shibata Y, Showers KA, Simon JM, Vales T, Wang T, Winter D, Zhang Z, Clarke ND, Birney E, Iyer VR, Crawford GE, Lieb JD, Furey TS (2011) Open chromatin defined by DNaseI and FAIRE identifies regulatory elements that shape cell-type identity. Genome Res 21:1757–1767
Boyle AP, Song L, Lee BK, London D, Keefe D, Birney E, Iyer VR, Crawford GE, Furey TS (2011) High-resolution genome-wide in vivo footprinting of diverse transcription factors in human cells. Genome Res 21:456–464
Hesselberth JR, Chen X, Zhang Z, Sabo PJ, Sandstrom R, Reynolds AP, Thurman RE, Neph S, Kuehn MS, Noble WS, Fields S, Stamatoyannopoulos JA (2009) Global mapping of protein-DNA interactions in vivo by digital genomic footprinting. Nat Methods 6:283–289
Neph S, Vierstra J, Stergachis AB, Reynolds AP, Haugen E, Vernot B, Thurman RE, John S, Sandstrom R, Johnson AK, Maurano MT, Humbert R, Rynes E, Wang H, Vong S, Lee K, Bates D, Diegel M, Roach V, Dunn D, Neri J, Schafer A, Hansen RS, Kutyavin T, Giste E, Weaver M, Canfield T, Sabo P, Zhang M, Balasundaram G, Byron R, MacCoss MJ, Akey JM, Bender MA, Groudine M, Kaul R, Stamatoyannopoulos JA (2012) An expansive human regulatory lexicon encoded in transcription factor footprints. Nature 489:83–90
Pepke S, Wold B, Mortazavi A (2009) Computation for ChIP-seq and RNA-seq studies. Nat Methods 6:S22–S32
Baek S, Sung MH, Hager GL (2012) Quantitative analysis of genome-wide chromatin remodeling. Methods Mol Biol 833:433–441
Tsompana M, Buck MJ (2014) Chromatin accessibility: a window into the genome. Epigenetics Chromatin 7:33
Sung MH, Guertin MJ, Baek S, Hager GL (2014) DNase footprint signatures are dictated by factor dynamics and DNA sequence. Mol Cell 56:275–285
Koohy H, Down TA, Hubbard TJ (2013) Chromatin accessibility data sets show bias due to sequence specificity of the DNase I enzyme. PLoS One 8, e69853
Boyle AP, Guinney J, Crawford GE, Furey TS (2008) F-Seq: a feature density estimator for high-throughput sequence tags. Bioinformatics 24:2537–2538
Song L, Crawford GE (2010) DNase-seq: a high-resolution technique for mapping active gene regulatory elements across the genome from mammalian cells. Cold Spring Harb Protoc 2010:pdb.prot5384
Grant CE, Bailey TL, Noble WS (2011) FIMO: scanning for occurrences of a given motif. Bioinformatics 27:1017–1018
Bailey TL, Williams N, Misleh C, Li WW (2006) MEME: discovering and analyzing DNA and protein sequence motifs. Nucleic Acids Res 34:W369–W373
Bailey TL (2011) DREME: motif discovery in transcription factor ChIP-seq data. Bioinformatics 27:1653–1659
Heinz S, Benner C, Spann N, Bertolino E, Lin YC, Laslo P, Cheng JX, Murre C, Singh H, Glass CK (2010) Simple combinations of lineage-determining transcription factors prime cis-regulatory elements required for macrophage and B cell identities. Mol Cell 38:576–589
Gupta S, Stamatoyannopoulos JA, Bailey TL, Noble WS (2007) Quantifying similarity between motifs. Genome Biol 8:R24
He HH, Meyer CA, Hu SS, Chen MW, Zang C, Liu Y, Rao PK, Fei T, Xu H, Long H, Liu XS, Brown M (2014) Refined DNase-seq protocol and data analysis reveals intrinsic bias in transcription factor footprint identification. Nat Methods 11:73–78
Lazarovici A, Zhou T, Shafer A, Dantas Machado AC, Riley TR, Sandstrom R, Sabo PJ, Lu Y, Rohs R, Stamatoyannopoulos JA, Bussemaker HJ (2013) Probing DNA shape and methylation state on a genomic scale with DNase I. Proc Natl Acad Sci U S A 110:6376–6381
Neph S, Stergachis AB, Reynolds A, Sandstrom R, Borenstein E, Stamatoyannopoulos JA (2012) Circuitry and dynamics of human transcription factor regulatory networks. Cell 150:1274–1286
Stergachis AB, Neph S, Sandstrom R, Haugen E, Reynolds AP, Zhang M, Byron R, Canfield T, Stelhing-Sun S, Lee K, Thurman RE, Vong S, Bates D, Neri F, Diegel M, Giste E, Dunn D, Vierstra J, Hansen RS, Johnson AK, Sabo PJ, Wilken MS, Reh TA, Treuting PM, Kaul R, Groudine M, Bender MA, Borenstein E, Stamatoyannopoulos JA (2014) Conservation of trans-acting circuitry during mammalian regulatory evolution. Nature 515:365–370
Piper J, Elze MC, Cauchy P, Cockerill PN, Bonifer C, Ott S (2013) Wellington: a novel method for the accurate identification of digital genomic footprints from DNase-seq data. Nucleic Acids Res 41, e201
Pique-Regi R, Degner JF, Pai AA, Gaffney DJ, Gilad Y, Pritchard JK (2011) Accurate inference of transcription factor binding from DNA sequence and chromatin accessibility data. Genome Res 21:447–455
Vierstra J, Wang H, John S, Sandstrom R, Stamatoyannopoulos JA (2014) Coupling transcription factor occupancy to nucleosome architecture with DNase-FLASH. Nat Methods 11:66–72
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2016 Springer Science+Business Media New York
About this protocol
Cite this protocol
Baek, S., Sung, MH. (2016). Genome-Scale Analysis of Cell-Specific Regulatory Codes Using Nuclear Enzymes. In: Mathé, E., Davis, S. (eds) Statistical Genomics. Methods in Molecular Biology, vol 1418. Humana Press, New York, NY. https://doi.org/10.1007/978-1-4939-3578-9_12
Download citation
DOI: https://doi.org/10.1007/978-1-4939-3578-9_12
Published:
Publisher Name: Humana Press, New York, NY
Print ISBN: 978-1-4939-3576-5
Online ISBN: 978-1-4939-3578-9
eBook Packages: Springer Protocols