
Abstract
Microvesicles and exosomes are released to the extracellular milieu and are detectable in body fluids. They act as unique vehicles for cargo transfer to other cells/tissues. They contain a set of membrane receptors, intracellular proteins and nucleic acids. Here, we describe basic techniques for their isolation and characterization from cell culture media or body fluids. We also describe critical techniques for characterizing their cargo (miRNAs) and their protein content. Finally, we present labeling methods for their use in in vitro procedures and for their in vivo delivery and subsequent analysis of their cargo transfer to multiple cell types.
Access provided by CONRICYT – Journals CONACYT. Download protocol PDF
Similar content being viewed by others


Key words
1 Introduction
Exosomes are small (30–150 nm) membrane vesicles of endocytic origin that are released by a variety of cells to the extracellular milieu by fusion of multivesicular bodies with the plasma membrane [1, 2]. The composition of exosomes depends on the cell of origin, although the membrane composition remains distinct from its parental cell as a result of a selective process of assembly, enabling specialized functions [3]. Exosomes contain a set of nucleic acids including miRNAs and mRNAs which could be transferred to target cells [4] inducing regulatory changes [5] under physiological conditions. Current estimate of microvesicle concentration in the peripheral blood of healthy individuals is 5–50 μg/ml; the majority of it has its origin in platelets [6]. Yet, this process could be deeply altered in different pathological settings such as immune response, inflammation, tumor progression and tissue repair. Exosome-mediated cargo transfer to adjacent or remote cells could deeply influence disease progression. This mechanism of intercellular communication is especially relevant in cancer. Accumulated evidence indicates the potential role of exosomes as non-invasive biomarkers with prognostic or diagnostic role, as they can be detected in body fluids [7, 8].
A lot of information about exosome-specific signatures, especially disease related can be obtained from the ExoCarta complendium of exosomal proteins and RNA [9].
In this chapter we have assembled and adapted several protocols for (a) the isolation of exosomes from conditioned medium, (b) the characterization of their nucleic acid cargo and hallmark proteins, (c) the measurement of their size and particle number, and (d) the fluorescent labeling for in vitro and in vivo tracking purposes [10].
2 Materials
-
1.
Ultracentrifuge plastic tubes were 25 × 89 mm, Ref. #326823 for a SW40 rotor of a Beckman Optima™ LE-80K ultracentrifuge. For the recovery of exosomes we used conical 35 ml ultracentrifuge tubes (Beckman Ref #358126) with their adaptors Ref #358156.
-
2.
The transmission electron microscope was from Philips CM120 BioTwin. The copper grids carbon coated were EMS CF300-Cu from Aname.
-
3.
The 4 μm latex beads were purchased from Invitrogen (Ref# A37304).
-
4.
Prepare all solutions using ultrapure water (prepared by purifying deionized water to attain a resistivity of 18 MΩ cm at 25 °C) and analytical grade reagents.
-
5.
DEPC water was obtained by dissolving for 12 h at 37 °C 0.1 g in 100 ml of deionized water and subsequently autoclaved.
-
6.
PBS and molecular grade BSA were purchased from Sigma. To prepare PBS/5 % BSA solution, 5 g of BSA were dissolved in 100 mL of PBS prewarmed at 37 °C by gently stirring and filtered by 0.22 μm.
-
7.
Penicillin/streptomycin solution (Sigma, Ref # P0781-100 ML).
-
8.
RIPA lysis buffer 1×: for 50 mL: 5 ml of 10 % NP-40 detergent solution, 1.5 ml of 5 M NaCl, 2.5 ml of 1 M Tris pH:7.4, 200 μl of 0.25 M EDTA and ~41 ml of ultrapure H2O. Add protease inhibitors as follows: 10 μl of PMSF (20 mg/ml), 10 μl of 0.5 M NaF, 5 μl of 200 mM NaVO3, and a Protease inhibitor cocktail tablet (Roche, Ref# 11 836 170 001).
-
9.
TRIZOL LS Reagent® was from Life Technologies.
-
10.
Red Blood Lysis Buffer , MINI26 cell linker kit and PKH67 Green Fluorescent Cell Linker Mini Kit (ref #MINI67) were purchased from Sigma.
-
11.
Glycogen was from Ambion.
-
12.
ExoQuick™ was from Systems Biosciences.
-
13.
Phycoeritrin was purchased from Jackson Inmuno Research, (cat# 016110-084).
-
14.
Anti-CD63 anti-human antibody was purchased from BD Biosciences (Ref #556019).
-
15.
Anti-IG2 goat biotinylated anti-mouse antibody was from Dako.
-
16.
INMMUNO solution was contained PBS, 1 % BSA, 0.1 % Tween 20 and 0.2 % DAPI.
-
17.
Vectashield® was purchased from Vector Laboratories.
-
18.
Automacs buffer was obtained by dissolving 2.5 ml FBS, 2.5 ml of 0.5 M EDTA, 5 ml of penicillin/streptomycin solution (Sigma, Ref # P0781-100 ML) in 500 ml of PBS.
3 Methods
3.1 Isolation of Exosomes from Cells in Culture
3.1.1 Preparation of Exosome-Depleted Medium
Work under sterile conditions all along these procedures (see Note 1 ).
-
1.
Thaw 50 ml aliquot of complement inactive fetal calf serum (FCS) and mix it with 160 ml of regular serum-free medium used in cell culture. This will allow filling six tubes (35 ml per tube).
-
2.
Use the Beckman plastic tubes.
-
3.
Complete to 35 ml each tube and equilibrate with a balance.
-
4.
Centrifuge 110,000 × g at 4 °C overnight (12–16 h) on an ultracentrifuge (see Note 2 ).
-
5.
Recover carefully the supernatant. The pellet is invisible.
-
6.
Filter through a 0.22 μm filter on sterile conditions.
-
7.
Add serum-free medium and antibiotics (if needed) to obtain the regular complete medium used in cell culture.
This complete medium is used to culture the cells and can be stored at 4 °C for 1 month.
3.1.2 Purification of Exosomes from Cultured Cells (Fig. 1) (See Note 3 )
-
1.
Cells should be growing exponentially until 80 % confluence. The medium should contain exosome-free FCS prepared as previously described in Subheading 3.1.1. Use the best culture conditions to minimize cellular debris as much as possible (see Notes 4 and 5 ).
-
2.
Collect 48–96 h -supernatant after the last medium change performed in complete medium. We use 10 ml per T-75 flask. Usually we collect six T-75 flasks to obtain 60 ml of conditioned medium (see Note 6 ).
-
3.
Centrifuge supernatant at 2000 × g for 20 min at 4 °C to spin down cell debris and death cells.
-
4.
Recover supernatant.
-
5.
Ultracentrifuge supernatant at 25,000 × g for 40 min at 4 °C to eliminate large microvesicles on an SW40 rotor or similar (see Note 7 ).
-
6.
Recover and filter the supernatant through 0.22 μm pore filters.
-
7.
Ultracentrifuge supernatant at 110,000 × g, 90 min, 4 °C using conical tubes and adaptors. This is crucial to ensure a good recovery of the invisible pellet.
-
8.
Discard supernatant and wash the pellet with PBS, first resuspend in 1 ml and then add 29 ml. Note that the pellet is not visible.
-
9.
Ultracentrifuge supernatant at 110,000 × g, 90 min, at 4 °C.
-
10.
Discard supernatant and resuspend pellet in 50–100 μl of sterile PBS in each tube and the collect the content of exosomes from six tubes on a regular 1.5 ml tube.
- 11.
3.1.3 Micorvesicle Imaging by TEM
-
1.
Use transmission electronic microscopy (TEM) copper grids carbon coated.
-
2.
Perform vacuum electrical discharge (glow discharge technique) so grids become temporarily hydrophilic.
-
3.
Put a 5 μl drop of each sample at the right concentration on a parafilm and invert grid on it.
-
4.
Leave 1 min at room temperature to allow adherence.
-
5.
Dry the drop with a Whatman paper and invert the grid over a 5 μl drop of 2 % uranyl acetate.
-
6.
Leave 30 s at room temperature.
-
7.
Repeat dye if necessary.
-
8.
Remove uranyl acetate and let the grid dry.
-
9.
Samples were assessed using a transmission electron microscope. Images were captured by a digital Olympus SIS camera.
3.1.4 Assessment of Particle Size and Number
A couple of techniques are important to measure the number and the size of the particles. It is important to carefully measure the exact number of cells and/protein from which exosomes were isolated for normalization purposes. These measurements are becoming mandatory in any research involving microvesicles or exosomes.
Step 1: Dynamic light scattering (DLS) and zeta potential determinations can be performed using a Zetasizer nanoseries instrument (Malvern Nano-Zetasizer). Add always same exosome concentration diluted in PBS (around 50 ng/μl). * Pick should appear between 30 and 120 nm (Fig. 1).

Procedure for exosome isolation and characterization techniques. (a) Schematic of exosome isolation. (b) Basic techniques required for the physical characterization and their miRNA and protein cargo
Step 2: Nanoparticle tracking analysis (NTA) measurements can be performed with a NanoSight LM20 instrument (NanoSight). The samples should be diluted at 2–5 ng/ ml and injected with a sterile syringe until the liquid reaches the tip of the nozzle.
3.2 Exosome Characterization
3.2.1 By Flow Cytometry: CD63
This protocol should be performed at 4 °C (see Note 9 ).
-
1.
5 μl exosomes + 5 μl latex beads 4 % w/v. *Less amount of exosomes is possible. Keep [exosome: bead] at 1:1 ratio.
-
2.
Incubate 30 min and resuspend every 5 min by gentle pipetting.
-
3.
Add 400 μl of [PBS/5 % BSA] solution.
-
4.
Incubate 30 min and resuspend every 5 min by gentle pipetting.
-
5.
Centrifuge 1000 × g for 10 min at 4 °C.
-
6.
Discard supernatant and resuspend pellet in 50 μl de [PBS/5 % BSA] solution.
-
7.
Incubate with first antibody diluted at 1/50 1 h, 4 °C (anti-CD63 anti-human antibody).
-
8.
Wash twice with 400 μl [PBS/5 % BSA] solution.
-
9.
Centrifuge at 1000 × g for 10 min at 4 °C.
-
10.
Discard supernatant and leave exosomes with beads resuspended in 200 μl of PBS/5 % BSA.
-
11.
Incubate with secondary biotinylated antibody diluted at 1/200 for 30 min at 4 °C (anti-IG2 goat biotinylated anti-mouse antibody).
-
12.
Wash twice with 400 μl [PBS/5 % BSA] solution.
-
13.
Centrifuge at 1000 × g for 10 min at 4 °C.
-
14.
Discard supernatant and resuspend pellet in 50 μl solution [PBS/5 % BSA].
-
15.
Add streptavidin with phycoerythrin (×28) 1/50 and incubate for 30 min in darkness.
-
16.
Wash twice with 400 μl PBS (without BSA).
-
17.
Centrifuge at 1000 × g for 10 min at 4 °C.
-
18.
Resuspend in 300 μl PBS and perform FACS analysis.
3.2.2 By Western Blot
Use 25 μg of proteins from the isolated exosomes and add 10× RIPA buffer to a final 1× concentration. Add protein loading buffer, boil the samples, and run a regular SDS-PAGE gel.
3.2.3 miRNA Extraction from Conditioned Media
Before engaging into large experiments, we suggest comparing the protocol below with other commercially available extraction methods such as ExoQuick™ (see Note 10 )
Similarly, the analysis of total miRNAs in the conditioned medium should be compared with the miRNA cargo of isolated exosomes. In our case, the free miRNA is negligible as compared to the miRNA cargo in exosomes.
Here, we describe the analysis in the conditioned media.
-
1.
Use 15 ml tubes for the miRNA extraction of 48–96-h cultured cells as previously stated. Use three tubes per cultured sample, and add 1.6 ml conditioned medium in each tube.
-
2.
Add 2 ml of TRIZOL LS Reagent®. Vortex and leave 5 min at room temperature.
-
3.
Add 2 μl spike for each miRNA (each miRNA at 25 fmol concentration) of three different miRNAs per sample for normalization purposes. A total 6 μl per tube.
-
4.
Add two volumes of chloroform (400 μl per tube) and leave for 3 min at room temperature.
-
5.
Centrifuge at 3500 × g for 15 min at 4 °C.
-
6.
Carefully take the upper aqueous phase (~2 ml) to another clean 15 ml tube.
-
7.
Add 3 μl of glycogen at a concentration of 5 mg/ml in RNase-free water.
-
8.
Add one volume of isopropanol (2 ml).
-
9.
Mix and distribute in 1.5 ml tubes.
-
10.
Centrifuge 12,000 × g for 10 min at 4 °C.
-
11.
Wash the pellet with 1 ml of cold ethanol 75 % and spin for 5 min, 7500 × g at 4 °C.
-
12.
Discard the supernatant.
-
13.
Dry the pellet at room temperature.
-
14.
For each original sample (which is distributed in three tubes), resuspend in 30 μl of DEPC water and store at −80 °C (see Note 11 ).
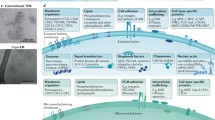
3.3 Tracking Microvesicles in Functional Assays (Fig. 2)
3.3.1 Fluorescence Microvesicle Labeling
3.3.1.1 Method 1: Exosome Labeling with PKH 26
For in vitro applications, we use the MINI26 cell linker kit, which incorporates a red fluorescent dye into lipid regions of the cell membrane.
Perform the protocol after step 7, Subheading 3.1.2, right before PBS washing step (see Note 12 ).
-
1.
Add diluent C to exosomes to reach a final concentration 2 × 107 particles/ml, i.e., derived from 1 × 106 cells and add 50 μl diluent C.
-
2.
Immediately prior to staining, prepare a volume of 2× PKH26 (4 × 10−6 M) in diluent C.
-
3.
Rapidly add volume of 2× Dye to exosomes.
-
4.
Final concentration: particles: 2 × 107 exosomes/ml PKH26: 2 × 10−6 M.
-
5.
Immediately mix the sample by pipetting. Rapid and homogeneous mixing is critical for uniform labeling because staining is nearly instantaneous.
-
6.
Incubate at room temperature for 7 min in the darkness.
-
7.
Stop the staining reaction by adding an equal volume of serum or compatible protein solution (i.e., 1 % BSA). Incubate 1 min.
-
8.
Add complete medium to the top of the tube.
-
9.
Centrifuge exosomes at 11,000 × g for 90 min at 4 °C.
-
10.
Discard supernatant and wash pellet with PBS.
-
11.
Centrifuge exosomes at 11,000 × g for 90 min at 4 °C.
-
12.
Discard supernatant and resuspend stained exosomes in 50 μl de PBS.
Keep exosomes at −80 °C.
3.3.1.2 Method 2: Exosome Labeling with PKH67
We use the PKH67 Green Fluorescent Cell Linker Mini Kit.
Perform the protocol after step 7, Subheading 3.1.2, right before PBS washing step (see Note 12 ).
-
1.
Take 100 μl resuspended exosomes in PBS.
-
2.
Add 500 μl of solution C [2 μl PKH67 + 448 μl solution C].
-
3.
Add PKH67 to exosomes and mix by pipetting up and down.
-
4.
Incubate for 2 min at room temperature.
-
5.
Neutralize with 1 ml [PBS/20 % BSA].
-
6.
Incubate for 1 min at room temperature.
-
7.
Add PBS to fill the centrifuge tube (~25 ml) in conical tubes and adaptors recommended.
-
8.
Centrifuge 11,000 × g for 90 min at 4 °C.
-
9.
Remove supernatant.
-
10.
Wash with PBS.
-
11.
Centrifuge at 11,000 × g for 90 min at 4 °C.
-
12.
Remove supernatant.
-
13.
Resuspend exosomes in 100 μl.
-
14.
Store exosomes at −80 °C.
3.3.2 Evaluation of In Vitro Uptake of Exosomes
3.3.2.1 Day 1
-
1.
Treat a rounded cover slide for a p-24 with 0.2 % gelatin over 20 min at 37 °C.
-
2.
Remove gelatin and let dry under the hood.
-
3.
Plate 2 × 105 of recipient cells per well.
3.3.2.2 Day 2
-
1.
Treat cells with 5–10 μg exosomes for 24 h (or other timing: 1, 5, 24 h).
3.3.2.3 Day 3
-
1.
Carefully wash the cells twice with PBS by slowly dropping the PBS through the tissue culture wall and fix them in 1 % paraformaldehyde for 15 min.
-
2.
Wash twice with PBS and plate can be kept for 1 week at 4 °C.
-
3.
Incubate for 30 min at 4 °C with INMUNO solution in dark 40 μl Immuno solution drops on a parafilm®.
-
4.
Wash cover slide twice with PBS.
-
5.
Mount with Vectashield®. Evaluate experiment on a fluorescence microscope.
3.3.3 Evaluation of In Vivo Uptake of Exosomes
After in vivo injection of exosomes, we used this protocol to evaluate the fluorescent in vivo uptake into several cell subpopulations of the bone marrow (Fig. 2) (see Note 13 ).

In vitro and in vivo functional assays for donor–acceptor transfer
-
1.
Label exosomes with PKH67, as detailed previously.
-
2.
Quantify exosome protein of isolated exosomes.
-
3.
Intravenously inject (i.v.) the mice with 10 μg in 100 μl fluorescently labeled exosomes and wait 4 h. Control mice will be i.v. injected with 100 μl PBS.
-
4.
Sacrifice mice and isolate the bone marrow by bone marrow “flushing” technique as follows:
-
(a)
Damped briefly death mice in ethanol 70 %.
-
(b)
Excised long bones from the hind limbs, perfectly clean from surrounding tissue and keep bones on α-MEM with no FCS.
-
(c)
Cut with scissors both bone ends. Handle the bones with tweezers.
-
(d)
Use a 27 G needle for flushing the bone marrow with a syringe containing 5 ml of serum-free α-MEM medium and collect it on a 50 ml tube. The bone will appear as a vivid white color.
-
(e)
Spin cells to 600 × g for 10 min at 4 °C and discard supernatant.
-
(f)
Gently resuspend the pelleted cells in complete medium [α-MEM/10 % FBS/1 % penicillin/streptomycin solution] ten times with a 18 G needle to disaggregate cell clumps, and filter the cell suspension through a 70 μm cell strainer to retain cellular debris.
-
(g)
Spin cells at 600 × g for 10 min at 4 °C.
-
(a)
-
5.
Resuspend cells on 5 mL of red blood lysis buffer and leave 5 min at 37 °C.
-
6.
Neutralize lysing buffer with 5 mL complete medium.
-
7.
Spin cells at 600 × g for 10 min at 4 °C.
-
8.
Resuspend cells in PBS and count the cells.
-
9.
Spin cells to 900 × g for 10 min at 4 °C.
-
10.
Resuspend in Automacs Buffer and maintain at 4 °C until use.
-
11.
For different subpopulation sorting, plate 100 μl (2 × 106 cells) in a U-bottom 96-well plate.
-
12.
Add diluted antibody that works for flow cytometry (see Note 14 ).
-
13.
Incubate for 15 min at 4 °C in the dark.
-
14.
Centrifuge at 800 × g for 2 min at 4 °C.
-
15.
Discard supernatant and resuspend pellet in 100 μl Automacs buffer.
-
16.
Take volume to a cytometer tube and raise volume to 300 μl.
-
17.
Use flow cytometer.
4 Notes
-
1.
Sterilize ultracentrifuge tubes and adaptors under UV light for 15 min. To avoid contamination be careful with the tissue culture technique. Serum used to complement cell medium used all along this procedure must be exosome-depleted.
-
2.
Before operating the centrifuge, operators should be carefully instructed on its use. Several key points should scrupulously be observed. For instance, the way of handling the rotor, the type of tubes to be used, filling the tubes almost to the top, and the equilibration of the tubes using a balance are critical measures to be taken before each ultracentrifugation.
-
3.
Before choosing the cell line of study it is worthy to check the amount of exosomes secreted by each cell line. Please note that the amount of secreted exosomes is cell-line dependent.
-
4.
Since stress conditions could increase the exosome secretion, it is important to keep and regularly check that all cell culture conditions (CO2, humidity, pH, mycoplasma-free, etc.) are exquisitely maintained all along different extraction procedures.
-
5.
We use a six conical 35 ml ultracentrifuge tubes (Beckman Ref #358126) with their adaptors Ref #358156. We distributed 60 ml of CM, leaving 30 ml of conditioned medium per tube.
-
6.
The amount of T-75 flasks will depend on the amount of exosomes secreted by the chosen cell line. For some cell lines, one flask could be enough.
-
7.
It is recommended to check the quality of the exosome purification with other methods commercially available such as ExoQuick® or Dynabeads®.
-
8.
It is recommended to perform protein quantification with small aliquot in each round of isolation (we used 3 μl of exosome suspension).
-
9.
Use a negative control running the same protocol but skipping step 7.
-
10.
Before starting to work with RNA, clean all the bench surfaces with ethanol 70 % and an RNase solution (RNase away, Molecular BioProducts). Special care should be put during isolation and analysis of miRNA. All solutions should be prepared using RNase-free glassware, autoclaved water and chemicals reserved to work with RNA. Disposable gloves should be worn all along these procedures. Diligently follow all waste disposal regulations when disposing waste materials. DEPC should be handled with care in a fume hood.
-
11.
For each sample, add the DEPC water in the first tube, resuspend and transfer to the second tube, and resuspend and pass to the third tube.
-
12.
Work in low light conditions since fluorescent dyes are photosensitive.
-
13.
This protocol could be adapted to other cell types as long as the antibody specific of this cellular phenotype works on flow cytometry.
-
14.
The dilution of each antibody should experimentally adjusted.
References
Simons M, Raposo G (2009) Exosomes: vesicular carriers for intercellular communication. Curr Opin Cell Biol 21(4):575–581. doi:10.1016/j.ceb.2009.03.007
Mittelbrunn M, Sanchez-Madrid F (2012) Intercellular communication: diverse structures for exchange of genetic information. Nat Rev Mol Cell Biol 13(5):328–335. doi:10.1038/nrm3335
Muralidharan-Chari V, Clancy JW, Sedgwick A, D’Souza-Schorey C (2010) Microvesicles: mediators of extracellular communication during cancer progression. J Cell Sci 123(Pt 10):1603–1611. doi:10.1242/jcs.064386
Valadi H, Ekstrom K, Bossios A, Sjostrand M, Lee JJ, Lotvall JO (2007) Exosome-mediated transfer of mRNAs and microRNAs is a novel mechanism of genetic exchange between cells. Nat Cell Biol 9(6):654–659. doi:10.1038/ncb1596
Ratajczak J, Miekus K, Kucia M, Zhang J, Reca R, Dvorak P, Ratajczak MZ (2006) Embryonic stem cell-derived microvesicles reprogram hematopoietic progenitors: evidence for horizontal transfer of mRNA and protein delivery. Leukemia 20(5):847–856. doi:10.1038/sj.leu.2404132
Hunter MP, Ismail N, Zhang X, Aguda BD, Lee EJ, Yu L, Xiao T, Schafer J, Lee ML, Schmittgen TD, Nana-Sinkam SP, Jarjoura D, Marsh CB (2008) Detection of microRNA expression in human peripheral blood microvesicles. PLoS One 3(11):e3694. doi:10.1371/journal.pone.0003694
Yu L, Todd NW, Xing L, Xie Y, Zhang H, Liu Z, Fang H, Zhang J, Katz RL, Jiang F (2010) Early detection of lung adenocarcinoma in sputum by a panel of microRNA markers. Int J Cancer 127(12):2870–2878. doi:10.1002/ijc.25289
Zen K, Zhang CY (2012) Circulating microRNAs: a novel class of biomarkers to diagnose and monitor human cancers. Med Res Rev 32(2):326–348. doi:10.1002/med.20215
Mathivanan S, Fahner CJ, Reid GE, Simpson RJ (2012) ExoCarta 2012: database of exosomal proteins, RNA and lipids. Nucleic Acids Res 40(Database issue):D1241–D1244. doi:10.1093/nar/gkr828
Valencia K, Luis-Ravelo D, Bovy N, Anton I, Martinez-Canarias S, Zandueta C, Ormazabal C, Struman I, Tabruyn S, Rebmann V, De Las RJ, Guruceaga E, Bandres E, Lecanda F (2014) miRNA cargo within exosome-like vesicle transfer influences metastatic bone colonization. Mol Oncol 8(3):689–703. doi:10.1016/j.molonc.2014.01.012
Acknowledgment
This work was supported by the Spanish Ministry of Economy and Competitiveness SAF2012-40056 to F.L, “UTE project FIMA” agreement, the Cancer Research Thematic Network of the Health Institute Carlos III (RTICC RD12/0036/0066), European Regional Development Fund (ERDF) “Una manera de hacer Europa.”
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2016 Springer Science+Business Media New York
About this protocol
Cite this protocol
Valencia, K., Lecanda, F. (2016). Microvesicles: Isolation, Characterization for In Vitro and In Vivo Procedures. In: Medarova, Z. (eds) RNA Imaging. Methods in Molecular Biology, vol 1372. Humana Press, New York, NY. https://doi.org/10.1007/978-1-4939-3148-4_14
Download citation
DOI: https://doi.org/10.1007/978-1-4939-3148-4_14
Publisher Name: Humana Press, New York, NY
Print ISBN: 978-1-4939-3147-7
Online ISBN: 978-1-4939-3148-4
eBook Packages: Springer Protocols