
Abstract
Recent annotation of the human transcriptome revealed that only 2 % of the genome encodes proteins while the majority of human genome is transcribed into noncoding RNAs. Although we are just beginning to understand the diverse roles long noncoding RNAs (lncRNAs) play in molecular and cellular processes, they have potentially important roles in human development and pathophysiology. However, targeting of RNA by traditional structure-based design of small molecule inhibitors has been difficult, due to a lack of understanding of the dynamic tertiary structures most RNA molecules adopt. Antisense oligonucleotides (ASOs) are capable of targeting specific genes or transcripts directly through Watson–Crick base pairing and thus can be designed based on sequence information alone. These agents have made possible specific targeting of “non-druggable targets” including RNA molecules. Here we describe how ASOs can be applied in preclinical studies to reduce levels of lncRNAs of interest.
Access provided by CONRICYT – Journals CONACYT. Download protocol PDF
Similar content being viewed by others


Key words
- Antisense oligonucleotide
- Control ASO
- Non-druggable targets
- Off-target
- RNA therapeutics
- Target reduction
- RNase H
- qRT-PCR
- Transfection
- Free uptake
1 Introduction
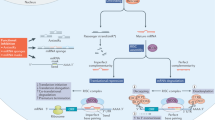
Targeted drug discovery efforts have mostly focused on proteins, particularly enzymes, secretary factors, and G-protein-coupled receptors [1]. Many proteins are considered “non-druggable” targets because closely related protein family members exist, making specificity difficult [2]. The “non-druggable” category of proteins includes transcription factors, structural proteins, and RNAs [3]. Indeed, direct targeting of RNAs, including both protein-encoding genes and noncoding transcripts, would potentially allow modulation of all transcriptional products, such as specific splice variant forms [4], eRNAs [5], long noncoding RNAs (lncRNA), and all protein coding RNAs [6, 7]. Antisense oligonucleotides is a technology that enables the direct targeting of RNA and greatly expands the freedom of drug target selection for the treatment of human diseases.
LncRNAs, arbitrarily defined as RNA transcripts longer than 200 nucleotides that do not encode proteins, have been proposed to modulate diverse biological functions . Although the functional roles and mechanisms of actions for the majority of lncRNAs remain unknown, recent studies have revealed that lncRNAs are involved in chromosome dosage compensation, modulation of chromatin status, and cell differentiation among other cellular processes [8, 9]. Moreover, mutation or dysregulation of lncRNAs have been linked to many human diseases including diabetics, cardiovascular diseases, central nervous system disease, and cancer (reviewed in [10, 11]). Thus, the selective depletion of specific lncRNA will allow us to both experimentally explore lncRNA functions and to pursue the most attractive of these as therapeutic targets to the diseases. The selective depletion of lncRNAs has posed a common challenge in lncRNA research [9]. Knocking down lncRNA by RNAi is a well-established approach. However, the presence and activities of RNAi machinery in nucleus is not thought to be robust and its existence in this compartment has been under intense debate. It is possibly for this reason that RNAi is limited in its ability to target nuclear-retained lncRNAs [12]. The difficulty of knocking down nuclear-retained lncRNA may be overcome by ASOs, another nucleic acid-based technology which enables specific targeting of any gene in human transcriptome. ASOs rely on RNAse H to cleave target RNAs irrespective of their subcellular localization due to RNase H’s ubiquitous presence in both cytoplasm and the nucleus [13, 14]. Importantly, ASOs’ efficacy in man has recently led to the FDA approval of Kynamro®, an ASO drug targeting ApoB, that lowers cholesterol in patients with homozygous familial hypercholesterolemia (HoFH) [15]. More than 30 ASO drugs are currently in preclinical or clinical testing [16, 17]. In this chapter we discuss the design of ASOs to target lncRNA and methods employed to evaluate ASOs in both cell-based assays and animals.
1.1 Design of ASOs
ASO, as we discuss here, refers to a synthetic molecule comprising a string of nucleotides or nucleotide analogs that bind to complementary RNA sequences with high specificity through Watson–Crick base pairing. ASOs can modulate levels of the targeted RNA through several mechanisms: (1) ASOs with properties of deoxyoligonucleotides may recruit RNase H to the DNA–RNA heteroduplex to degrade RNA [14]. (2) Binding of ASOs to target sequences may inhibit biogenesis or translation of the transcript of gene [3, 18]. Splicing [19], 3′ polyadenylation [20], RNA localization [7] are some examples where ASOs were demonstrated to achieve potential therapeutic goals. ASOs acting through RNAse H mechanism are typically 12–20 nucleotides in length, because approximately 12 nucleotides are required to recognize a unique sequence in the genome given the size of human genome. Unlike siRNAs , which are duplexes, ASOs are single-stranded molecules. Compared to double-stranded nucleotide compounds including siRNAs that are rigid, hydrophilic and have average molecular weight of 13,300 Da, ASOs are on average 5000–8000 Da, amphiphilic in nature and are more flexible, which allows efficient binding to target RNA.
Through chemical alterations of the natural nucleotides, ASOs have been designed to have drug-like properties. Naturally occurring nucleic acids are composed of ribonucleotides or deoxynucleotides linked with phosphodiester bonds. One of the first modifications made to ASOs was the phosphorothioate modification of the linkage. This modification protects ASOs from degradation by nucleases and increases half-life in serum, while still supporting RNase H activities [21]. These so-called first-generation ASOs were typically 20 nucleotides in length and are composed solely of deoxy residues [14]. However, due to low metabolic stability and suboptimal target binding affinity, the application of these early generation ASOs was limited in clinics [22]. Second-generation ASOs contain a central region of 8–10 phosphorothioate DNA nucleotides flanked by nucleotides modified at the sugar; this is called “gapmer” design [23]. Over the years, numerous nucleotide modifications have been tested in attempts to enhance binding affinity [24]. The bulky 2′-O-methoxyethyl (2′-MOE modification) improved metabolic stability of ASOs and prevented nonspecific protein interactions and thus improved overall safety profile relative to the first-generation ASOs [25]. Kynamro® is a systematically delivered 2′-MOE-modified 20-mer [26]. A more recently developed ASO chemistry incorporates the next generation 2′, 4′-constrained ethyl (cEt modification) in the residues flanking the deoxy central region. Because of the enhanced affinity provided by the cEt modification relative to the 2nd-generation ASOs, the cEt ASOs can be shorter; this contributes to higher ASO potency as the smaller molecular weight ASOs are more efficiently released in a cell [27]. Furthermore, ASOs of the same chemical class all have very similar pharmacodynamics, pharmacokinetic, and tissue accumulation features, making the overall drug development process more predictable and efficient for a given ASO drug. STAT3-Rx (AZD9150) is the first Gen 2.5 cEt ASO to enter clinical trials. It has shown single agent efficacy in patients with diffuse large B-cell lymphoma at modest doses [28].
Like many other drug classes, ASOs potentially have both “on-target” and “off-target ” effects. “Off-target ” effects occur due to ASOs’ binding to unintended sequences in non-target RNAs or through direct interactions with proteins independent of target hybridization [29, 30]. Binding to off-target transcripts can be avoided in large part with the use of computational algorithms that ensure that ASO sequences have little homology to genomic sequence other than that of the desired target. Off target effects have been further minimized with careful choice of chemical modifications and by extensive screening in vitro and in animal models.
The ability of ASOs to inhibit target RNAs does not depend on the abundance of the transcript. Levels of both rare transcripts such as enhancer RNAs, a class of relatively short noncoding RNAs that function to enhance gene expression , as well as very abundant transcripts such as metastasis associated lung adenocarcinoma transcript 1 (MALAT1) can be reduced equally well by ASOs that activate RNase H [5, 31, 32]. However, certain RNAs have proven difficult to target efficiently with ASOs despite repeated efforts. We speculate that specific features of the RNA such as transcript half-life, transcript secondary structures, and rates of RNA processing and nuclear export may contribute to such difficulties. On the other hand, targeting repeated sequences unique to the RNA transcript has been shown to greatly increase ASO potency [33]. Importantly, ASOs are capable of distinguishing between transcripts that differ by a single nucleotide, allowing for allele-specific suppression of a mutant gene while sparing the wild-type form [34, 35]. Such exclusive specificity achieved by ASOs has the potential for therapeutic targeting of otherwise essential genes.
1.2 Applications of ASO Designs with Different Mechanisms of Actions
ASOs decrease gene expression or disrupt the action of a functional RNA through two general mechanisms: RNase H-mediated and occupancy-based [3, 18]. ASOs that direct RNA degradation by RNase H bind to pre-mRNA or processed RNA through Watson–Crick base pairing, followed by RNase H1 recruitment to initiate cleavage of the target RNA. RNase H1 is a ubiquitously expressed nuclease that cleaves the RNA strand of an RNA–DNA hybrid. The enzyme is found in the cell nucleus , mitochondria, and, to a lesser extent, the cytoplasm. This makes ASO action different from the nucleic acid-based siRNA technology. The siRNA activity is mediated by the actions of the RNA-induced silencing complex (RISC), which is mainly localized to the cytoplasm. ASOs that mediate RNase H cleavage efficiently reduce levels of transcripts localized exclusively in the nucleus [7, 31, 32].
ASOs have also been shown to act through occupancy-based mechanisms, such as those designed to alter RNA splicing events. These ASOs bind to splicing regulatory sequences and function by hindering access of the splicing machinery. For an example, an ASO alters splicing of mutant SMN2 pre-mRNA to generate the exon-incorporating productive form in mammalian cells and in mouse models of spinal muscular atrophy (SMA) [19]; the splice-altering SMN2 ASO is currently being evaluated in clinical trials in SMA patients [36]. Additionally, ASOs have been designed to induce nonsense-mediated decay [37], to affect 3′ polyadenylation [20], to alter RNA localization [7], and to affect other RNA processing events [18]. In this chapter, we focus on applications of RNase H-mediated ASO and describe procedures to inhibit the expression of target RNAs.
2 Materials
-
1.
ASOs targeting MALAT1 in various species ISIS399479, ISIS395240, ISIS556089.
Ultraviolet–visible spectrophotometer.
-
2.
Cell lines:
-
4T1, a mouse mammary carcinoma cell line; LNCaP, a human prostate cancer cell line; THP-1, a human myeloid leukemia cell line. All cells were obtained from American Type Culture Collection (ATCC) and were maintained in RPMI1460 supplemented with 10 % fetal bovine serum and antibiotics.
-
ASO solutions adjusted to 200 μM.
-
RNAiMAX (Life Technologies).
-
Opti-MEM (Life Technologies).
-
96-well electroporation manipulator (BTX Harvard Apparatus).
-
High-throughput electroporation plates (BTX Harvard Apparatus).
-
-
3.
Male 6–8 weeks CD.1 mice (Charles River Laboratories, USA).
3 Methods for Validation of ASO Activity
In this chapter, we describe protocols to reduce levels of a lncRNA MALAT1 with Gen 2.0 ISIS399479, ISIS395240 or the more potent Gen 2.5 ISIS556089 ASOs. These protocols can be applied to additional Gen 2.0 and Gen 2.5 ASOs for in vitro and in vivo preclinical studies.
3.1 Preparation of ASOs
Dissolve ASOs directly in PBS to approximately 10 mg/ml (see Note 1 ). Like all nucleic acids, ASOs absorb ultraviolet light. The extinction coefficient of an oligonucleotide depends on base composition in the ASO sequence [38]. The extinction coefficients for the ASOs used in this protocol are listed in Table 1. ASO concentrations are determined by measuring the absorbance of the solution at 260 nM in a UV-visible light spectrometer after 1:1000 dilutions and calculating using the formula:
ASOs can be stored in aqueous buffer at 4 °C for ~1 month or at −20 °C for years without loss of activities. Occasionally some ASO solutions precipitate after extended storage at 4 °C. If precipitation is observed, filter the solution and recalculate the concentration.
3.2 Evaluation of ASO Activity In Vitro
Many cancer cell lines take up ASOs under physiological conditions without lipid-mediated transfection reagent (referred to as “free uptake ” hereinafter) [31, 39]. This process is independent of clathrin or caveolin pathways but specific receptors have not been identified yet [39]. In our extensive efforts to test free uptake in cancer cell line panels, we have identified at least one cell line in each cancer cell origin that takes up ASO very efficiently without the need for lipid transfection . Primary and early passage cells used for patient-derived xenograft models have higher propensity to take up ASOs for a particular type of cancer. This observation implies that the loss of free uptake abilities may be an artifact during cell line establishment. We have observed that the ability of a given cell line to take up ASO correlates with the ASO pharmacodynamics in the tumor models established from the same cell line [31]. Thus when possible, we test ASOs in cell lines in vitro by free uptake.
3.2.1 Delivery of ASOs to Cells by Free Uptake
-
1.
Log phase 4T1 cells are plated at 2–5 × 103 cells per well into 96-well plates and are incubated for 16 h in 95 μl of culture medium (see Note 2 ).
-
2.
Pre-diluted ASOs (5 μl of appropriate concentration stock) are added to cells to the desired final concentrations. A typical dose response analysis involves testing of the final concentrations at 80 nM, 400 nM, 2 μM, and 10 μM (see Note 3 ).
-
3.
Cells are harvested 24 h after addition of ASO, and RNA is prepared. ASO activity is examined by qRT-PCR using Taqman assays with the primers and probe sequences designed to amplify the RNA of interest, normalized to the expression of a housekeeping gene; the primers and probe used to amplify mouse MALAT1 are:
-
Forward primer: 5′-AGGCGGGCAGCTAAGGA-3′;
-
Reverse primer: 5′-CCCCACTGTAGCATCACATCA-3′;
-
Probe: 5′-FAM-TTCCTCTGCCGGTCCCTCGAAAG-TAMRA-3′ (see Note 4 ). Primers and probe sequences for housekeeping gene mouse Cyclophilin A are:
-
Forward primer: 5′-TCGCCGCTTGCTGCA-3′;
-
Reverse primer: 5′-ATCGGCCGTGATGTCGA-3′;
-
Probe: 5′-FAM-CCATGGTCAACCCCACCGTGTTC-TAMRA-3′ [26].
-
Typical data are shown in Fig. 1. ISIS399479 reduces levels of MALAT1 RNA with an IC50 of ~70 nM in 4T1 cells (see Note 5 ).
Fig. 1 
Mouse mammary tumor 4T1 cells were treated with ISIS399479, a Gen 2.0 ASO targeting mouse MALAT1 along with a control ASO for 24 h. RNA was harvested, and target reduction was examined by qRT-PCR . ISIS399479 caused dose-dependent inhibition of target gene expression , whereas the control ASO had little effect. UTC: Untreated cells
-

3.2.2 Delivery of ASOs into Cells Using Transfection Reagents
It is not always possible to find a cell line model that is amenable to free uptake . To obtain proof of concept data in vitro, ASOs can be delivered to cells by lipid-mediated transfection. The following is a protocol for 96-well format. Reagents can be scaled up proportionally for other plate formats.
-
1.
To examine the proliferation of LNCaP cells after ASO treatments, LNCaP are plated on 96-well plates at 2,000 cells per well in 100 μl 24 h prior to experiments.
-
2.
For each well, 0.15 μl of RNAiMax is mixed with 12.5 μl of Opti-MEM by brief vortexing.
-
3.
Five minutes later, 200 nM ASO diluted in 12.5 μl Opti-MEM is mixed with RNAiMAX by brief vortexing.
-
4.
The 25-μl ASO-RNAiMAX solution is incubated at room temperature for 15 min and is subject to fivefold stepwise dilution in Opti-MEM to 20, 4, and 0.8 nM.
-
5.
The 25-μl aliquot of ASO-RNAiMAX solution is added to each well containing cells to yield final concentrations of 20, 4, 0.8, and 0.16 nM.
-
6.
Cells are harvested after 24 h for RNA analyses and 5–6 days later for cell proliferation assays (see Note 6 ). The primers and probe used to amplify human MALAT1 are:
-
Forward primer: 5′-AAAGCAAGGTCTCCCCACAAG-3′;
-
Reverse primer: 5′-TGAAGGGTCTGTGCTAGATCAAAA-3′;
-
Probe: 5′-FAM-TGCCACATCGCCACCCCGT-TAMRA-3′.
-
Human β-actin gene is used to normalize RNA amounts and sequences for primers and probe are:
-
Forward primer: 5′-CGGACTATGACTTAGTTGCGTTACA-3′;
-
Reverse primer: 5′-GCCATGCCAATCTCATCTTGT-3′;
-
Probe: 5′-FAM-CCTTTCTTGACAAAACCTAACTTGCGCAGA-TAMRA-3′. Representative data is shown in Fig. 2, where ASOs were introduced to cell by transfection (a) or by free uptake (b).
Fig. 2 
Human prostate cancer LNCaP cells were treated with ISIS556089, a Gen 2.5 ASO targeting human MALAT1 along with a control ASO . (a) ASOs were transfected by transfection using RNAiMax reagents as described and RNA was harvested after 24 h. (b) ASOs were delivered to the cells by free uptake and RNA was collected after 48 h. Target knockdown was evaluated by qRT-PCR. UTC: Untreated cells
-

3.2.3 Delivery of ASOs to Cells by Electroporation
Some cell lines, including many suspension cells, are recalcitrant to lipid-mediated transfection . For analysis of ASO effects in these cells, we resort to electroporation in 96-well format.
-
1.
THP-1 cells proliferating in log-phase are collected and resuspended at 1 × 107 cells per ml in complete growth medium.
-
2.
Aliquots of 90 μl of cells are mixed with 10 μl ASOs at appropriate concentration; the solution is pipeted up and down (or vortexed gently) to mix (see Note 7 ). Samples are transferred to 96-well electroporation plate.
-
3.
Cell mixtures are pulsed at desired voltage for electroporation, typically 130 V for 6 ms. Cells are then collected from each well and washed twice with 120 μl of fresh medium.
-
4.
All cells are combined and plated at 50–100,000 cells per well for RNA extraction and 10,000 cells per well for analysis of proliferation.
3.3 Systemic Delivery of ASO in Mice
-
1.
ASOs are formulated in PBS containing Ca2+ and Mg2+ at 5 mg/ml and filtered through 0.45 μm sterile filters before use.
-
2.
ASOs can be administered via intraperitoneal (see Note 8 ), subcutaneous, or intravenous injection.
-
3.
To examine whether ASO is tolerated in normal animals, CD.1 mice are treated with the target-specific ASO (in this case, ISIS395240 and ISIS399479 for mouse MALAT1) at 50 mg/kg, twice weekly for 6 weeks (see Note 9 ). Body weights are recorded after each dose is given. Twenty-four hours after the last dose, animals are sacrificed, and blood is collected by cardiac puncture. Liver, kidney, and spleen are weighed, and liver pieces are collected to prepare RNA (see Note 10 ).
-
4.
Plasma is tested on a clinical analyzer for blood chemistry parameters, including alanine amino transferase (ALT), aspartate amino transferase (AST), total bilirubin, and blood urea nitrogen (see Note 11 ).
-
5.
RNA is prepared from liver or other relevant organs or tissues, and target reduction is evaluated by qRT-PCR . Typical data is shown in Fig. 3 (see Note 12 ).
Fig. 3 
Male CD.1 mice were treated intraperitoneally with indicated ASOs designed to target mouse MALAT1. (a) Blood chemistry markers were evaluated in plasma collected by cardiac puncture. (b) Mouse MALAT1 levels were measured in mouse livers using qRT-PCR . There were no notable changes in the blood chemistry from animals treated with two MALAT1 ASOs

4 Notes
-
1.
The chemically modified ASOs described here are typically soluble up to 50 mg/ml in water. Sometimes at high concentrations (>20 mg/ml), some compounds show slight yellow or green tint.
-
2.
ASO treatment affects the attachment of some cell lines, and thus we recommended that cells are incubated for at least 8 h after plating prior to ASO treatment. Cell plating densities between 10,000 and 100,000 cells per ml do not affect cells’ free uptake ability.
-
3.
Control ASOs designed to have no matches in human and mouse genome should also be included at the same concentrations. Some cell lines are especially sensitive to high concentrations of ASOs (>10 μM). In these lines, cell growth can be inhibited by ASOs in a sequence-independent manner. These ASO class effects can be better distinguished from on-target events when the control ASOs are included in the experiments in parallel.
-
4.
It is important to design qRT-PCR assays outside the ASO-hybridizing sequences. ASOs remain in RNAs purified from ASO-treated samples and would interfere with RT-PCR reactions if ASOs hybridize to the PCR products defined by the PCR primers, generating extremely low “false” signals, and misleadingly high degree of target knockdown.
-
5.
Typically cells are incubated with ASO-containing culture medium continuously for 24–96 h before cells are harvested for RNA analyses. Maximal RNA knockdown is observed in some non-dividing cells after 7–10 days. Incubation with MALAT1 ASO for merely 3 h is sufficient to initiate the necessary events leading to MALAT1 RNA downregulation 48 h later (Liang XH et al., manuscript in preparation). We observe that the incubation time required for the onset of ASO activities is cell line-specific. A careful time course study is necessary to reveal the dynamics of target RNA expression and inhibition for each ASO compound and each cell type.
-
6.
A control ASO that has no matching sequence in the human and mouse genome should always be included in transfection experiments to determine whether cell growth inhibition is due to the inhibition of specific target or general class effects of ASOs. Some cell lines are highly sensitive to lipid-mediated transfection and as little as 10 nM ASO leads to the inhibition of cell growth; in these cells, observed growth inhibition is not target-related as similar effects are typically observed with both targeted and control ASOs .
-
7.
Optimal experimental conditions are dependent on each cell line and electroporation apparatus used. We typically mix 200 μM of ASO with cell aliquots ranging from 0.5 × 106 to 2 × 106 cells. Tests are run with electroporation voltages ranging from 120 to 170 V. Cell viability is checked by trypan blue exclusion assay after electroporation. In order to ensure reliable data, we make sure at least 80 % cells survive the electric pulse. Efficiency of target reduction can be evaluated by comparing target RNA levels to levels in cells treated with control ASO and in mock electroporated cells. We always use the lowest voltage where >50 % target reduction is achieved.
-
8.
Intraperitoneal injection of ASOs results in greater target reduction in peritoneal macrophages than does subcutaneous dosing, presumably because intraperitoneal dosing allows direct access of ASOs to peritoneal monocyte/macrophage cells.
-
9.
The Gen 2.0 and Gen 2.5 ASOs discussed here demonstrate very similar tissue distribution profiles irrespective of their sequences. Target reduction in liver is observed 24 h after a single systemic administration with a peak in inhibition observed after 48–72 h. Generally a repeated dosing scheme is employed and we observe target inhibition in tumor cells in the 4T1 mouse model of breast cancer between 24–72 h after the last dose.
-
10.
ASOs distribute widely into tissues within 2 h after systemic administration [40, 41]. Organs of high ASO accumulation include kidney, liver, and spleen. Efficient downregulation of target RNA in fat, muscle, and small intestines has also been reported despite low concentrations of ASOs [32]. ASOs remain efficacious for 2–4 weeks in liver and more than 6 months in muscle [41]. ASOs do not cross the blood–brain barrier, thus need to be administered directly to cerebral spinal fluid to reduce target RNA levels in the central nervous system [19]. ASOs are carried by serum proteins in plasma and either taken up by tissues or gradually degraded by various nucleases and cleared in urine [41].
-
11.
ASOs are considered “well-tolerated” if mice treated with the ASOs show no significant changes in organ and body weights, no significant elevations in liver transaminases (ALT and AST) in serum, and no obvious signs of sickness. Different mouse strains may have different susceptibilities to ASOs’ non-target related toxicities. Therefore ASOs should be tested for tolerability under the same condition as the intended animal model. Toxicities in mice caused by off-target effects of ASOs can complicate interpretation of experimental results. Off-target effects may be distinguished from on-target pharmacology by dose–response experiments using two or more ASOs targeting the same target gene in the relevant animal models. We encourage the use of a second ASO designed to hybridize to a different region of the target RNA to confirm that observed pharmacology is not limited to one ASO compound. The relative potency of the two ASOs should be the same both in vitro and in vivo: the more potent ASO with a greater target reduction is predicted to demonstrate better efficacies in animals
-
12.
To ensure efficient target reductions in tumor-bearing mice, various ASO dosing schemes should be tested for each animal model. Depending on mouse strain, ASOs can be tolerated at 100–1000 mg/kg/week.
4.1 Conclusions
Antisense oligonucleotide drugs can be used to specifically and efficiently reduce levels of any RNA of interest, including many lncRNAs, both in cultured cells and in animals without the need for formulation with delivery vehicles. Antisense technology is a promising, versatile modality in preclinical studies for target validation, and for the therapeutic targeting of previously non-druggable targets to treat human diseases.
References
Rask-Andersen M, Almen MS, Schioth HB (2011) Trends in the exploitation of novel drug targets. Nat Rev Drug Discov 10:579–590
Overington JP, Al-Lazikani B, Hopkins AL (2006) How many drug targets are there? Nat Rev Drug Discov 5:993–996
Bennett CF, Swayze EE (2010) RNA targeting therapeutics: molecular mechanisms of antisense oligonucleotides as a therapeutic platform. Annu Rev Pharmacol Toxicol 50:259–293
Yamamoto Y, Loriot Y, Beraldi E, Zhang F, Wyatt AW, Al Nakouzi N et al (2015) Generation 2.5 antisense oligonucleotides targeting the androgen receptor and its splice variants suppress enzalutamide resistant prostate cancer cell growth. Clin Cancer Res 21:1675–1687
Lam MT, Cho H, Lesch HP, Gosselin D, Heinz S, Tanaka-Oishi Y et al (2013) Rev-Erbs repress macrophage gene expression by inhibiting enhancer-directed transcription. Nature 498:511–515
Meng L, Ward AJ, Chun S, Bennett CF, Beaudet al, Rigo F (2014) Towards a therapy for Angelman syndrome by targeting a long non-coding RNA. Nature 518(7539):409–12
Wheeler TM, Leger AJ, Pandey SK, MacLeod AR, Nakamori M, Cheng SH et al (2012) Targeting nuclear RNA for in vivo correction of myotonic dystrophy. Nature 488:111–115
Sauvageau M, Goff LA, Lodato S, Bonev B, Groff AF, Gerhardinger C et al (2013) Multiple knockout mouse models reveal lincRNAs are required for life and brain development. ELife 2:e01749
Li L, Chang HY (2014) Physiological roles of long noncoding RNAs: insight from knockout mice. Trends Cell Biol 24:594–602
Wapinski O, Chang HY (2011) Long noncoding RNAs and human disease. Trends Cell Biol 21:354–361
Li X, Wu Z, Fu X, Han W (2014) lncRNAs: insights into their function and mechanics in underlying disorders. Mutat Res Rev Mutat Res 762:1–21
Meister G (2013) Argonaute proteins: functional insights and emerging roles. Nat Rev Genet 14:447–459
Cerritelli SM, Frolova EG, Feng C, Grinberg A, Love PE, Crouch RJ (2003) Failure to produce mitochondrial DNA results in embryonic lethality in RNaseh1 null mice. Mol Cell 11:807–815
Wu H, Lima WF, Zhang H, Fan A, Sun H, Crooke ST (2004) Determination of the role of the human RNase H1 in the pharmacology of DNA-like antisense drugs. J Biol Chem 279:17181–17189
Lee RG, Crosby J, Baker BF, Graham MJ, Crooke RM (2013) Antisense technology: an emerging platform for cardiovascular disease therapeutics. J Cardiovasc Transl Res 6:969–980
Buller HR, Bethune C, Bhanot S, Gailani D, Monia BP, Raskob GE et al (2015) Factor XI antisense oligonucleotide for prevention of venous thrombosis. N Engl J Med 372:232–240
Gaudet D, Brisson D, Tremblay K, Alexander VJ, Singleton W, Hughes SG et al (2014) Targeting APOC3 in the familial chylomicronemia syndrome. N Engl J Med 371:2200–2206
Rigo F, Seth PP, Bennett CF (2014) Antisense oligonucleotide-based therapies for diseases caused by pre-mRNA processing defects. Adv Exp Med Biol 825:303–352
Rigo F, Chun SJ, Norris DA, Hung G, Lee S, Matson J et al (2014) Pharmacology of a central nervous system delivered 2′-O-methoxyethyl-modified survival of motor neuron splicing oligonucleotide in mice and nonhuman primates. J Pharmacol Exp Ther 350:46–55
Vickers TA, Wyatt JR, Burckin T, Bennett CF, Freier SM (2001) Fully modified 2′ MOE oligonucleotides redirect polyadenylation. Nucleic Acids Res 29:1293–1299
Baek MS, Yu RZ, Gaus H, Grundy JS, Geary RS (2010) In vitro metabolic stabilities and metabolism of 2′-O-(methoxyethyl) partially modified phosphorothioate antisense oligonucleotides in preincubated rat or human whole liver homogenates. Oligonucleotides 20:309–316
Henry SP, Geary RS, Yu R, Levin AA (2001) Drug properties of second-generation antisense oligonucleotides: how do they measure up to their predecessors? Current Opin Investig Drugs 2:1444–1449
Monia BP, Lesnik EA, Gonzalez C, Lima WF, McGee D, Guinosso CJ et al (1993) Evaluation of 2′-modified oligonucleotides containing 2′-deoxy gaps as antisense inhibitors of gene expression. J Biol Chem 268:14514–14522
Prakash TP (2011) An overview of sugar-modified oligonucleotides for antisense therapeutics. Chem Biodivers 8:1616–1641
Geary RS, Watanabe TA, Truong L, Freier S, Lesnik EA, Sioufi NB et al (2001) Pharmacokinetic properties of 2′-O-(2-methoxyethyl)-modified oligonucleotide analogs in rats. J Pharmacol Exp Ther 296:890–897
Lee RG, Fu W, Graham MJ, Mullick AE, Sipe D, Gattis D et al (2013) Comparison of the pharmacological profiles of murine antisense oligonucleotides targeting apolipoprotein B and microsomal triglyceride transfer protein. J Lipid Res 54:602–614
Seth PP, Vasquez G, Allerson CA, Berdeja A, Gaus H, Kinberger GA et al (2010) Synthesis and biophysical evaluation of 2′,4′-constrained 2′O-methoxyethyl and 2′,4′-constrained 2′O-ethyl nucleic acid analogues. J Org Chem 75:1569–1581
Hong DS, Kurzrock R, Kim Y, Woessner R, Younes A, Nemunaitis J, Fowler N, Zhou T, Schmidt J, Jo M, LeeSJ, Yamashita M, Hughes SG, Fayad L, Piha-Paul S, Nadella MVP, Mohseni M, Lawson D, Reimer C, Blakey DC, Xiao X, Hsu J, Monia BP, and MacLeod AR (2015). AZD9150, a nextgeneration antisense oligonucleotide Inhibitor of STAT3, with early evidence of clinical activity in lymphoma and lung cancer. Sci Transl Med (in press)
Senn JJ, Burel S, Henry SP (2005) Non-CpG-containing antisense 2′-methoxyethyl oligonucleotides activate a proinflammatory response independent of Toll-like receptor 9 or myeloid differentiation factor 88. J Pharmacol Exp Ther 314:972–979
Lima WF, Vickers TA, Nichols J, Li C, Crooke ST (2014) Defining the factors that contribute to on-target specificity of antisense oligonucleotides. PLoS One 9:e101752
Gutschner T, Hammerle M, Eissmann M, Hsu J, Kim Y, Hung G et al (2013) The noncoding RNA MALAT1 is a critical regulator of the metastasis phenotype of lung cancer cells. Cancer Res 73:1180–1189
Hung G, Xiao X, Peralta R, Bhattacharjee G, Murray S, Norris D et al (2013) Characterization of target mRNA reduction through in situ RNA hybridization in multiple organ systems following systemic antisense treatment in animals. Nucleic Acid Ther 23:369–378
Vickers TA, Freier SM, Bui HH, Watt A, Crooke ST (2014) Targeting of repeated sequences unique to a gene results in significant increases in antisense oligonucleotide potency. PLoS One 9:e110615
Skotte NH, Southwell AL, Ostergaard ME, Carroll JB, Warby SC, Doty CN et al (2014) Allele-specific suppression of mutant huntingtin using antisense oligonucleotides: providing a therapeutic option for all Huntington disease patients. PLoS One 9:e107434
Ostergaard ME, Southwell AL, Kordasiewicz H, Watt AT, Skotte NH, Doty CN et al (2013) Rational design of antisense oligonucleotides targeting single nucleotide polymorphisms for potent and allele selective suppression of mutant Huntingtin in the CNS. Nucleic Acids Res 41:9634–9650
Castro D, Iannaccone ST (2014) Spinal muscular atrophy: therapeutic strategies. Curr Treat Options Neurol 16:316
Ward AJ, Norrbom M, Chun S, Bennett CF, Rigo F (2014) Nonsense-mediated decay as a terminating mechanism for antisense oligonucleotides. Nucleic Acids Res 42:5871–5879
Cavaluzzi MJ, Borer PN (2004) Revised UV extinction coefficients for nucleoside-5′-monophosphates and unpaired DNA and RNA. Nucleic Acids Res 32:e13
Koller E, Vincent TM, Chappell A, De S, Manoharan M, Bennett CF (2011) Mechanisms of single-stranded phosphorothioate modified antisense oligonucleotide accumulation in hepatocytes. Nucleic Acids Res 39:4795–4807
Yu RZ, Grundy JS, Geary RS (2013) Clinical pharmacokinetics of second generation antisense oligonucleotides. Expert Opin Drug Metab Toxicol 9:169–182
Yu RZ, Lemonidis KM, Graham MJ, Matson JE, Crooke RM, Tribble DL et al (2009) Cross-species comparison of in vivo PK/PD relationships for second-generation antisense oligonucleotides targeting apolipoprotein B-100. Biochem Pharmacol 77:910–919
Acknowledgements
We thank XueHai Liang for discussion and sharing unpublished data. We are grateful to Lauren Elder for editorial assistance.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2016 Springer Science+Business Media New York
About this protocol
Cite this protocol
Zhou, T., Kim, Y., MacLeod, A.R. (2016). Targeting Long Noncoding RNA with Antisense Oligonucleotide Technology as Cancer Therapeutics. In: Feng, Y., Zhang, L. (eds) Long Non-Coding RNAs. Methods in Molecular Biology, vol 1402. Humana Press, New York, NY. https://doi.org/10.1007/978-1-4939-3378-5_16
Download citation
DOI: https://doi.org/10.1007/978-1-4939-3378-5_16
Published:
Publisher Name: Humana Press, New York, NY
Print ISBN: 978-1-4939-3376-1
Online ISBN: 978-1-4939-3378-5
eBook Packages: Springer Protocols