
Abstract
Healthy circadian rhythms are important for maintaining overall health. Several core clock genes, including Bmal1, Per, CLOCK, and Cry encode transcription factors that regulate gene expression in the kidney and in nearly all other organs and cell types. Modulation of clock genes can cause major physiological effects. Loss of any of the core clock genes in mice results in significant changes in blood pressure, indicating that the molecular clock is critical for regulation of blood pressure. The kidney regulates electrolyte and volume balance and is thus an important regulator of blood pressure. Several lines of evidence suggest a role for the kidney clock in blood pressure regulation.
Many aspects of renal function, including glomerular filtration rate and electrolyte excretion, are known to vary with a circadian rhythm. Multiple studies have demonstrated that the kidney is sensitive to food and light cues, consistent with a role for circadian rhythms in the regulation of renal function. In the kidney, clock genes are rhythmically expressed and thousands of genes that contribute to renal function are subject to transcriptional regulation by the core clock proteins. Indeed, several key circadian genes oscillate even in the fetal kidney. It is clear that the circadian clock is an important regulator of renal function and that a better understanding of how it functions can open up new avenues for the treatment of kidney disease and hypertension.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others

Keywords
15.1 Introduction
Since the formation of the earth, the sun has risen and set with a predictable rhythmicity giving the world day and night. This daily, circadian rhythm has been adapted by life in the form of rhythmic expression of genes and behaviors around a ~24 h day. Indeed, life forms from archaebacteria to humans have been shown to display circadian rhythms [reviewed in Richards and Gumz (2012)]. These rhythms were first noted by the French astronomer de Mairan three centuries ago with his observation that the opening and closing of the heliotrope plant occurred even in the absence of sunlight. Over a century ago, Vogel first reported circadian fluctuations in urine volume in humans. Twenty years ago, Dr. Joseph Takahashi and colleagues identified the CLOCK gene, one of the master regulators of the circadian clock in mammals (Vitaterna et al. 1994). Since then, there have been numerous advances in the field of circadian biology.
The circadian rhythms of the body are controlled by a master clock in the brain, which helps entrain numerous peripheral clocks throughout the body (Dibner et al. 2010). The master clock is located in the suprachiasmatic nucleus (SCN ) in the hypothalamus of the brain. This clock is entrained by light signals that enter through the retina and are subsequently relayed to the SCN. These signals are transmitted to peripheral clocks via neuronal and humoral signals.
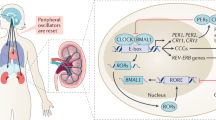
Through transcription-based feedback loops, a core group of clock genes forms a Transcription Translation Oscillating Loop (TTOL) , which is the mechanism for the function of the circadian clock. Briefly, circadian proteins CLOCK and Bmal1 form a heterodimer capable of binding to E-box elements in the promoters of circadian target genes (Fig. 15.1). When this heterodimer binds to E-box elements of Cryptochrome 1 and 2 (Cry1 and Cry2) and Period 1 and 2 (Per1 and Per2), Per and Cry transcription is activated in the positive arm of the loop. Then Per and Cry interact and inhibit CLOCK and Bmal1 activity, forming the negative arm of the loop and inhibiting their own transcription. CLOCK/Bmal1 also binds to E-box elements in the retinoid-related orphan receptor (ROR) and REV-ERBα promoters. ROR and REV-ERBα in turn mediate opposing action on Bmal1 transcription.

Transcription-translation oscillating loop. Bmal1 and CLOCK heterodimerize and bind to E-box elements on target genes to increase levels of transcription. They make up the positive arm of the loop while the other two core components of the clock, Cry and Per proteins, make up the negative loop. Once translated, Cry and Per inhibit the actions of CLOCK and Bmal1. Other integral parts of the clock machinery, ROR and Rev-erbα (encoded by the nr1d1 gene), also participate in the ~24 h oscillation of activation and inhibition of target gene transcription
15.2 Physiological Functions of the Kidney
The kidney is an important organ in the body because it not only removes waste but it is responsible for maintaining fluid and ion homeostasis . This is critical because even slight changes in ion balance can have deleterious effects. For example, alteration of sodium reabsorption can result in large changes in blood volume with subsequent effects on blood pressure. The kidney achieves blood filtration through the nephron, or functional unit of the kidney (Fig. 15.2). An adult human kidney contains about one million nephrons. Blood filtration starts when blood in capillaries comes into contact with the nephron via the glomerulus. Filtrate then passes into the proximal tubule (PT) where the bulk of sodium reabsorption occurs. The cells in this segment contain the sodium-glucose-like transporter 1 (SGLT1), which reabsorbs sodium and glucose and the sodium-hydrogen exchanger 3 (NHE3), which reabsorbs sodium and excretes hydrogen ions into the filtrate. Filtrate then passes into the Loop of Henle where active sodium reabsorption only occurs in the thick ascending loop (TAL). About 4 % of sodium reabsorption occurs in the next segment of the nephron, the distal convoluted tubule (DCT). The cells in this segment have the sodium-chloride co-transporter (NCC), which reabsorbs both sodium and chloride ions from the filtrate.

Model of a renal nephron and representative cell types. (This illustration is focused on mechanisms of sodium reabsorption that have been linked to the circadian clock.) Blood enters the nephron at the glomerulus and is filtered as it passes through the tubule segments. Filtrate first passes through (a) the proximal convoluted tubule (PT) followed by the Loop of Henle (cell diagram not shown), through (b) the distal convoluted tubule (DCT) and (c) finally the collecting duct (CD). Sodium reabsorption decreases as filtrate moves along the nephron as indicated by arrows with approximate percentages of sodium reabsorption indicated. Each portion of the nephron is characterized by different sodium transporters, shown in the representative cell models. Different sections of the nephron are also found in different regions of the kidney, indicated by the dashed lines to separate cortex, outer medulla and inner medulla
As filtrate enters the last part of the nephron, the collecting duct (CD), the final 4 % of sodium reabsorption from the filtrate occurs. This is a highly regulated process in order to avoid large changes in sodium reabsorption and blood pressure . All cell types in each segment of the nephron (PT, TAL, DCT, CD) have a sodium-potassium Na+, K+-ATPase on the basolateral side, which pumps sodium from the cells into the blood. In the principal cells of the collecting duct, the epithelial sodium channel (ENaC) facilitates the reabsorption of sodium from the filtrate. ENaC is regulated at many levels, one of which is by the hormone aldosterone. Aldosterone causes the increased transcription of the alpha subunit of ENaC via binding to mineralocorticoid receptors found inside the cell and subsequent translocation into the nucleus and binding to hormone response elements. The Gumz lab found that one of the core clock genes, Per1, actually regulates aldosterone thus showing that sodium reabsorption in the collecting duct of the kidney is at least partially under circadian control (Richards et al. 2013b).
Potassium is another electrolyte excreted in urine that has been shown to be regulated by the circadian clock as well (Gumz and Rabinowitz 2013). Numerous studies have shown that potassium excretion oscillates in a circadian rhythm with sodium in multiple species. Early microarray evidence accrued by Firsov et al. suggested that potassium transporters in the nephron are expressed in a circadian pattern (Zuber et al. 2009; Nikolaeva et al. 2012). Like sodium, potassium also seems to play a role in blood pressure. Not only does it appear that potassium excretion patterns are altered in disease states such as chronic kidney disease, potassium supplementation may be useful in treating hypertension (Zicha et al. 2011; Kanbay et al. 2013). It has been shown that potassium supplementation leads to increased sodium excretion and therefore decreased blood pressure. Potassium may also be beneficial in restoring a night time dip in blood pressure, lack of which is associated with higher risk of cardiovascular disease.
15.2.1 Evidence for a “Kidney Clock”
Like many other organs in the body, the kidney undergoes circadian fluctuations with respect to a number of aspects. In 1933, Robert Manchester reported on the diurnal rhythms of urine, sodium and potassium and showed that they peaked in the morning and reached a minimum at night (Manchester 1933). On a physiological level, many processes related to kidney function and blood pressure oscillate over the course of a day including glomerular filtration rate (GFR) (Koopman et al. 1989), sodium excretion and renal blood flow (Pons et al. 1996). At a transcriptional level, expression of clock genes including CLOCK, Bmal1, Cry1, Cry2, Per1 and Per2 oscillate with a ~24 h rhythm (Reppert and Weaver 2002).
The core clock proteins regulate the expression of up to 50 % of expressed genes in the body (Pizarro et al. 2013). The function of these clock genes is different depending on the tissue in which they are present. Wu et al. showed that a 30 min feeding stimulus was sufficient to not only significantly decrease transcript levels of the circadian gene Dbp but also to shift the peak of expression by 4 h in the heart but not in the kidney (Wu et al. 2012). The same study also showed decreased mRNA levels of Bmal1, Cry1, Per1 and Per2 in the heart with 30 min feeding stimulus while only Per1 mRNA was significantly decreased in the kidney.
Clock genes are ubiquitously expressed and dysregulation or loss of expression can cause profound physiological changes. In 2005, Okamura et al. provided the first evidence for the circadian control of a renal gene (Saifur Rohman et al. 2005). They showed that the Na+, H+ exchanger NHE3 is expressed in the membrane in a circadian manner and furthermore is regulated by CLOCK:Bmal1 heterodimers. This occurs in a similar fashion to that mentioned for aldosterone regulation. The CLOCK:Bmal1 heterodimers bind to E-box elements in the NHE3 gene to regulate its transcription. This group also showed that the circadian expression of NHE3 is severely blunted in Cry1/2 knockout (KO) mice, further supporting the notion of circadian control of this gene.
In 2006, Schibler et al. demonstrated the effects of knocking out three circadian transcription factors in mice (Dbp/Lef/Tcf triple KO) (Gachon et al. 2006). While single or double KOs of these factors do not produce much of a phenotype, triple KO mice usually do not live past 1 year. This may be due to the fact that the three factors have very conserved amino acid sequences and are able to compensate for the loss of one another. The triple KO caused changes in mRNA expression of numerous genes, many of which are related to drug metabolism or transport. Importantly, this group later showed that the Dbp/Lef/Tcf triple KO mice exhibited a phenotype of low blood pressure, reduced aldosterone levels and cardiac hypertrophy, providing strong evidence for the role of circadian proteins in cardiovascular physiology (Wang et al. 2010).
15.2.2 Circadian Clock Proteins and Blood Pressure
In 2007, Curtis et al. described the blood pressure phenotype of the Bmal1 KO mouse (Curtis et al. 2007). While the wild type mice have normal circadian variations in heart rate and blood pressure, KOs do not show these variations. The KO mice are also hypotensive, meaning they have low blood pressure, compared to the wild type mice. These results provided the first evidence that loss of a single circadian protein can dramatically alter blood pressure.
Per1 KO mice exhibit a lower blood pressure phenotype compared to wild type mice (Stow et al. 2012). This may be to due to increased renal endothelin 1 (ET-1, encoded by the Edn1 gene) as Per1 KOs have increased levels of ET-1 in the medulla of the kidney. While ET-1 is known as a potent vasoconstrictor in the vasculature, ET-1 acts to decrease blood pressure via inhibition of ENaC in the renal collecting duct (Lynch et al. 2013; Bugaj et al. 2008). The subsequent reduction in sodium reabsorption results in a decrease in blood volume, leading to decreased blood pressure.
As previously stated, Per2 is part of the negative arm of the Transcription Translation Loop of the circadian clock mechanism. Vukolic et al. characterized the Per2 mutant mouse, which contains a mutated, not knocked out, Per2 protein (Vukolic et al. 2010). These mutant Per2 proteins are shortened, missing possible dimerization sites. This group found that the mutant mice have a higher heart rate during the light period than wild type mice but have similar locomotor activity. The Per2 mutant mice also had shorter circadian periods of heart rate, mean arterial pressure and locomotion as compared to wild type mice. The mutant mice also had smaller variations in heart rate and blood pressure between light and dark. Examples of normal and aberrant circadian oscillation are described in Fig. 15.3.

Normal and aberrant circadian oscillation. A typical circadian cycle is depicted with the signal for gene expression or behavior on the y-axis and a 24 h period of time on the x-axis. The black curve represents a “normal” circadian period of ~24 h. In blue is a shortened period and in magenta is a lengthened period. A completely arrhythmic pattern is represented in the purple line
CLOCK KO mice are hypotensive and have mild diabetes insipidus but have normal 24 h blood pressure rhythms (Zuber et al. 2009). CLOCK KO mice also have altered circadian expression patterns of 20-HETE (20-hydroxyeicosatetraenoic acid), which is a regulator of blood pressure (Nikolaeva et al. 2012). 20-HETE can lead to both increases and decreases in blood pressure through actions on the kidney. If acting on preglomerular arterioles, the vasoconstrictive action of 20-HETE raises blood pressure (Nikolaeva et al. 2012). However, it is also capable of blocking channels and transporters that reabsorb sodium in the thick ascending limb and proximal tubule of the nephron, which leads to a decrease in blood pressure. Therefore, Firsov et al. have proposed the dysregulation of 20-HETE to be a contributing factor to the blood pressure phenotype of CLOCK KO mice. In 2014, Firsov elaborated on the role of Bmal1 by producing a mouse that has Bmal1 knocked out in renin-producing cells in the kidney (Tokonami et al. 2014). These Bmal1 mutant mice have decreased plasma aldosterone compared to control mice and also have significantly lower blood pressure compared to controls.
15.2.3 Development of the Kidney Clock
It has been known for decades that the fetal SCN is synchronized by the mother. After birth, maternal care helps to keep the clock entrained. Eventually, the neonatal SCN matures enough to take over its job as the central clock (Ohta et al. 2002). By using mice with luciferase-tagged Per2, Nishide et al. looked at the change in expression of Per2 in embryos to adult mice (Nishide et al. 2014). They showed that before birth, peak circadian rhythms of Per2 between the liver, kidney and lung were similar. However, after birth, these peaks changed between tissues. Furthermore, they showed that there is a phase shift in Per2 expression in lung and kidney from embryonic day 20 (E20) to adult mice. While there were shifts in these tissues, they found no significant change in the rhythms of the SCN, supporting the idea that it is not the sole source of entrainment for peripheral clocks.
One possible explanation for this shift in circadian expression irrespective of the SCN is that between 2 and 3 weeks after birth, pups switch from drinking milk to eating chow (Nishide et al. 2014). This change in nutrition composition may cause some alteration of digestive organs including the kidney. In addition, the pups are not fully mature after birth. Organs are still growing and changing which may also account for changes in circadian patterns due to loss or introduction of new factors because of the loss or change of cell phenotypes.
To probe even earlier stages of development, Dolatshad et al. looked at 10 day old embryos in conjunction with maternal rhythms (Dolatshad et al. 2010). They found that many core clock genes (CLOCK, Bmal1, Cry1 and Per2) were expressed as early as E10 but found little evidence that this expression was oscillating in a circadian manner up to E18. The maternal tissues, however, did express high levels of circadian oscillation. When embryonic tissues (heart, liver and kidney) were cultured, they did show circadian expression of the clock genes. While the authors determined that this rhythm was an artifact of the tissue culture process, it shows that the tissues are capable of being synchronized but something may prevent that from occurring. It is also possible that individual cells do have rhythmic expression but they are not synchronized within a tissue either due to interference by rapid mitosis of embryonic cells or some other factor.
Meszaros et al. explored this further by looking at clock genes specifically in rat embryonic kidneys (Meszaros et al. 2014). They saw that at E20 many clock (CLOCK, Per2) and clock-controlled genes (ENaC, Sgk1 and NHE3) exhibited circadian patterns of expressions whose amplitude was significantly increased later in life. At 1 week postpartum, even more genes were observed to display circadian patterns of expression including Bmal1, Cry1, Cry2, and Per1. These investigators also showed that by only allowing pups to nurse during the dark (active) period, their peak expression of CLOCK and Bmal1 was shifted by 12 h. This demonstrates how feeding can entrain clocks since normally the pups would feed during the mother’s inactive period during the day. By changing the feeding time, the phase of expression of clock genes was shifted as well. This may occur because the SCN is not fully developed in rats until postnatal day 10 (Sladek et al. 2004) so nutrient (as opposed to light) cues entrain the peripheral clock in the kidney.
15.2.4 Effect of Food and Light Cues on the Kidney Clock
As previously mentioned, one of the main entrainment signals for the peripheral clocks is feeding time. While this has been established for some time, Oike et al. explored the effect of certain diets, particularly high salt diets in mice. It has already been shown that the type of diet can alter gene expression to adapt to new needs (Ferraris 2001). These authors looked at how high salt diets can affect circadian gene expression and found that in the liver, kidney and lung high salt diets decrease expression of Bmal1 while increasing expression of Dbp over time.
While food cues are significant in the entrainment of peripheral clocks, it is important to remember that the central clock, which sends signals to the peripheral clocks, is entrained primarily by light. Wu et al. investigated this by looking at the effects of only reversing the light/dark (LD) cycle, restricting feeding to the daytime (the inactive period of the rat) or reversing both the LD cycle and feeding time in rat kidneys (Wu et al. 2010). LD reversal did not alter the expression pattern of Bmal1, Cry1, CLOCK or Per2 but did delay the peak expression of Per1 by 4 h. In addition, the expression levels of Per1, Cry1, CLOCK and Bmal1 were altered only with the reversal of the LD cycle.
Feeding time reversal caused peak expression shifts of 8–12 h for CLOCK, Cry1 and Bmal1 after 7 days while causing 4 h shifts in Per1 and Per2. Combining LD and feeding time reversal resulted in a total reversal of the circadian expression of all five genes studied within 7 days. This evidence points to the cumulative effects of light and food cues regarding the entrainment of peripheral clocks.
15.2.5 Circadian Regulators Dec1 and Dec2 in the Kidney
In addition to Per and Cry homologs, Dec1 and Dec2 are also negative regulators of the clock transcription translation loop. They are expressed in a circadian rhythm in most tissues. Recently, Wu et al. studied their expression in the kidney and the heart of rats (Wu et al. 2011). Both Dec1 and Dec2 showed a pattern of daily oscillation in the heart while only Dec2 showed a similar oscillation in the kidney. By restricting feeding to daytime, as opposed to the normal night time feeding of rats, they showed that in the heart, Dec1 and Dec2 expression was shifted about 8 h. However, in the kidney, Dec2 expression was shifted 4 h and Dec1 expression became rhythmic.
They next reversed feeding and the light/dark schedule for the rats. After 7 days, peak expression was shifted for Dec1 in the heart and Dec1 and Dec2 in the heart and kidney. Expression of Dec1 in the kidney did not change significantly. Noshiro et al. showed that Dec1 exhibits a circadian rhythm of expression in mice, indicating that this clock protein acts in tissue- and species-specific ways (Noshiro et al. 2005). Along with the fact that Dec1 and Dec2 have similar DNA binding domains, this evidence suggests that Dec1 may be redundant in the kidney. This is one of many indications of the importance of maintaining circadian clock function.
15.2.6 Per1 in the Kidney
A role for Per1 in the regulation of renal function was first conceived when it was identified as a novel aldosterone target gene in a murine model of the inner medullary collecting duct (IMCD) (Gumz et al. 2003). Per1 was subsequently shown to be induced by aldosterone treatment in vivo and to mediate downstream action on the well-known aldosterone target gene, the alpha subunit of the renal epithelial sodium channel (αENaC) (Gumz et al. 2009).
In order for Per1 to get into the nucleus and affect its target genes, it has to be phosphorylated. This occurs through the actions of Casein Kinase 1 isoforms δ/ε (CK1δ/ε) (Takano et al. 2004). The Gumz lab showed that a CK1δ/ε inhibitor (PF670462) does inhibit the entry of Per1 into the nucleus in mpkCCDc14 cells, which are a model of the murine CCD (Richards et al. 2012). This inhibition therefore prevents the interaction of Per1 with the E-box element from the promoter of the αENaC gene (Scnn1a). Furthermore, inhibition of Per1 nuclear entry decreases basal Scnn1a expression, indicating that Per1 not only increases ENaC transcription but is also involved in basal regulation of expression. This effect was observed at the protein level, as cells treated with the inhibitor show a 60 % decrease in αENaC membrane protein levels.
In addition to regulating Scnn1a, the Gumz lab investigated other genes that may be regulated by Per1 as well (Stow et al. 2012). Using mpkCCDc14 cells , Per1 was knocked down using siRNA. Per1 knockdown caused changes in mRNA expression of four genes associated with sodium transport. Fxyd5 mRNA levels decreased in response to Per1 knockdown whereas Ube2e3, Cav-1 and Edn1 mRNA levels all increased. Fxyd5 increases the activity of the Na, K-ATPase, which pumps sodium reabsorbed by ENaC on the apical side of cells through the basolateral side back into the blood (Lubarski et al. 2005). Ube2e3 is an E3 ubiquitin ligase, a class of enzymes that are known to ubiquitinate the ENaC subunits, thus targeting these proteins for proteasomal degradation (Debonneville and Staub 2004). This action removes ENaC from the membrane, thereby decreasing sodium reabsorption. Loss of Per1 increases the mRNA levels of this ligase, which could lead to higher protein levels and increased ENaC degradation. Cav-1 is a lipid raft protein that has been shown to participate in removal of ENaC from the membrane (Lee et al. 2009). Finally, ET-1 causes blockade of ENaC through decreasing the open probability of the channels via a mechanism involving the ETB receptor and nitric oxide (Bugaj et al. 2008; Gallego and Ling 1996). In order to investigate the role of Per1 in the regulation of these ENaC-modulating proteins, Per1 expression was knocked down using siRNA in mpkCCDc14 cells. ET-1 mRNA expression increased nearly fourfold after Per1 knockdown (Stow et al. 2012). After Per1 knockdown, mRNA and membrane αENaC protein levels were reduced (Richards et al. 2012). Cav-1 membrane protein levels were also increased, as were ET-1 protein levels. These results support a role for Per1 in the coordinate regulation of a number of genes that contribute to the regulation of ENaC in the kidney (Fig. 15.4).

Model for the action of Per1 in principal cells of the renal CD. Sodium derived from the filtrate enters a principal cell in the collecting duct through the epithelial sodium channel (ENaC). Sodium is exported from the cell through the action of the Na+, K+-ATPase and passes into the blood. ENaC is negatively regulated at the membrane by caveolin-1 (Cav-1) and endothelin (ET-1) while the Na+, K+-ATPase is positively regulated by Fxyd5. The genes encoding these proteins are regulated on a transcriptional level in the nucleus by Per1. Per1 activates transcription of the α subunit of ENaC (αENaC) and Fxyd5 but negatively regulates transcription of ET-1 and Cav-1. In other words, Per1 inhibits inhibitors of sodium reabsorption and activates activators of sodium reabsorption
Additional studies with Per1 KO mice showed that higher ET-1 protein levels were present in the inner medulla and cortex of the kidney compared to wild type mice (Stow et al. 2012). In wild type and Per1 KO mice, ET-1 levels were higher at noon (during their inactive period) when blood pressure is lower, showing an inverse relationship between blood pressure and renal ET-1 levels. This is consistent with the established action of ET-1 to inhibit ENaC activity, decreasing sodium reabsorption and therefore causing a decrease in blood pressure as well. Indeed, these Per1 KO mice exhibited an 18 mmHg decrease in mean arterial pressure compared to wild type mice. The contribution of ET-1 to this phenotype has not been directly tested.
Aldosterone regulates αENaC expression and Per1 appears to play a role in this regulation. Interaction of Per1 with E-box response elements in the αENaC promoter increases in aldosterone treated mpkCCDc14 cells (Richards et al. 2013c). Aldosterone treatment also resulted in increased binding of RNA pol II to the ENaC promoter, which is indicative of increased transcription. This regulation is also evidenced by the fact that Per1 knockdown decreases the aldosterone-mediated induction of αENaC expression (Gumz et al. 2009). This effect is paralleled with use of the CK1δ/ε inhibitor (Richards et al. 2012). In mpkCCDc14 cells, mRNA expression of αENaC is increased approximately 6× after addition of aldosterone compared to vehicle treated cells. In the presence of the CK1δ/ε inhibitor, αENaC mRNA levels drop below vehicle levels. With the CK1δ/ε inhibitor and aldosterone, mRNA levels increase about 1.5× compared to 6× with aldosterone treatment alone. Importantly, CK1δ/ε inhibitor treatment resulted in decreased ENaC activity in mpkCCDc14 cells and in A6 cells, a model of the amphibian kidney .
In addition to aldosterone regulating Per1, Per1 appears to regulate aldosterone levels. The Gumz lab investigated this possibility using Per1 heterozygous mice, which have an approximate 50 % reduction in Per1 expression (Richards et al. 2013a). Like all mice, they are nocturnal and are active at night. Wild type and Per1 heterozygous mice were investigated at noon and midnight, the midpoint of their rest and active phases, respectively. Per1 heterozygous mice do not exhibit the normal increase in plasma aldosterone during their active phase like wild type mice and their plasma aldosterone levels are lower than wild type mice as well. This may be due to decreased 3β-HSD expression, which was observed in Per1 heterozygous mice in a pattern similar to plasma aldosterone levels (decreased at both noon and midnight compared to wild type mice with a blunted circadian pattern).
3β-HSD is produced in the adrenal glands, which are also responsible for the production of aldosterone, among other hormones. Using a human adrenal cell line (NCI-H295R), it was shown that Per1 knockdown causes a 58 % decrease in 3β-HSD mRNA levels (Richards et al. 2013b). This result was confirmed in vivo in wild type mice using the CK1δ/ε inhibitor. Mice injected with the inhibitor every 12 h for 2.5 days showed a decrease in 3β-HSD mRNA of around 60–70 % compared to wild type mice.
In contrast to Per1 heterozygous mice, Cry1/2 KO mice have increased levels of 3β-HSD and an accompanying increase in plasma aldosterone levels (Doi et al. 2010). Indeed Per1 and Cry1/2 appear to exhibit opposing actions on target gene expression (Richards et al. 2013a). Per1 causes repression of Cry2 and knockdown of Per1 with shRNA in mpkCCDc14 cells does indeed result in increased Cry2 protein levels. This result was supported by the evidence that nuclear blockade of Per1 by the CK1δ/ε inhibitor increases cytosolic and nuclear Cry2 levels in AML12 (mouse liver) cells.
Similar results were found in mpkCCDc14 cells and AML12 cells with regard to the effect of Per1 and Cry2 on Per1 target genes (Richards et al. 2013a). Per1 mRNA expression was decreased with Per1 siRNA and increased with Cry2 siRNA. CLOCK mRNA expression also decreased with Per1 siRNA and increased with Cry2 siRNA. Cry2 mRNA expression increased with Per1 siRNA while it decreased with Cry2 siRNA. These results are consistent with the hypothesis that Per1 suppresses Cry2 in the Transcription-Translation Oscillating Loop, which inhibits Cry2 from suppressing CLOCK/Bmal1. Further support for the opposing actions of Per and Cry comes from the observation that Per1/Cry1 KO mice have a normalization of the phenotypes observed in single Per1 and Cry1 KO mice (Oster et al. 2003). Oster et al. showed that while Cry1 KO mice have a shortened circadian period (of about 22.5 h) (van der Horst et al. 1999), knocking out Per1 in Cry1 KO mice restores the normal circadian period of about 24 h.
Per1 also regulates the NaCl cotransporter (NCC) in the kidney (Richards et al. 2014). The thiazide sensitive NCC is expressed in the apical membrane of cells in the distal convoluted tubule (DCT) of the kidney where it mediates sodium and chloride entry into the cell. Using a model of the DCT, mDCT15 cells, the Gumz lab demonstrated that either Per1 knockdown or treatment with the CK1δ/ε inhibitor resulted in decreased NCC expression. NCC mRNA levels were decreased in the renal cortex of Per1 heterozygous mice compared to wild type controls. Likewise, NCC expression was reduced in CK1δ/ε inhibitor-treated wild type mice compared to vehicle treated controls. Importantly, CK1δ/ε inhibitor treatment resulted in decreased NCC activity in mDCT15 cells, supporting a role for Per1 in the regulation of a key sodium transporter in the kidney.
15.3 Role of the Kidney Clock in the Regulation of Arterial Blood Pressure
The main function of the kidney is to filter wastes out of blood and maintain homeostasis of fluid and ions in the body. This maintenance helps maintain blood pressure within a healthy range. Hypertension , or high blood pressure, can cause a myriad of problems in the body including stroke, heart failure and renal disease. It is the main risk factor for developing cardiovascular disease, which is the cause of death of about one third of Americans every year (Go et al. 2014).
There have been numerous studies which show a correlation between the kidney and blood pressure. Grisk et al. (2002) transplanted kidneys from normotensive rats into spontaneously hypertensive rats (SHRs). This resulted in a decrease in MAP in over 50 mmHg compared to sham operated SHRs. When normotensive rats received a kidney from SHRs, the MAP increased by around 40 mmHg. These findings give strong evidence for the importance of the kidney in maintaining healthy blood pressure.
Almost all forms of Mendelian (inherited) hypertension can be traced to some defect in the Renin-Angiotenson-Aldosterone System (RAAS) (Lifton et al. 2001). Briefly, when low blood pressure is sensed, the juxtaglomerular cells release the enzyme renin. In the blood stream, renin converts angiotensinogen to angiotensin I which is then converted to angiotensin II by the angiotensin-converting enzyme (ACE). Angiotensin II (AngII) is itself a vasoconstrictor, which causes an increase in blood pressure due to decreased volume for blood to occupy. AngII also stimulates aldosterone secretion, which as previously stated, leads to an increase in αENaC transcription. Increased ENaC in the cell membrane allows for increased sodium reabsorption back into the blood and a consequent increase in blood pressure. There is increasing evidence for an intrarenal RAAS system as well as a whole-body system (Moon 2013). This further implicates the kidney in the development of hypertension.
Further evidence for the kidney’s role in blood pressure maintenance has been provided by Coffman and colleagues [reviewed in Crowley et al. (2007)]. Dahl and Heine showed that a hypertensive phenotype tracks with the kidney by cross-transplantation studies using rats (Dahl and Heine 1975). They cross-transplanted kidneys from hypertension-resistant rats into hypertension-prone rats and found that it resulted in a significant drop in blood pressure. This phenomenon has been observed in human kidney transplant recipients as well (Curtis et al. 1983).
AngII mediates its actions primarily through the angiotensin receptor type 1 (AT1R) (Timmermans et al. 1993). In mice, there are two subtypes of this receptor (A and B) and it is widely accepted that type A (AT1AR) is the human homologue. Activation of AT1 by AngII causes release of aldosterone and vasoconstriction in the kidney, both of which lead to an increase in blood pressure [reviewed in Crowley et al. (2007)]. Coffman et al. also did cross-transplantation studies using four groups of mice: WT, Systemic KOs having AT1AR only in the kidney, Kidney KOs having AT1AR everywhere but the kidney and finally Total KOs lacking AT1AR everywhere (Crowley et al. 2007). These mice were generated by transplanting kidneys from either WT or AT1AR KO animals to respective recipients. For example, the Systemic KO was achieved through transplantation of a kidney from a WT animal into an AT1AR KO animal. The Kidney KO group had a significantly lower MAP compared to WT mice, demonstrating the impact the kidney has on blood pressure. It is important to note that the systemic KO animals had almost identical MAP values and similar circadian variation in MAP, demonstrating the importance of the systemic tissues in determining blood pressure as well.
Hypertension treatments such as ACE inhibitors aim to interrupt the RAAS system and decrease the amount of AngII being made, therefore reducing the hypertensive effects of the RAAS. Another possible target for hypertension treatment was observed in Cry1/Cry2 KO mice. These mice lacking integral circadian clock genes have salt-sensitive hypertension and increased plasma aldosterone (Doi et al. 2010). Microarray analysis identified an enzyme in the aldosterone synthesis pathway (Hsd3b6/3β-HSD) as having increased expression in these mice. This enzyme is involved in the conversion of pregnenolone to progesterone, which is an early step in aldosterone synthesis, and its expression is restricted specifically to zona glomerulosa cells of the adrenal gland. This enzyme could be a possible target to treat hypertension.
15.4 Role for Chronotherapy in Hypertension Treatment
Another proposed way of treating hypertension is chronotherapy. This is the administration of medicines with respect to time, primarily circadian time. Many hypertension medications are taken in the morning, mostly for convenience. However, a recently completed study on the effects of chronotherapy has shown that taking at least one hypertension medication at night not only significantly increases its effectiveness but also may increase its longevity. The MAPEC study also showed that nighttime hypertension medication dosing increased nocturnal dipping by about 30 % which, as previously mentioned, is associated with decreased risk of cardiovascular disease (Hermida et al. 2010; Kario and Shimada 2004).
15.5 Perspectives
In conclusion, it is quite apparent that the circadian clock is important for maintaining overall health and that modulation of clock genes can cause major physiological effects. Loss of any of the core clock genes in mice results in significant changes in blood pressure, indicating that these genes are important in its regulation. The kidney is also an important regulator of blood pressure and has been shown to display circadian rhythms of clock gene expression in addition to having specific genes under control of clock proteins themselves. This is important because it implicates the circadian clock in the proper function of the kidney which functions in a circadian manner as evidenced by the circadian rhythm of sodium excretion, renal blood flow and urine volume excretion. This further shows that blood pressure is at least partially under circadian control and provides new ideas for targets of hypertension treatments. It also sheds light on the fact that timing of treatment is important with regard to drug efficacy and patient well-being. It is clear that the circadian clock is an important regulator of renal function and that a better understanding of how it functions can lead to better treatments for kidney-related diseases.
References
Bugaj V, Pochynyuk O, Mironova E, Vandewalle A, Medina JL, Stockand JD (2008) Regulation of the epithelial Na+ channel by endothelin-1 in rat collecting duct. Am J Physiol Renal Physiol 295(4):F1063–F1070
Crowley SD, Gurley SB, Coffman TM (2007) AT(1) receptors and control of blood pressure: the kidney and more. Trends Cardiovasc Med 17(1):30–34. doi:10.1016/j.tcm.2006.11.002
Curtis JJ, Luke RG, Dustan HP et al (1983) Remission of essential hypertension after renal transplantation. N Engl J Med 309(17):1009–1015. doi:10.1056/NEJM198310273091702
Curtis AM, Cheng Y, Kapoor S, Reilly D, Price TS, Fitzgerald GA (2007) Circadian variation of blood pressure and the vascular response to asynchronous stress. Proc Natl Acad Sci U S A 104(9):3450–3455
Dahl LK, Heine M (1975) Primary role of renal homografts in setting chronic blood pressure levels in rats. Circ Res 36(6):692–696
Debonneville C, Staub O (2004) Participation of the ubiquitin-conjugating enzyme UBE2E3 in Nedd4-2-dependent regulation of the epithelial Na+ channel. Mol Cell Biol 24(6):2397–2409
Dibner C, Schibler U, Albrecht U (2010) The mammalian circadian timing system: organization and coordination of central and peripheral clocks. Annu Rev Physiol 72:517–549. doi:10.1146/annurev-physiol-021909-135821
Doi M, Takahashi Y, Komatsu R et al (2010) Salt-sensitive hypertension in circadian clock-deficient Cry-null mice involves dysregulated adrenal Hsd3b6. Nat Med 16(1):67–74
Dolatshad H, Cary AJ, Davis FC (2010) Differential expression of the circadian clock in maternal and embryonic tissues of mice. PLoS One 5(3), e9855. doi:10.1371/journal.pone.0009855
Ferraris RP (2001) Dietary and developmental regulation of intestinal sugar transport. Biochem J 360(Pt 2):265–276
Gachon F, Olela FF, Schaad O, Descombes P, Schibler U (2006) The circadian PAR-domain basic leucine zipper transcription factors DBP, TEF, and HLF modulate basal and inducible xenobiotic detoxification. Cell Metab 4(1):25–36
Gallego MS, Ling BN (1996) Regulation of amiloride-sensitive Na+ channels by endothelin-1 in distal nephron cells. Am J Physiol 271(2 Pt 2):F451–F460
Go AS, Mozaffarian D, Roger VL et al (2014) Executive summary: heart disease and stroke statistics--2014 update: a report from the American Heart Association. Circulation 129(3):399–410. doi:10.1161/01.cir.0000442015.53336.12
Grisk O, Kloting I, Exner J et al (2002) Long-term arterial pressure in spontaneously hypertensive rats is set by the kidney. J Hypertens 20(1):131–138
Gumz ML, Rabinowitz L (2013) Role of circadian rhythms in potassium homeostasis. Semin Nephrol 33(3):229–236. doi:10.1016/j.semnephrol.2013.04.003
Gumz ML, Popp MP, Wingo CS, Cain BD (2003) Early transcriptional effects of aldosterone in a mouse inner medullary collecting duct cell line. Am J Physiol Renal Physiol 285(4):F664–F673. doi:10.1152/ajprenal.00353.2002
Gumz ML, Stow LR, Lynch IJ et al (2009) The circadian clock protein Period 1 regulates expression of the renal epithelial sodium channel in mice. J Clin Invest 119(8):2423–2434. doi:10.1172/JCI36908
Hermida RC, Ayala DE, Mojon A, Fernandez JR (2010) Influence of circadian time of hypertension treatment on cardiovascular risk: results of the MAPEC study. Chronobiol Int 27(8):1629–1651. doi:10.3109/07420528.2010.510230
Kanbay M, Bayram Y, Solak Y, Sanders PW (2013) Dietary potassium: a key mediator of the cardiovascular response to dietary sodium chloride. J Am Soc Hypertens 7(5):395–400. doi:10.1016/j.jash.2013.04.009
Kario K, Shimada K (2004) Risers and extreme-dippers of nocturnal blood pressure in hypertension: antihypertensive strategy for nocturnal blood pressure. Clin Exp Hypertens 26(2):177–189
Koopman MG, Koomen GC, Krediet RT, de Moor EA, Hoek FJ, Arisz L (1989) Circadian rhythm of glomerular filtration rate in normal individuals. Clin Sci (Lond) 77(1):105–111
Lee IH, Campbell CR, Song SH et al (2009) The activity of the epithelial sodium channels is regulated by caveolin-1 via a Nedd4-2-dependent mechanism. J Biol Chem 284(19):12663–12669
Lifton RP, Gharavi AG, Geller DS (2001) Molecular mechanisms of human hypertension. Cell 104(4):545–556
Lubarski I, Pihakaski-Maunsbach K, Karlish SJ, Maunsbach AB, Garty H (2005) Interaction with the Na, K-ATPase and tissue distribution of FXYD5 (related to ion channel). J Biol Chem 280(45):37717–37724. doi:10.1074/jbc.M506397200, M506397200 [pii]
Lynch IJ, Welch AK, Kohan DE, Cain BD, Wingo CS (2013) Endothelin-1 inhibits sodium reabsorption by ET(A) and ET(B) receptors in the mouse cortical collecting duct. Am J Physiol Renal Physiol 305(4):F568–F573. doi:10.1152/ajprenal.00613.2012
Manchester RC (1933) The diurnal rhythm in water and mineral exchange. J Clin Invest 12(6):995–1008
Meszaros K, Pruess L, Szabo AJ, Gondan M, Ritz E, Schaefer F (2014) Development of the circadian clockwork in the kidney. Kidney Int. doi:10.1038/ki.2014.199
Moon JY (2013) Recent update of renin-angiotensin-aldosterone system in the pathogenesis of hypertension. Electrolyte Blood Press 11(2):41–45. doi:10.5049/EBP.2013.11.2.41
Nikolaeva S, Pradervand S, Centeno G et al (2012) The circadian clock modulates renal sodium handling. J Am Soc Nephrol 23(6):1019–1026. doi:10.1681/ASN.2011080842
Nishide SY, Hashimoto K, Nishio T, Honma K, Honma S (2014) Organ-specific development characterizes circadian clock gene Per2 expression in rats. Am J Physiol Regul Integr Comp Physiol 306(1):R67–R74. doi:10.1152/ajpregu.00063.2013
Noshiro M, Furukawa M, Honma S et al (2005) Tissue-specific disruption of rhythmic expression of Dec1 and Dec2 in clock mutant mice. J Biol Rhythms 20(5):404–418
Ohta H, Honma S, Abe H, Honma K (2002) Effects of nursing mothers on rPer1 and rPer2 circadian expressions in the neonatal rat suprachiasmatic nuclei vary with developmental stage. Eur J Neurosci 15(12):1953–1960
Oster H, Baeriswyl S, Van Der Horst GT, Albrecht U (2003) Loss of circadian rhythmicity in aging mPer1−/−mCry2−/− mutant mice. Genes Dev 17(11):1366–1379. doi:10.1101/gad.256103
Pizarro A, Hayer K, Lahens NF, Hogenesch JB (2013) CircaDB: a database of mammalian circadian gene expression profiles. Nucleic Acids Res 41(Database issue):D1009–D1013. doi:10.1093/nar/gks1161
Pons M, Forpomes O, Espagnet S, Cambar J (1996) Relationship between circadian changes in renal hemodynamics and circadian changes in urinary glycosaminoglycan excretion in normal rats. Chronobiol Int 13(5):349–358
Reppert SM, Weaver DR (2002) Coordination of circadian timing in mammals. Nature 418(6901):935–941
Richards J, Gumz ML (2012) Advances in understanding the peripheral circadian clocks. FASEB J 26(9):3602–3613. doi:10.1096/fj.12-203554
Richards J, Greenlee MM, Jeffers LA et al (2012) Inhibition of alphaENaC expression and ENaC activity following blockade of the circadian clock-regulatory kinases CK1delta/epsilon. Am J Physiol Renal Physiol 303(7):F918–F927. doi:10.1152/ajprenal.00678.2011
Richards J, All S, Skopis G et al (2013a) Opposing actions of Per1 and Cry2 in the regulation of Per1 target gene expression in the liver and kidney. Am J Physiol Regul Integr Comp Physiol 305(7):R735–R747. doi:10.1152/ajpregu.00195.2013
Richards J, Cheng KY, All S et al (2013b) A role for the circadian clock protein Per1 in the regulation of aldosterone levels and renal Na+ retention. Am J Physiol Renal Physiol 305(12):F1697–F1704. doi:10.1152/ajprenal.00472.2013
Richards J, Jeffers LA, All SC, Cheng KY, Gumz ML (2013c) Role of Per1 and the mineralocorticoid receptor in the coordinate regulation of alphaENaC in renal cortical collecting duct cells. Front Physiol 4:253. doi:10.3389/fphys.2013.00253
Richards J, Ko B, All S, Cheng KY, Hoover RS, Gumz ML (2014) A role for the circadian clock protein Per1 in the regulation of the NaCl co-transporter (NCC) and the with-no-lysine kinase (WNK) cascade in mouse distal convoluted tubule cells. J Biol Chem 289(17):11791–11806. doi:10.1074/jbc.M113.531095
Saifur Rohman M, Emoto N, Nonaka H et al (2005) Circadian clock genes directly regulate expression of the Na(+)/H(+) exchanger NHE3 in the kidney. Kidney Int 67(4):1410–1419
Sladek M, Sumova A, Kovacikova Z, Bendova Z, Laurinova K, Illnerova H (2004) Insight into molecular core clock mechanism of embryonic and early postnatal rat suprachiasmatic nucleus. Proc Natl Acad Sci U S A 101(16):6231–6236. doi:10.1073/pnas.0401149101
Stow LR, Richards J, Cheng KY et al (2012) The circadian protein period 1 contributes to blood pressure control and coordinately regulates renal sodium transport genes. Hypertension 59(6):1151–1156. doi:10.1161/HYPERTENSIONAHA.112.190892
Takano A, Isojima Y, Nagai K (2004) Identification of mPer1 phosphorylation sites responsible for the nuclear entry. J Biol Chem 279(31):32578–32585. doi:10.1074/jbc.M403433200
Timmermans PB, Wong PC, Chiu AT et al (1993) Angiotensin II receptors and angiotensin II receptor antagonists. Pharmacol Rev 45(2):205–251
Tokonami N, Mordasini D, Pradervand S et al (2014) Local renal circadian clocks control fluid-electrolyte homeostasis and BP. J Am Soc Nephrol 25(7):1430–1439. doi:10.1681/ASN.2013060641
van der Horst GT, Muijtjens M, Kobayashi K et al (1999) Mammalian Cry1 and Cry2 are essential for maintenance of circadian rhythms. Nature 398(6728):627–630. doi:10.1038/19323
Vitaterna MH, King DP, Chang AM et al (1994) Mutagenesis and mapping of a mouse gene, Clock, essential for circadian behavior. Science 264(5159):719–725
Vukolic A, Antic V, Van Vliet BN, Yang Z, Albrecht U, Montani JP (2010) Role of mutation of the circadian clock gene Per2 in cardiovascular circadian rhythms. Am J Physiol Regul Integr Comp Physiol 298(3):R627–R634
Wang Q, Maillard M, Schibler U, Burnier M, Gachon F (2010) Cardiac hypertrophy, low blood pressure, and low aldosterone levels in mice devoid of the three circadian PAR bZip transcription factors DBP, HLF, and TEF. Am J Physiol Regul Integr Comp Physiol 299(4):R1013–R1019. doi:10.1152/ajpregu.00241.2010, ajpregu.00241.2010 [pii]
Wu T, Ni Y, Dong Y et al (2010) Regulation of circadian gene expression in the kidney by light and food cues in rats. Am J Physiol Regul Integr Comp Physiol 298(3):R635–R641
Wu T, Ni Y, Zhuge F et al (2011) Significant dissociation of expression patterns of the basic helix-loop-helix transcription factors Dec1 and Dec2 in rat kidney. J Exp Biol 214(Pt 8):1257–1263. doi:10.1242/jeb.052100
Wu T, Fu O, Yao L, Sun L, Zhuge F, Fu Z (2012) Differential responses of peripheral circadian clocks to a short-term feeding stimulus. Mol Biol Rep 39(10):9783–9789. doi:10.1007/s11033-012-1844-0
Zicha J, Dobesova Z, Behuliak M, Kunes J, Vaneckova I (2011) Preventive dietary potassium supplementation in young salt-sensitive Dahl rats attenuates development of salt hypertension by decreasing sympathetic vasoconstriction. Acta Physiol 202(1):29–38. doi:10.1111/j.1748-1716.2010.02248.x
Zuber AM, Centeno G, Pradervand S et al (2009) Molecular clock is involved in predictive circadian adjustment of renal function. Proc Natl Acad Sci U S A 106(38):16523–16528
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2015 American Physiological Society
About this chapter
Cite this chapter
Solocinski, K., Gumz, M.L. (2015). The Circadian Clock in the Mammalian Kidney. In: Hyndman, K., Pannabecker, T. (eds) Sodium and Water Homeostasis. Physiology in Health and Disease. Springer, New York, NY. https://doi.org/10.1007/978-1-4939-3213-9_15
Download citation
DOI: https://doi.org/10.1007/978-1-4939-3213-9_15
Publisher Name: Springer, New York, NY
Print ISBN: 978-1-4939-3212-2
Online ISBN: 978-1-4939-3213-9
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)