
Abstract
The sensitivity and specificity of tandem mass spectrometers have made targeted proteomics the method of choice for the precise simultaneous measurement of many proteins in complex mixtures. Its application to the relative quantification of proteins in high-density lipoproteins (HDL) that have been purified from human plasma has revealed potential mechanisms to explain the atheroprotective effects of HDL. We describe a moderate throughput method for isolating HDL from human plasma that uses sequential density gradient ultracentrifugation, the traditional method of HDL purification, and subsequent trypsin digestion and nanoflow liquid chromatography-tandem mass spectrometry to quantify 38 proteins in the HDL fraction of human plasma. To control for the variability associated with digestion, matrix effects, and instrument performance, we normalize the signal from endogenous HDL protein-associated peptides liberated during trypsin digestion to the signal from peptides liberated from stable isotope-labeled apolipoprotein A-I spiked in as an internal standard prior to digestion. The method has good reproducibility and other desirable characteristics for preclinical research.
Access provided by CONRICYT – Journals CONACYT. Download protocol PDF
Similar content being viewed by others


Key words
- Multiple reaction monitoring
- Tandem mass spectrometry
- Nanoflow liquid chromatography
- Stable isotope-labeled internal standard protein
- Skyline
- High-density lipoprotein
- Protein quantification
- Quantitative proteomics
- Density gradient ultracentrifugation
1 Introduction
Elevated plasma concentrations of cholesterol bound to high-density lipoproteins (HDL-C) are associated with a reduced risk for developing cardiovascular disease, which has been well established in large epidemiological studies [1–3]. It has been proposed that HDL particles serve several anti-atherosclerotic functions, namely: reverse cholesterol transport from peripheral tissues, including from lipid plaques in the vascular intima of arteries [4, 5]; prevention or reduction of inflammation that mitigates endothelial cell activation [6, 7]; and prevention or reduction of the oxidation of low-density lipoproteins (LDL) [8, 9]. Analysis of the HDL proteome using shotgun mass spectrometry has demonstrated a complex heterogeneous composition of proteins of humoral and cellular origin with a vast array of functions [10–15]. The interplay of these proteins in the development of atherosclerosis and which proteins would serve as effective therapeutic targets is currently an intense area of research [14, 16]. Targeted mass spectrometry (MRM/SRM /PRM) has been demonstrated to be an effective method of quantifying apolipoproteins associated with HDL using stable isotope-labeled peptides and protein internal standards [17–19].
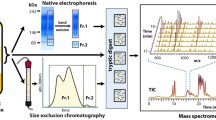
The protocol outlined here employs sequential potassium bromide density gradient ultracentrifugation to isolate the high-density lipoprotein (ρ = 1.063-1.210 g/mL) fraction of human plasma. Proteins are dialyzed to remove the potassium bromide and the protein concentration is determined using the Bradford method. Isolated HDL proteins are reduced, alkylated, and digested with trypsin. The resulting peptides are dried under vacuum and then reconstituted in an acidic slightly organic solvent for analysis using nanoflow liquid chromatography-tandem mass spectrometry. Peptides derived from the proteins of interest are identified by their retention time on the chromatographic column, their precursor mass (detected as the mass-to-charge ratio by the mass spectrometer), and the mass-to-charge ratio of specific fragments generated by collision-induced dissociation of the precursor peptide. Stable isotope-labeled internal standard protein (apolipoprotein A-I, apoA-I) spiked prior to digestion is used to control for digestion variability, matrix effects, and fluctuations in instrument performance. The chromatographic peak areas corresponding to the unlabeled endogenous peptides and the 15N-labeled apoA-I peptides are determined in the Skyline software package [20]. The relative abundance of each protein in HDL is then calculated as sum of the peak area of two to six of the most intense precursor-fragment pairs (i.e., transitions) of the endogenous peptide normalized by the peak area of one of the stable isotope-labeled internal standard peptides.
2 Materials
All buffers used for HDL isolation should be prepared with ultrapure deionized water and analytical grade reagents. All solvents used in MS analysis should be LC-MS grade purity. All reagents and solvents should be stored at room temperature unless otherwise noted. Waste reagents and solvents should be appropriately disposed of according to local regulations.
2.1 Plasma HDL Isolation
-
1.
Potassium bromide (KBr)-containing microcentrifuge tubes: Add 400 μL of 1.1863 g/mL (at 25 °C) KBr stock solution to 0.6 mL microcentrifuge tubes and dry down in vacuum centrifuge. Each microcentrifuge tube should contain 108.7 mg of dry KBr (see Note 1 ).
-
2.
Saline solution: 0.9 % (w/v) NaCl in H2O with 500 mM EDTA.
-
3.
Potassium phosphate/DTPA (K2HPO4/DTPA) buffer: 20 mM potassium phosphate, pH 7.4, 100 μM diethylene triamine pentaacetic acid (DTPA) in H2O.
-
4.
Potassium bromide (KBr) solution 1: density = 1.21 g/mL or 10.17 M in H2O (at 25 °C).
-
5.
Potassium bromide (KBr) solution 2: density = 1.063 g/mL or 8.93 M in H2O (at 25 °C).
-
6.
Dialysis cups: 3.5 kDa molecular weight cut-off (e.g., Thermo P/N 69552).
-
7.
Microcentrifuge tube float, 500 mL beaker, and stir plate at 5 °C.
-
8.
Ultracentrifuge with appropriate rotors and tubes (tube dimensions: 8 mm × 34 mm).
-
9.
Metal (aluminum or stainless steel) ultracentrifuge tube rack.
-
10.
Hamilton syringe with Chaney adapter to set volume at 125 μL.
-
11.
Microcentrifuge capable of 15k × g.
-
12.
Rocker table or similar agitation device.
2.2 Bradford Assay
-
1.
Coomassie Plus Protein Assay Reagent (Thermo Pierce). Store at 4 °C. Warm to RT before use. The solution is light sensitive and exposure must be kept to a minimum.
-
2.
Potassium phosphate/DTPA (K2HPO4/DTPA) buffer: 20 mM potassium phosphate, pH 7.4, 100 μM diethylene triamine pentaacetic acid (DTPA) in H2O. Can use solution prepared in Subheading 2.1, item 3.
-
3.
Bovine serum albumin (BSA) standard at 2 mg/mL in K2HPO4/DTPA buffer. Store at 4 °C. Warm to RT before use.
-
4.
Clear flat-bottom 96-well plate.
-
5.
Ultraviolet (UV) plate reader.
2.3 Stable Isotope-Labeled Proteins
-
1.
15N-labeled apoA-I internal standard protein (e.g., Cambridge Isotopes Laboratories, Tewksbury, MA).
2.4 Trypsin Digestion
-
1.
100 mM Ammonium bicarbonate buffer (AmBic): Dissolve 395.3 mg AmBic in 50 mL H2O. AmBic buffer should be prepared and used within 12 h.
-
2.
250 mM Dithiothreitol (DTT): Dissolve 50 mg of DTT in 648 mL of 100 mM AmBic. Prepare fresh solution every 12 h.
-
3.
0.2 % RapiGest SF Surfactant (Waters): Dissolve contents of 1 mg vial in 500 μL of 100 mM AmBic. Solution is stable for 1 week at 4 °C.
-
4.
RapiGest/DTT solution: Combine 675 μL of 0.2 % RapiGest and 27 μL of 250 mM DTT. Sufficient for 24 digestions.
-
5.
500 mM Iodoacetamide (IAA): Dissolve 50 mg of IAA in 541 μL of 100 mM AmBic. Note: IAA is extremely light sensitive, prepare just prior to use (≤2 min). Discard after use.
-
6.
1 mM Hydrochloric acid (HCl): Add 30 μL of 500 mM HCl stock to 15 mL of H2O to obtain a 1 mM HCl solution to dissolve lyophilized trypsin.
-
7.
0.05 μg/μL Trypsin: Just prior to digestion, add 400 μL of 1 mM HCl to 20 μg of lyophilized trypsin (e.g., Promega Gold sequencing grade trypsin). Trypsin should be dissolved by gently re-pipetting solution until dissolved. Store on ice until use. Do not vortex.
-
8.
All digestions should be performed in low protein binding microcentrifuge tubes, e.g., Eppendorf SafeLock LoBind 1.5 mL, in a Thermomixer that has been calibrated at 37 °C and speed of 1400 rpm.
-
9.
0.25 μg/μL 15N-labeled apoaA-I ISprot: Dissolve 250 μg of 15N-labeled apoaA-I in 1000 μL of 100 mM AmBic. Keep on wet ice. Aliquot as necessary and store at −20 °C.
-
10.
10 % FA in H2O (10 % FA): Add 455 μL of 88 % FA to a total volume of 4 mL in 15 mL conical tube using H2O (see Note 2 ).
-
11.
1 % FA in Acetonitrile (1 % FA/ACN): Add 250 μL of 88 % FA to a total volume of 22 mL in a 50 mL conical tube using acetonitrile (ACN) (see Note 2 ).
-
12.
Other equipment necessary for trypsin protein digestion includes: 1.5 mL low protein retention microcentrifuge tubes (e.g., Fisher Scientific P/N02-681-320), microcentrifuge capable of achieving 15k × g, speedvac, vortexer, phospholipid removal plate (e.g., Phenomenex Phree), and a positive pressure manifold, e.g., Biotage Pressure +96. The methods outlined here have been optimized for Phenomenex Phree 96-well plates.
2.5 Nano-LC-MRM-MS Solvents, Mobile Phases, and Columns
-
1.
Sample suspension solvent: 95 % H2O, 5 % acetonitrile, 0.1 % formic acid (FA).
-
2.
Sample dilution solvent: 0.1 % FA in H2O.
-
3.
Mobile phase A: 98:1 H2O to acetonitrile with 0.1 % FA. Mobile phase B: 98:1 acetonitrile to H2O with 0.1 % FA.
-
4.
Weak needle wash: H2O with 0.1 % FA. Strong needle wash: 2-propanol with 0.1 % FA.
-
5.
Trap column: XBridge BEH C18, 5 μm, 100 Å silica beads (Waters, MA) (or equivalent C18 packing material) are packed into a 0.1 mm i.d. fritted fused silica capillary (New Objective, MA) (or equivalent) to 30 × 0.1 mm column and fused silica capillary is cut to length of ~15 cm.
-
6.
Analytical column: XBridge BEH C18, 3.5 μm, 100 Å silica beads (Waters, MA) (or equivalent C18 packing material) are packed into a pulled tip fused silica capillary (75 μm i.d.) to 120 length.
-
7.
Capillary column oven capable of maintaining temperature of 50 °C for analytical column.
3 Methods
All procedures should be performed at room temperature unless otherwise noted. When handling blood products, always take universal precautions such as appropriate PPE and proper use of microcentrifuges to minimize aerosols.
3.1 HDL Isolation from Human Plasma
-
1.
Pre-chill ultracentrifuge and rotors to 5 °C.
-
2.
If necessary, rapidly thaw plasma at 37 °C, then briefly centrifuge (≤30 s at ~2000 × g). Place plasma on wet ice.
-
3.
Add 335 μL of plasma to microcentrifuge tubes containing 108.9 mg KBr, then place on rocker table to dissolve KBr. Briefly centrifuge and place tubes on wet ice.
-
4.
Add 350 μL of plasma/KBr solution from step 3 to ultracentrifuge tube using forward pipetting. Place tubes in metal rack (on wet ice) to keep the tubes chilled when not in the centrifuge.
-
5.
Add 150 μL of KBr solution 1 (ρ = 1.21 g/mL at 25 °C) to ultracentrifuge using reverse pipetting.
-
6.
Place ultracentrifuge tubes into chilled rotor. Spin samples at 120k rpm (625,698 × g) for 4.5 h at 5 °C.
-
7.
Remove 120 μL from meniscus. Dispense into the bottom of a new ultracentrifuge tube and rinse 1 time (see Note 3 ).
-
8.
Add 239 μL saline solution to ultracentrifuge tube (see Note 4 ).
-
9.
Add 140.8 μL KBr solution 2 (ρ = 1.063 g/mL at 25 °C) to the ultracentrifuge tube.
-
10.
Place ultracentrifuge tube into chilled rotor. Spin samples at 120k rpm (625,698 × g) for 2.5 h at 5 °C.
-
11.
Using the Hamilton syringe with Chaney adapter set to 125 μL, carefully remove 125 μL from the bottom of the ultracentrifuge tube, wipe the tip to remove the lipids that became adhered from passing through the meniscus, and dispense into 0.6 mL microcentrifuge tube. Place samples on wet ice (~5 °C).
3.2 Dialysis of Isolated HDL
-
1.
Test dialysis cups for leaks by placing a tube float with empty dialysis cups in beaker containing K2HPO4/DTPA buffer for 10 min at 5 °C with gentle stirring. Check for signs of leakage. No liquid should be in the dialysis cup when the membrane is intact, although the membrane will be wet.
-
2.
Remove float with dialysis cups from beaker. Dispense 125 μL of HDL from step 11 above onto the dialysis membrane. Carefully place the float back in the beaker and incubate for 3 h with stirring at 5 °C. Buffer should be changed three times during dialysis for a total of three, 3-h incubations.
-
3.
Blot outer portion of dialysis membrane on a clean paper towel to remove excess dialysis buffer. Place the dialysis cup in a labeled 1.5 mL microcentrifuge tube, briefly spin to maximum rpm (~15k × g) to rupture membranes and transfer dialyzed HDL to microcentrifuge tube.
-
4.
Place tubes on wet ice (~5 °C). Once the protein concentration has been determined, the samples can be aliquoted as necessary.
3.3 Bradford Assay to Determine Protein Concentration of Isolated HDL
-
1.
Dilute standard to make a 7-point calibration curve from 1000 to 15 μg/mL by making a 1:1 mixture of 2 mg/mL BSA standard with K2HPO4/DTPA buffer (1000 μg/mL) and serial dilute (twofold) to 15.6 μg/mL.
-
2.
In a 0.6 mL tube containing 27 μL of the K2HPO4/DTPA buffer (1:10 dilution), add 3 μL of the dialyzed HDL from Subheading 3.2, step 4.
-
3.
In duplicate, pipette 10 μL of standard or sample into the appropriate wells of the clear, flat-bottom 96-well plate. Do not allow bubbles to form in well when pipetting.
-
4.
In each well, add 150 μL of the Coomassie Plus Protein assay reagent to each well. Again, do not introduce bubbles into the well.
-
5.
Gently agitate the plate for 1 min and incubate for a total of 10 min (see Note 5 ).
-
6.
Read plate at λ = 595 nm.
-
7.
Once the protein concentrations of the HDL samples have been determined (be sure to account for 1:10 dilution), prepare aliquots containing 5 μg HDL protein in Eppendorf LoBind SafeLock Tubes.
3.4 Digestion of Isolated HDL Using Trypsin
-
1.
To each 5 μg HDL protein aliquot, add 4 μL of the 0.25 μg/μL 15N-labeled apoA-I ISprot working stock solution.
-
2.
Denaturation and reduction of proteins is performed by adding 26 μL of RapiGest/DTT solution, vortex briefly and incubate for 1 h in Thermomixer at 37 °C and 1400 rpm. Then briefly centrifuge to collect condensation.
-
3.
Prepare 500 mM IAA stock solution (see Subheading 2.4, item 5). Add 1.5 μL of the 500 mM IAA stock solution (C final = 15 mM IAA) to each sample. Vortex briefly, then incubate in the dark for 15 min. During the last 5 min of the incubation, prepare the trypsin working stock as described above (see Subheading 2.4, item 7).
-
4.
Add 5 μL of 0.05 μg/μL trypsin solution to each sample and incubate on Thermomixer at 37 °C and 1400 rpm for 3 h. Be sure to place trypsin back on ice.
-
5.
Briefly vortex HDL digests to collect condensate. Then add another 5 μL of 0.05 μg/μL trypsin to each digestion and incubate for 17 h on Thermomixer at 37 °C and 1400 rpm.
-
6.
After incubation, briefly vortex digests to collect condensate. Then add 62.4 μL of 10 % FA solution to cleave RapiGest and stop digestion. Incubate for 45 min in Thermomixer at 37 °C and 1400 rpm.
-
7.
Centrifuge samples at ≥15k × g for 15 min. Then verify order of samples in regard to orientation on the SPE plate.
-
8.
Add 390 μL of 1 % FA/ACN to each well. Then add 110 μL of HDL digest to each well. Seal plate with clear sealing tape and place on plate shaker for 5 min.
-
9.
Orient SPE plate over collection plate, remove plate seal tape, and mount plates on positive pressure displacement manifold.
-
10.
To collect eluent, set initial pressure at 4–5 psi for 5 min. Then increase pressure to 7–8 psi and hold for 5 min. Finally, increase pressure to 10–11 psi for 5 min.
-
11.
Transfer eluent to 1.5 mL low protein binding microcentrifuge tubes and speed vac without heat (~RT) to dryness; approximately 12 h.
-
12.
Store dried samples at −80 °C.
3.5 Nano-LC-MRM-MS Analysis of Tryptic Peptides
An example configuration of the nano-LC-MRM-MS system is a nanoACQUITY UPLC (Waters) coupled with TSQ Vantage (Thermo Scientific). However, the method can be adapted to other triple-quadrupole mass spectrometers. Alternatively, the method could also be used in the form of parallel reaction monitoring (PRM) on hybrid instruments (e.g., quadrupole time-of-flight or quadrupole-Orbitrap). Skyline could facilitate the transfer of methods described here to another instrument in a vendor-blind manner (see below). Capillary column and trap column are maintained at the temperature of 50 °C by means of a column heater.
3.5.1 Sample Suspension for Nano-LC-MRM Analysis
-
1.
Remove samples from −80 °C and allow them to warm to room temperature.
-
2.
Centrifuge samples at ≥15k × g for 5 min to ensure peptide pellet is in the bottom of the tube.
-
3.
Add 15 μL of 95:5 H2O/ACN with 0.1 % FA sample suspension solvent to each sample and mix at 1400 rpm for 2 h at RT in a Thermomixer.
-
4.
Then add 15 μL of 0.1 % FA dilution solvent to each sample and briefly vortex.
-
5.
Centrifuge samples at ≥15k × g for 15 min, then transfer to compatible vials for the auto-sampler coupled to the nano-LC unit. Ensure that no bubbles are present in the solution.
3.5.2 Chromatographic Conditions
-
1.
Injected sample is trapped and washed on the trapping column for 5 min with Mobile phase A at the flow rate 4 μL/min.
-
2.
Peptides are eluted from the trap column onto the analytical column and separated by the gradient detailed in Table 1.
Table 1 Chromatography schedule and flow rate for nano-LC-MRM analysis of HDL peptides
3.5.3 Mass Spectrometer Acquisition Conditions
-
Capillary temp = 325 °C.
-
Spray voltage = 2200 V.
-
Q1 peak width = 0.7.
-
Q3 peak width = 0.7.
-
Collision gas: 1.5 mTorr.
-
Collision energy: Calculated in Skyline.
-
Dwell time: 10 ms (except selected low abundance peptides where dwell set to 30 ms).
3.6 Develop MRM Transition Lists and Perform Data Analysis Using Skyline
Download and install Skyline from MacCoss laboratory Skyline webpage at: https://skyline.gs.washington.edu/labkey/project/home/software/Skyline/begin.view (see Note 6 ).
3.6.1 Skyline Settings for MRM Experiment
-
1.
Under the Settings option, open the Peptide Settings window. Choose the Digestion tab and in the Enzyme dropdown menu, select Trypsin [KR|P]. Select 0 in the Max missed cleavages dropdown menu. The Background proteome should be set to none.
-
2.
In the Filter tab, set the minimum length to 6 and maximum length to 25. Auto-select all matching peptides should be selected.
-
3.
Under the Modifications tab, in the Structural modifications box, select Carbamidomethyl Cysteine and Oxidation (M). Then select OK.
-
4.
Open Transition Settings and click on the Full-Scan tab. In the MS1 Filtering box, choose none from the Isotope peaks included dropdown menu. Below, in the MS/MS filtering box, choose none from the Acquisition method dropdown menu.
3.6.2 Preparing the Transition List
-
1.
Copy and paste each peptide from Table 2 Skyline file Targets list on the left side of the window. Alternatively, import FASTA files for proteins being analyzed and cull the list of peptides to those listed in Table 2 by right-clicking on the protein name and selecting Pick Children (see Note 7 ).
Table 2 List of HDL proteins, peptides, and corresponding transitions -
2.
For each apoA-I peptide that has a stable isotope-labeled internal standard counterpart, include the modified residues in the peptide by right-clicking on the precursor m/z and select Modify. In the Edit Modifications window, in the Isotope heavy column, select the dropdown menu that corresponds to each peptide and choose Label: 15N. Then select OK.
-
3.
Select the optimized transitions for each peptide from Table 2. When selecting transitions for peptides that include an internal standard, be sure to check the box that synchronizes selection for the stable isotope-labeled peptide (see Note 8 ).
3.6.3 Export Transition List
-
1.
From the menu bar, navigate to Export then choose Transition List. In the Export Transition List window, select the instrument from the Instrument type dropdown menu. Select Single Method with no optimization and a standard method type.
-
2.
Import transition list directly into MS method software. Verify that all precursor ion, transition ion, declustering potentials, collision energy, and any other necessary method parameters required by the instrument were correctly imported into MS method (see Note 9 ).
3.6.4 Analyzing MRM Data in Skyline
-
1.
Open the Skyline file that was used to prepare the transition list and save it as an analysis file specifically for these MS data and this experiment (see Note 10 ).
-
2.
To import MS data into Skyline, from the menu bar, navigate to Import then choose Results. In the Import Results window, select Add single-injection replicates in files and none from the Optimizing dropdown menu. Then select OK.
-
3.
Navigate to the data file containing the MS data to be analyzed. Select one or more MS spectra files for import into Skyline and select Open. Save the Skyline document after the spectral data has been imported (see Note 11 ).
-
4.
Verify that the transitions assigned to each peptide by Skyline are correct. Adjust integration limits for the transition peak areas as necessary. Export the results in .csv file format for analysis in Microsoft Excel (or similar software) under File in the menu bar, then navigate to Export and select Report. In the Export Report window, select Transition Results, then Export. Name the file and select Save.
4 Notes
-
1.
KBr stock solution at a density of 1.1863 g/mL is prepared by completely dissolving 124 g KBr in 400 mL of H2O. Intermittent heat may be required to fully dissolve KBr. Allow temperature of KBr solution equilibrate to 25 °C, and then adjust density down to 1.1863 g/mL using water at 25 °C. Aliquot 400 μL of KBr stock solution into 0.6 mL microcentrifuge tubes. Dry down using vacuum centrifugation with heat at ~80 °C for 40 min with a total spin time of 3 h. Cap tubes immediately upon drying. We generally prepare 100 tubes per batch. To determine the variability of the amount of KBr added to the tubes, number and tare 10 of the tubes prior to adding KBr stock solution and weigh each tube again after drying down. Each tube should contain 108.7 mg of KBr. The variability of the amount of KBr added is typically <0.5 % CV.
-
2.
Solutions are prepared by adding the total volume of H2O (4.0 mL) or acetonitrile (22.0 mL) to the tube, then removing the volume of FA, 455 and 250 μL respectively, from the tube and discarding. Then add back the required volume of 88 % FA to H2O or acetonitrile and mix well. For example, to make the 10 % FA solution, add 4.00 mL of H2O to the tube. Then, using a micropipettor, remove 455 μL of H2O and discard. Add back 455 μL of 88 % FA to the tube and mix well.
-
3.
Dispense the 120 μL of lipoproteins into a 0.6 mL tube. Re-aspirate and dispense a second time to rinse the walls of the tip. Discard tip after second dispense and use a fresh tip for the next sample.
-
4.
Dispense 239 μL normal saline near the top inside of the ultracentrifuge tube to rinse the lipoproteins down off the side.
-
5.
The Bradford reaction develops at different rates for BSA and for HDL proteins. Maintaining constant incubation times is vital for reproducible results between batches.
-
6.
Detailed Skyline tutorials that outline MRM method development and other features of the software not included in this section can be downloaded from the MacCoss laboratory website at: https://skyline.gs.washington.edu/labkey/wiki/home/software/Skyline/page.view?name=tutorials.
-
7.
The peptides and transitions in Table 2 represent proteotypic peptides for these 38 proteins. Proteotypic in this context means the peptide sequence has been determined to be unique in the human proteome (i.e., no splice isoforms, polymorphisms, or isobaric peptides being reported) and that it is readily detected using tandem MS. Furthermore, these peptide sequences: (1) do not contain methionine and (2) the peak area ratios of peptides from the same protein are highly correlated across a population of human samples.
-
8.
If spectral libraries obtained from DDA data or another source are being used to aid in MS peak identification, please visit the Skyline software tutorial page (see Note 5 ) to find detailed tutorials that outline how to implement spectral libraries in Skyline.
-
9.
Retention time scheduling for precursor ions can be added to the transition list .csv file in Microsoft Excel (or similar software) after exporting. Scheduled methods can be exported from Skyline using iRT retention time prediction. See Note 5 .
-
10.
Using a detailed file name that includes the acquisition data and pertinent information regarding the samples analyzed will make it easier to locate the Skyline data analysis file in the future.
-
11.
If the MS file names have a common prefix, an Import Results pop-up window will appear indicating the common prefix and the option to retain or remove the prefix. Selecting to remove the prefix will not affect the original file name.
References
Castelli WP, Anderson K, Wilson PWF et al (1992) Lipids and risk of coronary heart disease. The Framingham Study. Ann Epidemiol 2:23–28
Mahmood SS, Levy D, Vasan RS et al (2014) The Framingham Heart Study and the epidemiology of cardiovascular disease: a historical perspective. Lancet 383:999–1008
Boden WE (2000) High-density lipoprotein cholesterol as an independent risk factor in cardiovascular disease: assessing the data from Framingham to the veterans affairs high-density lipoprotein intervention trial. Am J Cardiol 86:19–22
Khera AV, Cuchel M, de la Llera-Moya M et al (2011) Cholesterol efflux capacity, high-density lipoprotein function, and atherosclerosis. N Engl J Med 364:127–135
Kontush A (2014) HDL-mediated mechanisms of protection in cardiovascular disease. Cardiovasc Res 103:341–349
Cheng AM, Rizzo-DeLeon N, Wilson CL et al (2014) Vasodilator-stimulated phosphoprotein protects against vascular inflammation and insulin resistance. Am J Physiol Endocrinol Metab 307:E571–E579
Cheng AM, Handa P, Tateya S et al (2012) Apolipoprotein A-I attenuates palmitate-mediated NF-κB activation by reducing toll-like receptor-4 recruitment into lipid rafts. PLoS One 7, e33917
Parthasarathy S, Raghavamenon A, Garelnabi M et al (2010) Oxidized low-density lipoprotein. In: Uppu RM, Murthy SN, Pryor WA et al (eds) Free radicals and antioxidant protocols SE-24. Humana Press, New York, pp 403–417
Navab M, Berliner JA, Subbanagounder G et al (2001) HDL and the inflammatory response induced by LDL-derived oxidized phospholipids. Arterioscler Thromb Vasc Biol 21:481–488
Karlsson H, Leanderson P, Tagesson C et al (2005) Lipoproteomics II: Mapping of proteins in high-density lipoprotein using two-dimensional gel electrophoresis and mass spectrometry. Proteomics 5:1431–1445
Vaisar T, Pennathur S, Green PS et al (2007) Shotgun proteomics implicates protease inhibition and complement activation in the antiinflammatory properties of HDL. J Clin Invest 117:746–756
Davidson WS, Silva RAGD, Chantepie S et al (2009) Proteomic analysis of defined HDL subpopulations reveals particle-specific protein clusters: relevance to antioxidative function. Arterioscler Thromb Vasc Biol 29:870–876
Gordon SM, Deng J, Lu LJ et al (2010) Proteomic characterization of human plasma high density lipoprotein fractionated by gel filtration chromatography. J Proteome Res 9:5239–5249
Hoofnagle AN, Heinecke JW (2009) Lipoproteomics: using mass spectrometry-based proteomics to explore the assembly, structure, and function of lipoproteins. J Lipid Res 50:1967–1975
Shah AS, Tan L, Long JL et al (2013) Proteomic diversity of high density lipoproteins: our emerging understanding of its importance in lipid transport and beyond. J Lipid Res 54:2575–2585
Birner-Gruenberger R, Schittmayer M, Holzer M et al (2014) Understanding high-density lipoprotein function in disease: recent advances in proteomics unravel the complexity of its composition and biology. Prog Lipid Res 56C:36–46
Agger SA, Marney LC, Hoofnagle AN (2010) Simultaneous quantification of apolipoprotein A-I and apolipoprotein B by liquid-chromatography-multiple-reaction-monitoring mass spectrometry. Clin Chem 56:1804–1813
Hoofnagle AN, Becker JO, Oda MN et al (2012) Multiple-reaction monitoring-mass spectrometric assays can accurately measure the relative protein abundance in complex mixtures. Clin Chem 58:777–781
Ronsein GE, Pamir N, von Haller PD et al (2015) Parallel reaction monitoring (PRM) and selected reaction monitoring (SRM) exhibit comparable linearity, dynamic range and precision for targeted quantitative HDL proteomics. J Proteomics 113:388–399
MacLean B, Tomazela DM, Shulman N et al (2010) Skyline: an open source document editor for creating and analyzing targeted proteomics experiments. Bioinformatics 26:966–968
Acknowledgements
This work was supported by NIH grants: HL111375, DK035816, HL089504 and NIH training grant T32HL007028 and AHA grants 0830231N and 14GRNT18410022. We thank Jennifer Wallace for her contributions to this chapter.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2016 Springer Science+Business Media New York
About this protocol
Cite this protocol
Henderson, C.M., Vaisar, T., Hoofnagle, A.N. (2016). Isolating and Quantifying Plasma HDL Proteins by Sequential Density Gradient Ultracentrifugation and Targeted Proteomics. In: Sechi, S. (eds) Quantitative Proteomics by Mass Spectrometry. Methods in Molecular Biology, vol 1410. Humana Press, New York, NY. https://doi.org/10.1007/978-1-4939-3524-6_7
Download citation
DOI: https://doi.org/10.1007/978-1-4939-3524-6_7
Published:
Publisher Name: Humana Press, New York, NY
Print ISBN: 978-1-4939-3522-2
Online ISBN: 978-1-4939-3524-6
eBook Packages: Springer Protocols