
Abstract
Antibody technology has gone through a long evolution. This review summarizes crucial events during the development of this expanding field, with emphasis on recent advances in the design and manipulation of functional Ig fragments for therapeutic applications. Currently, the challenge is to create next generation types of antibodies that offer improvements over classical MAbs with regard to biomolecular attributes and clinical performance. Bispecifics such as BiTEs and DARTs offer high potency due to a novel mode of action but involve complicated construction. Apart from established Ig fragments such as Fabs, scFvs and domain antibodies, alternative binding proteins are increasingly exploited as therapeutics. These engineered “protein scaffolds”, which are based on non-immunoglobulin proteins furnished with novel binding sites, offer improved manufacturability, facilitate new functional combinations and, thus, likely enable unique therapeutic modes of action to yield promising biobetters. The four most advanced scaffold technologies, Adnectins, Affibodies, Anticalins and DARPins, which were developed beyond the academic research stage and have reached clinical studies, constitute another focus of this review.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others

Keywords
- Diabetic Macular Edema
- Certolizumab Pegol
- scFv Fragment
- Interchain Disulfide Bond
- Recombinant Antibody Fragment
These keywords were added by machine and not by the authors. This process is experimental and the keywords may be updated as the learning algorithm improves.
Introduction: The Route to Antibody Alternatives as Biobetters
Proposed as a new type of medicine more than a century ago by Emil von Behring, initially as “antitoxins” and only later dubbed “antibodies” (Lindenmann 1984), monoclonal antibodies (MAbs) reached regulatory approval in 1986 and subsequently, in a humanized or human format, have become the most successful and fastest growing class of biopharmaceuticals with more than 30 marketed drugs today (Reichert 2012, 2014; Strohl and Strohl 2012). Antibody technology has gone through a long evolution, starting with animal sera for passive immunization against toxins. The first breakthrough with regard to developing monoclonal as opposed to polyclonal antibodies was the invention of hybridoma technology in 1975, thus allowing the preparation of monospecific antibodies obtained via immunization of rodents (Köhler and Milstein 1975). However, only one MAb derived from mouse hybridoma cell culture was successful in the clinic: Muromonab-CD3/Orthoclone (OKT3®), approved in 1986 (discontinued in 2010) as an immunosuppressant drug to reduce acute rejection in patients after organ transplantation (Strohl and Strohl 2012).
The recognition that MAbs from animal sources elicit an anti-drug immune response in patients prompted efforts to make them more alike their human counterparts. The first strategy was the construction of chimeric MAbs by combining the pair of murine variable domains, carrying the antigen-binding site, with the constant region of a human immunoglobulin (Ig) (Morrison et al. 1984). Since antigenic sites predominantly occur in the xenogenic constant regions, the resulting chimeric Ig proteins were well tolerated (although somewhat biased by their nature as immune suppressants), as first demonstrated with Rituximab (Rituxan®; anti-CD20) and Daclizumab (Zenapax®; anti-IL-2Rα), both approved by the FDA in 1997 for treating Non-Hodgkin’s lymphoma (NHL) / rheumatoid arthritis (RA) and to prevent transplant rejection, respectively (Strohl and Strohl 2012).
Following this major breakthrough, the technology of CDR-grafting was invented (Jones et al. 1986), allowing more complete humanization of MAbs by further exchanging the framework regions within the variable domains with those from a human Ig. One of the first and most successful examples has been Trastuzumab (Herceptin®) (Carter et al. 1992), a humanized antibody directed against the HER2 cell surface receptor overexpressed on metastasizing breast cancer cells, which was approved in 1998. However, as the conformation of the six CDRs is also influenced by the framework region, this type of humanization is a tricky approach that usually necessitates introduction of additional affinity-preserving mutations via protein engineering (Carter et al. 1992). This caveat eventually stimulated the development of alternative methods for MAb humanization, for example by resurfacing (Strohl and Strohl 2012).
With the advent of phage display techniques, the age of so-called “human” MAbs arose, initially by selecting recombinant antibody fragments from naïve variable gene/cDNA libraries that were cloned from non-immunized human donors. The first MAb that emerged from this technology was Adalimumab (Humira®), which neutralizes tumor necrosis factor α (TNFα) and received approval for the treatment of RA and Crohn’s disease in 2002 (Bain and Brazil 2003; Osbourn et al. 2005). Various other experimental strategies, both in vitro and in vivo, have followed, culminating in the breeding of transgenic animals that have their own Ig gene loci replaced by corresponding human chromosomal segments, thus directly allowing the biosynthesis of human MAbs—e.g. followed by cloning of the relevant V-genes from antibody producing B-cells (Lee et al. 2014; Lonberg 2005). However, it should be kept in mind that in spite of the commonly used term “human”, each MAb developed by one of these technologies is still foreign to the body of an individual who receives corresponding treatment since it exhibits novel CDR sequences (Harding et al. 2010). Hence, immunogenicity in the patient cannot be fully prevented and, in fact, with regard to immunological tolerability, it is clear that the early chimeric antibody concept already had the strongest impact (Getts et al. 2010; Strohl and Strohl 2012).
Far from over, technology development in the antibody field continues apace (cf. the Chapters in Part II of this monograph). Apart from searching for innovative MAbs directed against new targets and/or novel epitopes, having greater affinities and specificities, or even agonistic activities, there are numerous ongoing approaches to improve the generic features and the Ig format as a whole (Strohl and Strohl 2012). In particular, these efforts aim at the following features: enhancing immune effector functions (usually associated with the Fc region) such as antibody-dependent cellular cytotoxicity (ADCC) and complement-dependent cytotoxicity (CDC); prolonging (or adjusting) circulation half-life; achieving more homogeneous and functional glycosylation; arming with drugs or radioactive payloads; and creating bispecific MAbs, resulting in so-called second or third generation antibodies (Beck et al. 2010). Application of these strategies to marketed MAbs, while retaining the original validated epitope specificity, results in “biobetters” in the true sense. One recent example is the antibody Ado-Trastuzumab Emtansine (T-DM1, Kadcyla™), a toxic drug conjugate of Trastuzumab (Dirix et al. 2013).
Generally, the term biobetter refers to a therapeutic protein that is functionally similar (not identical) to an established biopharmaceutical but has improvements over the original in critical attributes and clinical performance (Beck 2011). However, in the case of antibodies this rather narrow definition deserves broader interpretation, especially if the focus is on the tunable antigen-binding function as dominating feature (Skerra 2003). In fact, the past two decades have given evidence that full size Igs with their characteristic Y-shaped molecular architecture are not the only suitable platform for the generation of protein reagents with novel ligand-binding properties for applications in biomedical research and human therapy. Once the technical challenges of engineering recombinant antibody fragments had been mastered, so-called alternative protein scaffolds were identified which, too, can provide specific binding sites with high affinities against a wide range of biochemical or cellular target structures (Skerra 2000a). Consequently, biobetters may be understood in this context also as advancements in the molecular format of Ig-based biologics as a class.
Functional Ig Fragments: from Fab to Domain Antibody
MAbs differ from classical biologics such as hormones or cytokines insofar as the generation of new molecular entities with differing target specificities and varying mode of action is possible on the basis of the same molecular format, the one of Igs. These proteins owe their remarkable ability for molecular recognition of almost any kind of antigen as well as their utility for diverse medical applications to their unique modular format (Bork et al. 1994; Padlan 1994; Strohl and Strohl 2012). On the first level, their molecular architecture comprises two types of domains which are either of constant or of variable character. Both rest upon the same basically conserved Ig fold which is characterized by a sandwich of two β-sheets with altogether 7 or 9 strands, respectively. Several constant domains assemble to form the Fc part at the stem of the Y-shaped protein, which is responsible for most effector functions, in particular binding and/or activation of cellular receptors or complement. The two arms of the Y are called antigen-binding fragments, Fabs, which are mutually identical in natural Igs and flexibly linked to the Fc stem via the hinge region. At each tip of the Y a pair of variable domains, VH from the heavy and VL from the light chain, forms the antigen-binding site.
Within these paired variable domains, there is another level of separation between structurally conserved and variable polypeptide segments, the so-called framework region(s) on the one hand and the hypervariable region(s) on the other. As regards the hypervariable regions there are in total six hypervariable loops, three per variable domain, also known as complementarity-determining regions (CDRs), which—upon tight association between VH and VL via their inner β-sheets—together form the combining site that intimately contacts the antigen during complex formation. Within each V-domain the three hypervariable loops are supported by the β-sandwich fold, which provides the structural framework. This discontinuous region shows high similarity in its backbone conformation, not only among the human antibody repertoire but also across species, a feature that provides the basis of the CDR-grafting technology for antibody humanization mentioned above.
This modular format explains the success of MAbs as almost universal protein tools for the tight and highly specific binding of a vast number of biomedically relevant molecular structures. Obviously, the β-sheet framework is sufficiently stable to structurally support a more or less unlimited number of CDR peptide sequences and to impose a defined three-dimensional conformation on them by providing the proper geometrical constraints. As a consequence, the number of possible CDR backbone conformations, called canonical structures, seems to be restricted (Al-Lazikani et al. 1997). However, the conformational stability of the Ig fold is not indefinite. In fact, it has been shown that the folding energy of the Ig framework statistically varies about some consensus sequence, which appears to constitute the thermodynamic minimum (Steipe et al. 1994), whereas the stability of the variable domain as a whole is clearly influenced by the individual sequences of the hypervariable loops (Jung and Plückthun 1997). Interestingly, these long known structural properties of Igs also match well with observations made for the alternative protein scaffolds discussed further below.
Nevertheless, with the ever increasing application of antibodies during the last two decades, several disadvantages have become apparent. For example, MAbs have a very large size and complicated composition, comprising four polypeptide chains, glycosylation of the heavy chains, at least, as well as multiple structurally relevant disulfide bonds. Thus, intact antibodies require manufacturing in eukaryotic expression systems, usually involving stably transfected mammalian cell lines, whose optimization and fermentation is laborious and costly (Steinmeyer and McCormick 2008; Werner 2004). Consequently, the exploration of alternative protein reagents that are capable of specifically recognizing and tightly binding ‘antigens’ has been stimulated, which led to the investigation of different types of antibody fragments and, eventually, even to the use of isolated Ig domains (Holliger and Hudson 2005; Skerra 1993).
Initially, the development of methods for the production and manipulation of functional Ig fragments in the bacterial host cell Escherichia coli (Better et al. 1988; Skerra and Plückthun 1988) enormously stimulated the field of antibody engineering (Humphreys 2003; Skerra 1993), not only with regard to research and discovery but also concerning biological drug development (Holliger and Hudson 2005; Nelson and Reichert 2009). Two major types of recombinant antibody fragments carrying the intact antigen-binding site of a MAb are commonly in use: first, the Fab fragment, which consists of the entire light chain and the N-terminal half of the heavy chain and has been traditionally prepared by selective proteolysis of full size Igs and, second, the Fv fragment; this smallest functional antibody fragment comprises just the two variable domains and is readily accessible only via recombinant DNA technology (Riechmann et al. 1988; Skerra and Plückthun 1988).
The conventional Fv fragment is made of the paired variable regions from both Ig chains which are held together just by non-covalent forces such that its stability is limited, in particular at elevated temperatures (like body temperature), low protein concentration and in the absence of antigen. Chain dissociation may be prevented by introducing Cys residues at appropriate locations into the framework of VH and VL, resulting in a disulfide crosslink (Glockshuber et al. 1990; Reiter et al. 1996) and yielding so-called disulfide-stabilized Fv fragments (dsFv). However, the gain in stability is often accompanied by poorer expression yields, as typically observed when introducing additional Cys residues and/or disulfide bridges into recombinant proteins.
A more widely applied approach is the construction of a fusion protein comprising the two variable domains artificially linked via a flexible peptide segment, yielding the so-called single chain Fv fragment (scFv) (Bird and Walker 1991). In this manner, VH and VL are not only covalently associated, but also the difficulty on the genetic level of simultaneously coexpressing two different polypeptide chains is avoided. Nevertheless, several disadvantages have been observed with this strategy, most notably the frequently reduced antigen-binding activity as a result of connecting one N-terminus—usually close to the combining site—with the peptide linker, which can lead to conformational changes or steric hindrance during antigen binding. This is especially the case for scFvs that are originally derived from full length MAbs and not directly selected from V-gene libraries (e.g. via phage display).
The scFv format also shows other undesired properties, such as low folding efficiency upon expression in E. coli, enhanced aggregation and, importantly, a pronounced tendency to form oligomers (Arndt et al. 1998; Demarest and Glaser 2008; Power et al. 2003), which can significantly hamper bioprocess development. Although numerous attempts were made to vary the length and amino acid sequence of the peptide linker and to alter the mutual order of the VH and VL domains within the fusion protein, these caveats have not been fundamentally alleviated and, therefore, laborious individual optimization is needed (Strohl and Strohl 2012). This may explain why, despite their flourishing in biomedical research for more than 25 years, no scFv has been approved as a biopharmaceutical to date (Nelson 2010). The currently most advanced scFv under clinical development (beside an immunotoxin fusion protein discussed below) is a PEG-linked conjugate with doxorubicin-charged liposomes targeting HER2, MM-302 (Merrimack, http://merrimackpharma.com), that has entered phase II/III studies for the treatment of breast cancer (Reichert 2015; Nielsen et al. 2002; Wickham et al. 2010).
Still, there were attempts to generate scFvs with elevated stability, for example using the so-called Immuna® screening platform (ESBAtech, http://www.esbatech.com) which involves the generation of stable humanized single-chain Fv fragments from rabbit monoclonal antibodies (Borras et al. 2010). A few scFv candidates, directed against TNFα (ESBA105) and VEGF were developed up to clinical phase II for topical application on the eye (Strohl and Strohl 2012; Thiel et al. 2013). Further development was pursued in the form of the PENTRA®body technology (Delenex, http://www.delenex.com) for local application in dermatology, where the anti-TNFα scFv (DLX105) appeared to induce a clinical response in psoriasis patients after intradermal administration (Tsianakas et al. 2014).
In the area of antimicrobial therapy, Efungumab (Mycograb™) (Pachl et al. 2006), which binds to the heat shock protein 90 of Candida albicans, and Aurograb™ (NCT00217841), an scFv directed against a surface protein of methicillin resistant Staphylococcus aureus, were brought to advanced clinical stages before their development was discontinued. Efungumab formed aggregates in solution, was contaminated with host proteins, and administration was associated with cytokine release while no treatment benefit was seen (EMEA 2007). In fact, reevaluation of its in vitro activity indicated that the presumed antifungal potentiation of amphotericin B was a nonspecific effect that was also seen with a wide range of unrelated proteins (Richie et al. 2012). Aurograb was discontinued following a review of phase II data showing lack of efficacy (Nelson 2010).
Other scFvs were clinically exploited as part of fusion proteins, either with distinct functional modules or in bispecific form, as will be discussed in greater detail further below. For example, Darleukin (L19-IL2) is a fusion protein between the scFv L19 selected from a phage display library against the extra-domain B (ED-B) of oncofetal fibronectin (a splice form specific for neovasculature described below) and the cytokine interleukin-2 (IL-2) to trigger immunopotentiating and antineoplastic activities. This immunocytokine has been developed as combination therapy with dacarbazine for the treatment of melanoma in several clinical studies up to phase II (Philogen, http://www.philogen.com). The same L19 scFv moiety is also under clinical investigation in different formats and/or combination therapies, for example as TNFα fusion protein, as well as for radio-immunotherapy (RIT). While initially L19 was described as a high-affinity scFv having a remarkable KD in the low picomolar range (Pini et al. 1998), a significantly diminished potency was measured when reformatted to a Fab (Gebauer et al. 2013). Indeed, detailed inspection of the scFv revealed its propensity to form a stable homodimer, and this form showed much enhanced ED-B binding activity (most likely due to an avidity effect) in a head to head comparison with both the isolated monomeric scFv and the inherently monovalent Fab.
In a similar fusion protein approach, an immunotoxin (VB4-845) has been developed by combining a humanized anti-EpCAM scFv with a truncated fragment of Pseudomonas exotoxin A (Premsukh et al. 2011). This biological drug candidate is in phase II clinical trials (Viventia, http://www.viventia.com) and has shown promising results in the treatment of loco-regional tumors such as non-invasive-bladder cancer (Vicinium™) as well as recurrent refractory cancer of the head and neck (Proxinium™). Notably, to allow repeated systemic dosing, VB4-845 was recently reformatted as a Fab fragment fused to a deimmunised version of the plant proteotoxin bouganin via a furin-cleavable linker (VB6-845) (Cizeau et al. 2009; Entwistle et al. 2012).
The currently most advanced recombinant immunotoxin utilizing an Fv moiety is Moxetumomab pasudotox (CAT-8015 or HA22), which is in phase III clinical trials (Reichert 2013). This fusion protein comprises PE38, a catalytically active fragment of Pseudomonas exotoxin A, fused to an anti-CD22 Fv. Originally, this program had started with an scFv fragment derived from the anti-CD22 MAb RFB4; however, to increase protein stability an interchain disulfide bond was introduced instead of the peptide linker, thus converting the Fv moiety into the dsFv format (Kreitman and Pastan 2011). In a phase I trial for refractory hairy-cell leukemia (HCL), Moxetumomab pasudotox showed a high response rate and a remarkable number of complete remissions, despite some residual immunogenicity (Kreitman et al. 2012).
A clearly more robust functional antibody fragment is the Fab, comprising the entire light chain and the variable domain and first constant domain of the heavy chain (also known as Fd fragment). Due to the additional tight packing between the pair of constant domains from both chains, compared with the association between VH and VL alone, and an interchain disulfide bond that is usually located close to the C-termini, the Fab is a structurally well-defined protein with high stability similar to a full size Ig. Thus, Fabs were considered early on for medical applications wherein either an Fc effector region is not needed or one of the usually beneficial features of intact MAbs is detrimental in the clinical context, for example the long circulation or the bivalency which may cause cellular signaling via receptor clustering. The first Fabs for clinical use were prepared according to the classical route by proteolysis from polyclonal antibodies elicited in immunized sheep and have served as antidotes to treat acute intoxications since decades (Holliger and Hudson 2005). Also, several Fabs were approved as diagnostic reagents for radio-immuno imaging of tumors via single-photon emission computed tomography (SPECT) in vivo: 99mTc-Sulesomab (LeukoScan) and 99mTc-Bectumomab (LymphoScan) (Mendler et al. 2014).
The first Fab that was approved for therapy by the FDA in 1994 was Abciximab (RheoPro®), which binds to the platelet glycoprotein IIb/IIIa (integrin α2b/β3) on blood platelets (Knight et al. 1995), thus inhibiting their aggregation in cardiovascular disease. Notably, this Fab is produced by papain cleavage from a full length chimeric MAb. In contrast, Ranibizumab (Lucentis®) was the first fully recombinant Fab produced in E. coli approved by the FDA in 2006 for treating wet age-related macular degeneration (AMD) by intravitreal injection (Rosenfeld et al. 2006). Ranibizumab (Y0317), which binds and neutralizes vascular endothelial growth factor (VEGF-A), was derived from the same parent mouse antibody as the humanized Ig Bevacizumab (Avastin®), previously developed for antiangiogenic cancer therapy, in a process involving in vitro affinity maturation via CDR mutagenesis and phage display (Chen et al. 1999).
The strictly monovalent nature and the lack of immunological effector functions are usually considered advantages of the Fab format, in particular if just the blocking of a biomolecular disease target or use as antidote is intended. However, the much shorter plasma half-life compared with full size Mabs, due to the smaller size and lack of endosomal recycling as explained in the next Chapter of this book, is a general disadvantage which hampers the broader clinical application of this protein class. One approach to circumvent this problem is PEGylation, as exemplified by Certolizumab pegol (CDP-870, Cimzia®), a bacterially produced Fab directed against TNFα conjugated to a branched PEG chain of 40 kDa via a single Cys residue that was introduced into the hinge region (Melmed et al. 2008; Rose-John and Schooltink 2003). Cimzia is approved since 2008 for the treatment of Crohn’s disease and RA (Blick and Curran 2007; Goel and Stephens 2010).
The number of marketed Fabs is still rather limited (Nelson 2010), despite previous optimistic expectations (Nelson and Reichert 2009). However, there is a robust clinical pipeline for this class of Ig fragments, often as PEGylated versions, with examples from major pharma and biotech companies: anti-TNFα Afelimomab (AZD9773, MAK195F, CytoFab™) for the treatment of severe sepsis; a PEGylated Fab directed against the type III secretion system of Pseudomonas aeroginosa (KB-001); anti-Factor D Lampalizumab (RG-7417) for treatment of dry AMD; anti-C5 Fab LFG316A against the same condition as well as other inflammatory eye diseases; the PEGylated humanised Fab CDP7657 developed as a monovalent CD40L antagonist for the treatment of systemic lupus erythematosus; and a humanized Fab (Idarucizumab, BI655075) destined as an antidote for dabigatran etexilate mesylate (Pradaxa®) to reverse its anticoagulation effect (Brennan 2014; Reichert 2015; Strohl and Strohl 2012).
Apart from scFv and Fab, another class of small Ig fragments known as domain antibodies (dAbs) or Nanobodies has attracted attention (Holliger and Hudson 2005). These artificial binding proteins comprise an isolated variable domain and exhibit a binding site made of just three CDRs. Consequently, such single-domain Ig fragments (sdIgs) cannot be derived from intact MAbs but must be generated by different methods. This approach is complicated by the fact that an unpaired variable Ig domain usually exposes a significant hydrophobic surface area to solvent, which is otherwise shielded by association either with the second variable domain from the other Ig chain or, as often seen for light chains, via formation of so-called Bence Jones homo-dimers (Stevens et al. 1991). Especially in the case of isolated heavy chain variable domains (VH) this fundamental property normally causes aggregation (Davies and Riechmann 1994; Glockshuber et al. 1990) and/or non-specific adsorption.
Natural antibodies that are devoid of light chains were initially identified as an Ig subclass in camels (Hamers-Casterman et al. 1993; Muyldermans et al. 2001; Wiecek 2010). In such “heavy-chain” antibodies, which are composed of a pair of heavy chains that lack the CH1 domain and sometimes have an extended hinge region, the VH domains have apparently evolved to stay soluble without hetero- or homo-dimerization. These camelid VH domains (dubbed VHH), which are found in camels, dromedaries, and also in llamas, reveal elevated surface hydrophilicity in the region that faces the VL domain in ordinary Igs. In addition, they have a much longer CDR-H3, which often participates in an additional disulfide bridge within the VH domain, thus partially shielding this interface region from solvent (Conrath et al. 2005; Muyldermans 2013).
Engineered camelid VHH domains (detached from the constant region) are exploited for disease diagnosis and therapy (Siontorou 2013) as Nanobodies® (Ablynx, http://www.ablynx.com). As these sdIgs intrinsically have a very short plasma half-life, the corresponding drug candidates are usually developed as fusion proteins to enlarge size and retard kidney filtration. This involves combining either several copies of the same domain or domains with different specificities, including one that confers affinity to serum albumin in order to prolong circulation via endosomal recycling, a mechanism further explained in the following Chapter. Notably, the latter strategy also provides a general targeting activity in the case of cancer and inflammatory diseases as these tissues are known to actively take up albumin (Merlot et al. 2014; Tijink et al. 2008).
Two advanced clinical candidates are Caplacizumab and Ozoralizumab (Ablynx). Caplacizumab (ALX-0081 and ALX-0681, respectively, for i.v. and s.c. administration) is a bivalent Nanobody, with two domains fused in tandem, directed against the A1 domain of von Willebrand Factor (vWF) that has successfully completed phase II trials in hematology to treat the rare disorder thrombotic thrombocytopenic purpura (TTP) (Bartunek et al. 2013; Holz 2012). Ozoralizumab (ATN103 or PF-5230896) is a bivalent Nanobody comprising two anti-TNFα modules which are linked to a third Nanobody that binds to human serum albumin to extend plasma half-life. A phase IIa study demonstrated a statistically significant improvement of disease scores compared with placebo in patients with active RA (Comer et al. 2012). Similarly, a monovalent anti-IL-6 receptor (IL-6R) Nanobody (ALX-0061) comprising a bispecific fusion protein with a second Nanobody that confers binding activity towards albumin has completed phase IIa studies and is developed for the treatment of inflammatory diseases like RA (Van Beneden et al. 2014). Two additional Nanobodies are part of phase I programs: ALX-0171 is a trivalent Nanobody (three identical copies consecutively fused to each other) that neutralizes Respiratory Syncytial Virus (RSV) and offers potential for inhalational administration to treat pulmonary infections; ALX-0761 constitutes a bispecific half-life extended Nanobody that neutralizes both IL-17A and IL-17F—comprising an N-terminal IL-17F specific moiety, a C-terminal moiety that binds both IL-17A and F and a central module conferring albumin affinity—which has shown efficacy in a cynomolgus monkey model of RA (Vanheusden et al. 2013).
Beyond therapeutic applications, Nanobodies have attracted attention for in vivo imaging (Chakravarty et al. 2014; Vaneycken et al. 2011). In this setting, their small size and rapid renal elimination allows high tumor to blood contrast already shortly after administration. The lead optimized HER2-specific Nanobody 2Rs15d was prepared in current-good-manufacturing-practice grade, labeled with 68Ga via a 1,4,7-triazacyclononane-1,4,7-triacetic acid (NOTA) derivative and assessed in biodistribution and positron emission tomography/computed tomography (PET/CT) studies using a murine SK-OV-3 tumor xenograft model, resulting in high-specific-contrast imaging of HER2-positive tumors with no observed toxicity (Xavier et al. 2013). 68Ga-NOTA-2Rs15d is under development for first-in-human clinical trials.
Another type of sdIg has been derived from the human immune system as so-called domain antibodies, dAbs (Connelly 2005), by employing cloned repertoires of human V-genes together with stringent selection protocols to achieve high protein solubility, apart from target-binding activity (Holt et al. 2003). So far, two products have advanced to the clinical trial stage. An anti-TNFα dAb-Fc fusion protein (Placulumab, ART-621, CEP-37247) (Gay et al. 2010) was tested in a phase II clinical study (NCT00928317) for the treatment of stable plaque psoriasis and RA (Strohl and Strohl 2012). Another dAb (dubbed AlbudAb™), which was selected to bind with affinities in the mid nM range to serum albumin from various species, was initially employed to create a fusion protein with the interleukin-1 receptor antagonist (IL-1Ra, also known as Anakinra), thus resulting in an immunomodulator for RA treatment that showed extended plasma half-life in a mouse model (Holt et al. 2008). AlbudAb was also combined with some bioactive peptides, in particular the orexigenic peptide YY (PYY) and Exendin-4, a clinically approved GLP-1 mimetic (Bao et al. 2013). A recombinant fusion protein of AlbudAb with Exendin-4 (GSK2374697) was investigated in a phase I study in normal and obese healthy volunteers (Hodge et al. 2013).
An alternative source of naturally occurring sdIgs are the so-called Ig novel antigen receptors (IgNARs) of shark (Kovaleva et al. 2014). Antigen-specific VNAR fragments of these receptors have similar structure and properties as VHH from camels and llamas (Stanfield et al. 2004; Streltsov et al. 2004), and they can be cloned as isolated proteins either from immunized animals or selected in vitro from combinatorial libraries. However, despite some efforts to exploit the shark VNAR format for therapeutic applications by biotech companies, e.g. AdAlta (http://adalta.com.au) and Haptogen/Wyeth (http://www.wyeth.co.za), clinical applications have not thus far been publicized (Zielonka et al. 2015).
The Concept of Alternative Protein Scaffolds
sdIgs offer a kind of conservative approach for generating MAb biobetters insofar as the methods for their selection (in vivo or in vitro) as well as their biophysical properties are related to conventionally engineered antibody fragments, except that they do not rely on the characteristic light/heavy chain association of natural human Igs. However, it is still not fully clear whether all sdIgs behave as truly monomeric proteins (Schiefner et al. 2011). At least in some cases, X-ray structural analysis has indicated a mode of dimerization in the crystal packing that resembles either the VH/VL pairing of classical Igs or the structurally analogous light chain homo-association that is typical for Bence-Jones proteins (George et al. 2014; Jespers et al. 2004b). Thus, like for scFvs, intrinsic oligomerization and aggregation propensities may affect bioprocess development and/or biological drug formulation and must be carefully assessed in each instance.
Interestingly, however, proteins with non-Ig fold can also be furnished with novel binding sites. This has been achieved using the methodology of combinatorial biotechnology, that is site-directed random mutagenesis in combination with phage display or other molecular selection techniques (Rothe et al. 2006; Skerra 2003). The resulting class of novel alternative binding reagents has also become known as engineered “protein scaffolds” (Skerra 2000a), illustrating the fact that a structurally rigid natural protein is utilized either to modify an existing binding site towards a prescribed target or to implement a new one. This development has triggered a paradigm shift since antibodies have no longer to be considered as a unique class of binding reagents in biomedical research or biotechnology (Sheridan 2007; Skerra 2003) and, eventually, it has enabled the concept of biobetters discussed in this Chapter.
Usually, a protein scaffold suitable for therapeutic applications may be derived from either of two sources: (1) a robust and small monomeric soluble protein (e.g. a lipocalin or a Kunitz protease inhibitor—for details on the latter, see (Fiedler and Skerra 2014; Nixon et al. 2014)) or (2) a stably folded domain excised from a cell surface (e.g. protein A or fibronectin) or cytoskeletal protein (e.g. ankyrin). Compared with antibodies or their recombinant fragments, such protein scaffolds promise practical advantages, in particular elevated stability and high production yield in microbial expression systems, together with an independent intellectual property situation (Fiedler and Skerra 2014).
As these novel binding proteins are obtained via biomolecular engineering in vitro, with the primary goal to achieve tight and highly specific target-binding activity, they can also be subjected to additional selection schemes in order to address other desired properties such as solubility, thermal stability and protease resistance (Jermutus et al. 2001; Jespers et al. 2004a; Jung et al. 1999; Sieber et al. 1998). Resulting protein reagents are expected to provide beneficial properties for subsequent drug development and manufacturing as a built-in feature. Since the effort to generate and/or develop such alternative binding proteins still is greater than that for preparation of a conventional antibody (or a recombinant Ig fragment), most of the ongoing activities in this area are directed towards therapeutic use—instead of aiming at research or in vitro diagnostic reagents—hence offering the chance of a high return on investment from a business perspective. This has led to a series of corresponding biobetter formats developed by biotech companies, some of which have already been adopted by the pharmaceutical industry, as will be described in detail in the following sections.
So far, more than 50 different protein scaffolds have been proposed during the past two decades and these approaches have been extensively summarized in several recent reviews (Binz et al. 2005; Gebauer and Skerra 2009; Gill and Damle 2006; Hey et al. 2005; Mintz and Crea 2013; Nuttall and Walsh 2008; Wurch et al. 2012). This field emerged in the late 1990s with research on three different protein scaffolds, initially: (1) the Z-domain of staphylococcal protein A, a three-helix bundle of 58 residues, providing an interface for protein binding on two of its α-helices, leading to the Affibodies® (Nord et al. 1995); (2) the 10th extracellular domain of human fibronectin III (10FN3), which adopts an Ig-like β-sandwich fold (94 residues) with two to three exposed loops but lacks the central disulfide bridge, leading to the Monobodies (Koide et al. 1998), subsequently also dubbed Adnectins™; (3) the lipocalins, comprising a diverse family of soluble β-barrel proteins that naturally form binding sites for small ligands with four structurally variable loops, resulting in the Anticalins® (Beste et al. 1999).
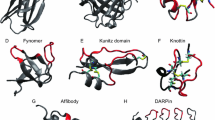
Whereas the Affibodies were initially derived from a bacterial protein, Adnectins and Anticalins are both based on endogenous human proteins. In contrast, the designed ankyrin repeat proteins, DARPins, were more recently proposed as an alternative scaffold as result of a protein design effort. In this case an artificial consensus sequence was derived from a series of known ankyrin repeat proteins, which are present in virtually all species and provide a rigid interface arising from modules with typically two anti-parallel α-helices and a β-turn as the underlying principle (Binz et al. 2003). Together, these four scaffolds (Fig. 1) currently constitute the most advanced approaches in this field, each with multiple target specificities exemplified in basic research as well as preclinical studies and at least one drug candidate tested in the clinic (Gebauer and Skerra 2009).

Structural comparison between single domain Ig fragments and advanced alternative protein scaffolds (shown from left to right in different colors), each in complex with a biomedically relevant target protein (light grey): Nanobody (green) and EGFR fragment (PDB entry: 4KRL); Nanobody (green) and ricin (4LHQ); AdNectin (violet) and IL23 heterodimer (3QWR); AdNectin (violet) and EGFR extracellular region (3QWQ); DARPin (orange) and HER2 fragment (4HRL); Affibody (albumin binding domain; blue) and albumin (1TF0); Affibody (blue) and HER2 extracellular region (3MZW); Anticalin (red) and CTLA-4 extracellular region (3BX7); Anticalin (red) and ED-B as part of a three-domain fibronectin fragment (4GH7); DARPin (orange) and caspase 3 (2XZT)
Otherwise, the field of engineered protein scaffolds has undergone some consolidation and appears to focus on those few that may potentially yield products with commercial value. In particular, technical demands at the outset of biopharmaceutical drug development left behind many protein scaffolds that were once proposed in an academic setting, but never matured beyond an initial model case study (Gebauer and Skerra 2009). As result, only few scaffold technologies, in particular those mentioned above, have successfully expanded after first in vitro proof of concept and now see increasing application toward medical use, as discussed in the following sections.
Adnectins
The fibronectin type III domain (FN3), a small (10 kDa) autonomous folding unit found in the abundant extracellular matrix proteins fibronectin and tenascin, as well as in a large variety of other multidomain cell adhesion proteins (Hohenester and Engel 2002), was one of the first protein scaffolds employed for generating novel binding sites. The FN3 structure closely resembles the Ig domain fold, exhibiting three exposed loops, termed BC, DE, and FG, analogous to the CDRs described further above. In contrast, the FN3 domain lacks the central disulfide bond which normally connects the sandwich of β-sheets. Despite their conserved structure, individual FN3 domains show considerable sequence divergence, which hinted at their tolerance for mutations introduced in order to implement desired binding functions (Koide et al. 1998).
FN3 random libraries were based on the 10th type III (10FN3) domain from human fibronectin to generate antibody (or, more precisely, sdIg) mimics initially by randomizing the BC and FG loops and, later, also the DE loop (Koide et al. 1998; Lipovsek et al. 2007; Xu et al. 2002). The first variant of this scaffold that had a significant, though still moderate affinity (KD ≈ 5 μM) for ubiquitin as a model target was selected from a phagemid display library and called “Monobody”. In combination with mRNA display technology, this scaffold was subsequently commercialized as “Trinectin” and continued as “Adnectin™” (Lipovsek 2011).
The design of the 10FN3-based random libraries was successively improved using insights from synthetic antibody libraries, including loop length variation and bias in amino acid composition within the binding site, which eventually allowed the generation of high-affinity binding proteins against various targets (with KD values down to the pM range) (Hackel et al. 2008, 2010; Hackel and Wittrup 2010; Koide et al. 2007). Furthermore, the 10FN3 scaffold was investigated for the potential to utilize all six loops—at both ends of the β-sandwich structure—to implement binding activities (Batori et al. 2002). No consensus has yet emerged as to which loop design strategy is most effective, although some studies indicate that the BC and FG loops, which are topologically equivalent to the CDRs H1 and H3 of a VH domain, are most important for target binding (Koide et al. 2007). Recently, an alternative recognition mode was described that utilizes a concave binding site on the surface of the β-sandwich (Gilbreth and Koide 2012; Ramamurthy et al. 2012). Hence, the wedge-shaped 10FN3 scaffold offers the possibility to address either cleft-like epitopes, e.g. on interleukin 23 (IL-23), or those with a convex shape, e.g. on epidermal growth factor receptor (EGFR) (Fig. 1), depending on the interface.
In 2006 the first Adnectin, Pegdinetanib (CT-322, BMS-844203, Angiocept), which specifically inhibits the vascular endothelial growth factor receptor 2 (VEGF-R2), i.e. the primary receptor in mediating tumor angiogenesis, entered clinical trials (Bloom and Calabro 2009; Dineen et al. 2008; Mamluk et al. 2010). Pegdinetanib, whose penultimate Cys residue (no. 93) is modified with a doubly methoxy-PEG-derivatized (40 kDa) maleimide, binds VEGF-R2 in vitro with a KD value of 11 nM (Mamluk et al. 2010). The first in human study confirmed the favorable pharmacokinetics of the PEGylated protein scaffold, showing slow plasma clearance with τ½ of 3–4 days (Tolcher et al. 2011). Notably, the majority of patients developed anti-drug antibodies (ADAs), which appeared to be specifically directed against the engineered binding loops, as no cross-reactivity with the original 10FN3 scaffold was seen. However, no adverse events associated with the ADA response appeared in this phase I trial (Tolcher et al. 2011). Nevertheless, a novel design of the Adnectin libraries aiming at eliminating immunogenic hot spots was recently published (Davis et al. 2013). In this approach, key areas within the β-strand B, the BC loop and β-strand C were kept as wild-type sequence while modifications were introduced into other regions of the scaffold to achieve high affinity target binding. Pegdinetanib was subsequently evaluated in multiple phase II trials to assess its applicability in a range of cancers including non-small cell lung cancer, glioblastoma multiforme and metastatic colorectal cancer.
Recently, a phase I clinical study of a second Adnectin (BMS-986089), a myostatin inhibitor developed to enhance muscle growth in indications such as muscular dystrophy or sarcopenia in older people (Cload et al. 2014), was initiated (BMS, http://www.b-ms.co.uk/research/pipeline). Myostatin is a secreted protein and member of the TGF-β family that is produced primarily in skeletal muscle cells, preventing muscle cell growth and differentiation, thus being of great interest as a therapeutic target in myopathies (Dschietzig 2014).
Other Adnectins in preclinical development include a fusion protein between fibroblast growth factor 21 (FGF21) and an “Adnectin pharmacokinetic enhancer” (AdPKE) as a potential therapeutic agent in diabetes. In FGF21-AdPKE the Adnectin moiety confers binding activity towards human as well as monkey serum albumin, thus extending the very short circulation half-life of human FGF21 from 4 to 96 h in cynomolgus monkeys (Klöhn et al. 2013), potentially offering once weekly subcutaneous dosing in human patients (Mukherjee 2013). The second Adnectin in advanced preclinical stage, BMS-938790 (likely in a PEGylated form), is a potent inhibitor of the inflammatory cytokine IL-23, whose increased levels are associated with several autoimmune diseases such as RA, multiple sclerosis (MS) and Crohn’s disease (Das Gupta 2014).
Apart from this 10FN3 scaffold, similar domains derived from the same fibronectin or related extracellular matrix proteins were also employed to generate scaffold libraries. For example, so-called Pronectins™ were developed on the basis of the 14th type III domain of fibronectin utilizing a bioinformatics approach (Mintz and Crea 2013). The Pronectin phage display library (Cappuccilli et al. 2014) was designed after analysis of the natural loop diversity found in thousands of non-redundant human FN3 sequences and comprises a combination of three sublibraries, each having mutations in only two of the exposed BC, DE and FG loops (Protelica, http://www.protelica.com).
Another related concept was recently adopted during development of the Centyrins (Jacobs et al. 2012). In this case, random libraries were built on an artificial consensus sequence derived from the tenascin FN3 domains. Not only the CDR-like loops but also exposed side chains on some of the β-strands were randomized to generate novel binding proteins, e.g. against human hepatocyte growth factor receptor (HGFR, c-MET), murine IL-17A and rat TNFα (Diem et al. 2014). Centyrins also offer potential as bispecific reagents (for broader discussion see further below), for example c-METxEGFR, engineered as a fusion protein of two Centyrin modules that are additionally linked to an albumin-binding domain (Klein et al. 2013).
Affibodies
Affibody molecules are small (7 kDa) artificial binding proteins originally derived from protein Z (Nilsson et al. 1987), an isolated engineered IgG-binding domain of the “protein A” found on the bacterial cell surface of Staphylococcus aureus. Combinatorial phage display libraries were generated by targeting random mutagenesis at residues exposed on the first two α-helices of the three-helix bundle scaffold in a region that is naturally involved in binding to the Fc part of antibodies (Nord et al. 1995, 1997). To date, a large number of Affibodies have been described, not only as research or purification reagents, but also exerting binding activities towards various medically relevant protein targets, e.g. insulin, fibrinogen, transferrin, TNFα, IL-8, gp120, CD28, IgA, IgE, IgM, EGFR, HER2 (Löfblom et al. 2010) and, recently, ErbB3/HER3 (Kronqvist et al. 2011). One Affibody, ZHER2:342, specific for the breast cancer target HER2, constitutes probably the most intensely studied protein of this class (>200 hits in PubMed; http://www.ncbi.nlm.nih.gov). This Affibody, which after extensive in vitro maturation shows extraordinary affinity for its target (KD = 22 pM) (Orlova et al. 2006), will be discussed further below.
Affibodies have been predominantly exploited for use in diagnostic settings, especially molecular imaging, where they offer beneficial features. The small size of this scaffold ensures rapid tissue penetration, fast blood clearance and, moreover, allows production via solid-phase peptide synthesis, permitting the direct site-specific incorporation of various chemical functionalities such as fluorescent probes or chelating groups for radioactive metal ions. The generally high stability, independent of disulfide bonds, in concert with the high solubility of most Affibodies further enable these molecules to resist even harsh chemical labelling conditions while retaining binding activity. Nevertheless, some Affibodies may adopt molten globule structures in the absence of their target proteins, probably due to the extensive mutagenesis of secondary structural elements (Wahlberg et al. 2003). Also, one Affibody was described that surprisingly adapts to the hairpin conformation of its target, the Alzheimer amyloid-β peptide, by dimerizing and forming a four-stranded intermolecular β-sheet, thus giving rise to an extended groove (Hoyer et al. 2008).
Up to now, Affibodies were developed in the clinic mainly as tracers for HER2-specific molecular imaging. In 2010, the 68Ga- or 111In-radiolabeled chelator-functionalized Affibody ABY-002 (DOTA-ZHER2:342-pep2) demonstrated for the first time that it is possible to localize via SPECT or PET/CT imaging even very small HER2-positive cancer lesions in patients with recurrent metastatic breast cancer and, notably, as early as 2 h after injection of the tracer (Baum et al. 2010). Imaging with ABY-002 was also successful in a patient that had already received Herceptin treatment. Indeed, ABY-002 binds to an epitope distinct from those of Trastuzumab (Herceptin®) and Pertuzumab (Perjeta®) (Eigenbrot et al. 2010) and, hence, this tracer may be useful for monitoring changes in HER2 expression during therapeutic intervention with these antibodies.
Compared to the current standard of tumor imaging, 18F-FDG PET, which indicates increased glucose metabolism, the uptake of ABY-002 in some metastatic lesions was distinct, either due to differing levels of HER2 expression or to the limited sensitivity of 18F-FDG PET/CT in early-stage cancer (Baum et al. 2010). However, this study also showed that the Affibody was mainly cleared via the liver, which obscured imaging of metastases in this organ. To address this issue, the scaffold of ABY-002 was reengineered by substituting surface-exposed side chains outside of the target interface to increase hydrophilicity, to achieve higher thermal and chemical stability, and to reduce unwanted binding to Ig as well as normal tissues, thus effecting elimination predominantly via the kidney (Feldwisch et al. 2010). The resulting anti-HER2 Affibody, ZHER2:2891, had an optimized surface that, despite the considerable deviation from the parental scaffold, fully retained tumor targeting functionality in vitro and in vivo in a mouse xenograft model (Ahlgren et al. 2010).
The clinical safety and efficacy of ZHER2:2891 for HER2 imaging in metastatic breast cancer was recently evaluated (Sörensen et al. 2014). In contrast to the parental molecule ABY-002 (DOTA-ZHER2:342-pep2), the new lead candidate ABY-025 ([MMA-DOTA-Cys61]-ZHER2:2891-Cys) can also be produced (apart from peptide synthesis) by recombinant expression in E. coli (Feldwisch et al. 2010) and, via a C-terminally introduced Cys residue (Z HER2:2891-Cys), coupled to the chelator maleimido-1,4,7,10-tetraazacyclododecane-1,4,7,10-tetraacetic acid (DOTA). The resulting radiolabeled molecule, 111In-ABY-025, allowed diagnosis of HER2 expression in seven patients while lacking the disadvantage of ABY-002, as the highest uptake of the new Affibody was seen in the kidneys (Sörensen et al. 2014). Probably as a benefit of the reengineered scaffold of ABY-025, and in good agreement with previous animal experiments (Ahlgren et al. 2010), no anti-ABY-025 antibodies were observed after single administration (100 μg). In one patient, molecular imaging with ABY-025 in conjunction with 18F-FDG PET enabled detection of a converted receptor status on metastases despite a HER2-positive primary tumor, and, in another patient, a brain lesion was diagnosed that was not seen via previous 18F-FDG PET alone (Sörensen et al. 2014).
Anti-HER2 Affibodies were also labeled with radionuclides suitable for RIT. Generally, the small size and rapid pharmacokinetics (Orlova et al. 2009), which is compatible with very short-lived positron emitting radionuclides like 68Ga, is particularly advantageous for molecular imaging, especially if compared to imaging with 89Zr-Trastuzumab, whose optimal time point to assess tumor accumulation is 4–5 days post injection (Dijkers et al. 2010). However, pronounced renal reabsorption of the Affibody radioconjugates and, thus, high levels of kidney exposure constitute a potential safety problem for therapeutic applications. Two approaches were evaluated to alleviate this problem for Affibodies: first, reducing renal uptake by modifying the pharmacokinetics via fusion to a second Affibody module that effects reversible binding to albumin (Orlova et al. 2013; Tolmachev et al. 2007, 2009b) and, second, decreasing renal retention of radioactivity by means of modified labelling chemistry (Orlova et al. 2010; Tolmachev et al. 2009a).
Notably, use of serum albumin as a drug carrier is often not only paired with longer circulation half-lives but also favors tumor targeting as albumin is known to preferentially localize to malignant versus normal tissue (Kratz 2008; Merlot et al. 2014). In the novel Affibody construct, 111In-ABY-027, the same target-specific molecule used for ABY-025 was fused to a small, high-affinity albumin-binding domain, ABD035, an engineered bacterial three-helix bundle protein with similar structure as the Z-domain (Jonsson et al. 2008), and site-specifically radiolabeled. In a mouse xenograft model 111In-ABY-027 showed a prolonged kinetic profile and significantly lower renal accumulation of radioactivity together with an increase in the delivered dose to the tumor compared to previous anti-HER2 Affibody reagents (Orlova et al. 2013). The impact of the nature and position of the radionuclide chelator within the protein molecule on the biodistribution of Affibodies was demonstrated using ZHER2:342 with different radiolabels, including mercaptoacetyl-containing peptide-based chelators for 99mTc-labelling. In this case, the use of more hydrophilic chelators resulted in a switch from predominantly hepatobiliary excretion to renal excretion, which significantly improved image contrast in the abdominal area (Tolmachev and Orlova 2009).
While most of the molecular imaging studies were performed with Affibodies specific for HER2, this approach was recently complemented by Affibodies targeting other important members of the transmembrane receptor tyrosine-kinase family such as EGFR (Malmberg et al. 2011; Tolmachev et al. 2010) and PDGF-Rβ (Strand et al. 2014; Tolmachev et al. 2014), which are both overexpressed in many malignancies.
Anticalins
Anticalins are antibody mimetics with designed ligand-binding properties derived from the natural scaffold of the lipocalins, a family of compact, soluble proteins that are abundant in human plasma and other body fluids and serve to transport or scavenge low molecular weight molecules. Lipocalins are found in many organisms including vertebrates, insects, plants and even bacteria (Åkerström et al. 2006). These functionally diverse small proteins, with a single polypeptide chain of 160–180 residues, generally form complexes with chemically sensitive biological compounds, in particular, vitamins, steroids, signaling molecules, lipids and other secondary metabolites. In humans, more than 12 different lipocalins have been identified thus far (Breustedt et al. 2006; Schiefner and Skerra 2015). Due to their high abundance in blood and their capacity for binding various physiologically active compounds, even some natural lipocalins have been considered for therapeutic use. In this regard, not only endogenous human lipocalins attracted attention, but also lipocalins from blood-sucking insects with medically relevant binding activities as well as low immunogenicity in humans.
For example, human neutrophil gelatinase-associated lipocalin (NGAL, also known as lipocalin 2, Lcn2, or as siderocalin), which tightly binds the catecholate-type siderophore FeIII · enterochelin/enterobactin (Goetz et al. 2002) as well as other siderophores of mycobacteria, was proposed for anti-infective drug development (Holmes et al. 2005). Apart from its potential use as antibacterial agent, its therapeutic application for renal protection from ischemia-reperfusion injury was suggested—based on the notion that NGAL also mediates iron trafficking in kidney epithelial cells (Mori et al. 2005). Furthermore, two insect lipocalins have attracted interest for clinical use: the histamine-binding protein (HBP) from the saliva of the tick Rhipicephalus appendiculatus (Paesen et al. 1999) and the C5 complement inhibitor (OmCI) from the soft tick Ornithodoros moubata (Barratt-Due et al. 2011; Fredslund et al. 2008).
In vivo efficacy was demonstrated for recombinant HBP (rEV131) in preclinical studies, where it was found to inhibit murine allergic asthma (Couillin et al. 2004), and this lipocalin was further evaluated up to a phase II clinical trial (NCT00353964). Preclinical studies of recombinant OmCI, initially termed rEV576, showed effective inhibition of complement and significantly reduced weakness in vivo in two rat models of experimentally acquired myasthenia gravis (Soltys et al. 2009). Other preclinical studies of OmCI demonstrated a protective effect against neural injury in an in vitro mouse model of the Miller Fisher syndrome (Halstead et al. 2008) as well as reduction of excessive inflammatory reactions associated with severe forms of Influenza A virus infections in mice (Garcia et al. 2013). OmCI, meanwhile named Coversin (Varleigh Immuno Pharmaceuticals, http://www.vipimmunopharma.com), has been evaluated in a phase I clinical study (Weston-Davies et al. 2013).
The central folding motif of the lipocalins is a structurally highly conserved β-barrel: eight antiparallel β-strands wind around a central axis and form an almost cylindrical cup-shaped structure. The bottom of this β-barrel is closed by short loops, and a hydrophobic core is formed there by densely packed amino acid side chains. At the open end, a set of four loops forms the entrance to the ligand pocket. While in general the amino acid sequence homology among the lipocalins is extremely low (Flower 1996), these four loops additionally exhibit large differences in their lengths and backbone conformations, in line with the diverse ligand specificities seen for the different family members (Skerra 2000b; Schiefner and Skerra 2015). Thus, in principle, the ligand-binding sites of lipocalins with their four structurally hypervariable loops resemble those of Igs with their set of six CDRs. However, a major advantage of the lipocalin scaffold is its simple composition of just one rather short polypeptide chain. Thus, biotechnological production and purification of lipocalins and their engineered variants is much easier and more cost-effective than antibody production, especially in connection with an E. coli expression system.
The peculiar ligand-binding site of the lipocalins with its (outside the Ig superfamily) unprecedented structural plasticity has prompted the engineering of novel antigen-binding proteins with antibody-like properties that were named “Anticalins” (Beste et al. 1999; Skerra 2001). Employing the methods of combinatorial protein design to alter the four hypervariable loops at the entrance to the ligand pocket as well as adjoining β-strand regions in a directed manner, artificial lipocalins with novel ligand specificities for prescribed targets can be generated (Gebauer and Skerra 2012). Meanwhile, several different natural lipocalin scaffolds for which three-dimensional structures became available have been utilized to generate Anticalins (Richter et al. 2014): e.g. the bilin-binding protein (BBP) from the butterfly Pieris brassicae, the human apolipoprotein D (ApoD), human lipocalin 1 (Lcn1, also known as tear lipocalin, Tlc) and human lipocalin 2 (Lcn2, NGAL).
In the case of BBP, a blue pigment protein that naturally protects insects from oxidative stress (Schmidt and Skerra 1994), the first Anticalins with novel specificities were selected via phage display from a semi-synthetic random library to bind the dye fluorescein (Beste et al. 1999), the plant steroid digoxigenin (Schlehuber et al. 2000) and a phthalic ester plasticiser (Mercader and Skerra 2002). These findings demonstrated the high tolerance of the lipocalin scaffold for introduction of artificial side chains into the binding site on a wider scale and, with the help of site-directed mutagenesis studies and crystal structure analyses (Korndörfer et al. 2003a, b; Vopel et al. 2005), confirmed the fundamental similarity with the combining site of antibodies.
Building on this knowledge, Anticalins were subsequently designed on the basis of human lipocalins, in particular Lcn1 and Lcn2, with the goal to ensure high tolerability to patients upon therapeutic application (Mendler and Skerra 2013). Again, these Anticalins were generated via combinatorial protein engineering in an in vitro process that in principle resembles the humoral immune response against an antigen (Gebauer and Skerra 2012). One notable aspect of this technology is that due to the large differences between natural members of the lipocalin family each scaffold involves an individually optimized random library design. This pertains to choosing the most suitable set of residues to be targeted for mutagenesis, whereby the total number of randomized positions is generally limited according to theoretical considerations (Richter et al. 2014). In case of the Lcn2 scaffold it was nicely demonstrated that the library design can be optimized in a few iterative cycles, also taking advantage of X-ray structural information (Gebauer et al. 2013). The third generation Lcn2-based Anticalin library resulting from this endeavor is highly potent in yielding binding proteins against a wide range of biomolecular targets, showing high affinities in the nM to pM range directly after phage display selection from the naive library (Richter et al. 2014) while still offering the potential for further improvement via in vitro affinity maturation.
Anticalins have been selected against a series of disease-relevant protein antigens. An early example was the extracellular domain of cytotoxic T-lymphocyte-associated antigen 4 (CTLA-4, CD152), which has attracted considerable interest as target of immune-checkpoint inhibitors (Pardoll 2012). In agreement with its function as a negative regulatory T-cell co-receptor, the lead Anticalin showed potent blocking activity and, consequently, an immunostimulatory effect on the T-cell response in an animal model of infectious disease (Schönfeld et al. 2009). Furthermore, this Anticalin has shown potential for the immunotherapy of cancer in preclinical studies (Pieris, http://www.pieris-ag.com). Another protein-specific Anticalin recognizes a disease-specific splice variant of the extracellular matrix protein fibronectin (Fn) exhibiting the extra-domain B (ED-B) (Gebauer et al. 2013). Since in adults ED-B-positive, so-called oncofetal Fn is almost exclusively expressed during tumor angiogenesis, apart from wound healing and some other pathophysiological states that involve neovascularization, it has emerged as a promising marker for various cancers and constitutes a validated target for in vivo imaging (Kaspar et al. 2006), as already mentioned further above. Cognate Anticalins show specific staining of ED-B positive cells in immunofluorescence microscopy and, notably, allow the detection of primary glioblastoma multiforme in human patients (Albrecht et al. submitted).
Beside Lcn2-based Anticalins, other human lipocalin scaffolds have been successfully applied to generate highly specific and functionally active binding proteins against medically relevant targets. The human Lcn1/Tlc scaffold served to develop an Anticalin that effectively blocks VEGF-A (Chekhonin et al. 2013) and constitutes a drug candidate for the treatment of solid cancers. This Anticalin, dubbed Angiocal® (PRS-050; conjugated with 40 kDa branched PEG), has been investigated as inhibitor of tumor angiogenesis in a first-in-human phase I trial, demonstrating safety and high tolerability as well as a terminal half-life (τ½) ranging from 5.5 to 7.0 days (Mross et al. 2013). Importantly, PRS-050 appeared to lack immunogenicity, based on the absence of an ADA response in 24 patients with post-baseline samples available, including six patients who received biweekly dosing and one who had received altogether 17 doses. Based on the encouraging data from this repeat dose escalating study, Angiocal shows promise for phase II clinical trials.
Recently, another Anticalin based on the Lcn1/Tlc scaffold (PRS-110; conjugated with 40 kDa branched PEG) was shown to act as a highly potent and specific c-Met antagonist with both ligand-dependent and ligand-independent activity in mouse xenograft models (Olwill et al. 2013). Moreover, a radiolabeled version of this PEGylated Anticalin, 89Zr-PRS-110, allowed visualization of c-Met tumor expression via in vivo imaging in mice (Terwisscha van Scheltinga et al. 2014).
Like the natural lipocalins and as mentioned above for the BBP platform, Anticalins derived from a human scaffold, in particular Lcn2, can also tightly bind low molecular weight substances, i.e. hapten-like targets. This has been shown for an Anticalin developed with pM affinity against a metal-chelate complex comprising a lanthanide ion, e.g. YIII, and a derivative of diethylenetriamine pentaacetic acid (DTPA) (Eggenstein et al. 2013; Kim et al. 2009), which offers a promising reagent for in vivo pretargeting radioimmuno therapy and diagnostics. Hapten-specific Anticalins based on the BBP scaffold have been investigated for therapeutic applications as well. The fluorescein-specific Anticalin FluA was used, after genetic fusion to an scFv fragment recognizing the Fn extra-domain A as angiogenesis marker, for pretargeted payload delivery in a mouse tumor xenograft model (Steiner et al. 2013). Also, an affinity-improved digoxigenin-binding Anticalin (DigiCal) showed promising results in a rat study as antidote to treat digitalis intoxication (Eyer et al. 2012).
High affinity Anticalins with blocking activity were also selected against peptides, for example the Aβ peptide (Rauth et al. in preparation), which plays a crucial role in the pathophysiology of Alzheimer’s disease, and hepcidin (Hohlbaum et al. 2011), a negative regulator of iron homeostasis relevant in anemia (Ruchala and Nemeth 2014). The development of Anticalins against the hepcidin peptide has been funded by the European Commission via the EUROCALIN Consortium (EUROCALIN, http://www.eurocalin-fp7.eu) to promote clinical investigation. PRS-080, an anti-hepcidin Anticalin designed to treat anemia, has entered a phase I clinical trial in 2014 (Pieris). Generally, the lipocalin scaffold appears to offer the most versatile platform to engineer binding proteins against a wide range of target molecules with different sizes, shapes and biomolecular properties (Fig. 1), including the potential to create multispecific or multifunctional fusion proteins (e.g. so-called Duocalins). Furthermore, it is the only type of scaffold for which natural representatives have been subjected to biopharmaceutical development.
DARPins
The so-called designed ankyrin repeat protein (DARPin) scaffold emerged from a consensus design approach (Binz et al. 2004; Forrer et al. 2004) based on the highly abundant natural ankyrin repeat proteins, ARPs (Grove et al. 2008; Mosavi et al. 2002). ARPs are found, for example, as cytoskeletal proteins, transcriptional initiators, cell cycle regulators and signal transducers mostly in the cellular cytoplasm, where they mediate protein-protein interactions. This protein class exhibits a rigid and also modular fold comprising characteristic repeat units of 33 amino acids, each with two anti-parallel α-helices that are mutually connected by a short loop and are linked to the next unit by an extended β-turn. Natural ARPs usually contain four to six of these repeats (while this number can rise up to ≥24, e.g. for ankyrin itself), which are stacked on each other to form a compact, overall rod-shaped domain, also called linear solenoid (Fig. 1). To generate DARPins, random amino acid substitutions are introduced into the β-turn and the first α-helix of each module, usually employing three repeat units (depending on the DARPin library), together with N- and C-terminal capping units. Typically, six potential target interaction positions per internal repeat are mutated in the artificial ARP consensus sequence.
From such combinatorial libraries DARPins with high affinities have been selected, normally via ribosome display, against a variety of proteins, including several medically relevant targets: for example, HIV gp120 (Mann et al. 2013; Schweizer et al. 2008), EpCAM (Stefan et al. 2011), Alzheimer amyloid-β peptide (Aβ) (Hanenberg et al. 2014) and, as will be described in greater detail below, VEGF-A (Stahl et al. 2013). Since the advantageous biophysical properties of the parental ankyrin scaffold such as high-level expression, solubility, and stability are often retained in these DARPins, they are also well suited for the generation of multispecific constructs (Molecular Partners, http://www.molecularpartners.com). Indeed, the therapeutic potential of DARPins in the area of cancer therapy is supported by the development of bispecific and/or biparatopic DARPins (Jost et al. 2013) as well as toxin fusions (Simon et al. 2014).
Furthermore, methods were established to extend the intrinsically very short plasma half-life of DARPins (Zahnd et al. 2010), which appears to be only few minutes in cynomolgus monkeys (Klöhn et al. 2013). To this end, DARPins were site-specifically functionalized using unique Cys residues or, alternatively, through incorporation of non-natural amino acids via metabolic pressure in order to allow selective conjugation with PEG, serum albumin and, optionally, with cytotoxins (Moody et al. 2014; Simon et al. 2013, 2014). An interesting concept of DARPins directed against tumor-specific cellular surface receptors such as HER2, EGFR and EpCAM aims at gene therapy: different DARPin-targeted viruses were shown to efficiently discriminate between tissues, potentially offering cell type-specific in vivo gene delivery (Friedrich et al. 2013; Munch et al. 2013). Notably, viral envelope proteins fused with DARPins showed superior incorporation to corresponding fusion proteins with scFvs, which are typically used for this kind of approach (Munch et al. 2011), probably owing to the more robust nature of this non-Ig scaffold.
The lead clinical candidate DARPin, Abicipar pegol (initially developed as MP0112, after reformulation named AGN-150998), is a potent antagonist of VEGF-A and inhibits all relevant subtypes with IC50 ≤ 10 pM (Binz et al. 2014; Klöhn et al. 2013; Stahl et al. 2013). Abicipar pegol is currently tested in two human eye diseases related to neovascularization: age-related macular degeneration (AMD), a painless eye condition that leads to the gradual loss of central vision, and diabetic macular edema (DME) (Campochiaro et al. 2013). In two phase I/II clinical trials, MP0112 was shown to be well tolerated and to efficiently neutralize VEGF-A after a single intravitreal injection. However, application of the DARPin was also associated with adverse events such as ocular inflammation in most of the patients, probably caused by impurities from the bacterially produced drug. MP0112 was subsequently reformulated into AGN-150998 (EudraCT 2011). Although the sponsor has not yet publicized which modifications were made, it appears that the formulation of AGN-150998 comprises a DARPin covalently linked to a single molecule of methoxy maleimide polyethylene glycol 20 (mPEG20), possibly even containing a modified Fc part of human Ig (CROSS 2011; INN 2012).
Safety and efficacy of AGN-150998 were recently tested in a phase II clinical trial (REACH study), where the reformulated drug proved to be at least as effective as monthly dosed Ranibizumab (Lucentis), the standard of care for wet AMD, while showing longer duration of action (NCT01397409; Reuters 2014). However, in contrast to Ranibizumab, a humanized high affinity anti-VEGF Fab produced in E. coli (Rosenfeld et al. 2006) mentioned already further above, patients treated with AGN-150998 still experienced ocular inflammation, although to a lesser extent than in phase I (Campochiaro et al. 2013). Improvements to the manufacturing process for AGN-150998 are expected before commencing phase III studies (Bloomberg 2013).
Apart from this monospecific protein drug, the dual-antagonistic DARPin MP0260, which is directed both against VEGF-A and platelet-derived growth factor B (PDGF-B), is under development for the treatment of wet AMD and related conditions (Molecular Partners). Considering that another anti-VEGF/anti-PDGF combination therapy of wet AMD, i.e. Fovista® with Ranibizumab, had already shown superior efficacy in a phase II trial compared to Lucentis monotherapy (Boyer 2013), a bispecific “all-in-one” drug such as MP0260 might prove beneficial. However, in light of the potential complications and burden for the patient associated with repeated intravitreal drug injections for the treatment of chronic ocular diseases, an alternative route of administration would be highly desirable. Yet, only preclinical data were published so far with regard to the potential application of DARPins as eye-drops (Stahl et al. 2013).
Bispecific Constructs
Engineered protein scaffolds are no longer regarded as alternatives to conventional MAbs just for the sake of intellectual property or other commercial aspects; rather, there is increasing recognition that they, together with robust Ig fragments, may enable unique therapeutic modes of action more easily owing to their small size and facile manipulation, possibly extending to bioprocess development. One area of great interest (Garber 2014) is the preparation of bispecific binding proteins, in particular by way of the modular fusion approaches mentioned above. In fact, despite the long history of bispecific MAbs (Milstein 2000)—starting with hybrid hybridoma (quadroma) technology—this classical format still poses a challenge with regard to biomanufacturing, even though several strategies were recently developed to drive heterodimerization of the two halves of the Y-shaped Ig molecule more efficiently (Strohl and Strohl 2012).
One successful strategy that makes use of scFv fragments is the bispecific T-cell engager format, BiTE® (Bäuerle and Reinhardt 2009), directing the patient’s T-cell immune response to cancer cells by creating a physical link. This concept arose from the notion that bispecific MAbs which redirect non-cognate T-lymphocytes to tumor cells were found to be generally more efficient in triggering cell killing or elimination than conventional MAbs of the same target specificity (Riethmüller 2012; Weiner and Hillstrom 1991). To provide a simpler molecular format, BiTEs are composed of two flexibly linked scFvs, fused in tandem, one binding to CD3 as part of the T-cell receptor (TCR) complex and the other one to a predefined surface antigen on the tumor cell, for example CD19 on B-cell lymphomas. The small size of BiTEs allows bringing target and effector cells in close proximity, thereby enabling the formation of a cytolytic synapse and triggering tumor cell killing—a process that is normally seen only between T-cells and antigen presenting cells (Dreier et al. 2003; Hoffmann et al. 2005; Offner et al. 2006).
In the BiTE approach, systemic cytokine release and toxicities are low or negligible, probably as a result of the monovalent format, which does not activate the CD3 receptor on non-engaged T-cells in the absence of target cells (Brischwein et al. 2007). So far, BiTEs have been generated against several tumor-associated target molecules such as CD19, CD33, EpCAM, HER2, CEA, ephrin A2 (EphA2), and melanoma-associated chondroitin sulfate proteoglycan (MCSP) (Bäuerle et al. 2009).
Blinatumomab (AMG103, formerly MT103 or MEDI-538) (Löffler et al. 2000) was the first genetically engineered bispecific antibody that entered clinical trials and constitutes the most advanced BiTE (Garber 2014). Besides CD3, it targets the B-cell surface marker CD19 which is ubiquitously expressed throughout all stages of B-cell differentiation and, in particular, present on virtually all tested malignant B-lineage cells in acute lymphoblastic leukemia (ALL) (Raponi et al. 2011). This biological drug candidate has been tested in combination with chemotherapy in a phase III trial in ALL patients who were not cured by chemotherapy alone and were given a poor prognosis (NCT02013167). A preceding large phase II study had confirmed antileukemic activity of Blinatumomab alone in a difficult-to-treat population with relapsed/refractory ALL (Topp et al. 2014). Considering these encouraging results, the FDA designated Blinatumomab in July 2014 as breakthrough therapy for ALL and, furthermore, this compound received orphan drug designation for the treatment of non-Hodgkin’s lymphoma (NHL) (Viardot et al. 2013). Blinatumomab was approved by the FDA in the end of 2014 as Blincyto™ for treating a rare form of B-cell ALL (Sheridan 2015).
Another BiTE (AMG330) targets CD33, a cell surface marker of acute myeloid leukemia (AML), i.e. the most frequent form of acute leukemia in adults. Ex vivo studies of AMG330 with primary AML cells revealed potent cytotoxicity, showing efficient T-cell activation and expansion even in samples with low CD33 expression. Moreover, AMG330 did not modulate surface expression of CD33, suggesting that long-term exposure to the BiTE, as in the clinical application of Blinatumomab, should not diminish expression of the receptor (Krupka et al. 2014; Laszlo et al. 2014). Given the fact that all previous attempts to introduce novel targeted therapies for AML have failed to date—ultimately including the anti-CD33 antibody-drug conjugate (ADC) Mylotarg®, which had been approved in 2000 but was subsequently withdrawn from the market due to safety issues and limited benefit (Strohl and Strohl 2012)—it will be interesting to see if this BiTE may offer a more potent treatment.
The success of this bispecific format triggered the design of other scFv-based constructs with similar size and valence. A competing technology is the dual-affinity re-targeting (DART®) molecule (Johnson et al. 2010), a heterodimer composed of two polypeptide chains, each comprising a VH domain from one Fv fused to a VL domain from another one, thus resembling the classical Diabody format (Holliger et al. 1993). However, DARTs are covalently linked by a C-terminal interchain disulfide bond, which makes them independent of the stability of the natural VL/VH association and avoids domain exchange, which is known to affect conventional Diabodies (Johnson et al. 2010).
In head-to-head in vitro experiments comparing the DART and BiTE formats with the same Fv sequences, the CD19xCD3 DART was 4–60 fold more potent in redirecting cell killing (Moore et al. 2011). This was surprising insofar as the DART protein displayed only a twofold higher affinity for each target molecule. Consequently, this platform has been developed for different targets (MacroGenics, http://www.macrogenics.com). The first DART that entered phase I clinical study is MGD006 which binds the tumor target CD123 and redirects, via its CD3 specificity, T-cells against corresponding leukemic cells, resulting in the clearance of AML blasts in vitro and in vivo (Al Hussaini et al. 2013). Furthermore, the DART MGD007, directed against gpA33, is under development for the treatment of colorectal cancer and other gastrointestinal tumors (MacroGenics).
The relatively short biological half-life of BiTEs and DARTs, which is only a few hours in humans (Hoffman and Gore 2014), requires an appropriate dosing schedule and/or formulation to ensure lasting activation of T-cells against target cells. Blinatumomab, for instance, is administered as continuous intravenous infusion over four weeks to achieve sufficient steady-state plasma levels of the drug.
In comparison, a more favorable pharmacokinetic profile has been described for TandAb®s (Kipriyanov 2009; Kipriyanov et al. 1999; Klöhn et al. 2013). In this format, two pairs of scFvs with different specificities are genetically fused in tandem to self-assemble in a head-to-tail fashion, thus forming a tandem Diabody structure (Kontermann 2012). By individually engineering the lengths and compositions of the linker sequences that join the four Fv moieties, the individual V regions can be forced to specifically pair with their counterparts in the second polypeptide chain, thus stabilizing the bispecific protein (Le Gall et al. 2004). With their higher molecular weight (110 kDa) the molecular size of TandAbs is slightly above the renal threshold for first-pass kidney clearance and their half-life in non-human primates is between 3 and 23 h, depending on the dosage (Klöhn et al. 2013). Exhibiting binding activity for both the T-cell effector CD3 and the disease target CD19, the TandAb AFM11 was demonstrated to induce killing of tumor cells in preclinical studies, without any cytotoxicity toward target-negative cells (Zhukovsky et al. 2012b, 2014). Also, compared to similar BiTE and DART reagents, the tetravalent AFM11 showed increased avidity for both CD19 and CD3 as well as higher potency in preclinical experiments (McAleese and Eser 2012; Zhukovsky et al. 2014).
As in classical MAbs, bivalent binding activity is expected to lead to higher avidity and also efficacy, but this must be evaluated for each target with great care. Interestingly, application of elevated concentrations of AFM11 to T-cells in the absence of target-positive cells caused down-modulation of the CD3/TCR-complex following an initial T-cell boost (Zhukovsky et al. 2012a, b), a phenomenon that has not been reported for monovalent BiTEs and DARTs. A phase I clinical study with AFM11 in patients with CD19-positive B-cell malignancies was started in 2014 (NCT02106091). The most clinically advanced TandAb AFM13 (CD30xCD16A), which recruits host natural killer cells and macrophages via CD16A (FcγRIIIa) to kill CD30-positive cells in both B- and T-cell malignancies (Rajkovic et al. 2012), has been scheduled for a phase II study after it was demonstrated to be well tolerated and had shown anti-tumor activity in advanced Hodgkin lymphoma patients (Affimed, http://www.affimed.com). Like BiTEs and DARTs, TandAbs are also under development for the treatment of solid tumors (e.g. AFM21; EGFRvIIIxCD3).
In the context of bispecific biobetters, beyond the Ig fragments and alternative scaffolds discussed so far, there are also some miscellaneous approaches that deserve mention, especially as these have already reached the clinic or are close to it. For example, Fcab™s are engineered (homo-dimeric) Fc fragments that carry mutated loop sequences at the C-terminal tip of the CH3 domain, thus conferring target specificity while retaining FcRn binding as well as immunological effector functions (Wozniak-Knopp et al. 2010). A HER2-specific drug candidate, FS102 (Batey et al. 2013), is expected to reach a phase I investigation in the near future (F-star, http://www.f-star.com). Notably, this technology forms a link between antibody fragments and alternative scaffolds insofar as the binding site in the Fc fragment is generated via combinatorial design in a manner typical for non-Ig proteins. In an extension of the Fcab technology the mAb2™ format comprises a full-length MAb in which the natural Fc region has been replaced by an Fcab (Woisetschläger et al. 2013). Such bispecific MAbs can bind (bivalently) two different antigens at the same time, with corresponding drug candidates in the discovery to preclinical stage in immuno-oncology.
Furthermore, Fynomers® are non-Ig scaffolds based on the SH3 domain of the human intracellular Fyn tyrosine kinase, which carries two flexible loops suitable for randomization (Grabulovski et al. 2007). The FynomAb® technology platform involves fusion of several Fynomers with an Ig Fc region, thus creating a hybrid homodimeric construct. The lead product COVA322, a bispecific anti-TNFα/anti-IL17A FynomAb (Covagen, http://covagen.com), is in a phase Ib/IIa study for psoriasis (NCT02243787).
Conclusions and Outlook
Both recombinant antibody fragments and engineered binding proteins based on non-Ig scaffolds have emerged as biobetters which complement and, in the future, may even supersede MAbs for various applications in medical therapy (Table 1). With regard to Ig-derived binding proteins, either as recombinant fragments of existing MAbs or directly selected from cloned V-gene libraries, the Fab format is currently most successful, with several established biopharmaceuticals and a growing number of drug candidates in the clinical pipeline (Nelson and Reichert 2009). While their stably monovalent behavior provides a clear mechanistic benefit in a range of therapeutic settings, the predominant disadvantage of Fabs is their short circulation in the blood, which they share with all other platforms discussed in this Chapter. With Cimzia there is already a clinically approved version that shows an in vivo half-life comparable to MAbs, achieved with the help of PEGylation. Further conjugates of this kind are under development, both for Fabs and for most of the alternative scaffolds. However, PEGylation, with its potential burden of a non-biodegradable synthetic polymer, will likely be replaced by more advanced technologies, for example by albumin-binding peptides (Dennis et al. 2002) or by PASylation (Schlapschy et al. 2013), as will be discussed in the next Chapter.
Despite its long history and broad use in basic science, the scFv format is less likely to become a successful therapeutic drug class by itself, especially with regard to systemic application. The reason lies in the generally poor biochemical behavior and inferior stability at the level of protein folding and solubility (Demarest and Glaser 2008). The scFv format appears more promising as part of fusion proteins such as bispecific antibody reagents, e.g. the BITEs, or if linked to cytotoxic enzymes as recombinant immunotoxins. Nevertheless, even in these settings the labile VH/VL pairing and the presence of two crucial disulfide bonds often cause problems during biosynthesis and bioprocess development. In this context the advantages of truly single-domain binding proteins, including sdIgs and alternative scaffolds, are brought to light, which likely will replace scFv fragments as target-specific fusion partners in the long run.
Currently, Nanobodies are the most broadly developed class of sdIgs with regard to molecules in the clinic and disease-relevant target specificities. In contrast to dAbs, Nanobodies are generated from immunized animals and, although it is claimed that their immunogenic potential in patients is low, still a certain risk prevails due to the set of conserved mutations that are necessary to maintain solubility and folding stability of the camelid VHH format but that are distinct from the human germline V-gene sequences. Also, it is interesting to note that, at least for therapeutic applications, Nanobodies are not employed as single domain proteins per se but are usually fused with other Nanobody modules. Apart from achieving higher avidity for the biomedical target in case of the biparatopic constructs, the resulting larger size generally slows down kidney filtration, even if not incorporating an albumin-specific Nanobody module to further extend plasma half-life.
In some sense, sdIgs of this kind bridge conventional antibody technology, including their antigen-binding fragments, to the alternative scaffolds with their fully synthetic binding sites. The non-Ig scaffold that is structurally most closely related to dAbs or Nanobodies is the Adnectin, whose fold still remotely belongs to the Ig superfamily but shows important deviations, in particular the missing central disulfide bond. Nevertheless, Adnectins exhibit three CDR-like loops in homologous regions at one end of the wedge-like β-sandwich, which can be randomized in order to create Adnectin libraries suitable for in vitro selection against prescribed targets. Consequently, Adnectins are likely to show a similar preference for pocket-like epitopes as has been discussed for Nanobodies (Gebauer and Skerra 2009). While the number of publicized development programs has remained low, there is still considerable academic research on this protein scaffold in progress (Gilbreth and Koide 2012; Hackel and Wittrup 2010). Of note, the Adnectin scaffold constitutes an isolated domain from the abundant extracellular matrix protein fibronectin and, thus, it does not occur as such in the body. Consequently, there has been concern about the exposure of immunological neoepitopes in Adnectins. However, in clinical trials conducted so far, ADA responses, though detected, were mostly directed against the engineered loops (Tolcher et al. 2011).
Like the FN3 format, the scaffold for Affibodies is an isolated domain excised from the larger extracellular region of a cell surface protein, in this case the Z domain of protein A from Staphylococcus aureus. As this small three-helix bundle does not offer a set of spatially contiguous loops, exposed side chains in two neighboring α-helices were chosen for random mutagenesis. This leads to a rather flat interface for complex formation, fixed by the underlying secondary structure. A problem inherited by this scaffold is its bacterial origin, which poses a risk with regard to ADA formation. Even though a deimmunized version has been developed recently (Feldwisch et al. 2010), it is probably wise to apply this class of alternative binding proteins preferentially either to in vivo imaging, where only minute protein amounts are administered and multiple doses are not required, or to highly immune-suppressed patients.
DARPins are based on an engineered consensus sequence from the ankyrin repeat proteins and, albeit the fold differs, this scaffold is characterized by a conformationally rigid secondary structure not unlike Affibodies. This may be considered an advantage and explains the stability and beneficial biochemical behavior of many DARPins. Generally, it appears that this scaffold with its rather flat binding site is optimally suited to recognize protein targets (Binz et al. 2004; Boersma and Plückthun 2011); in fact, similar to Nanobodies, DARPins have found use as reagents for co-crystallisation of sensitive proteins to promote their X-ray structural analysis (Bukowska and Grütter 2013). To make DARPins also amenable to small molecule or peptide targets, so-called loop-DARPins were recently designed (Schilling et al. 2014). However, it remains to be seen if this concept is useful for wider application, especially with regard to biopharmaceutical development. Notably, so far, DARPins have only been studied in phase II clinical trials for local application in the eye, which is a relatively immune-privileged organ.
Anticalins distinguish themselves from the other non-Ig scaffolds discussed here as they are derived from natural soluble plasma proteins abundant in the human body. This is not the only feature they share with Igs. In addition, their natural binding site, which is made of four structurally hypervariable loops mounted on a sandwich-like, almost circular arrangement of β-strands, shows fundamental similarity to the CDRs of MAbs. However, there are also two key differences: first, natural lipocalins prefer small molecules as ligands and, second, they are not subject to genetic mechanisms of somatic recombination and hypermutation like antibodies. Nevertheless, the methodology of combinatorial protein design has closed this gap, and Anticalins have convincingly demonstrated the ability to address the full spectrum of conceivable medically relevant targets, i.e. proteins (both soluble and as part of cell surface receptors), peptides and, naturally, haptens, similar to their endogenous counterparts.
The preparation of Fc fusion proteins, which is discussed more broadly in the next Chapter of this book, has become popular for most of the non-Ig scaffolds and even for scFv fragments, in this case yielding so-called small modular immunopharmaceuticals (SMIPs) (Hayden-Ledbetter et al. 2009). This concept has been carried further by creating fusions between engineered binding proteins specific for one target and a full length MAb that carries its own specificity for another target, as exemplified by the mAb2 platform (Woisetschläger et al. 2013). These approaches form another link between the more conservative antibody engineering field and the presumably more innovative area of alternative scaffolds, indicating that at least from the industry perspective, both territories are on their way to optimize novel biopharmaceuticals by incorporating the best properties of each platform. However, with increasing complexity of the resulting constructs (Kontermann 2012), which also creates challenges for bioprocess development and quality control, the rationale behind the molecular design is not always clear, especially if compared to the classical bispecific antibody format (Milstein 2000).
In fact, it has to be questioned whether the diverse constructs that are currently in discussion really provide for biobetters in a functional sense. From the protein design perspective, it may be worthwhile examining more critically the features as well as benefits and drawbacks of the individual concepts to generate binding proteins before combining them in a hasty manner. A fast and successful biological drug development program is most likely based on a simple molecular design. This is probably easiest to realize for the construction of antagonists which just block a cell surface receptor or a signaling molecule, and there are already numerous examples of that in the alternative scaffold field. Another area with future potential for biobetters could be their combination with toxic drugs, as an alternative to the currently flourishing ADCs. Here, the non-Ig scaffolds may provide decisive advantages, including (1) the capability for site-directed conjugation with the drug in defined stoichiometry while retaining high target-binding activity, (2) possibly better endosomal escape into the cytoplasm of the target cell, enhancing cytotoxic effects, and (3) lower liver toxicity due to distinct routes of clearance.
References
Ahlgren S, Orlova A, Wållberg H, Hansson M, Sandström M, Lewsley R, Wennborg A, Abrahmsen L, Tolmachev V, Feldwisch J (2010) Targeting of HER2-expressing tumors using 111In-ABY-025, a second-generation affibody molecule with a fundamentally reengineered scaffold. J Nucl Med 51:1131–1138
Åkerström B, Borregaard N, Flower DA, Salier J-S (2006) Lipocalins. Landes Bioscience, Georgetown
AL Hussaini MH, Ritchey J, Rettig MP, Eissenberg L, Uy GL, Chichili G, Moore PA, Johnson S, Collins L, Bonvini E, DiPersio JF (2013) Targeting CD123 in leukemic stem cells using dual affinity re-targeting molecules (DARTs®). Blood 122:360
Albrecht V, Richter A, Pfeiffer S, Gebauer M, Lindner S, Gieser E, Schüller U, Schichor C, Gildehaus FJ, Bartenstein P, Tonn JC, Skerra A, Glass R. Anticalins directed against the fibronectin extra domain B (ED-B) as diagnostic tracers for glioblastomas. Submitted
Al-Lazikani B, Lesk AM, Chothia C (1997) Standard conformations for the canonical structures of immunoglobulins. J Mol Biol 273:927–948
Arndt KM, Müller KM, Plückthun A (1998) Factors influencing the dimer to monomer transition of an antibody single-chain Fv fragment. Biochemistry 37:12918–12926
Bain B, Brazil M (2003) Adalimumab. Nat Rev Drug Discov 2:693–694
Bao W, Holt LJ, Prince RD, Jones GX, Aravindhan K, Szapacs M, Barbour AM, Jolivette LJ, Lepore JJ, Willette RN, DeAngelis E, Jucker BM (2013) Novel fusion of GLP-1 with a domain antibody to serum albumin prolongs protection against myocardial ischemia/reperfusion injury in the rat. Cardiovasc Diabetol 12:148
Barratt-Due A, Thorgersen EB, Lindstad JK, Pharo A, Lissina O, Lambris JD, Nunn MA, Mollnes TE (2011) Ornithodoros moubata complement inhibitor is an equally effective C5 inhibitor in pigs and humans. J Immunol 187:4913–4919
Bartunek J, Barbato E, Heyndrickx G, Vanderheyden M, Wijns W, Holz JB (2013) Novel antiplatelet agents: ALX-0081, a Nanobody directed towards von Willebrand factor. J Cardiovasc Transl Res 6:355–363
Batey S, Leung K, Rowlands R, Isaac S, Carvalho J, Weller S, Wydro M, Gaspar M, Medcalf M, Pegram R, Drewett V, Tuna M, Haurum J, Sun HH (2013) Preclinical evaluation of FS102: a HER2-specific Fcab with a novel mechanism of action. Mol Cancer Ther 12:B123
Batori V, Koide A, Koide S (2002) Exploring the potential of the monobody scaffold: effects of loop elongation on the stability of a fibronectin type III domain. Protein Eng 15:1015–1020
Bäuerle PA, Reinhardt C (2009) Bispecific T-cell engaging antibodies for cancer therapy. Cancer Res 69:4941–4944
Bäuerle PA, Kufer P, Bargou R (2009) BiTE: teaching antibodies to engage T-cells for cancer therapy. Curr Opin Mol Ther 11:22–30
Baum RP, Prasad V, Muller D, Schuchardt C, Orlova A, Wennborg A, Tolmachev V, Feldwisch J (2010) Molecular imaging of HER2-expressing malignant tumors in breast cancer patients using synthetic 111In- or 68Ga-labeled affibody molecules. J Nucl Med 51:892–897
Beck A (2011) Biosimilar, biobetter and next generation therapeutic antibodies. mAbs 3:107–110
Beck A, Wurch T, Bailly C, Corvaia N (2010) Strategies and challenges for the next generation of therapeutic antibodies. Nat Rev Immunol 10:345–352
Beste G, Schmidt FS, Stibora T, Skerra A (1999) Small antibody-like proteins with prescribed ligand specificities derived from the lipocalin fold. Proc Natl Acad Sci U S A 96:1898–1903
Better M, Chang CP, Robinson RR, Horwitz AH (1988) Escherichia coli secretion of an active chimeric antibody fragment. Science 240:1041–1043
Binz HK, Stumpp MT, Forrer P, Amstutz P, Plückthun A (2003) Designing repeat proteins: well-expressed, soluble and stable proteins from combinatorial libraries of consensus ankyrin repeat proteins. J Mol Biol 332:489–503
Binz HK, Amstutz P, Kohl A, Stumpp MT, Briand C, Forrer P, Grütter MG, Plückthun A (2004) High-affinity binders selected from designed ankyrin repeat protein libraries. Nat Biotechnol 22:575–582
Binz HK, Amstutz P, Plückthun A (2005) Engineering novel binding proteins from nonimmunoglobulin domains. Nat Biotechnol 23:1257–1268
Binz HK, Forrer P, Stumpp MT (2014) Modified binding proteins inhibiting the VEGF-A receptor interaction. Patent publication US20140221295A1
Bird RE, Walker BW (1991) Single chain antibody variable regions. Trends Biotechnol 9:132–137
Blick SK, Curran MP (2007) Certolizumab pegol: in Crohn’s disease. BioDrugs 21:195–201
Bloom L, Calabro V (2009) FN3: a new protein scaffold reaches the clinic. Drug Discov Today 14:949–955
Bloomberg (2013) Allergan shares fall after CEO says two studies delayed. http://www.bloomberg.com/news/2013-05-01/allergan-shares-fall-after-ceo-says-drug-trial-will-be-delayed.html
Boersma YL, Plückthun A (2011) DARPins and other repeat protein scaffolds: advances in engineering and applications. Curr Opin Biotechnol 22:849–857
Bork P, Holm L, Sander C (1994) The immunoglobulin fold. Structural classification, sequence patterns and common core. J Mol Biol 242:309–320
Borras L, Gunde T, Tietz J, Bauer U, Hulmann-Cottier V, Grimshaw JP, Urech DM (2010) Generic approach for the generation of stable humanized single-chain Fv fragments from rabbit monoclonal antibodies. J Biol Chem 285:9054–9066
Boyer D (2013) A phase 2b study of Fovista™, a platelet derived growth factor (PDGF) inhibitor in combination with a vascular endothelial growth factor (VEGF) inhibitor for neovascular age-related macular degeneration (AMD). Invest Ophthalmol Vis Sci 54:2175
Brennan FR (2014) Monoclonal antibodies in phase 1 and 2 studies for immunological disorders. In: Dübel S, Reichert JM (eds) Handbook of therapeutic antibodies, 2nd edn. Wiley, Weinheim
Breustedt DA, Schönfeld DL, Skerra A (2006) Comparative ligand-binding analysis of ten human lipocalins. Biochim Biophys Acta 1764:161–173
Brischwein K, Parr L, Pflanz S, Volkland J, Lumsden J, Klinger M, Locher M, Hammond SA, Kiener P, Kufer P, Schlereth B, Baeuerle PA (2007) Strictly target cell-dependent activation of T cells by bispecific single-chain antibody constructs of the BiTE class. J Immunother 30:798–807
Bukowska MA, Grütter MG (2013) New concepts and aids to facilitate crystallization. Curr Opin Struct Biol 23:409–416
Campochiaro PA, Channa R, Berger BB, Heier JS, Brown DM, Fiedler U, Hepp J, Stumpp MT (2013) Treatment of diabetic macular edema with a designed ankyrin repeat protein that binds vascular endothelial growth factor: a phase I/II study. Am J Ophthalmol 155:697–704
Cappuccilli G, Crea R, Shen R, Hokanson CA, Kirk PB, Liston DR (2014) Universal fibronectin type III binding-domain libraries. Patent publication US8680019B2
Carter P, Presta L, Gorman CM, Ridgway JB, Henner D, Wong WL, Rowland AM, Kotts C, Carver ME, Shepard HM (1992) Humanization of an anti-p185HER2 antibody for human cancer therapy. Proc Natl Acad Sci U S A 89:4285–4289
Chakravarty R, Goel S, Cai W (2014) Nanobody: the “magic bullet” for molecular imaging? Theranostics 4:386–398
Chekhonin VP, Shein SA, Korchagina AA, Gurina OI (2013) VEGF in tumor progression and targeted therapy. Curr Cancer Drug Targets 13:423–443
Chen Y, Wiesmann C, Fuh G, Li B, Christinger HW, McKay P, de Vos AM, Lowman HB (1999) Selection and analysis of an optimized anti-VEGF antibody: crystal structure of an affinity-matured Fab in complex with antigen. J Mol Biol 293:865–881
Cizeau J, Grenkow DM, Brown JG, Entwistle J, MacDonald GC (2009) Engineering and biological characterization of VB6-845, an anti-EpCAM immunotoxin containing a T-cell epitope-depleted variant of the plant toxin bouganin. J Immunother 32:574–584
Cload S, Engle L, Lipovsek D, Madireddi M, Rakestraw GC, Swain J, Zhao W (2014) Fibronectin based scaffold domain proteins that bind to myostatin. Patent publication US20140105896A1
Comer G, Hegen M, Sharma A, Shields K (2012) Methods of treating immune disorders with single domain antibodies against TNF-alpha. Patent publication WO2012131053A1
Connelly R (2005) Fully human domain antibody therapeutics: the best of both worlds. Innov Pharm Technol 2005:42–45
Conrath K, Vincke C, Stijlemans B, Schymkowitz J, Decanniere K, Wyns L, Muyldermans S, Loris R (2005) Antigen binding and solubility effects upon the veneering of a camel VHH in framework-2 to mimic a VH. J Mol Biol 350:112–125
Couillin I, Maillet I, Vargaftig BB, Jacobs M, Paesen GC, Nuttall PA, Lefort J, Moser R, Weston-Davies W, Ryffel B (2004) Arthropod-derived histamine-binding protein prevents murine allergic asthma. J Immunol 173:3281–3286
CROSS (2011) N189084: the tariff classification of AGN-150998 from Germany. http://rulings.cbp.gov
Das Gupta R (2014) Preclinical development of an anti-IL-23 Adnectin and advancement into the clinic. Abstract; IBC’s 9th annual next generation protein therapeutics Summit, June 4–6. San Francisco, CA
Davies J, Riechmann L (1994) ‘Camelising’ human antibody fragments: NMR studies on VH domains. FEBS Lett 339:285–290
Davis J, Lipovsek D, Camphausen R (2013) Fibronectin binding domains with reduced immunogenicity. Patent publication WO2013067029A2
Demarest SJ, Glaser SM (2008) Antibody therapeutics, antibody engineering, and the merits of protein stability. Curr Opin Drug Discov Devel 11:675–687
Dennis MS, Zhang M, Meng YG, Kadkhodayan M, Kirchhofer D, Combs D, Damico LA (2002) Albumin binding as a general strategy for improving the pharmacokinetics of proteins. J Biol Chem 277:35035–35043
Diem MD, Hyun L, Yi F, Hippensteel R, Kuhar E, Lowenstein C, Swift EJ, O’Neil KT, Jacobs SA (2014) Selection of high-affinity Centyrin FN3 domains from a simple library diversified at a combination of strand and loop positions. Protein Eng Des Sel 27:419–429
Dijkers EC, Oude Munnink TH, Kosterink JG, Brouwers AH, Jager PL, de Jong JR, van Dongen GA, Schroder CP, Lub-de Hooge MN, de Vries EG (2010) Biodistribution of 89Zr-trastuzumab and PET imaging of HER2-positive lesions in patients with metastatic breast cancer. Clin Pharmacol Ther 87:586–592
Dineen SP, Sullivan LA, Beck AW, Miller AF, Carbon JG, Mamluk R, Wong H, Brekken RA (2008) The Adnectin CT-322 is a novel VEGF receptor 2 inhibitor that decreases tumor burden in an orthotopic mouse model of pancreatic cancer. BMC Cancer 8:352
Dirix LY, Rutten A, Huget P, Dirix M (2013) Trastuzumab emtansine in breast cancer. Expert Opin Biol Ther 13:607–614
Dreier T, Baeuerle PA, Fichtner I, Grün M, Schlereth B, Lorenczewski G, Kufer P, Lutterbüse R, Riethmüller G, Gjorstrup P, Bargou RC (2003) T cell costimulus-independent and very efficacious inhibition of tumor growth in mice bearing subcutaneous or leukemic human B cell lymphoma xenografts by a CD19-/CD3-bispecific single-chain antibody construct. J Immunol 170:4397–4402
Dschietzig TB (2014) Myostatin – from the mighty mouse to cardiovascular disease and cachexia. Clin Chim Acta 433:216–224
Eggenstein E, Eichinger A, Kim HJ, Skerra A (2013) Structure-guided engineering of Anticalins with improved binding behavior and biochemical characteristics for application in radio-immuno imaging and/or therapy. J Struct Biol 185:203–214
Eigenbrot C, Ultsch M, Dubnovitsky A, Abrahmsen L, Hard T (2010) Structural basis for high-affinity HER2 receptor binding by an engineered protein. Proc Natl Acad Sci U S A 107:15039–15044
EMEA (2007) Refusal CHMP assessment report for Mycograb. http://www.ema.europa.eu/docs/en_GB/document_library/EPAR_-_Public_assessment_report/human/000658/WC500070523.pdf
Entwistle J, Brown JG, Chooniedass S, Cizeau J, MacDonald GC (2012) Preclinical evaluation of VB6-845: an anti-EpCAM immunotoxin with reduced immunogenic potential. Cancer Biother Radiopharm 27:582–592
EudraCT (2011) No 2011-002526-43: Single and repeat dose study of the safety and efficacy of AGN-150998 in patients with exudative age-related macular degeneration. http://www.clinicaltrialsregister.eu/ctr-search/trial/2011-002526-43/DE
Eyer F, Steimer W, Nitzsche T, Jung N, Neuberger H, Müller C, Schlapschy M, Zilker T, Skerra A (2012) Intravenous application of an anticalin dramatically lowers plasma digoxin levels and reduces its toxic effects in rats. Toxicol Appl Pharmacol 263:352–359
Feldwisch J, Tolmachev V, Lendel C, Herne N, Sjoberg A, Larsson B, Rosik D, Lindqvist E, Fant G, Hoiden-Guthenberg I, Galli J, Jonasson P, Abrahmsen L (2010) Design of an optimized scaffold for affibody molecules. J Mol Biol 398:232–247
Fiedler M, Skerra A (2014) Non-antibody scaffolds as alternative therapeutic agents. In: Dübel S, Reichert JM (eds) Handbook of therapeutic antibodies, 2nd edn. Wiley, Weinheim
Flower DR (1996) The lipocalin protein family: structure and function. Biochem J 318:1–14
Forrer P, Binz HK, Stumpp MT, Plückthun A (2004) Consensus design of repeat proteins. ChemBioChem 5:183–189
Fredslund F, Laursen NS, Roversi P, Jenner L, Oliveira CL, Pedersen JS, Nunn MA, Lea SM, Discipio R, Sottrup-Jensen L, Andersen GR (2008) Structure of and influence of a tick complement inhibitor on human complement component 5. Nat Immunol 9:753–760
Friedrich K, Hanauer JR, Prüfer S, Münch RC, Völker I, Filippis C, Jost C, Hanschmann KM, Cattaneo R, Peng KW, Plückthun A, Buchholz CJ, Cichutek K, Mühlebach MD (2013) DARPin-targeting of measles virus: unique bispecificity, effective oncolysis, and enhanced safety. Mol Ther 21:849–859
Garber K (2014) Bispecific antibodies rise again. Nat Rev Drug Discov 13:799–801
Garcia CC, Weston-Davies W, Russo RC, Tavares LP, Rachid MA, Alves-Filho JC, Machado AV, Ryffel B, Nunn MA, Teixeira MM (2013) Complement C5 activation during influenza A infection in mice contributes to neutrophil recruitment and lung injury. PLoS One 8:e64443
Gay RD, Clarke AW, Elgundi Z, Domagala T, Simpson RJ, Le NB, Doyle AG, Jennings PA (2010) Anti-TNFα domain antibody construct CEP-37247: full antibody functionality at half the size. mAbs 2:625–638
Gebauer M, Skerra A (2009) Engineered protein scaffolds as next-generation antibody therapeutics. Curr Opin Chem Biol 13:245–255
Gebauer M, Skerra A (2012) Anticalins: small engineered binding proteins based on the lipocalin scaffold. Methods Enzymol 503:157–188
Gebauer M, Schiefner A, Matschiner G, Skerra A (2013) Combinatorial design of an Anticalin directed against the extra-domain B for the specific targeting of oncofetal fibronectin. J Mol Biol 425:780–802
George J, Compton JR, Leary DH, Olson MA, Legler PM (2014) Structural and mutational analysis of a monomeric and dimeric form of a single domain antibody with implications for protein misfolding. Proteins 82:3101–3116
Getts DR, Getts MT, McCarthy DP, Chastain EM, Miller SD (2010) Have we overestimated the benefit of human(ized) antibodies? mAbs 2:682–694
Gilbreth RN, Koide S (2012) Structural insights for engineering binding proteins based on non-antibody scaffolds. Curr Opin Struct Biol 22:413–420
Gill DS, Damle NK (2006) Biopharmaceutical drug discovery using novel protein scaffolds. Curr Opin Biotechnol 17:653–658
Glockshuber R, Malia M, Pfitzinger I, Plückthun A (1990) A comparison of strategies to stabilize immunoglobulin Fv-fragments. Biochemistry 29:1362–1367
Goel N, Stephens S (2010) Certolizumab pegol. mAbs 2:137–147
Goetz DH, Holmes MA, Borregaard N, Bluhm ME, Raymond KN, Strong RK (2002) The neutrophil lipocalin NGAL is a bacteriostatic agent that interferes with siderophore-mediated iron acquisition. Mol Cell 10:1033–1043
Grabulovski D, Kaspar M, Neri D (2007) A novel, non-immunogenic Fyn SH3-derived binding protein with tumor vascular targeting properties. J Biol Chem 282:3196–3204
Grove TZ, Cortajarena AL, Regan L (2008) Ligand binding by repeat proteins: natural and designed. Curr Opin Struct Biol 18:507–515
Hackel BJ, Wittrup KD (2010) The full amino acid repertoire is superior to serine/tyrosine for selection of high affinity immunoglobulin G binders from the fibronectin scaffold. Protein Eng Des Sel 23:211–219
Hackel BJ, Kapila A, Wittrup KD (2008) Picomolar affinity fibronectin domains engineered utilizing loop length diversity, recursive mutagenesis, and loop shuffling. J Mol Biol 381:1238–1252
Hackel BJ, Ackerman ME, Howland SW, Wittrup KD (2010) Stability and CDR composition biases enrich binder functionality landscapes. J Mol Biol 401:84–96
Halstead SK, Humphreys PD, Zitman FM, Hamer J, Plomp JJ, Willison HJ (2008) C5 inhibitor rEV576 protects against neural injury in an in vitro mouse model of Miller Fisher syndrome. J Peripher Nerv Syst 13:228–235
Hamers-Casterman C, Atarhouch T, Muyldermans S, Robinson G, Hamers C, Songa EB, Bendahman N, Hamers R (1993) Naturally occurring antibodies devoid of light chains. Nature 363:446–448
Hanenberg M, McAfoose J, Kulic L, Welt T, Wirth F, Parizek P, Strobel L, Cattepoel S, Spani C, Derungs R, Maier M, Plückthun A, Nitsch RM (2014) Amyloid-β peptide-specific DARPins as a novel class of potential therapeutics for Alzheimer disease. J Biol Chem 289:27080–27089
Harding FA, Stickler MM, Razo J, DuBridge RB (2010) The immunogenicity of humanized and fully human antibodies: residual immunogenicity resides in the CDR regions. mAbs 2:256–265
Hayden-Ledbetter MS, Cerveny CG, Espling E, Brady WA, Grosmaire LS, Tan P, Bader R, Slater S, Nilsson CA, Barone DS, Simon A, Bradley C, Thompson PA, Wahl AF, Ledbetter JA (2009) CD20-directed small modular immunopharmaceutical, TRU-015, depletes normal and malignant B cells. Clin Cancer Res 15:2739–2746
Hey T, Fiedler E, Rudolph R, Fiedler M (2005) Artificial, non-antibody binding proteins for pharmaceutical and industrial applications. Trends Biotechnol 23:514–522
Hodge RJ, O’Connor-Semmes RL, Lin J, Chism JP, Andrews SM, Gaddy JR, Nunez DJ (2013) GSK2374697, a long-acting GLP-1 mimetic: first use of an AlbudAb™ in humans – pharmacokinetics, pharmacodynamics, safety, and tolerability in healthy volunteers. Abstract No. 60-LB; American Diabetes Association 73rd scientific sessions, June 21–25. Chicago, IL
Hoffman LM, Gore L (2014) Blinatumomab, a bi-specific anti-CD19/CD3 BiTE® antibody for the treatment of acute lymphoblastic leukemia: perspectives and current pediatric applications. Front Oncol 4:63
Hoffmann P, Hofmeister R, Brischwein K, Brandl C, Crommer S, Bargou R, Itin C, Prang N, Baeuerle PA (2005) Serial killing of tumor cells by cytotoxic T cells redirected with a CD19-/CD3-bispecific single-chain antibody construct. Int J Cancer 115:98–104
Hohenester E, Engel J (2002) Domain structure and organisation in extracellular matrix proteins. Matrix Biol 21:115–128
Hohlbaum AM, Trentman S, Gille H, Allersdorfer A, Belaiba RS, Huelsmeyer M, Christian J, Sandal T, Matschiner G, Jensen K, Skerra A, Audoly LP (2011) Discovery and preclinical characterization of a novel hepcidin antagonist with tunable PK/PD properties for the treatment of anemia in different patient populations. Blood 118: Abstract 687
Holliger P, Hudson PJ (2005) Engineered antibody fragments and the rise of single domains. Nat Biotechnol 23:1126–1136
Holliger P, Prospero T, Winter G (1993) “Diabodies”: small bivalent and bispecific antibody fragments. Proc Natl Acad Sci U S A 90:6444–6448
Holmes MA, Paulsene W, Jide X, Ratledge C, Strong RK (2005) Siderocalin (Lcn 2) also binds carboxymycobactins, potentially defending against mycobacterial infections through iron sequestration. Structure 13:29–41
Holt LJ, Herring C, Jespers LS, Woolven BP, Tomlinson IM (2003) Domain antibodies: proteins for therapy. Trends Biotechnol 21:484–490
Holt LJ, Basran A, Jones K, Chorlton J, Jespers LS, Brewis ND, Tomlinson IM (2008) Anti-serum albumin domain antibodies for extending the half-lives of short lived drugs. Protein Eng Des Sel 21:283–288
Holz JB (2012) The TITAN trial – assessing the efficacy and safety of an anti-von Willebrand factor Nanobody in patients with acquired thrombotic thrombocytopenic purpura. Transfus Apher Sci 46:343–346
Hoyer W, Grönwall C, Jonsson A, Ståhl S, Härd T (2008) Stabilization of a β-hairpin in monomeric Alzheimer’s amyloid-β peptide inhibits amyloid formation. Proc Natl Acad Sci U S A 105:5099–5104
Humphreys DP (2003) Production of antibodies and antibody fragments in Escherichia coli and a comparison of their functions, uses and modification. Curr Opin Drug Discov Devel 6:188–196
INN (2012) Proposed International Nonproprietary Names for Pharmaceutical Substances (INN): List 108. WHO Drug Inf 26:401–471
Jacobs SA, Diem MD, Luo J, Teplyakov A, Obmolova G, Malia T, Gilliland GL, O’Neil KT (2012) Design of novel FN3 domains with high stability by a consensus sequence approach. Protein Eng Des Sel 25:107–117
Jermutus L, Honegger A, Schwesinger F, Hanes J, Plückthun A (2001) Tailoring in vitro evolution for protein affinity or stability. Proc Natl Acad Sci U S A 98:75–80
Jespers L, Schon O, Famm K, Winter G (2004a) Aggregation-resistant domain antibodies selected on phage by heat denaturation. Nat Biotechnol 22:1161–1165
Jespers L, Schon O, James LC, Veprintsev D, Winter G (2004b) Crystal structure of HEL4, a soluble, refoldable human VH single domain with a germ-line scaffold. J Mol Biol 337:893–903
Johnson S, Burke S, Huang L, Gorlatov S, Li H, Wang W, Zhang W, Tuaillon N, Rainey J, Barat B, Yang Y, Jin L, Ciccarone V, Moore PA, Koenig S, Bonvini E (2010) Effector cell recruitment with novel Fv-based dual-affinity re-targeting protein leads to potent tumor cytolysis and in vivo B-cell depletion. J Mol Biol 399:436–449
Jones PT, Dear PH, Foote J, Neuberger MS, Winter G (1986) Replacing the complementarity-determining regions in a human antibody with those from a mouse. Nature 321:522–525
Jonsson A, Dogan J, Herne N, Abrahmsén L, Nygren P-Å (2008) Engineering of a femtomolar affinity binding protein to human serum albumin. Protein Eng Des Sel 21:515–527
Jost C, Schilling J, Tamaskovic R, Schwill M, Honegger A, Plückthun A (2013) Structural basis for eliciting a cytotoxic effect in HER2-overexpressing cancer cells via binding to the extracellular domain of HER2. Structure 21:1979–1991
Jung S, Plückthun A (1997) Improving in vivo folding and stability of a single-chain Fv antibody fragment by loop grafting. Protein Eng 10:959–966
Jung S, Honegger A, Plückthun A (1999) Selection for improved protein stability by phage display. J Mol Biol 294:163–180
Kaspar M, Zardi L, Neri D (2006) Fibronectin as target for tumor therapy. Int J Cancer 118:1331–1339
Kim HJ, Eichinger A, Skerra A (2009) High-affinity recognition of lanthanide(III) chelate complexes by a reprogrammed human lipocalin 2. J Am Chem Soc 131:3565–3576
Kipriyanov SM (2009) Generation of bispecific and tandem diabodies. Methods Mol Biol 562:177–193
Kipriyanov SM, Moldenhauer G, Schuhmacher J, Cochlovius B, Von der Lieth CW, Matys ER, Little M (1999) Bispecific tandem diabody for tumor therapy with improved antigen binding and pharmacokinetics. J Mol Biol 293:41–56
Klein D, Jacobs S, Sheri M, Anderson M, Attar R, Barnakov A, Brosnan K, Bushey B, Chevalier K, Chin D, Cornejo C, Diem M, Hyun L, Kuhar E, McCabe F, Picha K, Spinka-Doms T, Swift E, O’Neil K (2013) Bispecific Centyrin simultaneously targeting EGFR and c-Met demonstrates improved activity compared to the mixture of single agents. Cancer Res 73(8 Suppl): Abstract nr LB-312
Klöhn PC, Wuellner U, Zizlsperger N, Zhou Y, Tavares D, Berger S, Zettlitz KA, Proetzel G, Yong M, Begent RH, Reichert JM (2013) IBC’s 23rd annual antibody engineering, 10th annual antibody therapeutics international conferences and the 2012 annual meeting of the antibody society, San Diego, 3–6 Dec 2012. mAbs 5, 178–201
Knight DM, Wagner C, Jordan R, McAleer MF, DeRita R, Fass DN, Coller BS, Weisman HF, Ghrayeb J (1995) The immunogenicity of the 7E3 murine monoclonal Fab antibody fragment variable region is dramatically reduced in humans by substitution of human for murine constant regions. Mol Immunol 32:1271–1281
Köhler G, Milstein C (1975) Continuous cultures of fused cells secreting antibody of predefined specificity. Nature 256:495–497
Koide A, Bailey CW, Huang X, Koide S (1998) The fibronectin type III domain as a scaffold for novel binding proteins. J Mol Biol 284:1141–1151
Koide A, Gilbreth RN, Esaki K, Tereshko V, Koide S (2007) High-affinity single-domain binding proteins with a binary-code interface. Proc Natl Acad Sci U S A 104:6632–6637
Kontermann R (2012) Dual targeting strategies with bispecific antibodies. mAbs 4:182–197
Korndörfer IP, Beste G, Skerra A (2003a) Crystallographic analysis of an “anticalin” with tailored specificity for fluorescein reveals high structural plasticity of the lipocalin loop region. Proteins 53:121–129
Korndörfer IP, Schlehuber S, Skerra A (2003b) Structural mechanism of specific ligand recognition by a lipocalin tailored for the complexation of digoxigenin. J Mol Biol 330:385–396
Kovaleva M, Ferguson L, Steven J, Porter A, Barelle C (2014) Shark variable new antigen receptor biologics – a novel technology platform for therapeutic drug development. Expert Opin Biol Ther 14:1527–1539
Kratz F (2008) Albumin as a drug carrier: design of prodrugs, drug conjugates and nanoparticles. J Control Release 132:171–183
Kreitman RJ, Pastan I (2011) Antibody fusion proteins: anti-CD22 recombinant immunotoxin moxetumomab pasudotox. Clin Cancer Res 17:6398–6405
Kreitman RJ, Tallman MS, Robak T, Coutre S, Wilson WH, Stetler-Stevenson M, Fitzgerald DJ, Lechleider R, Pastan I (2012) Phase I trial of anti-CD22 recombinant immunotoxin moxetumomab pasudotox (CAT-8015 or HA22) in patients with hairy cell leukemia. J Clin Oncol 30:1822–1828
Kronqvist N, Malm M, Göstring L, Gunneriusson E, Nilsson M, Hoiden Guthenberg I, Gedda L, Frejd FY, Ståhl S, Löfblom J (2011) Combining phage and staphylococcal surface display for generation of ErbB3-specific Affibody molecules. Protein Eng Des Sel 24:385–396
Krupka C, Kufer P, Kischel R, Zugmaier G, Bögeholz J, Köhnke T, Lichtenegger FS, Schneider S, Metzeler KH, Fiegl M, Spiekermann K, Baeuerle PA, Hiddemann W, Riethmüller G, Subklewe M (2014) CD33 target validation and sustained depletion of AML blasts in long-term cultures by the bispecific T-cell-engaging antibody AMG 330. Blood 123:356–365
Laszlo GS, Gudgeon CJ, Harrington KH, Dell’Aringa J, Newhall KJ, Means GD, Sinclair AM, Kischel R, Frankel SR, Walter RB (2014) Cellular determinants for preclinical activity of a novel CD33/CD3 bispecific T-cell engager (BiTE) antibody, AMG 330, against human AML. Blood 123:554–561
Le Gall F, Reusch U, Little M, Kipriyanov SM (2004) Effect of linker sequences between the antibody variable domains on the formation, stability and biological activity of a bispecific tandem diabody. Protein Eng Des Sel 17:357–366
Lee EC, Liang Q, Ali H, Bayliss L, Beasley A, Bloomfield-Gerdes T, Bonoli L, Brown R, Campbell J, Carpenter A, Chalk S, Davis A, England N, Fane-Dremucheva A, Franz B, Germaschewski V, Holmes H, Holmes S, Kirby I, Kosmac M, Legent A, Lui H, Manin A, O’Leary S, Paterson J, Sciarrillo R, Speak A, Spensberger D, Tuffery L, Waddell N, Wang W, Wells S, Wong V, Wood A, Owen MJ, Friedrich GA, Bradley A (2014) Complete humanization of the mouse immunoglobulin loci enables efficient therapeutic antibody discovery. Nat Biotechnol 32:356–363
Lindenmann J (1984) Origin of the terms ‘antibody’ and ‘antigen’. Scand J Immunol 19:281–285
Lipovsek D (2011) Adnectins: engineered target-binding protein therapeutics. Protein Eng Des Sel 24:3–9
Lipovsek D, Lippow SM, Hackel BJ, Gregson MW, Cheng P, Kapila A, Wittrup KD (2007) Evolution of an interloop disulfide bond in high-affinity antibody mimics based on fibronectin type III domain and selected by yeast surface display: molecular convergence with single-domain camelid and shark antibodies. J Mol Biol 368:1024–1041
Löfblom J, Feldwisch J, Tolmachev V, Carlsson J, Ståhl S, Frejd FY (2010) Affibody molecules: engineered proteins for therapeutic, diagnostic and biotechnological applications. FEBS Lett 584:2670–2680
Löffler A, Kufer P, Lutterbüse R, Zettl F, Daniel PT, Schwenkenbecher JM, Riethmüller G, Dörken B, Bargou RC (2000) A recombinant bispecific single-chain antibody, CD19 x CD3, induces rapid and high lymphoma-directed cytotoxicity by unstimulated T lymphocytes. Blood 95:2098–2103
Lonberg N (2005) Human antibodies from transgenic animals. Nat Biotechnol 23:1117–1125
Malmberg J, Tolmachev V, Orlova A (2011) Imaging agents for in vivo molecular profiling of disseminated prostate cancer – targeting EGFR receptors in prostate cancer: comparison of cellular processing of [111In]-labeled affibody molecule ZEGFR:2377 and cetuximab. Int J Oncol 38:1137–1143
Mamluk R, Carvajal IM, Morse BA, Wong H, Abramowitz J, Aslanian S, Lim AC, Gokemeijer J, Storek MJ, Lee J, Gosselin M, Wright MC, Camphausen RT, Wang J, Chen Y, Miller K, Sanders K, Short S, Sperinde J, Prasad G, Williams S, Kerbel R, Ebos J, Mutsaers A, Mendlein JD, Harris AS, Furfine ES (2010) Anti-tumor effect of CT-322 as an adnectin inhibitor of vascular endothelial growth factor receptor-2. mAbs 2:199–208
Mann A, Friedrich N, Krarup A, Weber J, Stiegeler E, Dreier B, Pugach P, Robbiani M, Riedel T, Moehle K, Robinson JA, Rusert P, Plückthun A, Trkola A (2013) Conformation-dependent recognition of HIV gp120 by designed ankyrin repeat proteins provides access to novel HIV entry inhibitors. J Virol 87:5868–5881
McAleese F, Eser M (2012) RECRUIT-TandAbs: harnessing the immune system to kill cancer cells. Future Oncol 8:687–695
Melmed GY, Targan SR, Yasothan U, Hanicq D, Kirkpatrick P (2008) Certolizumab pegol. Nat Rev Drug Discov 7:641–642
Mendler CT, Skerra A (2013) Anticalins: an emerging class of novel biologics to treat cancer and other severe diseases. Drug Future 38:169–179
Mendler CT, Friedrich L, Schlapschy M, Schwaiger M, Wester H-J, Skerra A (2015) High contrast tumor imaging with radio-labelled antibody Fab fragments tailored for optimized pharmacokinetics via PASylation. mAbs 7:96–109
Mercader JV, Skerra A (2002) Generation of anticalins with specificity for a nonsymmetric phthalic acid ester. Anal Biochem 308:269–277
Merlot AM, Kalinowski DS, Richardson DR (2014) Unraveling the mysteries of serum albumin – more than just a serum protein. Front Physiol 5:299
Milstein C (2000) With the benefit of hindsight. Immunol Today 21:359–364
Mintz CS, Crea R (2013) Protein scaffolds – the next generation of protein therapeutics? BioProcess Int 11:40–48
Moody P, Chudasama V, Nathani RI, Maruani A, Martin S, Smith ME, Caddick S (2014) A rapid, site-selective and efficient route to the dual modification of DARPins. Chem Commun (Camb) 50:4898–4900
Moore PA, Zhang W, Rainey GJ, Burke S, Li H, Huang L, Gorlatov S, Veri MC, Aggarwal S, Yang Y, Shah K, Jin L, Zhang S, He L, Zhang T, Ciccarone V, Koenig S, Bonvini E, Johnson S (2011) Application of dual affinity retargeting molecules to achieve optimal redirected T-cell killing of B-cell lymphoma. Blood 117:4542–4551
Mori K, Lee HT, Rapoport D, Drexler IR, Foster K, Yang J, Schmidt-Ott KM, Chen X, Li JY, Weiss S, Mishra J, Cheema FH, Markowitz G, Suganami T, Sawai K, Mukoyama M, Kunis C, D’Agati V, Devarajan P, Barasch J (2005) Endocytic delivery of lipocalin-siderophore-iron complex rescues the kidney from ischemia-reperfusion injury. J Clin Invest 115:610–621
Morrison SL, Johnson MJ, Herzenberg LA, Oi VT (1984) Chimeric human antibody molecules: mouse antigen-binding domains with human constant region domains. Proc Natl Acad Sci U S A 81:6851–6855
Mosavi LK, Minor DL Jr, Peng ZY (2002) Consensus-derived structural determinants of the ankyrin repeat motif. Proc Natl Acad Sci U S A 99:16029–16034
Mross K, Richly H, Fischer R, Scharr D, Buchert M, Stern A, Gille H, Audoly LP, Scheulen ME (2013) First-in-human phase I study of PRS-050 (Angiocal), an Anticalin targeting and antagonizing VEGF-A, in patients with advanced solid tumors. PLoS One 8:e83232
Mukherjee R (2013) FGF21-Adnectin-Pharmacokinetic Enhancer: a modified FGF21 protein with uniquely extended pharmacokinetic profile for the treatment of metabolic diseases. Abstract; Ramanbhai Foundation 6th international symposium, February 4–6. Ahmedabad, India
Munch RC, Muhlebach MD, Schaser T, Kneissl S, Jost C, Plückthun A, Cichutek K, Buchholz CJ (2011) DARPins: an efficient targeting domain for lentiviral vectors. Mol Ther 19:686–693
Munch RC, Janicki H, Volker I, Rasbach A, Hallek M, Buning H, Buchholz CJ (2013) Displaying high-affinity ligands on adeno-associated viral vectors enables tumor cell-specific and safe gene transfer. Mol Ther 21:109–118
Muyldermans S (2013) Nanobodies: natural single-domain antibodies. Annu Rev Biochem 82:775–797
Muyldermans S, Cambillau C, Wyns L (2001) Recognition of antigens by single-domain antibody fragments: the superfluous luxury of paired domains. Trends Biochem Sci 26:230–235
NCT00217841 Aurograb and Vancomycin in MRSA Infection. http://clinicaltrials.gov
NCT00353964 Safety and efficacy study of rEV131 in the treatment of ocular inflammation after cataract surgery. http://clinicaltrials.gov
NCT00928317 Dose ranging study of ART621 in subjects diagnosed with rheumatoid arthritis taking methotrexate. http://clinicaltrials.gov
NCT01397409 Evaluation of AGN-150998 in exudative age-related macular degeneration (AMD). http://clinicaltrials.gov
NCT02013167 Ph 3 trial of Blinatumomab vs investigator’s choice of chemotherapy in patients with relapsed or refractory ALL. http://clinicaltrials.gov
NCT02106091 Safety study to assess AFM11 in patients with relapsed and/or refractory CD19 positive B-cell NHL or B-precursor ALL. http://clinicaltrials.gov
NCT02243787 Safety and tolerability study of COVA322 in patients with stable chronic moderate-to-severe plaque psoriasis. http://clinicaltrials.gov
Nelson AL (2010) Antibody fragments: hope and hype. mAbs 2:77–83
Nelson AL, Reichert JM (2009) Development trends for therapeutic antibody fragments. Nat Biotechnol 27:331–337
Nielsen UB, Kirpotin DB, Pickering EM, Hong K, Park JW, Shalaby MR, Shao Y, Benz CC, Marks JD (2002) Therapeutic efficacy of anti-ErbB2 immunoliposomes targeted by a phage antibody selected for cellular endocytosis. Biochim Biophys Acta 1591:109–118
Nilsson B, Moks T, Jansson B, Abrahmsén L, Elmblad A, Holmgren E, Henrichson C, Jones TA, Uhlén M (1987) A synthetic IgG-binding domain based on staphylococcal protein A. Protein Eng 1:107–113
Nixon AE, Sexton DJ, Ladner RC (2014) Drugs derived from phage display – from candidate identification to clinical practice. mAbs 6:73–85
Nord K, Nilsson J, Nilsson B, Uhlén M, Nygren P-Å (1995) A combinatorial library of an α-helical bacterial receptor domain. Protein Eng 8:601–608
Nord K, Gunneriusson E, Ringdahl J, Ståhl S, Uhlén M, Nygren P-Å (1997) Binding proteins selected from combinatorial libraries of an α-helical bacterial receptor domain. Nat Biotechnol 15:772–777
Nuttall SD, Walsh RB (2008) Display scaffolds: protein engineering for novel therapeutics. Curr Opin Pharmacol 8:609–615
Offner S, Hofmeister R, Romaniuk A, Kufer P, Baeuerle PA (2006) Induction of regular cytolytic T cell synapses by bispecific single-chain antibody constructs on MHC class I-negative tumor cells. Mol Immunol 43:763–771
Olwill SA, Joffroy C, Gille H, Vigna E, Matschiner G, Allersdorfer A, Lunde BM, Jaworski J, Burrows JF, Chiriaco C, Christian HJ, Hülsmeyer M, Trentmann S, Jensen K, Hohlbaum AM, Audoly L (2013) A highly potent and specific MET therapeutic protein antagonist with both ligand-dependent and ligand-independent activity. Mol Cancer Ther 12:2459–2471
Orlova A, Magnusson M, Eriksson TL, Nilsson M, Larsson B, Hoiden-Guthenberg I, Widstrom C, Carlsson J, Tolmachev V, Ståhl S, Nilsson FY (2006) Tumor imaging using a picomolar affinity HER2 binding affibody molecule. Cancer Res 66:4339–4348
Orlova A, Wallberg H, Stone-Elander S, Tolmachev V (2009) On the selection of a tracer for PET imaging of HER2-expressing tumors: direct comparison of a 124I-labeled affibody molecule and trastuzumab in a murine xenograft model. J Nucl Med 50:417–425
Orlova A, Tran TA, Ekblad T, Karlström AE, Tolmachev V (2010) 186Re-maSGS-ZHER2:342, a potential Affibody conjugate for systemic therapy of HER2-expressing tumours. Eur J Nucl Med Mol Imaging 37:260–269
Orlova A, Jonsson A, Rosik D, Lundqvist H, Lindborg M, Abrahmsen L, Ekblad C, Frejd FY, Tolmachev V (2013) Site-specific radiometal labeling and improved biodistribution using ABY-027, a novel HER2-targeting affibody molecule-albumin-binding domain fusion protein. J Nucl Med 54:961–968
Osbourn J, Groves M, Vaughan T (2005) From rodent reagents to human therapeutics using antibody guided selection. Methods 36:61–68
Pachl J, Svoboda P, Jacobs F, Vandewoude K, van der Hoven B, Spronk P, Masterson G, Malbrain M, Aoun M, Garbino J, Takala J, Drgona L, Burnie J, Matthews R, Mycograb Invasive Candidiasis Study Group (2006) A randomized, blinded, multicenter trial of lipid-associated amphotericin B alone versus in combination with an antibody-based inhibitor of heat shock protein 90 in patients with invasive candidiasis. Clin Infect Dis 42:1404–1413
Padlan EA (1994) Anatomy of the antibody molecule. Mol Immunol 31:169–217
Paesen GC, Adams PL, Harlos K, Nuttall PA, Stuart DI (1999) Tick histamine-binding proteins: isolation, cloning, and three-dimensional structure. Mol Cell 3:661–671
Pardoll DM (2012) The blockade of immune checkpoints in cancer immunotherapy. Nat Rev Cancer 12:252–264
Pini A, Viti F, Santucci A, Carnemolla B, Zardi L, Neri P, Neri D (1998) Design and use of a phage display library. Human antibodies with subnanomolar affinity against a marker of angiogenesis eluted from a two-dimensional gel. J Biol Chem 273:21769–21776
Power BE, Doughty L, Shapira DR, Burns JE, Bayly AM, Caine JM, Liu Z, Scott AM, Hudson PJ, Kortt AA (2003) Noncovalent scFv multimers of tumor-targeting anti-Lewisy hu3S193 humanized antibody. Protein Sci 12:734–747
Premsukh A, Lavoie JM, Cizeau J, Entwistle J, MacDonald GC (2011) Development of a GMP phase III purification process for VB4-845, an immunotoxin expressed in E. coli using high cell density fermentation. Protein Expr Purif 78:27–37
Rajkovic E, Knackmuss S, Reusch U, Rothe A, Topp M, Younes A, Ravic M, Hucke C, Zhukvosky E, Little M (2012) RECRUIT-TandAb AFM13: overcoming limitations of monoclonal antibodies in Hodgkin lymphoma. Cancer Res 72(8 Suppl):Abstract nr 3521
Ramamurthy V, Krystek SR Jr, Bush A, Wei A, Emanuel SL, Das Gupta R, Janjua A, Cheng L, Murdock M, Abramczyk B, Cohen D, Lin Z, Morin P, Davis JH, Dabritz M, McLaughlin DC, Russo KA, Chao G, Wright MC, Jenny VA, Engle LJ, Furfine E, Sheriff S (2012) Structures of adnectin/protein complexes reveal an expanded binding footprint. Structure 20:259–269
Raponi S, De Propris MS, Intoppa S, Milani ML, Vitale A, Elia L, Perbellini O, Pizzolo G, Foa R, Guarini A (2011) Flow cytometric study of potential target antigens (CD19, CD20, CD22, CD33) for antibody-based immunotherapy in acute lymphoblastic leukemia: analysis of 552 cases. Leuk Lymphoma 52:1098–1107
Rauth S, Hinz D, Eichinger A, Schneider M, Uhrig M, Mayhaus M, Riemenschneider M, Skerra A. High-affinity Anticalins with aggregation-blocking activity directed against the Alzheimer β-amyloid peptide. In preparation
Reichert JM (2012) Marketed therapeutic antibodies compendium. mAbs 4:413–415
Reichert JM (2013) Antibodies to watch in 2013: mid-year update. mAbs 5:513–517
Reichert JM (2014) Antibodies to watch in 2014: mid-year update. mAbs 6:799–802
Reichert JM (2015) Antibodies to watch in 2015. mAbs 7:1–8
Reiter Y, Brinkmann U, Lee B, Pastan I (1996) Engineering antibody Fv fragments for cancer detection and therapy: disulfide-stabilized Fv fragments. Nat Biotechnol 14:1239–1245
Reuters (2014) Stage 3 phase 2 study of DARPin abicipar pegol (previously MP0112) supports progressing to phase III development program. http://uk.reuters.com/article/2014/07/01/molecular-partners-idUKnBw015546a+100+BSW20140701
Richie DL, Ghannoum MA, Isham N, Thompson KV, Ryder NS (2012) Nonspecific effect of Mycograb on amphotericin B MIC. Antimicrob Agents Chemother 56:3963–3964
Richter A, Eggenstein E, Skerra A (2014) Anticalins: exploiting a non-Ig scaffold with hypervariable loops for the engineering of binding proteins. FEBS Lett 588:213–218
Riechmann L, Foote J, Winter G (1988) Expression of an antibody Fv fragment in myeloma cells. J Mol Biol 203:825–828
Riethmüller G (2012) Symmetry breaking: bispecific antibodies, the beginnings, and 50 years on. Cancer Immun 12:12
Rose-John S, Schooltink H (2003) CDP-870 – Celltech/Pfizer. Curr Opin Investig Drugs 4:588–592
Rosenfeld PJ, Brown DM, Heier JS, Boyer DS, Kaiser PK, Chung CY, Kim RY, MARINA Study Group (2006) Ranibizumab for neovascular age-related macular degeneration. N Engl J Med 355:1419–1431
Rothe A, Hosse RJ, Power BE (2006) In vitro display technologies reveal novel biopharmaceutics. FASEB J 20:1599–1610
Ruchala P, Nemeth E (2014) The pathophysiology and pharmacology of hepcidin. Trends Pharmacol Sci 35:155–161
Schiefner A, Skerra A (2015) The menagerie of human lipocalins: a natural protein scaffold for molecular recognition of physiological compounds. Acc Chem Res 48:976–985
Schiefner A, Chatwell L, Korner J, Neumaier I, Colby DW, Volkmer R, Wittrup KD, Skerra A (2011) A disulfide-free single-domain VL intrabody with blocking activity towards Huntingtin reveals a novel mode of epitope recognition. J Mol Biol 414:337–355
Schilling J, Schöppe J, Plückthun A (2014) From DARPins to LoopDARPins: novel LoopDARPin design allows the selection of low picomolar binders in a single round of ribosome display. J Mol Biol 426:691–721
Schlapschy M, Binder U, Börger C, Theobald I, Wachinger K, Kisling S, Haller D, Skerra A (2013) PASylation: a biological alternative to PEGylation for extending the plasma half-life of pharmaceutically active proteins. Protein Eng Des Sel 26:489–501
Schlehuber S, Beste G, Skerra A (2000) A novel type of receptor protein, based on the lipocalin scaffold, with specificity for digoxigenin. J Mol Biol 297:1105–1120
Schmidt FS, Skerra A (1994) The bilin-binding protein of Pieris brassicae. cDNA sequence and regulation of expression reveal distinct features of this insect pigment protein. Eur J Biochem 219:855–863
Schönfeld D, Matschiner G, Chatwell L, Trentmann S, Gille H, Hülsmeyer M, Brown N, Kaye PM, Schlehuber S, Hohlbaum AM, Skerra A (2009) An engineered lipocalin specific for CTLA-4 reveals a combining site with structural and conformational features similar to antibodies. Proc Natl Acad Sci U S A 106:8198–8203
Schweizer A, Rusert P, Berlinger L, Ruprecht CR, Mann A, Corthesy S, Turville SG, Aravantinou M, Fischer M, Robbiani M, Amstutz P, Trkola A (2008) CD4-specific designed ankyrin repeat proteins are novel potent HIV entry inhibitors with unique characteristics. PLoS Pathog 4:e1000109
Sheridan C (2007) Pharma consolidates its grip on post-antibody landscape. Nat Biotechnol 25:365–366
Sheridan C (2015) Amgen’s bispecific antibody puffs across finish line. Nat Biotechnol 33:219–221
Sieber V, Plückthun A, Schmid FX (1998) Selecting proteins with improved stability by a phage-based method. Nat Biotechnol 16:955–960
Simon M, Frey R, Zangemeister-Wittke U, Plückthun A (2013) Orthogonal assembly of a designed ankyrin repeat protein-cytotoxin conjugate with a clickable serum albumin module for half-life extension. Bioconjug Chem 24:1955–1966
Simon M, Stefan N, Borsig L, Plückthun A, Zangemeister-Wittke U (2014) Increasing the antitumor effect of an EpCAM-targeting fusion toxin by facile click PEGylation. Mol Cancer Ther 13:375–385
Siontorou CG (2013) Nanobodies as novel agents for disease diagnosis and therapy. Int J Nanomedicine 8:4215–4227
Skerra A (1993) Bacterial expression of immunoglobulin fragments. Curr Opin Immunol 5:256–262
Skerra A (2000a) Engineered protein scaffolds for molecular recognition. J Mol Recognit 13:167–187
Skerra A (2000b) Lipocalins as a scaffold. Biochim Biophys Acta 1482:337–350
Skerra A (2001) ‘Anticalins’: a new class of engineered ligand-binding proteins with antibody-like properties. J Biotechnol 74:257–275
Skerra A (2003) Imitating the humoral immune response. Curr Opin Chem Biol 7:683–693
Skerra A, Plückthun A (1988) Assembly of a functional immunoglobulin Fv fragment in Escherichia coli. Science 240:1038–1041
Soltys J, Kusner LL, Young A, Richmonds C, Hatala D, Gong B, Shanmugavel V, Kaminski HJ (2009) Novel complement inhibitor limits severity of experimentally myasthenia gravis. Ann Neurol 65:67–75
Sörensen J, Sandberg D, Sandström M, Wennborg A, Feldwisch J, Tolmachev V, Aström G, Lubberink M, Garske-Román U, Carlsson J, Lindman H (2014) First-in-human molecular imaging of HER2 expression in breast cancer metastases using the 111In-ABY-025 affibody molecule. J Nucl Med 55:730–735
Stahl A, Stumpp MT, Schlegel A, Ekawardhani S, Lehrling C, Martin G, Gulotti-Georgieva M, Villemagne D, Forrer P, Agostini HT, Binz HK (2013) Highly potent VEGF-A-antagonistic DARPins as anti-angiogenic agents for topical and intravitreal applications. Angiogenesis 16:101–111
Stanfield RL, Dooley H, Flajnik MF, Wilson IA (2004) Crystal structure of a shark single-domain antibody V region in complex with lysozyme. Science 305:1770–1773
Stefan N, Martin-Killias P, Wyss-Stoeckle S, Honegger A, Zangemeister-Wittke U, Plückthun A (2011) DARPins recognizing the tumor-associated antigen EpCAM selected by phage and ribosome display and engineered for multivalency. J Mol Biol 413:826–843
Steiner M, Gutbrodt K, Krall N, Neri D (2013) Tumor-targeting antibody-anticalin fusion proteins for in vivo pretargeting applications. Bioconjug Chem 24:234–241
Steinmeyer DE, McCormick EL (2008) The art of antibody process development. Drug Discov Today 13:613–618
Steipe B, Schiller B, Plückthun A, Steinbacher S (1994) Sequence statistics reliably predict stabilizing mutations in a protein domain. J Mol Biol 240:188–192
Stevens FJ, Solomon A, Schiffer M (1991) Bence Jones proteins: a powerful tool for the fundamental study of protein chemistry and pathophysiology. Biochemistry 30:6803–6805
Strand J, Varasteh Z, Eriksson O, Abrahmsen L, Orlova A, Tolmachev V (2014) Gallium-68-labeled Affibody molecule for PET imaging of PDGFRβ expression in vivo. Mol Pharm 11:3957–3964
Streltsov VA, Varghese JN, Carmichael JA, Irving RA, Hudson PJ, Nuttall SD (2004) Structural evidence for evolution of shark Ig new antigen receptor variable domain antibodies from a cell-surface receptor. Proc Natl Acad Sci U S A 101:12444–12449
Strohl WR, Strohl LM (2012) Therapeutic antibody engineering: current and future advances driving the strongest growth area in the pharmaceutical industry. Woodhead, Cambridge, UK
Terwisscha van Scheltinga AG, Lub-de Hooge MN, Hinner MJ, Verheijen RB, Allersdorfer A, Hülsmeyer M, Nagengast WB, Schröder CP, Kosterink JG, de Vries EG, Audoly L, Olwill SA (2014) In vivo visualization of MET tumor expression and anticalin biodistribution with the MET-specific anticalin 89Zr-PRS-110 PET tracer. J Nucl Med 55:665–671
Thiel MA, Wild A, Schmid MK, Job O, Bochmann F, Loukopoulos V, Alcantara W, Schmidt A, Lichtlen P, Escher D (2013) Penetration of a topically administered anti-tumor necrosis factor alpha antibody fragment into the anterior chamber of the human eye. Ophthalmology 120:1403–1408
Tijink BM, Laeremans T, Budde M, Stigter-van Walsum M, Dreier T, de Haard HJ, Leemans CR, van Dongen GA (2008) Improved tumor targeting of anti-epidermal growth factor receptor Nanobodies through albumin binding: taking advantage of modular Nanobody technology. Mol Cancer Ther 7:2288–2297
Tolcher AW, Sweeney CJ, Papadopoulos K, Patnaik A, Chiorean EG, Mita AC, Sankhala K, Furfine E, Gokemeijer J, Iacono L, Eaton C, Silver BA, Mita M (2011) Phase I and pharmacokinetic study of CT-322 (BMS-844203), a targeted Adnectin inhibitor of VEGFR-2 based on a domain of human fibronectin. Clin Cancer Res 17:363–371
Tolmachev V, Orlova A (2009) Update on Affibody molecules for in vivo imaging of targets for cancer therapy. Minerva Biotecnol 21:21–30
Tolmachev V, Orlova A, Pehrson R, Galli J, Baastrup B, Andersson K, Sandström M, Rosik D, Carlsson J, Lundqvist H, Wennborg A, Nilsson FY (2007) Radionuclide therapy of HER2-positive microxenografts using a 177Lu-labeled HER2-specific Affibody molecule. Cancer Res 67:2773–2782
Tolmachev V, Mume E, Sjoberg S, Frejd FY, Orlova A (2009a) Influence of valency and labelling chemistry on in vivo targeting using radioiodinated HER2-binding Affibody molecules. Eur J Nucl Med Mol Imaging 36:692–701
Tolmachev V, Wållberg H, Andersson K, Wennborg A, Lundqvist H, Orlova A (2009b) The influence of Bz-DOTA and CHX-A″-DTPA on the biodistribution of ABD-fused anti-HER2 Affibody molecules: implications for 114mIn-mediated targeting therapy. Eur J Nucl Med Mol Imaging 36:1460–1468
Tolmachev V, Rosik D, Wallberg H, Sjoberg A, Sandstrom M, Hansson M, Wennborg A, Orlova A (2010) Imaging of EGFR expression in murine xenografts using site-specifically labelled anti-EGFR 111In-DOTA-ZEGFR:2377 Affibody molecule: aspect of the injected tracer amount. Eur J Nucl Med Mol Imaging 37:613–622
Tolmachev V, Varasteh Z, Honarvar H, Hosseinimehr SJ, Eriksson O, Jonasson P, Frejd FY, Abrahmsen L, Orlova A (2014) Imaging of platelet-derived growth factor receptor β expression in glioblastoma xenografts using Affibody molecule 111In-DOTA-Z09591. J Nucl Med 55:294–300
Topp MS, Goekbuget N, Stein AS, Bargou RC, Dombret H, Fielding AK, Ribera JM, Foà R, Zugmaier G, Holland C, Maniar T, Huber B, Nagorsen D, Kantarjian HM (2014) Confirmatory open-label, single-arm, multicenter phase 2 study of the BiTE antibody blinatumomab in patients (pts) with relapsed/refractory B-precursor acute lymphoblastic leukemia (r/r ALL). J Clin Oncol 32:5s (suppl; abstr 7005)
Tsianakas A, Brunner P, Ghoreschi K, Berger C, Loser K, Röcken M, Stingl G, Luger T, Jung T (2014) Topical administration of the single-chain anti-TNFα antibody DLX105 suppresses TNFα and Th17 cytokines in psoriatic skin. Abstract No. LB011; 44th annual meeting of the European Society for Dermatological Research (ESDR), Sept 10–13. Copenhagen, Denmark
Van Beneden K, Verschueren K, Willems W, Wouters H, D’Artois J, De Swert K, Arold G, De Bruyn S (2014) Impact of clinical remission on physical function in patients with rheumatoid arthritis treated with ALX-0061: post-hoc analysis of phase I/II data. Ann Rheum Dis 73:506
Vaneycken I, D’Huyvetter M, Hernot S, De Vos J, Xavier C, Devoogdt N, Caveliers V, Lahoutte T (2011) Immuno-imaging using nanobodies. Curr Opin Biotechnol 22:877–881
Vanheusden K, Detalle L, Hemeryck A, Vicari A, Grenningloh R, Poelmans S, Wouters H, Stöhr T (2013) Pre-clinical proof-of-concept of ALX-0761, a Nanobody neutralising both IL-17A and IL-17F in a cynomolgus monkey collagen induced arthritis model. Abstract No. 1287; annual meeting of the American College of Rheumatology (ACR), Oct 26–30. San Diego, CA
Viardot A, Goebeler M, Pfreundschuh M, Adrian N, Libicher M, Degenhard E, Stieglmaier J, Zhang A, Nagorsen D, Bargou RC (2013) Open-label phase 2 study of the bispecific T-cell engager (BiTE®) Blinatumomab in patients with relapsed/refractory diffuse large B-cell lymphoma. Blood 122:1811
Vopel S, Mühlbach H, Skerra A (2005) Rational engineering of a fluorescein-binding anticalin for improved ligand affinity. Biol Chem 386:1097–1104
Wahlberg E, Lendel C, Helgstrand M, Allard P, Dincbas-Renqvist V, Hedqvist A, Berglund H, Nygren P-Å, Härd T (2003) An affibody in complex with a target protein: structure and coupled folding. Proc Natl Acad Sci U S A 100:3185–3190
Weiner GJ, Hillstrom JR (1991) Bispecific anti-idiotype/anti-CD3 antibody therapy of murine B cell lymphoma. J Immunol 147:4035–4044
Werner RG (2004) Economic aspects of commercial manufacture of biopharmaceuticals. J Biotechnol 113:171–182
Weston-Davies W, Westwood JP, Nunn M (2013) Phase 1 clinical trial of novel complement C5 inhibitor coversin. Mol Immunol 56:264
Wickham TJ, Reynolds J, Drummond DC, Kirpotin DB, Lahdenranta J, Leonard S, Geretti E, Lee H, Klinz S, Hendriks BS, Olivier K, Eckelhofer I, Park JW, Benz CC, Moyo VM, Niyikiza C, Nielsen UB (2010) Preclinical safety and activity of MM-302, a HER2-targeted liposomal doxorubicin designed to have an improved safety and efficacy profile over approved anthracyclines. Cancer Res 70(24 Suppl):Abstract nr P3-14–09.
Wiecek AS (2010) Nanobodies: going single-domain. BioTechniques. http://www.biotechniques.com/news/Nanobodies-Going-single-domain/biotechniques-257771.html
Woisetschläger M, Rüker F, Mudde GC, Wozniak-Knopp G, Bauer A, Himmler G (2013) Modular antibody engineering: antigen binding immunoglobulin Fc CH3 domains as building blocks for bispecific antibodies (mAb2). In: Schmidt SR (ed) Fusion protein technologies for biopharmaceuticals (p. 583–589). Wiley, Chichester
Wozniak-Knopp G, Bartl S, Bauer A, Mostageer M, Woisetschläger M, Antes B, Ettl K, Kainer M, Weberhofer G, Wiederkum S, Himmler G, Mudde GC, Rüker F (2010) Introducing antigen-binding sites in structural loops of immunoglobulin constant domains: Fc fragments with engineered HER2/neu-binding sites and antibody properties. Protein Eng Des Sel 23:289–297
Wurch T, Pierré A, Depil S (2012) Novel protein scaffolds as emerging therapeutic proteins: from discovery to clinical proof-of-concept. Trends Biotechnol 30:575–582
Xavier C, Vaneycken I, D’Huyvetter M, Heemskerk J, Keyaerts M, Vincke C, Devoogdt N, Muyldermans S, Lahoutte T, Caveliers V (2013) Synthesis, preclinical validation, dosimetry, and toxicity of 68Ga-NOTA-anti-HER2 Nanobodies for iPET imaging of HER2 receptor expression in cancer. J Nucl Med 54:776–784
Xu L, Aha P, Gu K, Kuimelis RG, Kurz M, Lam T, Lim AC, Liu H, Lohse PA, Sun L, Weng S, Wagner RW, Lipovsek D (2002) Directed evolution of high-affinity antibody mimics using mRNA display. Chem Biol 9:933–942
Zahnd C, Kawe M, Stumpp MT, de Pasquale C, Tamaskovic R, Nagy-Davidescu G, Dreier B, Schibli R, Binz HK, Waibel R, Plückthun A (2010) Efficient tumor targeting with high-affinity designed ankyrin repeat proteins: effects of affinity and molecular size. Cancer Res 70:1595–1605
Zhukovsky E, Reusch U, Burkhardt C, Knackmuss S, Fucek I, Eser M, McAleese F, Ellwanger K (2012a) A T cell-engaging CD3 Recruit-Tandab potently kills CD19+ tumor B cells. Blood 120:3721
Zhukovsky E, Reusch U, Burkhardt C, Knackmuss S, Fucek I, Eser M, McAleese F, Ellwanger K, Little M (2012b) High affinity CD3 RECRUIT TandAb for T cell-mediated lysis of CD19+ tumor B cells. J Clin Oncol 30:8059
Zhukovsky E, Knackmuss S, Reusch U, Wall C, Ellwanger K, Fucek I, Burkhardt C (2014) Preclinical development, primary and secondary pharmacodynamics, of the CD19/CD3 Tandab (AFM11). J Clin Oncol 32:e19546
Zielonka S, Empting M, Grzeschik J, Könning D, Barelle CJ, Kolmar H (2015) Structural insights and biomedical potential of IgNAR scaffolds from sharks. mAbs 7:15–25
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2015 American Association of Pharmaceutical Scientists
About this chapter
Cite this chapter
Gebauer, M., Skerra, A. (2015). Alternative Protein Scaffolds as Novel Biotherapeutics. In: Rosenberg, A., Demeule, B. (eds) Biobetters. AAPS Advances in the Pharmaceutical Sciences Series, vol 19. Springer, New York, NY. https://doi.org/10.1007/978-1-4939-2543-8_13
Download citation
DOI: https://doi.org/10.1007/978-1-4939-2543-8_13
Publisher Name: Springer, New York, NY
Print ISBN: 978-1-4939-2542-1
Online ISBN: 978-1-4939-2543-8
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)