
Abstract
Many researchers use the social amoeba Dictyostelium discoideum as a model organism to study various aspects of the eukaryotic cell chemotaxis. Traditionally, Dictyostelium chemotaxis is considered to be driven mainly by branched F-actin polymerization. However, recently it has become evident that Dictyostelium, as well as many other eukaryotic cells, can also employ intracellular hydrostatic pressure to generate force for migration. This process results in the projection of hemispherical plasma membrane protrusions, called blebs, that can be controlled by chemotactic signaling.
Here we describe two methods to study chemotactic blebbing in Dictyostelium cells and to analyze the intensity of the blebbing response in various strains and under different conditions. The first of these methods—the cyclic-AMP shock assay—allows one to quantify the global blebbing response of cells to a uniform chemoattractant stimulation. The second one—the under-agarose migration assay—induces directional blebbing in cells moving in a gradient of chemoattractant. In this assay, the cells can be switched from a predominantly F-actin-driven mode of motility to a bleb-driven chemotaxis, allowing one to compare the efficiency of both modes and explore the molecular machinery controlling chemotactic blebbing.
Access provided by CONRICYT – Journals CONACYT. Download protocol PDF
Similar content being viewed by others



Key words
1 Introduction
Researchers in the field of eukaryotic cell chemotaxis have developed multiple experimental techniques to study the efficiency and molecular mechanisms of cellular gradient sensing , polarization , and motility . Most of these approaches are based on the use of devices where a steady-state gradient of chemoattractant is created by its diffusion through the liquid bridge connecting two reservoirs with a buffer containing different concentrations of a chemoattractant (Zigmond chamber [1, 2], Dunn chamber [3, 4], Insall chamber [5]). The cells are normally placed on the surface of a cover slip representing the bottom of this bridge and are therefore covered with a buffer. Although this system has proven itself as a powerful tool to study certain aspects of chemotaxis, in nature cells rarely happen to move through liquid environments and on 2D surfaces. More often, they migrate through resistive 3D structures—such as tissues, extracellular matrix, or soil. It has become evident in the last decade that the principles of cell migration , and especially the nature of the forces driving cell locomotion, differ dramatically between these two modes of migration [6–8]. Cells under a liquid buffer normally generate motile force by branched actin polymerization at the cell front, while in resistive environments intracellular hydrostatic pressure, which pushes the membrane forward in the form of blebs , becomes the main motor of cell motility . The set of techniques for studying three-dimensional chemotaxis has been limited so far mainly to transwell assays where the efficiency of 3D chemotaxis is evaluated by the number of cells that traverse the porous membrane separating two wells containing a buffer with different chemoattractant concentrations (Boyden chamber assay [9, 10], Transwell® invasion assays [11]). However, in these assays one cannot precisely observe cellular events accompanying the migration. In the other category of experiments, researchers observe the cells moving in vivo within natural three-dimensional tissues, but in this case they cannot control the chemoattractant gradients faced by the cells, which can also readily move out of the plane of focus.
Here we describe a method allowing the precise observation of Dictyostelium cells moving through a resistive environment and employing mainly the bleb-driven mode of migration . In this method the cells are attracted under a stiff agarose overlay by a cyclic-AMP gradient, which is formed across the agarose gel by diffusion from a cyclic-AMP-filled trough. Initially, an under-agarose migration assay was described by the Knecht group as a method to flatten the vegetative Dictyostelium cells for a better quality of microscopy [12, 13]. Thus, when applied for the developed Dictyostelium cells, this method not only switches them to the blebbing mode of migration but also allows the observation of the accompanying events with a high spatiotemporal resolution [14].
Since F-actin polymerization has been long considered the basic mechanism driving the chemotactic response , many researchers measured F-actin polymerization dynamics after a uniform saturating chemoattractant stimulation of cells [15, 16]. But given that blebbing is the other and similarly important component of the chemotactic response, there is a need to measure the blebbing response s as well. Here we describe a method for evaluating the levels of cellular blebbing induced by chemoattractant stimulation in Dictyostelium . By evaluating the number and dynamics of blebs in this “cyclic-AMP shock assay ” one can compare the blebbing responses in different conditions and between different strains of Dictyostelium, thus examining the environmental and molecular factors that control blebbing [14, 17].
2 Materials
-
1.
Axenic medium: HL5 plus glucose medium (Formedium), 200 μg/ml dihydrostreptomycin. Sterile.
-
2.
KK2 buffer: 16.5 mM KH2PO4, 3.9 mM K2HPO4, 2 mM MgSO4, 0.1 mM CaCl2, pH 6.1. If sterile, add magnesium and calcium after autoclaving to avoid precipitation.
-
3.
Cyclic-AMP solutions in KK2: 9 μM for cell pulsing, 15 μM for cyclic-AMP shock assay , and 4 μM for under-agarose chemotaxis . A 100 mM cyclic-AMP stock solution can be prepared from free acid by neutralization with KOH.
-
4.
Low-melting-point ultrapure agarose.
-
5.
Two- and eight-well chambered slides (Nunc Lab-Tek, Thermo Scientific), well area—4.0 and 0.7 cm2, respectively.
-
6.
Inverted microscope with a 40× or 63× objective lens.
3 Methods
3.1 Preparation of Developed Dictyostelium Cells
-
1.
Grow Dictyostelium cells in a shaking suspension at 22 °C in axenic medium to a density of 1–5 × 106 cells/ml (see Note 1 ). For this, we recommend using conical flasks and shaking the suspension at 180 rpm. Alternatively, the cells can be grown in association with bacteria on nutrient agar plates (see Note 2 ).
-
2.
Transfer the cell suspension into a 50 ml Falcon tube and harvest the cells by centrifugation in a bench-top centrifuge at 1200 rpm (300 × g), 2 min, room temperature.
-
3.
Discard the supernatant and wash the cells three times by resuspending them in KK2 and centrifuging at 1200 rpm (300 × g), 2 min. Every time discard the supernatant and resuspend the cells in fresh KK2.
-
4.
After the last wash, discard the supernatant, resuspend the cells in KK2, and count the cell density using a cell counter or a hemocytometer.
-
5.
Pellet the cells by centrifugation, discard the supernatant, and resuspend the cells in KK2 at 2 × 107 cells/ml.
-
6.
Transfer 10 ml of the cell suspension to a 50 ml conical flask (flask volume of at least three times the volume of the cell suspension is required for proper aeration).
-
7.
Place the flask with the cell suspension on a shaker at 180 rpm and incubate for 1 h at 22 °C (the cell starvation step).
-
8.
After 1-h starvation , start stimulating the cells with pulses of cyclic-AMP (using an automated pump) to drive developmental gene expression: every 6 min inject 100 μl of a 9 μM cyclic-AMP solution into the flask to produce a final concentration of 60–90 nM after each pulse (see Note 3 ). To get a good blebbing response , pulse the cells with cyclic-AMP for 4.5–5.5 h (see Note 4 ). After this time, small clumps of cells can be observed sticking to the walls of the flasks, giving a morphological check for adequate development.
-
9.
Pellet the developed cells by centrifugation (to get rid of cyclic-AMP present in the suspension and other secreted metabolites), and resuspend them in fresh KK2.
3.2 Uniform Chemoattractant Stimulation (“Cyclic-AMP Shock” Assay)
-
1.
Dilute the suspension of developed Dictyostelium cells to a final density of about 1 × 106 cells/ml (in KK2 buffer).
-
2.
Plate the cells in an eight-well chambered slide by placing 210 μl of the diluted cell suspension per well. This gives a cell density of ~2 × 105 cells per well. Let the cells settle for about 15 min, and move the slides under the microscope.
-
3.
Start time-lapse imaging of the cells in a particular well of the slide (see Note 5 ), and then gently add dropwise 15 μl of a 15 μM cyclic-AMP solution to the well to provide a final concentration of 1 μM (if the cells respond poorly to this stimulation, try increasing the final concentration of cyclic-AMP to 2–4 μM). Image the cells for the following 60–100 s. The time of cyclic-AMP addition is apparent as a disturbance in the image, and normally, intensive blebbing follows 10–50 s later (Fig. 1).
Fig. 1 
Blebbing response of Dictyostelium cells to a uniform cyclic-AMP stimulation (cyclic-AMP shock). (a) DIC images of a group of cells before and after the cyclic-AMP stimulation (cyclic-AMP is added immediately after the “0 sec” time point). (b) Two sample cells shown at higher magnification to illustrate the appearance of blebs : upper panels—DIC images, lower panels—confocal images of the ABD-GFP fluorescent reporter for F-actin [19]. The images were collected using Zeiss 710 laser scanning confocal microscope with a 63× oil emersion objective. For the experiment, Ax2 strain Dictyostelium discoideum cells were developed for 6 h and stimulated with 1 μM cyclic-AMP. Scale bars—5 μm
-
4.
To characterize and compare the blebbing response in different strains (or under different conditions), analyze the time-lapse movies and extract such quantitative characteristics of the response as the percentage of cells that demonstrate blebbing, average number of blebs per cell, or duration of blebbing.

3.3 Under-Agarose Chemotaxis Assay
-
1.
Preheat a two-well chambered slide to 50–60 °C by placing it on a heated metal thermoblock. This prevents the slide from cracking when the melted agarose is poured in.
-
2.
Carefully heat a 0.5–2 % suspension of low-melting-point ultrapure agarose in KK2 (see Note 6 ). During heating, mix it regularly until the agarose melts completely, and the solution becomes transparent and homogeneous. Optionally: add a fluorescent dye into the melted agarose (e.g., 0.5 mg/ml rhodamine B isothiocyanate-dextran) to negatively stain the cells. Use dyes that do not easily diffuse through or bind to the plasma membrane, or get endocytosed by the cell.
-
3.
Let the agarose cool down to ~50–60 °C and then pour a thin layer of melted agarose into each preheated two-well chambered slide. Use 800 μl of agarose per well to get a ~2 mm layer (see Note 7 ). Hereinafter keep the slides in a moist chamber (a closed box with a wet tissue in it) to prevent the agarose drying.
-
4.
When the agarose stiffens, gently cut rectangular troughs in it using a sharp scalpel (see Note 8 for important details). We normally cut three parallel troughs (~1.5 × 8 mm2 in size), about 5 mm apart, fill the middle one with chemoattractant , and put two different strains of Dictyostelium that we would like to compare in the side ones (Fig. 2). But if only one type of cell is to be analyzed two troughs will suffice.
Fig. 2 
Scheme of a two-well microscopy chambered slide prepared for an under-agarose chemotaxis experiment: upper image—top view, lower image—vertical cross section. Agarose is poured into the chambers, and then, after solidification, parallel troughs are cut in the agarose and filled either with 4 μM cyclic-AMP (central troughs) or cell suspension (side troughs)
-
5.
Fill one of the troughs (the central one if you have three troughs, or any one if you have two) with 4 μM cyclic-AMP. Fill the remaining one(s) with the developed Dictyostelium cells suspended in KK2 (~105 cells per trough).
-
6.
Wait for 30–40 min before starting imaging the cells. During this time the cyclic-AMP gradient will form in the agarose layer, the cells will settle on the slide, and start migrating under the agarose along this gradient.
-
7.
Start time-lapse imaging of the cells migrating under the agarose. For acquisition, use an inverted microscope with a sufficient temporal and spatial resolution to allow bleb detection (see Note 5 ). Make sure that the agarose gel does not dry during imaging (otherwise, this will not only interfere with the imaging because of the changes in optical properties, but also, when drying, the gel may squash the cells underneath and thereby affect their appearance and behavior). If necessary, add a few drops of KK2 into the troughs and onto the surface of the agarose gel to keep it moist. Figure 3 shows an example of a Dictyostelium cell migrating under the agarose in the blebbing mode.
Fig. 3 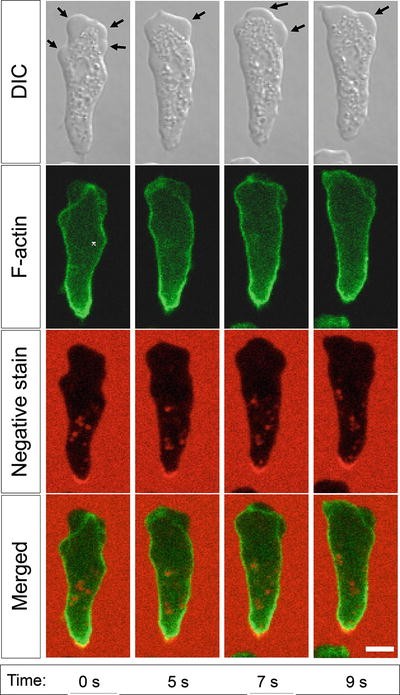
Blebs at the front edge of a Dictyostelium cell migrating under the agarose. ABD-GFP is used as a reporter for F-actin [19], and 0.5 mg/ml rhodamine B isothiocyanate-dextran is added into the agarose as a fluorescent dye for negative staining. The images were collected using Zeiss 780 laser scanning confocal microscope with a 63× oil emersion objective. For the experiment, Ax2 strain Dictyostelium discoideum cells were developed for 6 h and chemoattracted under 0.7 % agarose overlay. Scale bar—5 μm
-
8.
Analyze the movies of the cells chemotaxing under agarose to measure their overall blebbing activity, localization of the blebs on the cell surface, as well as speed and directionality of migration . When analyzing the high spatiotemporal resolution movies, it is important to use software optimized for accurate cell segmentation and tracking of small and short-lived cellular protrusions (for example, we use electrostatic contour migration-based program QuimP11 [18]).


4 Notes
-
1.
We recommend using at least 2 × 108 cells per experiment; otherwise the cell suspension gets too dilute during the cyclic-AMP pulsing step. Such a number of cells can be obtained from approximately 50 ml of cell suspension (grown to ~ 4 × 106 cells/ml), or from two clearing bacterial plates (see Note 2 ).
-
2.
Working stocks of Dictyostelium should be renewed from spores or frozen stocks every month to prevent genetic drift. If necessary (e.g., when using a strain of Dictyostelium that does not grow well in axenic medium), Dictyostelium cells can be grown on nutrient agar plates in association with an overnight culture of Klebsiella aerogenes bacteria—on so-called clearing plates. To set up a clearing plate, mix Dictyostelium cells with a bacterial suspension and plate them out together by spreading on SM agar plates (1.5 % agar with 1 % peptone, 0.1 % yeast extract, 0.22 % KH2PO4, 0.1 % Na2HPO4, 1 % glucose, 8 mM MgSO4). The bacteria initially outgrow the Dictyostelium amoebae and form a lawn but then they exhaust their nutrient and are consumed by the amoebae. When this happens the plate “clears” (goes translucent). Experiments are normally done with cells from plates that have started to look translucent, or have “half-cleared.” The initial Dictyostelium inoculum is adjusted so that this happens 42–48 h after plating (for example, for the Dictyostelium Ax2 strain, 2 × 105 cells are mixed with 200 μl of the overnight bacterial culture and evenly spread on an SM agar plate). The plates should be of a dryness such that all surface liquid dries after no more than about 1 h; excess liquid causes poor growth. Harvest the cells from clearing plates by scraping and resuspend in KK2 buffer. Repeatedly wash them with KK2 by low-speed centrifugation as above.
-
3.
If the cells are known or suspected to be defective in cyclic-AMP relay, use 400 nM cyclic-AMP pulses (final concentration after each pulse), instead of 70–90 nM.
-
4.
During development the cells become increasingly blebby [14]; therefore, slightly longer cyclic-AMP pulsing times are recommended for blebbing experiments, compared to traditional chemotaxis studies. However, with longer pulsing, the cells tend to become stickier and aggregate into large clumps, which makes the observation of individual cells more challenging. We therefore do not recommend pulsing the cells for more than 5.5 h.
-
5.
For time-lapse imaging of blebbing we recommend using an inverted microscope with a 40× or 63× objective lens and a frame rate of 0.5–2 frames per second, as the blebs are characterized by a ~1 μm size and a rapid expansion (the whole growth phase takes less than a second) [14].
-
6.
As the agarose concentration increases from 0.5 to 2 %, the Dictyostelium cells switch gradually from the hybrid mode of migration (blebbing + F-actin -driven pseudopodia ) to a predominantly bleb-driven motility [14].
-
7.
We normally use a ~2 mm layer of agarose because thicker layers absorb more laser light in the microscopy experiments and also may prevent oxygen diffusion to the cells that move underneath. At the same time, thinner agarose layers are harder to work with, especially, when cutting the wells (in particular, for lower agarose concentrations).
-
8.
Proper cutting of the troughs in the agarose gel is a critical step in this protocol. One must be careful not to scratch the bottom of the slide when making the cuts, as this may abolish cell migration . It is also important not to detach the gel from the slide’s bottom when removing the pieces of agarose from the troughs. Otherwise, the cells will easily migrate through the gap between the agarose and the bottom in the areas of detachment without meeting a sufficient resistance, and therefore without switching to the blebbing mode of migration . We normally use a curved scalpel blade to cut the long sides of the rectangular troughs with a rocking motion, and an elongated triangular one to cut the short sides and remove the pieces of agarose. The scalpel blade must be sharp to make the walls of the troughs clear-cut and avoid agarose detachment during cutting.
References
Zigmond SH (1977) Ability of polymorphonuclear leukocytes to orient in gradients of chemotactic factors. J Cell Biol 75:606–616
Zigmond SH (1988) Orientation chamber in chemotaxis. Methods Enzymol 162:65–72
Zicha D, Dunn GA, Brown AF (1991) A new direct-viewing chemotaxis chamber. J Cell Sci 99:769–775
Zicha D, Dunn G, Jones G (1997) Analyzing chemotaxis using the Dunn direct-viewing chamber. Methods Mol Biol 75:449–457
Muinonen-Martin AJ, Veltman DM, Kalna G, Insall RH (2010) An improved chamber for direct visualisation of chemotaxis. PLoS One 5:e15309
Petrie RJ, Gavara N, Chadwick RS, Yamada KM (2012) Nonpolarized signaling reveals two distinct modes of 3D cell migration. J Cell Biol 197:439–455
Vu LT, Jain G, Veres BD, Rajagopalan P (2015) Cell migration on planar and three-dimensional matrices: a hydrogel-based perspective. Tissue Eng Part B Rev 21:67–74
Charras G, Sahai E (2014) Physical influences of the extracellular environment on cell migration. Nat Rev Mol Cell Biol 15:813–824
Boyden S (1962) The chemotactic effect of mixtures of antibody and antigen on polymorphonuclear leucocytes. J Exp Med 115:453–466
Falasca M, Raimondi C, Maffucci T (2011) Boyden chamber. Methods Mol Biol 769:87–95
Marshall J (2011) Transwell(®) invasion assays. Methods Mol Biol 769:97–110
Laevsky G, Knecht DA (2001) Under-agarose folate chemotaxis of Dictyostelium discoideum amoebae in permissive and mechanically inhibited conditions. Biotechniques 31:1140–1149
Woznica D, Knecht DA (2006) Under-agarose chemotaxis of Dictyostelium discoideum. Methods Mol Biol 346:311–325
Zatulovskiy E, Tyson R, Bretschneider T, Kay RR (2014) Bleb-driven chemotaxis of Dictyostelium cells. J Cell Biol 204:1027–1044
Hall AL, Schlein A, Condeelis J (1988) Relationship of pseudopod extension to chemotactic hormone-induced actin polymerization in amoeboid cells. J Cell Biochem 37:285–299
Langridge PD, Kay RR (2007) Mutants in the Dictyostelium Arp2/3 complex and chemoattractant-induced actin polymerization. Exp Cell Res 313:2563–2574
Langridge PD, Kay RR (2006) Blebbing of Dictyostelium cells in response to chemoattractant. Exp Cell Res 312:2009–2017
Tyson RA, Zatulovskiy E, Kay RR, Bretschneider T (2014) How blebs and pseudopods cooperate during chemotaxis. Proc Natl Acad Sci U S A 111:11703–11708
Pang KM, Lee E, Knecht DA (1998) Use of a fusion protein between GFP and an actin-binding domain to visualize transient filamentous-actin structures. Curr Biol 8:405–408
Acknowledgements
We thank Richard Tyson and Till Bretschneider for discussions and the Medical Research Council (MRC file reference number U105115237) and a Herchel Smith Fellowship for support.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2016 Springer Science+Business Media New York
About this protocol
Cite this protocol
Zatulovskiy, E., Kay, R.R. (2016). Chemotactic Blebbing in Dictyostelium Cells. In: Jin, T., Hereld, D. (eds) Chemotaxis. Methods in Molecular Biology, vol 1407. Humana Press, New York, NY. https://doi.org/10.1007/978-1-4939-3480-5_7
Download citation
DOI: https://doi.org/10.1007/978-1-4939-3480-5_7
Published:
Publisher Name: Humana Press, New York, NY
Print ISBN: 978-1-4939-3478-2
Online ISBN: 978-1-4939-3480-5
eBook Packages: Springer Protocols