
Abstract
Through the work of photochemists extending over many decades, there now exists a wealth of information on the various reactions that photoexcited gas phase molecules undergo. Most of this information relates to the product molecules that are formed, either as the direct result of a primary photochemical act, such as photodissociation, or through subsequent secondary reactions, involving collisions with other molecules in the gas. Recently, there has been an extensive effort directed at determining the exact energy distributions of the primary products formed in photodissociation. With the use of nanosecond tunable-laser techniques, such as laser-induced fluorescence (LIF) and coherent anti-Stokes Raman spectroscopy (CARS), scientists have successfully determined the nascent electronic, vibrational, and rotational energy distributions of various diatomic fragments such as CN, OH, NO, and O2 that are directly formed in the photodissociation of many kinds of molecules. The ready availability of high-quality, tunable, nanosecond lasers has made determination of the above-mentioned collisionless energy distributions a relatively straightforward process. The determination of product translational energies has long effectively been handled by angularly resolved time-of-flight (TOF) spectroscopy, or by sub-Doppler resolution spectroscopy, including a recently improved version of the latter, velocity-aligned Doppler spectroscopy (Xu et al., 1986).
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others


Keywords
These keywords were added by machine and not by the authors. This process is experimental and the keywords may be updated as the learning algorithm improves.
1 Introduction
Through the work of photochemists extending over many decades, there now exists a wealth of information on the various reactions that photoexcited gas phase molecules undergo. Most of this information relates to the product molecules that are formed, either as the direct result of a primary photochemical act, such as photodissociation, or through subsequent secondary reactions, involving collisions with other molecules in the gas. Recently, there has been an extensive effort directed at determining the exact energy distributions of the primary products formed in photodissociation. With the use of nanosecond tunable-laser techniques, such as laser-induced fluorescence (LIF) and coherent anti-Stokes Raman spectroscopy (CARS), scientists have successfully determined the nascent electronic, vibrational, and rotational energy distributions of various diatomic fragments such as CN, OH, NO, and O2 that are directly formed in the photodissociation of many kinds of molecules. The ready availability of high-quality, tunable, nanosecond lasers has made determination of the above-mentioned collisionless energy distributions a relatively straightforward process. The determination of product translational energies has long effectively been handled by angularly resolved time-of-flight (TOF) spectroscopy, or by sub-Doppler resolution spectroscopy, including a recently improved version of the latter, velocity-aligned Doppler spectroscopy (Xu et al., 1986).
Of great interest, but until recently unobtainable, is detailed knowledge of the time sequences of the various internal conversions, rearrangements, dissociations, etc. that molecules typically undergo upon photoexcitation. To illustrate this point, let us consider Figure 8.1, which depicts an alkyl azide analog of the Curtius rearrangement for an acyl azide molecule. It is known that the weakest bond in the covalently bonded azide group lies between the two nitrogen atoms closest to the carbon atom. This fact explains the finding that molecular nitrogen is invariably produced in the photolysis of covalently bonded azides. The existence of the stable methylenimine product in the case of Figure 8.1 implies a breaking of the C—H bond and forming of an N—H bond. However, as to whether the above reactions actually happen sequentially in the order described, or whether the whole sequence occurs in a simultaneous, concerted manner, the relevant literature is quite contentious. It would appear that an advanced technique of kinetic spectroscopy,Footnote 1 having a spectral range that includes portions of the infrared and having a subpicosecond time resolution, could provide direct answers in the above example, provided that the time to form the final methylenimine product is no less than several hundred femtoseconds. Experimentally, one would monitor the times for disappearance of the azide symmetric or antisymmetric stretches and compare these with the appearance time for the N—H stretch.

Rearrangement mechanism proposed for the photolysis of methyl azide
While time-resolved broadband infrared probing of the vibrational modes of photoexcited molecules should thus give easily interpretable results, it appears, unfortunately, to be a rather difficult technique to develop and to apply. In general, the optical absorbances associated with purely vibrational transitions are a few orders of magnitude weaker than those associated with electronic transitions. Thus, signal-to-noise considerations become of paramount importance. Aside from questions of sensitivity, there is also the problem that only a very limited spectral range has thus far been demonstrated for subpicosecond kinetic spectroscopy in the infrared. For many photoexcited molecules one can, of course, determine exact times of dissociation by observing the appearance times of products of the photodissociation. Here one can fortunately utilize as monitors electronic transitions with their intrinsically high molar absorbances. Examples are given further on in this chapter. Electronic transitions between excited states of a molecule can also be utilized to monitor the motion of a molecule along an excited state potential surface, tracking the times when the molecule internally converts or fragments. This is illustrated by another example, discussed further on, in which an infrared subpicosecond kinetic spectroscopy probe is actually utilized.
Of course, the problem of spectroscopically monitoring in real time the unimolecular reactions of a photoexcited molecule can also be attacked in ways other than through kinetic spectroscopy. A.W. Zewail’s group uses a different time-resolved approach, which is illustrated by their recent study of ICN photodissociation (Dantus et al., 1987). After a subpicosecond UV pump pulse has initiated ICN fragmentation, a tunable LIF subpicosecond probe pulse induces the CN fragments to fluoresce. They recorded the CN* excitation spectrum as a function of pump-probe delay, providing new information about the photodissociation dynamics.
We have tried briefly in the preceding paragraphs to stress the potential utility of a subpicosecond kinetic spectroscopy approach to the real-time study of intramolecular photoinitiated reactions of gas-phase molecules. As is well known, subpicosecond kinetic spectroscopy has already been employed successfully in the condensed phase by several research groups to elucidate the dynamics of biological processes, to follow the approach to equilibrium in photoexcited dyes, to study ultrafast processes in semiconductors, etc. However, such studies, almost without exception, have relied on the use of broadband time-resolved spectroscopic probes in the visible. Our group has recently started to make advances toward developing equipment capable of providing a wider subpicosecond spectral probing range. In recent papers (Glownia et al., 1986a, 1987a, 1987b) we have described an apparatus capable of simultaneously generating both intense subpicosecond UV (308, 248.5 nm) excitation pulses and subpicosecond continua for probing photoexcited molecules via broadband absorption spectroscopy. Both UV (230 to 450 nm) and IR (2.2 to 2.7 μm) continua have thus far been produced. A method of upconverting the latter to the visible for ease of detection has also been demonstrated. In the present chapter this apparatus is described in detail. Also given is an account of some of the first experiments performed using this equipment.
The organization of this chapter is as follows. Since the pulses in both excitation and probe channels are derived from subpicosecond UV pulses amplified in XeCl gain modules, we begin, in Section 2, with a description of the apparatus we have built for producing intense subpicosecond 308-nm pulses. In Section 3 we show how these 308-nm pulses can be used to generate ultrafast UV supercontinuum pulses and also how the latter can be used to seed an amplification process in a KrF excimer gain module in order to produce intense subpicosecond pulses at 248 nm. Section 4 describes our technique for producing an ultrafast IR continuum, as well as the method we use for upconverting it to the visible for ease of detection. Exactly how the UV and IR continua are utilized in subpicosecond kinetic spectroscopy experiments is shown in the examples discussed in the balance of the chapter. Section 5 describes an IR experiment we have performed, the measurement of the \( \overset{\sim }{B}\to \tilde{A} \) internal conversion rate in 1,4-diazabicyclo[2.2.2]octane (DABCO) vapor, while Section 6 describes some preliminary results obtained in the case of two subpicosecond kinetic spectroscopy experiments recently attempted, photolysis of thallium halide vapors at 248 and 308 nm and photolysis of chlorine dioxide vapor at 308 nm. Section 7 concludes with a brief description of two promising directions than can now be taken in our approach to subpicosecond kinetic spectroscopy.
2 160-fs XeCl Excimer Amplifier System
The suitability of commercially available excimer gain modules for amplification of ultrafast UV pulses has been apparent for many years. Around 1982, various groups (Corkum and Taylor, 1982; Egger et al., 1982; Bucksbaum et al., 1982; Szatmári and Schäfer, 1984a) successfully utilized discharge-pumped excimer gain modules for amplification of UV pulses having durations of a few picoseconds. However, it was well known (e.g., see Corkum and Taylor, 1982) that the gain bandwidth of these systems is such that amplification of much shorter pulses can also be accomplished. The first published accounts of subpicosecond pulse amplification in excimers were published some four years later (Glownia et al., 1986b; Schwarzenbach et al., 1986). Glownia et al. (1986b) used a pair of XeCl gain modules to amplify 350-fs, 308-nm pulses to ∼10-mJ energies with <1-mJ amplified spontaneous emission (ASE) content. Seed pulses for the excimer amplifier were formed through the combined use of a synch-pumped mode-locked dye laser tuned to 616 nm, a single-mode fiber pulse compressor (Nakatsuka et al., 1981; Nikolaus and Grischkowsky, 1983), a four-stage Nd3+ : YAG-laser-pumped dye amplifier, and, finally a KDP frequency-doubling crystal. Schwarzenbach et al. (1986) used generally similar methods to produce subpicosecond seed pulses suitable for amplification at 248.5 nm in KrF gain modules.
In 1987 further significant advances in excimer-based UV subpicosecond amplification were made. Szatmári et al. (1987a) reported the generation of 220-fs pulses at 308 nm from an XeCl amplifier. Shortly afterwards, the same Göttingen group (Szatmári et al., 1987b) reported having obtained 15-mJ, 80-fs, 248.5-nm pulses from a KrF amplifier and having then amplified these pulses to 900 GW peak power in a wide-aperture KrF discharge amplifier. A remarkable feature of the above work is that the seed pulses in each case were formed directly with nanosecond excimer-pumped dye laser sources. A novel method discovered earlier (Szatmári and Schäfer, 1983, 1984b) of generating single, picosecond pulses through the combined use of an excimer-pumped, quenched, dye laser and a distributed feedback dye laser (DFDL) was improved on by Szatmári et al. (1987a, 1987b) to the degree that subpicosecond pulses were produced. These pulses were amplified and then frequency doubled prior to final amplification in an excimer gain module. One of the many advantages of the technique employed by the Göttingen group is that, due to the wide wavelength range accessible with DFDLs, all of the known rare-gas halide wavelengths can be reached through frequency doubling or mixing. Possible disadvantages of the Göttingen technique center around the inherent difficulties in adjusting and stabilizing the DFDL. It will be interesting to follow the development of this unique approach and to see also if it eventually benefits from commercial product engineering.
In 1987 a full description was published (Glownia et al., 1987b) of the XeCl excimer-based system our group currently employs, which generates bandwidth-limited, 160-fs, 308-nm pulses at a 10-Hz rate. Subpicosecond pulses at ∼616 nm are formed in a colliding-pulse mode-locked (CPM) laser (Fork et al., 1981; Valdmanis et al., 1985), amplified in a four-stage Nd3+ : YAG laser-pumped amplifier chain, and then frequency doubled in a 1-mm-long KDP crystal, forming seed pulses at 308 nm for further amplification in the XeCl excimer gain module. Since this sytem is the heart of our subpicosecond pump-probe apparatus, we summarize its main features here.
The design of the CPM laser is generally similar to that of Valdmanis et al. (1985), incorporating four dispersion-compensating prisms in the seven-mirror ring cavity arrangement of Fork et al. (1981). The pulse repetition rate is 116 MHz. An 80-mm focal-length lens is used to focus the 514.5-nm CW Ar+-ion laser pump beam into the Rhodamine 6G gain jet. Surprisingly, optimum mode locking at 616 nm, described below, requires only 1.4 W of pump power. Both the gain and DODCI absorber jets are standard Coherent dye laser nozzles (dye stream thickness ∼100 μm). A relatively dilute concentration of DODCI (50 mg in 21 ethylene glycol) was found to be optimum for operation at 616 nm. The CPM output power is about 20 mW in each arm when the laser is optimized at this frequency.
We generally tune the intraprism path length for minimum amplified laser-pulse duration (see below) while maintaining the peak of the output spectrum near 616 nm. With this adjustment, the CPM pulse width is about 240 fs. However, when measured after the beam has propagated through an additional 5 cm of H2O, the pulse width is 200 fs, showing that the laser operates with excess negative dispersion in the cavity. It should be noted that this CPM laser can readily produce much shorter pulse widths (∼70 fs) at longer wavelengths. However, the spectrum of the amplified and frequency-doubled pulses cannot then properly match the XeCl gain profile. The first three stages of the dye amplifier are excited transversely; the fourth, longitudinally. Kiton Red 620 dye is used in the first stage; Sulforhodamine 640 in the last three stages. The solvent used in all stages is H2O + 4% Ammonyx LO. Malachite Green bleachable absorber dye jets are used between the first three amplifier stages to control amplified spontaneous emission. The small-signal attenuations of the absorber jets are roughly 10× and 200×. Pumping of the dye amplifier chain is accomplished with ∼125 mJ of 532-nm light from a Quanta-Ray DCR-2A Nd3+ : YAG laser operating in the short pulse (2-ns central peak) mode. We typically measure a total amplified pulse energy of ∼0.6 mJ. The autocorrelation trace of the amplified CPM dye laser pulses at full power indicates a pulse width of ∼200 fs.
The ∼200-fs amplified 616-nm pulses are spatially compressed to a beam diameter of ∼2 mm, then frequency doubled in 1-mm-long KDP crystals to form seed pulses for amplification in the XeCl excimer gain module. In Glownia et al. (1987b) extensive frequency broadening of the second harmonic was noted when the size of the input beam in the KDP crystal was allowed to be less than 2 mm in diameter. This was attributed to self-phase modulation (SPM) occurring in the KDP crystals. Since the UV spectral width of the seed pulses was observed to be typically 10× the XeCl gain bandwidth, it was argued by Glownia et al. (1987b) that the positive frequency sweep associated with the most intense part of the pulse would result in a nearly in-phase excitation of all the frequencies lying under the XeCl gain curve. This is an exact prescription for forming bandwidth-limited pulses. This argument was used to explain the observation (see below) that the UV pulses amplified by the XeCl gain module are bandwidth limited.
The UV seed pulses are amplified in a single pass through a Lambda-Physik EMG101-MSC excimer gain module (45-cm-long discharge). Provided that the seed pulse spectrum is relatively flat over the XeCl gain profile, the spectrum of the amplified 308-nm pulse appears as shown in Figure 8.2. Figure 8.3 displays a typical amplified 308-nm pulse autocorrelation trace. (Two-photon ionization in DABCO vapor is used for the 308-nm pulse autocorrelation measurements.) Measurements made on this system over a period of more than one year have consistently shown the amplified 308-nm pulse width to be near 160 fs, a number very close to the bandwidth limit calculated (Glownia et al., 1987b) for the spectrum of Figure 8.2. This observed facile generation of bandwidth-limited UV pulses is rationalized by the argument mentioned in the preceding paragraph.

Spectrum of amplified 308-nm pulses

Autocorrelation trace of amplified 308-nm pulses
With the use of 1-mm-long KDP doubling crystals, pulse energies of 4 to 5 mJ are achieved in a single pass through the XeCl excimer gain module. With the scheme shown in Figure 8.4, however, a second UV pulse, having roughly the same energy, can be obtained during the same excimer discharge. Because the efficiency of second harmonic generation in the first 1-mm-long KDP crystal is only ∼10%, enough 616-nm light remains to generate a second UV seed pulse having almost the same energy as the first. If the two UV seed pulses are spaced apart by 2 or 3 ns, there is sufficient time for repumping the XeCl B state, according to Corkum and Taylor (1982). Thus amplification in the XeCl gain module occurs in the form of pairs of orthogonally polarized pulses, each pulse 4 to 5 mJ in energy. The 160-fs amplified UV pulse pairs are then separated by a Brewster polarizer into pump and probe channels (see Figure 8.14).

Scheme employed for multiplexing the 308-nm pulses (u, ultraviolet; r, red)
3 Ultraviolet Supercontinuum Generation
It was noted by Glownia et al. (1986b) that gentle focussing in air of the XeCl-amplified subpicosecond pulses resulted in a spectral broadening of the pulses by roughly a factor ten. Figure 8.5 shows a typical single-shot spectrum of an amplified 308-nm, 160-fs pulse after it has been focused in the laboratory atmosphere with a 1-m lens. The spectrum is almost 100 cm−1 wide, with the spectral enhancement being predominantly on the Stokes side. At the same time it was observed that the far-field pattern of the beam beyond the focal point of the 1-m lens usually contained bright spots in which the light was concentrated. These observations qualitatively suggested to us that SPM and self-focusing were involved in the above phenomena. However, due to the multimode spatial character of the beam, no direct steps were taken to verify these speculations. Instead, it was decided to experiment with a variety of gases (Ar, H2, N2, and CO2) under high pressure to see if any differences in spectral broadening could be discerned between the various gases and also whether spectral continua with widths in excess of ∼1000 cm−1 could be produced by this method. The results of our measurements were reported in Glownia et al. (1986a, 1986c). The main features are summarized below. Independently of us, P. Corkum’s group at National Research Council, Canada, observed the same basic phenomenon of supercontinuum generation from high-pressure gases (Corkum et al., 1986a, 1986b). In their case, both subpicosecond and picosecond amplified red pulses were successfully utilized as pump pulses. The basic physics of this newly discovered phenomenon is discussed by Corkum et al. (1986b). More recent observations and deductions about gas-phase supercontinuum generation are contained in Corkum and Rolland (Chapter 7 in the present volume).

Single-shot spectrum of amplified 308-nm pulse, recorded after the latter was focused in air with a 1-m lens
Figure 8.6 displays the spectrum of the energetic UV supercontinuum beam that emerges from a high-pressure Ar cell when high-power subpicosecond UV (308 nm) pulses are focused into the cell. Consistent with the finding of Corkum et al. (1986b), we observe nearly full transmission (>80%) of energy through the pressurized cell, with no significant degradation of the beam profile. There is thus adequate probe energy to pass through a photoexcited sample on to a high-dispersion visible-UV spectrograph, to be then recorded on an unintensified silicon diode array. With the supercontinuum source and spectrograph/detector system we normally employ, a spectral resolution of better than 0.3 Å is achieved. For probe continua at longer wavelengths, one must continue to rely on the various condensed-matter supercontinua (Alfano and Shapiro, 1970a, 1970b; Fork et al., 1982) or else on gas-phase supercontinua pumped by a red laser (Corkum et al., 1986a, 1986b), since all supercontinua peak at the pump wavelengths employed. It should be pointed out, however, that condensed-matter supercontinua peaking in the UV evidently cannot easily be generated. Our attempts to achieve this effect with the use of amplified subpicosecond 308-nm pulses as pump pulses were unsuccessful, possible due to nonlinear absorption in the various liquids tried.

Spectrum of UV supercontinuum beam emerging from Ar cell, p = 40 atm. Pump pulses (4 mJ, 308 nm, 160 fs) were focused into the middle of the 60-cm-long Ar cell with a 50-cm lens. Average of 64 shots
Because of the large spectral extent of supercontinuum pulses, they are broadened in time by group velocity dispersion (GVD). For condensed-matter visible supercontinua, Li et al. (1986) have measured spectral delays with the use of a streak camera and filters. Utilizing a cross-correlation technique, Fork et al. (1983) have measured the sweep of a supercontinuum generated in an ethylene glycol jet. We have ultilized a novel method, based on time-resolved absorption spectroscopy, to measure with subpicosecond resolution the frequency sweep of the supercontinuum displayed in Figure 8.6. This method, in the form in which it was originally demonstrated (Misewich et al., 1988a), is now briefly described.
Thallium chloride molecules, contained in a vapor cell at 450°C, were irradiated by 250-fs, 248-nm pump pulses derived (by a method to be described below) from 160-fs, 308-nm pulses. Supercontinuum probe pulses were simultaneously obtained from the same apparatus by focusing 4-mJ, 160-fs, 308-nm pulses into 40-atm Ar with a 50-cm lens. The probe pulses were directed into the TlCl vapor colinearly with the pump pulses, then dispersed in a spectrograph and recorded on an optical multichannel analyzer (OMA). Absorbances were computed by comparison of supercontinuum intensities recorded with and without the UV pump blocked.
Thallium chloride molecules irradiated at 248 nm undergo prompt dissociation into Tl and Cl atoms, with the former being distributed into ground 2 P 1/2 and first-excited 2 P 2/3 (7793 cm−1) states in roughly a 30:70% ratio (van Veen et al., 1981). Several allowed transitions, spanning a wide range of frequencies, connect the two states with various higher excited states. By plotting the individual rise times of the above atomic transitions as a function of pump-probe delay, one obtains the plot in Figure 8.7, which shows that the 40-atm Ar supercontinuum is characterized by a positive chirp of approximately 1340cm−1/ps.

Frequency sweep of 40-atm Ar supercontinuum beam whose spectrum is shown in Figure 8.6. Data points correspond to Tl absorptive transitions whose onsets were measured
Our technique utilizing TlCl photodissociation to measured the UV super-continuum frequency sweep has also revealed some interesting features of the photodissociation itself. We defer a discussion of the transient absorption spectra of photodissociating TlCl and TlI molecules until Section 6. From the results presented there, however, it can be stated that while the total duration of the gas-phase supercontinuum pulse emerging from the high-pressure cell is on the order of 10 ps, the effective time resolution is much better. As shown above, the probe continuum pulse has a fast red-to-blue linear sweep. Our spectral results (Section 6) indicate that the cross-correlation between the 160-fs, 308-nm pump pulse and a given wavelength interval of the swept probe pulse is stable to at least ±50 fs from shot to shot. It is this observed stability that gives the UV gas-phase supercontinuum pulse its good time resolution.
The UV supercontinuum pulses of Figure 8.6 can be directly used as seed pulses suitable for further amplification in KrF gain modules. From Figure 8.6 one sees that there is roughly a microjoule of energy in the supercontinuum between 248 and 249 nm, the wavelength range over which KrF amplification occurs. With this amount of input energy one easily obtains 6-mJ output pulses through single-pass amplification in a KrF gain module with a 45-cm-long discharge region (Glownia et al., 1986a, 1986c, 1987c). In Glownia et al. (1986a), the high-pressure cell used to form the KrF seed pulses contained H2 gas. We originally used hydrogen because of a coincidence between an anti-Stokes Raman wavelength and the wavelength at which maximum KrF gain occurs. Stimulated Raman scattering (SRS) occurs in high-pressure H2 gas when 160-fs, 308-nm pump pulses are applied, but not without the simultaneous occurrence of UV continuum generation. In several other molecular gases we failed to observe SRS with our 308-nm, 160-fs pump source, but we do observe UV continuum generation. Since SRS is known to be a ubiquitous phenomenon when high-power pulses of a few picoseconds duration are applied to high-pressure molecular gases (Mack et al., 1970), once must conclude that the conditions for its occurrence are made far less favorable as one proceeds to the subpicosecond domain, while the occurrence of supercontinuum generation becomes much more likely.
4 Subpicosecond Time-Resolved Infrared Spectral Photography
Time-resolved infrared spectral photography (TRISP) (Avouris et al., 1981; Bethune et al., 1981, 1983; Glownia et al., 1985) is a nonlinear optical technique by which a broadband (Δν ∼ 1000 cm−1) infrared absorption spectrum can be recorded in a single shot of a few nanoseconds duration. The IR spectral range that has thus far been convered with this technique is 2 to 11 μm (Bethune et al., 1983). Recently, we reported a successful extension of the TRISP technique to the subpicosecond time domain (Glownia et al., 1987a). The IR spectral region that can be probed with our present ultrafast apparatus is only 2.2 to 2.7 μm, but extension of subpicosecond capability to other IR ranges seems possible.
In this section we present details of the 2.2–2.7-μm subpicosecond TRISP apparatus. In the following section we describe the actual measurement of a subpicosecond photophysical event with the use of this apparatus.
In a TRISP apparatus, means for generating a broadband infrared sample probing pulse are combined with a method for upconverting and detecting this signal in the visible. Our ultrafast TRISP apparatus combines a new subpicosecond IR continuum generator with a standard TRISP upconverter. We first discuss formation of an ultrashort IR continuum. Powerful subpicosecond IR pulses at ∼2.4 μm are produced by stimulated electronic Raman scattering (SERS) in Ba vapor (Figure 8.8). The latter is contained at ∼10 torr pressure inside an Inconel pipe heated to ∼1050°C. The length of the heated region is ∼0.5 m. As pump pulses we directly utilize amplified 308-nm, 160-fs pulses. Although application of ∼20-ns XeCl laser pulses to Ba vapor is known (Burnham and Djeu, 1978; Cotter and Zapka, 1978) to produce SERS only on the 6s 2 1 S 0 → 6s5d 1 D 2 transition, with a Stokes output near 475 nm, we find, by contrast, that with ultrashort 308-nm excitation SERS occurs only on the 6s 2 1 S 0 → 6s7s 1 S 0 transition, with a Stokes output peaked near 2.4 μm. The 2.4-μm SERS output is highly photon efficient, with measured IR output pulse energies of ∼0.4 mJ for ∼5-mJ UV input pulses. The SERS threshold is lower than 1 mJ, even for an unfocused pump beam. Spectrally, the IR output is found to be a continuum, extending from 2.2 to 2.7 μm (Figure 8.9). Occurrence of the 2.4-μm SERS process is accompanied by the presence of a parametrically generated beam of light in the vicinity of the 535-nm Ba resonance line.

Diagram of the Ba SERS process

Spectrum of Raman Stokes light, recorded with the use of a scanning monochromator and PbS detector
We have also measured the ∼2.4-μm IR continuum pulse width by nonbackground-free autocorrelation, with the second harmonic being generated in a thin LiIO3 crystal. For the 250-fs, 308-nm pump pulses that were applied (Glownia et al., 1986d), the IR pulse width was determined to be ∼160 fs.
In Glownia et al. (1987a), a plausible argument was given to account for the switch of Stokes wavelength from 475 nm to 2.4 μm that occurs with ultrashort excitation. It was suggested that this switch could be attributed to the change in Raman gain regime (from stationary to transient) that occurs in going from ∼20 ns to subpicosecond 308-nm excitation pulses. In the stationary regime (t p > T 2), the intensity of the Stokes wave increases in accordance with the law
where Γ0 is the static gain, inversely proportional to the Raman linewidth. In the transient regime, the intensity of the Stokes wave assumes (for a square input pulse) the value (Akhmanov et al., 1972; Carman et al., 1970)
Since Γ0 is directly proportional to T 2, one sees that there is no dependence of Stokes gain on Raman linewidth in the transient regime. A possible explanation for the Stokes wavelength switch would thus be that the collisional linewidth of the 6s5d 1 D 2 state is sufficiently narrow compared with that of the 6s7s 1 S 0 state to favor Raman Stokes generation of 475 nm in the stationary case, even though the remaining factors in Γ0 favor Stokes generation at 2.4 μm. In the transient regime the gain is independent of T 2, and the above-mentioned remaining factors entirely determine the Stokes wavelength. No Ba linewidth data are available to support this contention. However, a crude calculation of the van der Waals interaction between a ground state Ba atom and a Ba atom in either the 6s7s 1 S 0 state or the 6s5d 1 D 2 state, with use of London’s general formula (Margenau, 1939), indicates a larger width for the 6s7s state.
We now discuss upconversion of the IR. With the use of a polished Si wafer, the horizontally polarized ultrashort IR pulses (ν IR ) are colinearly combined with the vertically polarized ∼15-ns pulses (ν L ) from a tunable narrowband furan 1 dye laser. With the timing between the two sources adjusted so that the subpicosecond IR pulses occur within the 15-ns-long dye laser pulses, both beams are sent into an Rb upconverter (Glownia et al., 1985) where the dye laser beam induces SERS on the Rb 5s → 6s Stokes transition, producing a narrowband vertically polarized Stokes wave ν S . Horizontally polarized, visible continuum pulses at ν L −ν S ± ν IR are then observed to emerge from the Rb cell when ν L is tuned to phase match either upconversion process.
Figure 8.10 shows a recording of the (lower-sideband) upconverted spectrum. In Figure 8.11 portions of two upconverted spectra are superimposed. In one case, the IR pulse was passed through an empty 20-cm cell; in the other case it was passed through the same cell filled with 200 torr of CO. The deduced absorbance is shown in Figure 8.12. A surprising finding is the observed increase in upconverted signal at the peaks of the CO 2–0 bands. This is explained as follows (Glownia et al., 1987a). Under the conditions of Figure 8.11 and 8.12, the upconverted signal was heavily saturated by the subpicosecond IR pulse; that is, too few photons at ν L and ν S were available during the actual IR pulse to allow efficient upconversion of the latter. Therefore a decrease in transmitted light due to molecular resonance absorption during the IR pulse did not result in a measurable decrease in upconverted signal. However, the coherently reemitted light of the molecules (Hartmann and Laubereau, 1984), occurring for a time the order of T 2 after the IR pulse, when the upconverter is no longer saturated, was able to be efficiently upconverted, resulting in the observed peaks. Positive IR absorption is observed in the upconverted spectra when the subpicosecond IR probe beam is sufficiently attenuated.

Upconverted TRISP spectrum (128-shot average, lower sideband). The spectrum is saturated (see text). The three absorptions are Rb excited state (5p) absorptions occurring at the visible wavelengths shown

Superimposed upconverted spectra (cell empty and filled with 200-torr CO gas). Each spectrum is the average of 128 shots

Absorbance formally deduced from Figure 8.11
5 Application of Subpicosecond TRISP: Measurement of Internal Conversion Rates in DABCO Vapor
We recently reported (Glownia et al., 1987c) the first application of subpicosecond time-resolved IR absorption spectroscopy to measure an ultrafast molecular process. This experiment combined subpicosecond 248-nm excitation with subpicosecond IR continuum probing to measure the \( \overset{\sim }{B}\to \tilde{A} \) internal conversion rate in DABCO vapor. A diagram of the photophysical processes involved is shown in Figure 8.13. The idea that the \( \overset{\sim }{B}\to \tilde{A} \) internal conversion rate in DABCO might be high enough to require ultrafast techniques for its measurement is contained in an earlier study (Glownia et al., 1985), in which the population of the \( \tilde{A} \) state was monitored following the application of a 30-ns, 248.5-nm KrF laser excitation pulse. A high \( \overset{\sim }{B}\to \tilde{A} \) internal conversion rate for DABCO was also implied in a recent two-color laser photoionization spectroscopy study (Smith et al., 1984). In the above-mentioned earlier study of DABCO by our group, nanosecond TRISP was used to monitor the \( \tilde{A} \) state population. This was because the \( \overset{\sim }{B}\leftarrow \tilde{A} \) transition (occurring at ∼2.5 μm) was found to have a much higher oscillator strength than all other transitions connecting the \( \tilde{A} \) state with higher electronic states (Glownia et al., 1985). Since the subpicosecond TRISP apparatus described in the last section monitors the region 2.2 to 2.7 μm, we decided to measure the DABCO \( \overset{\sim }{B}\to \tilde{A} \) internal conversion with the greatly improved time resolution this apparatus offers.

Diagram of photophysical processes involved in the subpicosecond DABCO experiment
Collimated 2-mJ, 250-fs, 248.5-nm pulses were sent unfocused (beam dimensions: 2 cm × 1cm) into a 60-cm-long cell containing DABCO at its ambient vapor pressure (∼0.3 torr) together with 100 torr of H2. The linear absorption of the DABCO at 248.5 nm (40,229 cm−1) was more than 50%, even though this wavelength lies near the point of minimum absorbance between the ν′ = 0 ← ν″ = 0 (39,807 cm−1) and next highest vibronic peaks of the lowest-energy, dipole-allowed, band (Halpern et al., 1968; Hamada et al., 1973).This band system has been assigned (Parker and Avouris, 1978, 1979) as \( {\overset{\sim }{B}}^{\kern-.2em 1}{E}^{\prime}\left[3{p}_{x,y}\left(+\right)\right]\leftarrow {\overset{\sim }{X}}^{\kern-.2em 1}{A}_1^{\prime}\left[n\left(+\right)\right] \). Optical transitions from the ground state \( \overset{\sim }{X} \) to the first excited state, the \( {\tilde{A}}^1{A}_1^{\prime}\left[3s\left(+\right)\right] \) (origin at 35,785 cm−1), are one-photon forbidden, two-photon allowed (Parker and Avouris, 1978, 1979).
The 160-fs IR continuum pulses that probe the \( \tilde{A} \) state population were directed through the vapor collinearly with the UV photoexcitation pulses, upconverted to the visible, and then dispersed in a spectrograph equipped with an unintensified OMA detection system. The pump-probe delay could be varied up to ±ns by means of an optical delay arm. Absorbances were computed by comparison of upconverted intensities recorded with and without the UV pump blocked.
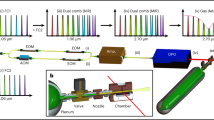
A block diagram of the experiment is shown in Figure 8.14. As described in Section 2, subpicosecond pulses at ∼616 nm formed in a CPM laser are amplified, then frequency doubled, forming seed pulses at ∼308 nm for further amplification in the XeCl excimer gain module. Amplification of the UV pulses in the latter occurs in the form of pairs of orthogonally polarized pulses, spaced 2 to 3 ns apart, formed in the multiplexer described also in Section 2. The 160-fs amplified UV pulse pairs are separated by a polarization-sensitive coupler into pump and probe channels. The pump channel 308-nm pulses are focused into high-pressure gas to form seed pulses for amplification at 248.5 nm in a KrF module (see Section 3). The probe channel pulses are Raman shifted in Ba vapor to form IR probe continuum pulses (see Section 4). The narrowband pulsed dye laser drives the Rb upconverter (see Section 4).

Diagram of experimental apparatus for the DABCO experiment
Figure 8.15a shows the absorbance recorded when the probe is delayed ∼4 ps with respect to the pump (point (a) in Figure 8.16), while Figure 8.15b displays the absorbance with the probe arriving just before the pump (point (b) in Figure 8.16). The absorbance recorded at 2.494 μm, as a function of probe delay, is shown in Figure 8.16. The large absorption band that develops represents transitions \( {\overset{\sim }{B}}^{\dagger}\leftarrow {\tilde{A}}^{\dagger } \) of vibrationally excited \( \tilde{A} \) state molecules, containing up to 4400 cm−1 of vibrational energy. Since the \( \overset{\sim }{B}\leftarrow \tilde{A} \) transition is one that occurs between Rydberg states, vertical (Δν = 0) transitions are expected. Thus it is not surprising that the peak of the band in Figure 8.15a appears very close to the \( \overset{\sim }{B}\leftarrow \tilde{A} \) peak for vibrationally equilibrated \( \tilde{A} \) state molecules, shown here in Figure 8.17 and described in detail in Glownia et al. (1985). However, the width of the \( \overset{\sim }{B}\leftarrow \tilde{A} \) band is obviously greater in the vibrationally excited case.

(a) Absorbance (base 10) with probe delayed ≈4 ps with respect to pump. (b) Absorbance with probe pulse preceding pump pulse

Peak \( {\overset{\sim }{B}}^{\dagger}\leftarrow {\tilde{A}}^{\dagger } \) absorbance as a function of probe pulse delay time with respect to pump pulse

Absorbance measured with probe pulse delayed 75 ns with respect to pump pulse. The latter was an 8-mJ, 25-ns pulse obtained by operating the KrF gain module as a conventional laser. 60-cm DABCO cell, 3-atm H2 pressure
The computer-generated curve in Figure 8.16 is a nonlinear least squares fit to the data. The fit indicates a rise time of ∼500 fs. Although the infrared and ultraviolet pulse widths were determined by autocorrelation to be ∼160 and ∼250 fs, respectively, the cross-correlation between these pulses has not been measured. Thus, the rise time in our experiment could be limited by the laser system cross-correlation. In any case, the process converting DABCO states accessed by the subpicosecond 248.5-nm pump beam into vibrationally excited \( \tilde{A} \) states is observed to occur on a time scale that is at least as fast as ∼500 fs. That internal conversion to vibrationally excited \( \tilde{A} \) state molecules is the dominant process for photoexcited DABCO molecules, even at UV pump intensities of ∼4GW/cm2, is also underscored by the fact that there is no apparent decrease in the integrated intensity of the 2.5-μm absorption band induced at these pump intensities, as compared with the 2.5-μm integrated intensity induced by 25-ns UV pulses at comparable fluence levels (compare Figures 8.15a and 8.17).
The DABCO experiment shows how subpicosecond TRISP can be used to monitor the internal conversion of photoexcited molecules in real time. In the sequence of spectra corresponding to the data points in Figure 8.16, it is clearly seen that the wavelength at which the peak absorbance occurs undergoes a definite blue-to-red shift as the magnitude of the absorbance grows in. It is tempting to attribute this to an intramolecular vibrational redistribution (IVR) process. However, heavy caution must be applied here, since the same apparent phenomenon could easily be induced by a red-to-blue sweep of the IR probe continuum. We have no information at present as to whether or not the IR continuum is swept.
6 Preliminary Results on the Application of the UV Supercontinuum Probe
In Section 3 we described a convenient method, based on photofragmentation of thallium halides, by which the sweep of the UV supercontinuum can be measured. In the process of measuring the rise times of the Tl absorption lines, we have consistently noted that the latter assume unusual line shapes, with enhanced integrated intensities, for a period lasting roughly a picosecond, beginning the moment the atomic absorption is first discerned and ending when the asymptotic, normal appearing, absorption line profile is finally attained. We present some of these preliminary spectral results in this section. These results are qualitatively discussed in terms of a model based on the transient behavior of the polarization induced by the subpicosecond swept UV continuum as the latter interacts with the time-varying population of two-level atoms produced by the photolysis pulse. A detailed description of our model will be presented elsewhere (Misewich et al., 1988b).
There has been broad interest for some time in the spectroscopy of the thallium halides. The ultraviolet absorption cross sections have been measured by Davidovits and Bellisio (1969). The UV absorption spectra comprise a number of well-defined bands whose conformity between the various halides is striking. In the most recent thallium halide photofragmentation study (van Veen et al., 1981), these bands are simply labeled A, B, C, and D. In that work, the time-of-flight spectra and angular distributions of photofragments were measured for the thallium halides at a variety of UV wavelengths, including 308 and 248 nm. We have now utilized subpicosecond pulses, at both 308 and 248 nm, to separately excite TlI and TlCl molecules. In the case of the former, 308 nm is very close to the peak of the C band, while 248 nm lies on the high-frequency side of the D band. For the latter, 308 nm is near the peak of the B band, while 248 nm excites the C band.
Figures 8.18, 8.19, and 8.20 show the appearance of time-resolved absorption spectra recorded in the vicinity of the 377.6-nm Tl 7S 1/2 ← 6P 1/2 resonance line following the application of 160-fs, 308-nm pump pulses to TlI vapor. It is seen that roughly 1 ps elapses from the moment the atomic transition appears to the point at which no further changes in the appearance of the atomic resonance line occur. From the TOF data presented in van Veen et al. (1981), iodine atoms produced by 308-nm photolysis of TlI have a relatively broad distribution of translational velocities peaking at ∼4.4 × 104 cm/s. Hence the average Tl–I separation at large distances must increase as ∼7.1 × 104 cm/s. However, at smaller distances the average rate of increase of separation is much smaller, because the atoms are accelerated from rest. We have been unable to mark the exact time of occurrence of the 308-nm pump pulse with respect to the times shown in Figures 8.18 to 8.20. However, if one assumes the pump pulse occurs somewhere in the vicinity of Figures 8.18a and b, one deduces that the asymptotic line shape must be attained well before the Tl-I separation has increased by 7.1 Å.

Time-resolved absorption spectra, recorded in the vicinity of the 377.6 -nm Tl 7S 1/2 ← 6P 1/2 resonance line, following the application of 160-fs, 308-nm pulses to TII vapor. Pump-probe separation increased by 100-fs between each spectrum shown. Each spectrum represents the average absorbance (base 10) deduced from dual 64-shot continuum accumulations, one with and one without the 308-nm pump blocked. The dashed line marks the position of the asymptotic resonance peak

Continuation of Figure 8.18, with pump-probe separation in (a) increased by 100 fs over that in Figure 8.18 (d)

The most striking feature of Figures 8.18 to 8.20 is, of course, the dispersion-like appearance of the atomic absorption feature for the first 600 or 700 fs, with evidence of a spectral region in which apparent gain prevails. A superficially similar phenomenon was recently observed by Fluegel et al. (1987) in their femtosecond studies of coherent transients in semiconductors. However, in that work the dispersive structure observed in the normalized differential transmission spectra in the region of the excition resonance, when pump and probe pulses overlapped in time, was attributed to the frequency shift of the exciton resonance, i.e., the optical Stark shift. In the case of Figures 8.18 to 8.20 there is no preexisting absorption line to be shifted when the pump is applied.
The appearance of the absorption spectra when the 308-nm pump intensity is reduced by roughly a factor 3 (0.5 ND filter inserted in the pump arm) is shown in Figures 8.21 and 8.22. From the entire sequence of spectra constituting this particular experiment, we have selected the eight consecutive spectra that most closely correspond with Figures 8.18 and 8.19. (Exact correspondence between the pump-probe delays of Figures 8.18 to 8.20 was not possible to maintain because of the insertion of the 0.5 ND filter in the pump arm.) The peak absorbances are clearly saturated. However, the integrated absorbances may not be so, since the spectra in Figures 8.21 and 8.22 are clearly narrower. This line broadening is a feature we have observed in all our Tl-halide spectra, with both 308- and 248-nm pumping and in both TlI and TlCl. Widths of all Tl absorption lines, even those measured at very long pump-probe separations, are dependent on the UV pump intensity applied. A reasonable explanation of this phenomenon is Stark broadening due to creation of ions or electrons in the vapor by the UV pump pulse.


Figures 8.23 to 8.26 show the appearance of the absorption spectra as a function of time in the vicinity of the 351.9-nm 6D 5/2 ← 6P 3/2 and 352.9-nm 6D 3/2 ← 6P 3/2 absorption lines, following application of a 308-nm, 160-fs excitation pulse to TlI vapor. These transitions thus monitor thallium atoms in the excited 6P 3/2 state (7793 cm−1). Note the absence of any evident spectral region with apparent gain during any part of the sequence. However, there are again strong transiently appearing asymmetries in the two line shapes.

Time-resolved spectra, recorded in the vicinity of the 351.9-nm 6P 5/2 ← 6P 3/2 and 352.9-nm 6P 3/2 ← 6P 3/2 Tl absorption lines, following the application of 160-fs, 308-nm pulses to TlI vapor. Pump-probe separation increased by 100 fs between spectra



Generally similar results were obtained in the case of 248-nm pump excitation of TlI, and with both 248- and 308-nm excitation of TlCl. In the case of TlI excited by a 248-nm pump, the time evolution of the line shapes of the two resonances near 352 and 353-nm is similar to that shown in Figures 8.23 to 8.26, with pronounced red wings during a period of again approximately a picosecond. The 377.6-nm line is much less intense and as a result comparatively difficult to minotor. These appears again to be a transiently appearing negative absorption on the high-frequency side of the line, but its magnitude is much less than the magnitude of the differential positive absorption appearing on the low-frequency side of the line. The latter absorbance monotonically grows to a final value of ∼0.1 for the same UV pump powers for which the 353-nm absorbance (the weak line in Figures 8.23 to 8.26) almost attains the value 0.4. Clearly, a large inversion on the 6P 3/2 − 6P 1/2 transition is produced by 248-nm photolysis of TlI.
For the TlCl, with 248-nm excitation, the 352-nm absorption develops a pronounced blue wing, in contrast with the cases discussed above. This blue wing is evident for roughly 1.2 ps, then quickly disappears as the final line shape is assumed. During the 1.2-ps interval, the magnitude of the integrated absorption of the 352-nm line is enhanced by at least a factor 2, relative to that for the asymptotically attained line shape. The 377.6-nm line appears to grow monotonically to its asymptotic value, without noticeable line shape distortions other than a slight negative differential absorption on the high-frequency side of the line. For TlCl with 308-nm pumping, there is no observed 352- or 353-nm absorption feature, in agreement with the specific finding of van Veen et al. (1981) that only one dissociative channel (either Tl + Cl or Tl + Cl*) is active when TlCl is pumped at 308 nm. Since this should be the simplest situation to analyze, we present in Figures 8.27 to 8.29 some of the observed spectra for this specific case. A region of negative differential absorption is again clearly seen in some of the spectra (Figures 8.28a–d, Figure 8.29a).

Time-resolved spectra, recorded near the 377.6-nm Tl line, following application of 160-fs, 308-nm pulses to TlCl vapor. 100-fs steps between successive spectra


We now present a qualitative explanation for the unusual spectral line shapes observed for the first picosecond following the photolysis pulse. Our model is based on the transient behavior of the polarization induced by the subpicosecond swept UV continuum pulse as the latter interacts with the time-varying (growing) population of two-level atoms produced by the photolysis pulse. As we will show below, in order to get a reasonable correspondence of the line shapes calculated in this manner with the observed line shapes, especially with those possessing the most unusual feature (e.g., Figures 8.18 to 8.20), one has to allow the created atoms to undergo continuous frequency shifts in time for a period after the photolysis pulse.
The numerical calculations we have performed are basically straightforward. An optically thin sample is assumed. There are two contributions to the output field from the resonant vapor, i.e., E tot(t, z) = E in(t, z) + E rad(t, z), where E in(t, z) is the swept UV continuum and E rad(t, z) is the field radiated by the polarization it induces in the medium. Various contributions to the latter are numerically calculated, based on solutions of Schrödinger’s equation for a two-level atom interacting with a swept continuum pulse
where a 1 and a 0 are the time-dependent amplitudes of the upper and lower atomic states, and ω a is the atomic resonance frequency, equal to ω 1 − ω 0. The rotating-wave approximation has been made in Eq. (3). We assume the swept continuum pulse to have the form
with
and
Equations (4) to (6) imply that the instantaneous frequency of the UV continuum sweeps across the frequency ω c at time t = 0. The value of b was taken to correspond with the value we measured for the supercontinuum sweep rate, 1340 cm−1/ps. Equation (6) shows the form of the swept continuum pulse amplitude that we assumed in our numerical integrations. We generally specified a width Δ on the order of a picosecond. The wave function for the atom is
and the polarization P is generally expressed as
where N is the atomic density.
We now outline the general procedure that was followed in obtaining numerical solutions. Let a 0 = R(t − t i ) be the amplitude of the lower state for an atom created at time t i . The solution to Eq. (3) can be formally expressed as
The contribution to the polarization (per atom) is
Equation (9) was numerically integrated with the use of a specific rise function
The numerical integration in Eq. (9) was combined with an additional integration over another distribution function: D(t) = dN(t)/dt, where N(t) represents the atomic population. We specified D(t) to be proportional to the quantity sech2 [(t − TSECH2)/WSECH2]. Here the quantity TSECH2 marks the time at which the growth rate of N(t) achieves its maximum, and WSECH2 characterizes the width of the growth period. Thus the total polarization P is given by
We are modeling the dissociation of a diatomic molecule to create two atoms. At early times in the dissociation, when the two atoms are close to one another, the atomic transition frequencies are perturbed by the bending of the potential surfaces. We allowed for the existence of a continuous red (or blue) shift of the atomic transition frequency by making ω a in the above equations a function of time relative to the creation of the atom:
where \( {\omega}_a^0 \) is the unperturbed atomic transition frequency.
In the slowly varying envelope approximation (SVEA), it is assumed that the total field E tot(t, z) and polarization P(t, z) can be written in the following forms:
and
where \( \mathfrak{e}\left(t,z\right) \) and \( \mathfrak{p}\left(z,t\right) \) are complex functions of z and t that vary little in an optical period or wavelength. Following the usual procedure of neglecting second derivatives of the slowly varying quantities \( \mathfrak{e}\left(t,z\right) \) and \( \mathfrak{p}\left(t,z\right) \), one obtains on substitution of (14) and (15) into the wave equation the well-known complex field self-consistency equation
If one defines a retarded time τ = t − z/c, one can rewrite Eq. (16) as
From Eqs. (9), (10), (12), and (15), \( \mathfrak{p}\left(z,\tau \right) \) is seen to have no explicit dependence on \( z:\mathfrak{p}\left(z,\tau \right)=\mathfrak{p}\left(\tau \right) \). Thus we can integrate Eq. (17) to obtain
Hence, in general,
with the last term of Eq. (19) representing the field E rad(z, t) radiated by the polarization induced in the medium by E in(z, t). The spectral dependence of the total field E tot(z, t) is given by the sum of the Fourier transforms \( {\overset{\sim }{\mathfrak{e}}}_{\mathrm{in}}\left(\omega \right) \) and \( {\overset{\sim }{\mathfrak{e}}}_{\mathrm{rad}}\left(\omega \right) \), and the spectral dependence of the collected intensity at the end of the vapor cell is given by
With the use of fast Fourier transform numerical computation techniques, and with the quantity ω c in Eq. (9) set equal to \( {\omega}_a^0 \), we obtained computer plots of the quantity \( { \log}_{10}\left[{I}_{\mathrm{out}}\left(\omega \right)/{\left|{\overset{\sim }{\mathfrak{e}}}_{\mathrm{in}}\left(\omega \right)\right|}^2\right] \) for various choices of the parameters introduced above.
An example of a sequence of calculated spectra is shown in Figures 8.30 to 8.32. The parameters used here were chosen in an attempt to approximate the shape of the observed spectra shown in Figures 8.18 to 8.20. It is seen that a qualitative agreement exists between theory and experiment, with the particular observed feature of a transient spectral region of negative absorbance clearly captured in the calculated spectra. In Figures 8.30 to 8.32, the quantity γ was chosen to correspond to a polarization dephasing time of T 2 = 1.3 ps. This value results in an asymptotic atomic linewidth (∼6cm−1 FWHM) that closely approximates the measured value.

Calculated transient absorption spectra for a sequence of pump-probe separations increasing by 100 fs between successive spectra. The following parameter values were used: γ = 0.0015 fs−1, WSECH2 = 150 fs, WTANH = 50 fs, RMAX = 40 cm−1, RTAU = 600 fs


Several general conclusions can be drawn by examining the shapes of the various calculated spectra. Most important, without the inclusion of a red shift RMAX very little asymmetry appears in the spectra, and there is no significant negative absorption.
The rise time of the absorption seems to be simply related to the quantity WSECH2, for all values of RMAX.
For a given T 2, even for a relatively large RMAX (e.g., 60 cm−1), decreasing RTAU below T 2/10 has the effect of reducing the height of the transiently appearing wings, so that the spectra are dominated at all times by a symmetric peak centered at \( {\omega}_a^0 \). To get more pronounced wings and a greater asymmetry for a given RMAX, one has to increase RTAU. However, if RTAU becomes comparable to T 2, additional oscillatory half-cycles of absorption and gain begin to appear in the absorption spectrum. For RTAU >> T 2, a simple growth of the absorption line at a frequency \( {\omega}_a^0 \) − RMAX is observed.
A blue atomic resonance frequency shift reverses the asymmetry, producing transient negative absorption in the region \( \omega <{\omega}_a^0 \). Changing the direction of the probe continuum sweep, however, does not appear to affect the appearance of the absorption spectrum, at least with the use of the b value appropriate for our case.
We generally used a single value (50 fs) for WTANH. The spectra were seen to be generally insensitive to the choice of this parameter, provided it was taken to be short enough.
To summarize very briefly, it appears that the transient absorption spectra we have obtained of photolytically produced atoms contain qualitative information regarding the “transition state” that occurs between the time a molecule has absorbed a UV photon and the time its constituent atoms have fully separated. Specifically, information can be obtained about the rise time of the atomic population and the frequency shift that the atom undergoes during dissociation. However, information about the latter tends to be diluted by the polarization dephasing time T 2 of the separating atoms.
We now describe a brief result obtained by applying the UV supercontinuum to the gas-phase molecule chlorine dioxide, OClO. For several decades it has been assumed that the sole result of applying light to the structured OClO (\( {\tilde{A}}^2{A}_2\leftarrow {\overset{\sim }{X}}^2{B}_1 \)) band system, which extends from ∼460 to 280 nm, is the photolytic production of ClO + O (Bethune et al., 1983). From OClO absorption spectra taken at very high spectral resolution, in which the widths of individual vibronic lines could be measured, it was inferred that OClO predissociates at a rate strongly dependent on how far above the origin one photoexcites the molecule (Michielson et al., 1981).
As an initial test and possible calibration of the UV gas-phase supercontinuum, we attempted an experiment to photolyze OClO with a 160-fs, 308-nm UV pulse and then probe the appearance of the ClO radical with the UV supercontinuum pulse. The ClO radical strongly absorbs from ∼310 to 250 nm, a region largely nonoverlapping with the OClO absorption band. With 308-nm excitation, subpicosecond predissociation rates for OClO were expected.
When the above spectral region was probed, no ClO was detected, with pump-probe delays of up to a nanosecond tried. The 160-fs, 308-nm excitation pulse was then replaced by a standard 25-ns, 308-nm pulse with the same 0.04J/cm2 fluence. (We sent 3 mJ through a 3-nm aperture into a 10-nm cell containing 5 torr OClO.) Again, no significant absorption was detected at relatively short delays (Figure 8.33a). However, if the probe was delayed by several hundred nanoseconds, the fully developed ClO spectrum was easily observed (Figure 8.33b). We thus conclude that 308-nm photolysis of OClO produces \( \mathrm{C}\mathrm{l}+{\mathrm{O}}_2^{\ast } \), with \( {\mathrm{O}}_2^{\ast } \) most likely being in high vibrational levels of the \( {}^1{\sum}_g^{+} \) state. The observed slow appearance of ClO results from the combination of Cl atoms with unphotolyzed parent OClO molecules

(a) Single-pass absorbance spectrum recorded through a 10-cm cell filled with 5-torr OClO with probe pulse occurring at the end of a 25-ns, 308-nm photolysis pulse. Pump energy of 3 mJ was sent through a 3-nm aperture. (b) Same as (a) but recorded 900 ns after the photolysis pulse
For 5 torr of unphotolyzed OClO present in the cell and with use of the known 300 K rate constant for (21), 6 × 10−11 cm3 molec−1 s−1, one predicts the ClO would appear in ∼100 ns, approximately what was observed. This OClO result should have a large impact on further understanding the interesting sequence of chemical reactions initiated by photolysis of OClO (Bethune et al., 1983).
7 Promising Directions for Subpicosecond Kinetic Spectroscopy
In the Introduction we presented an example of a unimolecular photochemical reaction, the rearrangement of methyl azide (Figure 8.1). The example was primarily meant to be illustrative. The IR subpicosecond continua required to probe the most relevant vibrational transitions have not yet been developed. However, through the use of the UV subpicosecond gas-phase continuum discussed in the preceding sections, an alternative method of attacking this problem, and many similar problems, now appears to be possible. Specifically, it should now be possible to observe momentarily in the UV the singlet spectrum of methylnitrene (CH3N) and then to watch it disappear as the molecule isomerizes to singlet methylenimine (CH2NH). Demuynck et al. (1980) have predicted little or no barrier for isomerization from singlet CH3N to singlet CH2NH, but they also predict a sizable barrier (53 kcal/mol) for isomerization of triplet CH3N. In recent years, searches for optical spectra of CH3N were made in several photolysis and pyrolysis studies, but no CH3N was ever observed in any of these studies, most likely because of rapid isomerization of singlet CH3N to CH2NH. (By the spin conservation selection rule, photolysis of methyl azide has to produce singlet nitrene, since the ground state of N2 is a singlet.) Recently, the \( {\tilde{A}}^3E-{\overset{\sim }{X}}^3{A}_2 \) ultraviolet emission spectrum of triplet CH3N was observed (Carrick and Engelking, 1984; Franken et al., 1970) by reacting methyl azide with metastable (\( {A}^3{\sum}_u^{+} \)) N2 in a flowing afterglow. This result qualitatively confirms the prediction of a high triplet isomerization barrier height made by Demuynck et al. (1980). The triplet CH3N (0, 0) band occurs at 314.3 nm, not too distant from the 336-nm origin of the A 3Π − X 3Σ− band system of the isoelectronic radical NH. Since NH has also an allowed singlet system (c 1Π − a 1Δ), with origin at 324 nm, one should expect an analogous singlet CH3N system to exist, with an origin somewhere in the region of 300 nm. Methyl azide can be photolyzed at either 248 or 308 nm. With the effective good time resolution of the swept UV supercontinuum demonstrated in Section 6, one should be able to observe the transient singlet band system of CH3N, even if the latter isomerizes in a time of half a picosecond.
We conclude by mentioning one improvement in the subpicosecond kinetic spectroscopy technique we employ that is scheduled to be tried soon. This is the incorporation of a reference arm for improved sensitivity. With the use of two matched spectrograph-OMA systems, it should be possible to cancel in every shot the effect of random spectral variations in the supercontinuum intensity. At present, we require two independent accumulations of several tens of shots, one with the pump blocked and one with the pump unblocked, from which the spectral variation of absorbance is electronically calculated. Incorporation of such a reference arm is, of course, simpler with the UV supercontinuum than in the case of the IR continua, due to the need to upconvert the latter. It will be interesting to see how much subpicosecond kinetic spectroscopy can benefit from such an improvement once it is implemented.
Acknowledgments We are indebted to J.E. Rothenberg for suggesting to us an initial conceptual framework for the calculations. We also thank M.M.T. Loy and D. Grischkowsky for helpful discussions. L. Manganaro assisted us with some of the instrumentation. This research was sponsored in part by the U.S. Army Research Office.
Notes
- 1.
The term “kinetic spectroscopy” was used by G. Porter, R. Norrish, and others, who pioneered the field of flash photolysis. Since the technique we describe in this chapter also relies on the use of a time-resolved, broadband, absorption spectroscopy probe, we choose to describe our work with the same term, with the addition, however, of the prefix “subpicosecond.”
References
Akhmanov, S.A., K.N. Drabovich, A.P. Sukhorukov, and A.K. Shchednova (1972) Combined effects of molecular relaxation and medium dispersion in stimulated Raman scattering of ultrashort light pulses. Sov. Phys. JETP 35, 279-286.
Alfano, R.R. and S.L. Shapiro (1970a) Emission in the region 4000 to 7000 Å via four-photon coupling in glass. Phys. Rev. Lett. 24, 584-587.
Alfano, R.R. and S.L. Shapiro (1970b) Observation of self-phase modulation and small scale filaments in crystals and glasses. Phys. Rev. Lett. 24, 592-594.
Avouris, Ph., D.S. Bethune, J.R. Lankard, J.A. Ors, and P.P. Sorokin (1981) Time-resolved infrared spectral photography: study of laser-initiated explosions in HN3. J. Chem. Phys. 74, 2304-2312.
Bethune, D.S., J.R. Lankard, P.P. Sorokin, R.M. Plecenik, and Ph. Avouris (1981) Time-resovled infrared study of bimolecular reactions between tert-butyl radicals. J. Chem. Phys. 75, 2231-2236.
Bethune, D.S., A.J. Schell-Sorokin, J.R. Lankard, M.M.T. Loy, and P.P. Sorokin (1983) Time-resolved study of photo-induced reactions of chlorine dioxide. In B.A. Garetz and J.R. Lombardi (eds.), Advances in Laser Spectroscopy, vol. 2, pp. 1-43. Wiley, New York.
Burksbaum, P.A., J. Bokor, R.H. Storz, and J.C. White (1982) Amplification of ultrashort pulses in krypton fluoride at 248 nm. Opt. Lett. 7, 399-401.
Burnham, R. and N. Djeu (1978) Efficient Raman conversion of XeCl-laser radiation in metal vapors. Opt. Lett. 3, 215-217.
Carman, R.L., F. Shimizu, C.S. Wang, and N. Bloembergen (1970) Theory of Stokes pulse shapes in transient stimulated Raman scattering. Phys. Rev. A 2, 60-72.
Carrick, P.G. and P.C. Engelking (1984) The electronic emission spectrum of methylnitrene. J. Chem. Phys. 81, 1661-1665.
Corkum, P.B. and R.S. Taylor (1982) Picosecond amplification and kinetic studies of XeCl. IEEE J. Quantum Electron. QE-18, 1962-1975.
Corkum, P.B., C. Rolland, and T. Srinivasan-Rao (1986a) Supercontinuum generation in gases: a high order nonlinear optics phenomenon. In G.R. Fleming and A.E. Siegman (eds.), Ultrafast Phenomena V, pp. 149-152. Springer-Verlag, New York.
Corkum, P.B., C. Rolland, and T. Srinivasan-Rao (1986b) Supercontinuum generation in gases. Phys. Rev. Lett. 57, 2268-2271.
Cotter, D. and W. Zapka (1978) Efficient Raman conversion of XeCl excimer laser radiation in Ba vapour. Opt. Commun. 26, 251-255.
Dantus, M., M.J. Rosker, and A.H. Zewail (1987) Real-time femtosecond probing of “transition states” in chemical reactions. J. Chem. Phys. 87, 2395-2397.
Davidovits, P. and J.A. Bellisio (1969) Ultraviolet absorption cross sections for the thallium halide and silver halide vapors. J. Chem. Phys. 50, 3560-3567.
Demuynck, J., D.J. Fox, Y. Yamaguchi, and H.F. Schaefer III (1980) Triplet methyl nitrene: an indefinitely stable species in the absence of collisions. J. Am. Chem. Soc. 102, 6204-6207.
Egger, H., T.S. Luk, K. Boyer, D.F. Muller, H. Pummer, T. Srinivasan, and C.K. Rhodes (1982) Picosecond, tunable ArF* excimer laser source. Appl. Phys. Lett. 41, 1032-1034.
Fluegel, B., N. Peyghambarian, G. Olbright, M. Lindberg, S.W. Koch, M. Joffre, D. Hulin, A. Migus, and A. Antonetti (1987) Femtosecond studies of coherent transients in semiconductors. Phys. Rev. Lett. 59, 2588-2591.
Fork, R.L., B.I. Greene, and C.V. Shank (1981) Generation of optical pulses shorter than 0.1 psec by colliding pulse mode locking. Appl. Phys. Lett. 38, 671-672.
Fork, R.L., C.V. Shank, R.T. Yen, and C. Hirlimann (1982) Femtosecond continuum generation. In K.B. Eisenthal, R.M. Hochstrasser, W. Kaiser, and A. Laubereau (eds.), Picosecond Phenomena III, pp. 10-13. Springer-Verlag, New York.
Fork, R.L., C.V. Shank, C. Hirlimann, R. Yen, and W.J. Tomlinson (1983) Femtosecond white-light continuum pulses. Opt. Lett. 8, 1-3.
Franken, Th., D. Perner, and M.W. Bosnali (1970) UV-absorptionsspektren von methyl- und äthylnitren mittels pulsradiolyse in der gasphase. Z. Naturforsch. A 25, 151-152.
Glownia, J.H., G. Arjavalingam, and P.P. Sorokin (1985) The potential of DABCO for two-photon amplification. J. Chem. Phys. 82, 4086-4101.
Glownia, J.H., J. Misewich, and P.P. Sorokin (1986a) Ultrafast ultraviolet pump-probe apparatus. J. Opt. Soc. Am. B 3, 1573-1579.
Glownia, J.H., G. Arjavalingam, P.P. Sorokin, and J.E. Rothenberg (1986b) Amplification of 350-fsec pulses in XeCl excimer gain modules. Opt. Lett. 11, 79-81.
Glownia, J.H., J. Misewich, and P.P. Sorokin (1986c) New excitation and probe continuum sources for subpicosecond absorption spectroscopy. In G.R. Fleming and A.E. Siegman (eds.), Ultrafast Phenomena V, pp. 153-156. Springer-Verlag, New York.
Glownia, J.H., J. Misewich, and P.P. Sorokin (1986d) Amplification in a XeCl excimer gain module of 200-fsec UV pulses derived from a colliding pulse mode locked (CPM) laser system. Proc. Soc. Photo-Opt. Instrum. Eng. 710, 92-98.
Glownia, J.H., J. Misewich, and P.P. Sorokin (1987a) Subpicosecond time-resolved infrared spectral photography. Opt. Lett. 12, 19-21.
Glownia, J.H., J. Misewich, and P.P. Sorokin (1987b) 160-fsec XeCl excimer amplification system. J. Opt. Soc. Am. B 4, 1061-1065.
Glownia, J.H., J. Misewich, and P.P. Sorokin (1987c) Subpicosecond IR transient absorption spectroscopy: measurement of internal conversion rates in DABCO vapor. Chem. Phys. Lett. 139, 491-495.
Halpern, A.M., J.L. Roebber, and K. Weiss (1968) Electronic structure of cage amines: absorption spectra of triethylenediamine and quinuclidine. J. Chem. Phys. 49, 1348-1357.
Hamada, Y., A.Y. Hirikawa, and M. Tsuboi (1973) The structure of the triethylenediamine molecule in an excited electronic state. J. Mol. Spectrosc. 47, 440-456.
Hartmann, H.-J. and A. Laubereau (1984) Transient infrared spectroscopy on the picosecond time-scale by coherent pulse propagation. J. Chem. Phys. 80,4663-4670.
Li, Q.X., T. Jimbo, P.P. Ho, and R.R. Alfano (1986) Temporal distribution of picosecond super-continuum generated in a liquid measured by a streak camera. Appl. Opt. 25, 1869-1871.
Mack, M.E., R.L. Carman, J. Reintjes, and N. Bloembergen (1970) Transient stimulated rotational and vibrational Raman scattering in gases. Appl. Phys. Lett. 16, 209-211.
Margenau, H. (1939) Van der Waals forces. Rev. Mod. Phys. 11, 1-35.
Michielson, S., A.J. Merer, S.A. Rice, F.A. Novak, K.F. Freed, and Y. Hamada (1981) A study of the rotational state dependence of predissociation of a polyatomic molecule: the case of ClO2. J. Chem. Phys. 74, 3089-3101.
Misewich, J., J.H. Glownia, and P.P. Sorokin (1988a) Measurement with subpicosecond resolution of the frequency sweep of an ultrashort supercontinuum. In Conference on Lasers and Electro-Optics Technical Digest Series 1988, vol. 7, pp. 420–421. Optical Society of America. Washington, D.C.
Misewich, J., J.H. Glownia, J.E. Rothenberg, and P.P. Sorokin (1988b) Subpicosecond UV kinetic spectroscopy; Photolysis of thallium halide vapors. Chem. Phys. Lett. 150, 374-379.
Nakatsuka, H., D. Grischkowsky, and A.C. Balant (1981) Nonlinear picosecond-pulse propagation through optical fibers with positive group velocity dispersion. Phys. Rev. Lett. 47, 910-913.
Nikolaus, B. and D. Grischkowsky (1983) 90-fsec tunable optical pulses obtained by two-stage pulse compression. Appl. Phys. Lett. 43, 228-230.
Parker, D.H. and Ph. Avouris (1978) Multiphoton ionization spectra of two caged amines. Chem. Phys. Lett. 53, 515-520.
Parker, D.H. and Ph. Avouris (1979) Multiphoton ionization and two-photon fluorescence excitation spectroscopy of triethylenediamine. J. Chem. Phys. 71, 1241-1246.
Schwarzenbach, A.P., T.S. Luk, I.A. McIntyre, V. Johann, A. McPherson, K. Boyer, and C.K. Rhodes (1986) Subpicosecond KrF* excimer-laser source. Opt. Lett. 11, 499-501.
Smith, M.A., J.W. Hager, and S.C. Wallace (1984) Two-color laser photoionization spectroscopy in a collisionless free-jet expansion: spectroscopy and excited-state dynamics of diazabicyclooctane. J. Phys. Chem. 88, 2250-2255.
Szatmári, S. and F.P. Schäfer (1983) Simple generation of high-power, picosecond, tunable excimer laser pulses. Opt. Commun. 48, 279-283.
Szatmári, S. and F.P. Schäfer (1984a) Generation of intense, tunable ultrashort pulses in the ultraviolet using a single excimer pump laser. In D.H. Auston and K.B. Eisenthal (eds.), Ultrafast Phenomena IV, pp. 56-59. Springer-Verlag, New York.
Szatmári, S. and F.P. Schäfer (1984b) Excimer-laser-pumped psec-dye laser. Appl. Phys. B 33, 95-98.
Szatmári, S., B. Racz, and F.P. Schäfer (1987a) Bandwidth limited amplification of 220 f. pulses in XeCl. Opt. Commun. 62, 271-276.
Szatmári, S., F.P. Schäfer, E. Muller-Horsche, and W. Mukenheim (1987b) Hybrid dye-excimer laser system for the generation of 80 fs, 900 GW pulses at 248 nm. Opt. Commun. 63, 305-309.
Valdmanis, J.A., R.L. Fork, and J.P. Gordon (1985) Generation of optical pulses as short as 27 femtoseconds directly from a laser balancing self-phase modulation, group-velocity dispersion, saturable absorption, and saturable gain. Opt. Lett. 10, 131-133.
van Veen, N.J.A., M.S. deVries, T. Beller, and A.E. deVries (1981) Photofragmentation of thallium halides. Chem. Phys. 55, 371-384.
Xu, Z., B. Koplitz, S. Buelow, D. Bauch, and C. Wittig (1986) High-resolution kinetic energy distributions via Doppler shift measurements. Chem. Phys. Lett. 127, 534-540.
Author information
Authors and Affiliations
Rights and permissions
Copyright information
© 2016 Springer Science+Business Media New York
About this chapter
Cite this chapter
Glownia, J.H., Misewich, J., Sorokin, P.P. (2016). Utilization of UV and IR Supercontinua in Gas-Phase Subpicosecond Kinetic Spectroscopy. In: The Supercontinuum Laser Source. Springer, New York, NY. https://doi.org/10.1007/978-1-4939-3326-6_8
Download citation
DOI: https://doi.org/10.1007/978-1-4939-3326-6_8
Published:
Publisher Name: Springer, New York, NY
Print ISBN: 978-1-4939-3324-2
Online ISBN: 978-1-4939-3326-6
eBook Packages: Physics and AstronomyPhysics and Astronomy (R0)