
Abstract
The term thalassemia was first applied to the anemias encountered frequently in people of the Italian and Greek coasts and nearby islands. The term now refers to a group of inherited disorders of globin chain synthesis. Thalassemia comprises of a group of hemoglobinopathies, which are classified according to the specific globin chain (α or β) whose synthesis is impaired. Thus, α-thalassemia and β-thalassemia are depression of synthesis of the respective chain.
Access provided by CONRICYT-eBooks. Download reference work entry PDF
Similar content being viewed by others


The term thalassemia was first applied to the anemias encountered frequently in people of the Italian and Greek coasts and nearby islands. The term now refers to a group of inherited disorders of globin chain synthesis. Thalassemia comprises of a group of hemoglobinopathies, which are classified according to the specific globin chain (α or β) whose synthesis is impaired. Thus, α-thalassemia and β-thalassemia are depression of synthesis of the respective chain.
Synonyms and Related Disorders
Alpha-thalassemia (Hb H disease or alpha-thalassemia intermedia, Hb Bart’s hydrops fetalis, or alpha-thalassemia major); Beta-thalassemia (beta-thalassemia major or Cooley anemia or homozygous beta-thalassemia, beta-thalassemia intermedia or compound heterozygote, beta-thalassemia minor or beta-thalassemia trait
Genetics/Basic Defects
-
1.
Inheritance
-
1.
Autosomal recessive.
-
2.
Most β-thalassemias are inherited in a Mendelian recessive fashion, but there is a subgroup of β-thalassemia alleles that behave as dominant negatives (Thein 2013).
-
1.
-
2.
α-Thalassemia syndromes (Kelly 2012)
-
1.
α-Thalassemia occurs when there is a defect or deletion in one or more of the four genes responsible for α-globin production, leading to insufficient or absent α-globin synthesis.
-
2.
A defect or deletion of one α-globin gene leads to a condition called silent trait that is completely asymptomatic.
-
3.
The presence of two defective α-globin genes causes α-thalassemia trait, manifesting as a mild microcytic, hypochromic anemia. Hemoglobin electrophoresis can aid in diagnosis, but only during the neonatal period, when hemoglobin Bart is detected at 3–10%. Because hemoglobin F predominates at birth, impaired production of α-globin chains results in excess unpaired γ-globin chains that form tetramers, making hemoglobin Bart. After birth, as the infant’s dominant hemoglobin transitions from F to A, γ-globin chain production subsides, and hemoglobin Bart is no longer detected. No treatment is necessary for α-thalassemia trait.
-
4.
The presence of three defective α-globin genes causes hemoglobin H disease. Hemoglobin Bart is detected (15–30%) on electrophoresis during the neonatal period. Later in life, when hemoglobin A should predominate, excess unpaired β-globin chains accumulate and form tetramers, making hemoglobin H. Unstable hemoglobin H precipitates in circulating red blood cells, leading to hemolysis. Patients have microcytic, hypochromic anemia and hepatosplenomegaly and also may have bony abnormalities resulting from marrow expansion, as well as cholelithiasis and icterus. Transfusion usually is not required, but occasionally patients require splenectomy.
-
5.
Hydrops fetalis occurs when all four α-globin genes are defective or absent. In the affected fetus, no α-globin chains are synthesized, and hemoglobin Bart is the predominant hemoglobin. Oxygen delivery to tissues is severely impaired, and the affected fetus has profound anemia, hepatosplenomegaly, and anasarca and usually is stillborn or dies shortly after birth. Lifelong transfusion therapy is required for those infants who survive.
-
1.
-
3.
α-Thalassemia (Vichinsky 2010)
-
1.
Normal fetal hemoglobin synthesis
-
1.
Early in gestation, embryonic hemoglobins (Gower1, Gower2, and Portland), which do not contain α-globin chains, are the predominant hemoglobins.
-
2.
They are rapidly replaced by fetal and then adult hemoglobin, which contain α-globin chains.
-
3.
Therefore, α-thalassemia mutations become phenotypically evident by 12 weeks of gestation.
-
1.
-
2.
Synthesis of α-chains is directed by four α-genes, two on each chromosome 16.
-
3.
α-Thalassemia represents a group of conditions with reduced or absent synthesis of one to all four of α-globin genes.
-
1.
α-Thalassemia-2 (α-/αα): resulting from deletion of one of the two α-globin genes.
-
2.
α-Thalassemia-1 (−−/αα): resulting from deletion of both α-globin genes.
-
3.
Hb H disease (−−/-α): resulting from deletion of three α-globin genes.
-
4.
Hb Bart’s hydrops fetalis (−−/−−): resulting from deletion of all four α-globin genes. Physiologically nonfunctional homotetramers γ4 and β4 make up most of the hemoglobin in the erythrocytes in infants with the Bart’s hydrops fetalis syndrome.
-
1.
-
4.
When both α-genes on a single chromosome are inactive, the designation αo-thalassemia is used. When there is some production of α-globin chains, α+-thalassemia is designated.
-
1.
-
4.
β-Thalassemia
-
1.
Synthesis of β-chains: directed by a single β-gene on each chromosome 11
-
2.
Caused by the reduced (β+) or absent (βo) synthesis of the β-globin chains of the hemoglobin tetramer which is made up of two α-globin and two β-globin chains (α2β2)
-
3.
Mutations in β-thalassemia
-
1.
In contrast to the α-thalassemias, most of the common β-thalassemia mutants are caused by point mutations rather than by gene deletion.
-
2.
Single base-pair mutations in the DNA alter processing of messenger RNA: most common.
-
3.
Chain terminator defects.
-
4.
Frameshift mutations.
-
5.
Polyadenylation mutations.
-
6.
Promoter mutations.
-
1.
-
4.
βo-Thalassemia
-
1.
The mutations prevent β-globin chain synthesis entirely.
-
2.
Absent β-globin synthesis in βo-thalassemia homozygotes.
-
3.
Accounts for about one-third of thalassemia patients.
-
1.
-
5.
β+-Thalassemia
-
1.
The mutations prevent β-chain synthesis partially.
-
2.
β-Globin synthesis reduces to 5–30% of normal levels in β+-thalassemia homozygotes.
-
1.
-
6.
β+/βo-Thalassemia compound heterozygotes
-
1.
-
5.
Pathogenesis (Yaish 2010)
-
1.
Basic defect in all types of thalassemia: imbalanced globin chain synthesis.
-
2.
A decrease in the rate of production of a certain globin chain (α, β, γ, δ) impedes hemoglobin (Hb) synthesis and creates an imbalance with the other globin chain that normally produce globin chains.
-
3.
Consequences of impaired production of globin chains.
-
1.
Result in less Hb deposit in each RBC, leading to hypochromasia.
-
2.
Deficiency in Hb causes RBCs to be smaller, leading to the classic hypochromic microcytic features of thalassemia.
-
1.
-
1.
-
6.
Phenotype-genotype correlation (Origa 2015)
-
1.
Homozygosity or compound heterozygosity for β-thalassemia most commonly results in the clinical phenotype of transfusion-dependent thalassemia major.
-
2.
However, a consistent proportion of homozygotes develop milder forms, called thalassemia intermedia, which range in severity from thalassemia major to the β-thalassemia carrier state.
-
3.
Ascertained molecular mechanisms leading to thalassemia intermedia.
-
1.
β-Thalassemia mutations
-
1.
Mild mutation
-
2.
Silent mutation
-
3.
Mild/silent mutation
-
1.
-
2.
Coinherited α-thalassemia
-
1.
Single α-globin gene deletion (-α/αα)
-
2.
Deletion of two α-globin gene (-α/-α or – /αα)
-
3.
Point mutations of the major α-2-globin gene
-
1.
-
3.
Genetic determinant of high Hb F production
-
1.
Due to the β-thalassemia mutation per se (δβ-thalassemia, β-promoter deletion)
-
2.
Coinherited Agamma or Ggamma promoter mutation (−158 Ggamma (A → T); −196 Agamma (C → T))
-
3.
Heterocellular HPFH, BCL11A on chromosome 2, and HBS1L-MYB region on chromosome 6
-
1.
-
1.
-
1.
Clinical Features
-
1.
α-Thalassemia (Harteveld and Higgs 2010; Origa et al. 2013)
-
1.
The most common genetic disorder of hemoglobin synthesis, affects up to 5% of the world’s population (Vichinsky 2010)
-
2.
Found in people of African descent, Indochina, Malaysia, and China
-
3.
Clinical classification of α-thalassemia (Galanello and Cao 2011)
-
1.
Silent carrier: clinically and hematologically normal
-
2.
Thalassemia trait
-
1.
Microcytosis
-
2.
Hypochromia
-
3.
Mild anemia
-
1.
-
3.
Hb H disease
-
1.
Moderate to severe microcytic, hypochromic, hemolytic anemia
-
2.
Mild jaundice
-
3.
Moderate hepatosplenomegaly
-
1.
-
4.
Hb Bart’s hydrops fetalis syndrome
-
1.
Severe anemia
-
2.
Generalized edema
-
3.
Ascites
-
4.
Marked hepatosplenomegaly
-
5.
Skeletal and cardiovascular malformations, usually death in utero
-
1.
-
1.
-
4.
Severity of resulting anemia quite variable ranging from asymptomatic carriers to a fatal in utero disease: depends on the number of functioning α-genes
-
1.
α-Thalassemia-2 or silent carrier α-thalassemia (α-/αα) (deficiency of only one globin gene)
-
1.
Twenty-five percent of African Americans
-
2.
3.4% of Greek Americans
-
3.
Slight microcytic red blood cells
-
4.
Borderline or minimal anemia
-
1.
-
2.
α-Thalassemia-1 or α-thalassemia trait (– /αα)(deficiency of two globin genes)
-
1.
Fifteen to twenty percent of Thai.
-
2.
Mildly anemic.
-
3.
Microcytic red blood cells.
-
4.
Slight variation in red blood cell size.
-
5.
Mild splenomegaly.
-
6.
Work capacity not impaired.
-
7.
Same phenotypic effect in the individual homozygous for α-thalassemia-2 (α-/α-): This form is much more common in black populations.
-
1.
-
3.
Hb H disease or α-thalassemia intermedia (−−/-α) (deficiency of three globin genes)
-
1.
One percent of Thai.
-
2.
Significant hypochromic anemia in the neonatal period.
-
3.
Microcytic anemia.
-
4.
Reticulocytosis.
-
5.
Jaundice.
-
6.
Splenomegaly (may be severe and occasionally complicated by hypersplenism).
-
7.
Growth retardation may be seen in children.
-
8.
Hemolysis precipitated by infections or oxidant drugs (e.g., iron, sulfonamides).
-
9.
Leg ulcers.
-
10.
Gallstones.
-
11.
Folic acid deficiency.
-
12.
Needs occasional transfusions, splenectomy, or avoidance of precipitating drugs.
-
13.
Severity of clinical features: related to the molecular basis of the disease. Patients with nondeletional types of Hb H disease are more severely affected than those with the common deletional types of Hb H disease.
-
1.
-
4.
Hb Bart’s hydrops fetalis or α-thalassemia major (−−/−−) (deficiency of all four globin genes)
-
1.
Most severe form of α-thalassemia
-
2.
Incompatible with life unless intrauterine blood transfusion is given because not enough functional hemoglobin is produced to sustain tissue oxygenation
-
3.
Profound oxygen deprivation in utero with resulting heart failure (hydrops fetalis)
-
4.
Stillborn or die soon after birth
-
5.
Pale edematous infant with signs of cardiac failure (edema and ascites) and prolonged intrauterine anemia
-
6.
Massive hepatosplenomegaly
-
7.
Severe erythroblastic anemia
-
8.
Retardation in brain growth
-
9.
Skeletal and cardiovascular anomalies
-
10.
Enlargement of the placenta
-
1.
-
1.
-
1.
-
2.
β-Thalassemia (Cao and Galantello 2010; Kelly 2012; Origa 2015)
-
1.
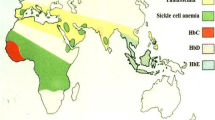
Found largely in people of African and Mediterranean descent, Far East, Middle East, and the Asian subcontinent
-
1.
Highest incidence
-
1.
Cyprus (14%)
-
2.
Sardinia (12%)
-
3.
Southeast Asia
-
1.
-
2.
The high gene frequency of beta-thalassemia in these regions
-
1.
Most likely related to the selective pressure from Plasmodium falciparum malaria, as it is indicated by its distribution quite similar to that of present or past malaria endemics. Carriers of beta-thalassemia are indeed relatively protected against the invasion of Plasmodium falciparum.
-
2.
However, because of population migration and, to a limited extent, slave trade, beta-thalassemia is, at present, also common in Northern Europe, North and South America, Caribbean, and Australia.
-
1.
-
1.
-
2.
Classification of beta-thalassemia (Galanello and Origa 2010)
-
1.
Beta-thalassemia
-
1.
Thalassemia major
-
2.
Thalassemia intermedia
-
3.
Thalassemia minor
-
1.
-
2.
Beta-thalassemia with associated Hb anomalies
-
1.
HbC/beta-thalassemia
-
2.
HbE/beta-thalassemia
-
3.
HbS/beta-thalassemia (clinical condition more similar to sickle cell disease than to thalassemia major or intermedia)
-
1.
-
3.
Hereditary persistence of fetal Hb and beta-thalassemia
-
4.
Beta-thalassemia associated with other manifestations
-
5.
Beta-thalassemia-trichothiodystrophy
-
6.
X-linked thrombocytopenia with thalassemia
-
1.
-
3.
Clinical features dependent on reduced (β+) or absent (βo) synthesis of β-globin
-
1.
β-Thalassemia major (also known as Cooley’s anemia or homozygous β-thalassemia)
-
1.
Due to inheritance of two β-thalassemia alleles, one on each copy of chromosome 11.
-
2.
Generally recognized to be a homozygous state for whichever thalassemia gene is involved.
-
3.
Clinically a severe disorder.
-
4.
Infants born free of significant anemia, protected by prenatal Hb F production.
-
5.
Onset: 6–12 months.
-
6.
Diagnosis: evident by 2 years of age.
-
7.
Manifestations in early childhood: anemia, pallor, growth retardation, tiredness, abdominal swelling due to hepatosplenomegaly, infection, and jaundice.
-
8.
Manifestations in childhood and adult: severe anemia, infection, tiredness, growth retardation, distinctive facies, hepatosplenomegaly, cardiomegaly, abdominal pain, leg ulcers, osteoporosis, and iron overload.
-
9.
At significant risk for developing overwhelming, often fatal infection after splenectomy (postsplenectomy syndrome).
-
10.
Severe anemia usually necessitating chronic blood transfusions.
-
11.
Craniofacial manifestation of β-thalassemia major (Javid and Said-Al-Naief 2015): The most characteristic oral and maxillofacial/head and neck skeletal changes are the upslanted eyes and the so-called chipmunk faces, with prominent frontal bossing, prominent cheekbones, and nasal bridge depression, accompanied by hypertrophied maxilla (often with exposure of the maxillary teeth) and mandible caused by excessive active bone marrow hyperplasia. In the maxilla, in particular, the large, widened medullary spaces and trabeculae radiographically mimic a “chicken wire” radiographic pattern.
-
12.
Prognosis: average survival of children with untreated thalassemia major (<4 years)
-
1.
-
2.
β-Thalassemia intermedia (compound heterozygotes)
-
1.
Due to inheritance of two β-thalassemia mutations (one mild and one severe or two mild mutations) or occasionally due to inheritance of complex combinations such as α-β-thalassemia.
-
2.
Affected individuals: either compound heterozygotes for two different thalassemia genes or heterozygotes for a structural variant plus a thalassemia gene. Homozygotes for the milder β+-thalassemia also have thalassemia intermedia.
-
3.
Less severe clinical phenotype: cardiomegaly, osteoporosis, fractures, arthritis, and splenomegaly in some patients.
-
4.
Neurological complications of beta-thalassemia: Cognitive impairment, abnormal findings on evoked potentials, complications due to extramedullary hematopoiesis, cerebrovascular disease, and peripheral neuropathy comprise the broad spectrum of neurological involvement (Nemtsas et al. 2015).
-
5.
Significant anemia but usually do not require chronic blood transfusions.
-
1.
-
3.
β-Thalassemia minor (also known as β-thalassemia trait or heterozygous β-thalassemia)
-
1.
Due to presence of a single β-thalassemia mutation and a normal β-globin gene on the other chromosome
-
2.
Clinically asymptomatic but may have mild or minimal anemia
-
1.
-
4.
Silent carrier β-thalassemia
-
1.
No symptoms except for possible low RBC indices.
-
2.
Mutation causing thalassemia is very mild and represents β + -thalassemia.
-
1.
-
1.
-
1.
-
3.
Practical guide to the diagnosis of thalassemia (Dumars et al. 1996)
-
1.
The first involves any patient with a low mean corpuscular volume (MCV) with or without anemia.
-
2.
The second is a neonatal screening result indicating possible presence of thalassemia.
-
3.
Finally, evaluation for thalassemia should be considered in the context of family planning or pregnancy in patients whose ethnicity indicates origin from high-risk geographic areas.
-
1.
Diagnostic Investigations
-
1.
Screening tests for thalassemias.
-
1.
Low MCV (mean corpuscular volume)
-
1.
Increased Hb A2 and/or Hb F: indicate β-thalassemia syndrome
-
2.
Normal Hb A2 and Hb F, and ferritin >20 ng/mL: suggest possible α-thalassemia syndrome
-
1.
-
2.
Normal MCV, normal electrophoresis: thalassemia syndrome unlikely
-
1.
-
2.
α-Thalassemia (Chen 1992; Origa et al. 2013): More difficult to diagnose because characteristic elevations in Hb A2 (α2δ2) or Hb F (α2γ2), seen in β-thalassemia, do not occur.
-
1.
α-Thalassemia-2
-
1.
Laboratory notation: FA + Bart’s
-
2.
Small amount of Hb Bart’s (1–2%) in affected infants at birth
-
3.
Remainder of the hemoglobin: Hb F and Hb A (α2β2)
-
4.
Hb F and Hb Bart’s: disappear after a few months of age
-
5.
Minimal microcytosis: remains
-
1.
-
2.
α-Thalassemia-1
-
1.
Laboratory notation: FA + Bart’s (impossible to differentiate the two forms of α-thalassemia from the electrophoretic pattern).
-
2.
Slightly larger amount of Hb Bart’s in cord blood (3–5%).
-
3.
Hb F and Hb Bart’s: disappear after birth.
-
4.
Unequivocal microcytosis: Mean red cell volume of 65–70 fl/cell remains.
-
5.
Abnormally low Hb A2 proportion.
-
1.
-
3.
Hb H disease
-
1.
Reduction in α-chain does not affect the production of β-chain which is produced in excess and tends to form tetramers (β4 or Hb H).
-
2.
Hb A + Hb H (β4) + Hb Bart’s (γ4).
-
3.
Presence of Heinz bodies: inclusions representing β-chain tetramers (Hb H), which are unstable and precipitate in the RBC, giving the appearance of a golf ball
-
1.
-
4.
Hb Bart’s hydrops fetalis
-
1.
Hypochromic macrocytes with nucleated red blood cells
-
2.
γ4 (Bart’s hemoglobin): nonfunctional as an oxygen transporter
-
1.
-
5.
Targeted mutation analysis
-
1.
Deletion of a single α-globin gene (α+-thalassemia mutations)
-
2.
Deletion of both α-globin genes on one chromosome (α°-thalassemia mutations)
-
3.
Sequence variant HbConstant Spring
-
1.
-
6.
Sequence analysis: used to identify point mutations (including rare termination codon mutations and hyperunstable α-globin variants) in the coding regions of HBA1 and HBA2 when an α-globin deletion is not identified and suspicion for α-thalassemia is high
-
7.
Deletion/duplication analysis can be used to detect common, rare, and/or novel deletions and duplications involving HBA1 and HBA2.
-
1.
-
3.
β-Thalassemia (Chen 1992)
-
1.
β-Thalassemia major
-
1.
Small, thin, and distorted red blood cells containing markedly reduced amounts of hemoglobin
-
2.
Peripheral blood smear
-
1.
Severe microcytic hypochromic anemia (no anemia at birth)
-
2.
Anisocytosis
-
3.
Poikilocytosis (speculated teardrop and elongated cells)
-
4.
Abundant nucleated red cells (i.e., erythroblasts)
-
5.
Occasional immature leukocytes
-
1.
-
3.
Hemolytic anemia
-
4.
Hemoglobin profile: predominant Hb F
-
5.
In patient with homozygous βo-thalassemia: absent Hb A with normal amounts of Hb A2
-
6.
In newborn with β+-thalassemia: Hb F (about 90%) decreases with advancing age but always considerably higher than normal (10–90%)
-
1.
-
2.
β-Thalassemia intermedia (Haddad et al. 2014)
-
1.
Intermediate Hb concentration (7–10 g/dL)
-
2.
Microcytic hypochromic anemia
-
3.
HPLC electrophoresis
-
1.
Hb A2 (>4%)
-
2.
Hb F (5–99%)
-
1.
-
4.
Molecular characteristics
-
1.
Mild/silent mutation.
-
2.
Coinheritance of α-thalassemia may be present.
-
3.
Hereditary persistence of Hb F, δβ-thalassemia, Gγ XMN1 polymorphism.
-
1.
-
1.
-
3.
β-Thalassemia minor
-
1.
Mild hypochromic microcytosis (Rucknagel 1992)
-
2.
Target cells
-
3.
Mild to minimal anemia (hemoglobin concentration average 1–2 g/dL below normal)
-
4.
Elevation of Hb A2 and Hb F (70–99%) during early years of life
-
5.
Anemia worsen during pregnancy
-
1.
-
1.
-
4.
Imaging studies
-
1.
Chest X-ray to evaluate cardiac size and shape
-
2.
Skeletal survey
-
1.
Skeletal response to marrow proliferation
-
1.
Expanding marrow space
-
2.
Thinning of cortical bone
-
3.
Resorption of cancellous bone
-
4.
Resulting in generalized loss of bone density
-
5.
Frequent small lucencies resulting from focal proliferation of marrow
-
6.
“Periosteal response” caused by perforation of the cortex by hypertrophic and hyperplastic marrow subperiosteally
-
1.
-
2.
Classic facies observed in thalassemia major
-
1.
Classic “hair-on-end” appearance of the skull
-
2.
Maxilla overgrowth resulting in maxillary overbite
-
3.
Prominent upper incisors
-
4.
Separation of the orbits
-
1.
-
3.
Various bone deformities seen in ribs, long bones, and flat bones
-
4.
Premature fusion of the epiphyses
-
1.
-
3.
MRI/CT scans to evaluate the amount of iron in the liver in patients on chelation therapy
-
1.
-
5.
Carrier screening
-
1.
α-Thalassemia (Vichinsky 2010)
-
1.
Commonly, microcytosis using an MCV <82 fL and/or hypochromia (MCH <27 pg) is often used as a population screening technique.
-
1.
Anemia is unreliable and is often not present in adults with two α-globin gene deletions.
-
2.
Once iron deficiency is ruled out or corrected, microcytosis should be further evaluated in the individual and his or her partner in order to determine their risk for hydrops.
-
1.
-
2.
Molecular diagnosis for α-globin mutations: essential for at-risk couples.
-
1.
-
2.
β-Thalassemia
-
1.
Ongoing in several at-risk populations in the Mediterranean (Cao et al. 2002).
-
2.
Carrier testing relies on hematological analysis: When the hematological analysis indicates a beta-thalassemia carrier state, molecular genetic testing of HBB can be performed to identify a disease-causing mutation.
-
1.
-
1.
-
6.
Molecular genetic testing for α- and β-thalassemias
-
1.
α-Thalassemia (Origa et al. 2013)
-
1.
HBA1, the gene encoding α1-globin, and HBA2, the gene encoding α2-globin, are the two genes most commonly associated with α-thalassemia.
-
2.
Molecular genetic testing of HBA1 and HBA2 detects deletions in about 90% and point mutations in about 10% of affected individuals.
-
1.
-
2.
β-Thalassemia (Origa 2015)
-
1.
Molecular genetic testing of the gene encoding the hemoglobin subunit beta (HBB): available clinically
-
1.
Targeted mutation analysis
-
2.
Sequence analysis
-
1.
-
1.
-
1.
Genetic Counseling
-
1.
Recurrence risk (Chen 1992)
-
1.
α-Thalassemia
-
1.
Patterns of inheritance of the α-thalassemia when both parents are α-thalassemia-1 (−−/αα).
-
1.
25% normal
-
2.
Fifty percent α-thalassemia-1
-
3.
Twenty-five percent Hb Bart’s (hydrops fetalis)
-
1.
-
2.
Patterns of inheritance of the α-thalassemia syndrome when one parent is α-thalassemia-1 (−−/αα) and the other parent is α-thalassemia-2 (α-/αα).
-
1.
Twenty-five percent normal
-
2.
Twenty-five percent α-thalassemia-1
-
3.
Twenty-five percent α-thalassemia-2
-
4.
Twenty-five percent Hb H disease
-
1.
-
3.
If the carrier of α+-thalassemia is a homozygote, clearly, the risk of Hb H disease is 1:2 (50%) (Harteveld and Higgs 2010).
-
4.
Patient’s offspring: not increased unless the spouse is a carrier.
-
5.
Twenty-five percent of African Americans have heterozygous α-thalassemia-2 (α-/αα). Thus, 1.5% are homozygotes for α-thalassemia-2 (α-/α-). But since the more severe α-thalassemia-1 is not present, subsequent children are not at risk of having Hb H or Bart’s hydrops fetalis.
-
1.
-
2.
β-Thalassemia (Galanello and Origa 2010)
-
1.
Patient’s sib: 25%
-
2.
Patient’s offspring: not increased unless the spouse is a carrier
-
1.
-
1.
-
2.
Prenatal diagnosis: available to all families at risk of either severe α-thalassemia or β-thalassemia
-
1.
α-Thalassemia
-
1.
Ultrasonography (Tongsong et al. 1996).
-
1.
Hepatosplenomegaly (>90%)
-
2.
Cardiomegaly (>90%)
-
3.
Edematous placenta (>90%)
-
4.
Ascites (>90%)
-
5.
Oligohydramnios (82%)
-
6.
Subcutaneous edema (75%)
-
7.
Decreased fetal movement (74%)
-
8.
Cord edema (63%)
-
9.
Enlarged umbilical vessel (62%)
-
10.
Pericardial or pleural effusion (15%)
-
1.
-
2.
Sonographic markers of fetal α-thalassemia major (Li et al. 2015).
-
1.
Classic sonographic characteristics of hydrops fetalis syndrome do not appear until the late second trimester (Tongsong et al. 1996), but it has been reported that some cases of hydrops fetalis can be identified by sonography in the early second trimester (Lam et al. 1997).
-
2.
Moreover, cardiovascular responses secondary to α-thalassemia major can take effect in the first trimester, bringing more blood flow to vital organs such as the heart and brain (Ghosh et al. 1994; Lam et al. 1999; Picklesimer et al. 2007).
-
3.
Therefore, many sonographic markers may be observed before the onset of fetal hydrops (Ghosh et al. 1987) and could be used to detect fetuses with α-thalassemia major.
-
4.
Among all markers, the cardiothoracic ratio, placental thickness, and middle cerebral artery PSV (peak systolic velocity) are the most studied ones.
-
1.
-
3.
Molecular hybridization technique to detect complete absence of α-genes in fetal amniocytes in a pregnancy at risk for homozygous α-thalassemia-1 and the hydrops fetalis syndrome (Dozy et al. 1979).
-
1.
The DNA obtained from cultured amniotic fluid cells was studied by hybridization with globin cDNA in solution and on filters (Southern technique).
-
2.
Both analyses demonstrated no alpha-globin structural genes.
-
3.
Following termination of the pregnancy, the diagnosis was established by the presence of only hemoglobins Bart’s (gamma 4) and Portland (zeta 2 gamma 2) in the fetal blood.
-
1.
-
4.
Quantitative polymerase chain reaction for the rapid prenatal diagnosis of homozygous α-thalassemia (Hb Bart’s hydrops fetalis).
-
5.
Preimplantation genetic diagnosis (PGD) may be an option for some families in which the pathogenic variants have been identified (Origa et al. 2013).
-
1.
-
2.
β-Thalassemia
-
1.
Available to couples who are carriers of β-thalassemia and their hemoglobin gene mutations have been identified by DNA analysis.
-
2.
Direct DNA analysis by molecular hybridization methods for the presence of the thalassemia mutation from fetal cells obtained by amniocentesis and chorionic villus biopsy.
-
3.
Detection of point mutations in single gene disorders by enriching fetal cells from maternal blood by magnetic cell sorting followed by isolation of pure fetal cells by microdissection. In two pregnancies at risk for sickle cell anemia and beta-thalassaemia, we successfully identified the fetal genotypes (Cheung et al. 1996). Thus, prenatal diagnosis of single gene disorders by recovering fetal cells from maternal circulation appears to be a feasible approach.
-
4.
Approach to prenatal diagnosis complicated by presence of the heterogeneity of thalassemia mutations.
-
5.
Preimplantation genetic diagnosis available for at-risk pregnancies requires prior identification of the disease-causing mutations in the family (Galanello and Origa 2010).
-
1.
-
3.
Preimplantation genetic diagnosis (Cao and Kan 2013)
-
4.
Preconceptional diagnosis (Cao and Kan 2013)
-
1.
Based on the analysis of the first polar body of unfertilized eggs followed by analysis of the second polar bodies after fertilization, which is performed to avoid misdiagnosis resulting from recombination during the first meiosis (Verlinsky et al. 1990).
-
2.
Diagnosis was obtained by multiple nested PCR analysis to detect the mutations as well as polymorphic alleles at the β-globin cluster (Kuliev et al. 2011; Zachaki et al. 2011).
-
3.
HLA typing of the embryo to select a nonaffected fetus HLA compatible with a previous affected sibling was recently proposed (Kuliev et al. 2011).
-
1.
-
1.
-
3.
Management
-
1.
α-Thalassemia (Segel et al. 2002)
-
1.
No therapy necessary for patients with α-thalassemia trait
-
2.
Avoid exposure to oxidant medications (e.g., iron, sulfonamides) which accelerate precipitation of Hb H and exacerbate hemolysis
-
3.
Prompt treatment of infection especially in postsplenectomy
-
4.
Hb H disease
-
1.
Folate supplementation
-
2.
Chronic transfusion therapy (consider iron chelation therapy to avoid iron overloading)
-
3.
Splenectomy in rare instances of hypersplenism
-
4.
Allogeneic bone marrow transplantation limited to the most severely affected patients
-
1.
-
5.
Bart’s hemoglobinopathy
-
1.
Usually results in neonatal death.
-
2.
Patients rarely salvaged by intrauterine transfusions and subsequent stem cell transplantation.
-
3.
The diagnosis, management, and prognosis of homozygous α-thalassemia/hydrops fetalis is changing; advances in antenatal diagnosis, intrauterine intervention (Carr et al. 1995), and postnatal therapy have resulted in long-term survival of children previously felt to have an invariably fatal disease (Singer et al. 2000; Weisz et al. 2009; Vichinsky 2010).
-
1.
-
6.
Homozygous α-thalassemia and hydrops fetalis (Vichinsky 2009)
-
1.
A complex, usually fatal disease that is devastating to the entire family.
-
2.
There is a diversity in the genotype and phenotype expression of this syndrome that presents challenges in at-risk couple counseling and population screening.
-
3.
Presently, counseling and testing of at-risk populations is inadequate.
-
4.
More cases are being diagnosed unexpectedly.
-
5.
Intrauterine transfusion therapy appears promising in minimizing the morbidity and mortality of homozygous α-thalassemia. However, most of these patients require lifetime transfusion therapy and chelation.
-
6.
Recent advances in stem cell transplantation have resulted in some patients being cured.
-
7.
Successful cases of related, unrelated, and mismatched stem cell transplantation for α-thalassemia major have been reported.
-
1.
-
7.
α-Thalassemia syndromes are common and have a wide range of clinical phenotypes (Vichinsky 2013). Hemoglobin H disease morbidity is often underappreciated. These patients require early diagnosis and ongoing monitoring. Hemoglobin Bart’s hydrops is a tragic, usually fatal complication that can be prevented by adequate screening and counseling. Improved outcome with intrauterine transfusions creates ethical issues for the family and health care providers.
-
1.
-
2.
β-Thalassemia (Galanello and Origa 2010; Origa 2015)
-
1.
No specific therapy required for β-thalassemia trait.
-
2.
Regular blood transfusions in thalassemia major.
-
1.
Correct the anemia.
-
2.
Suppress erythropoiesis.
-
3.
Inhibit increased gastrointestinal absorption of iron.
-
1.
-
3.
Treatment of individuals with thalassemia intermedia (Taher et al. 2012).
-
1.
Symptomatic treatment
-
2.
Blood transfusion
-
3.
Splenectomy
-
4.
Folic acid supplementation
-
5.
Iron chelation
-
6.
Treatment of extramedullary erythropoietic masses (radiotherapy, transfusions, or hydroxyurea in selected cases)
-
1.
-
4.
Iron chelation with desferrioxamine to eliminate the iron overload secondary to multiple blood cell transfusions and to increase iron absorption.
-
5.
Role of natural agents (Kukreja et al. 2013).
-
1.
Various natural agents like angelicin, rapamycin, FT, bergamot, romidepsin, wheatgrass, Curcuma comosa, Astragalus, apicidin, curcuminoid, piceatannol, and resveratrol have been reported to induce HbF level in beta-thalassemic patients.
-
2.
Various natural compounds like wheatgrass, desferrioxamine, and Tetracarpidium conophorum have also been known for their iron chelation property for the treatment of beta-thalassemia.
-
3.
More data are needed on the bioavailability of these natural compounds and their effects on human.
-
1.
-
6.
Bone marrow transplantation (Lucarelli et al. 1995; Giardini and Lucarelli 1999) remains to be the only definitive cure currently available for patients with thalassemia.
-
1.
From an HLA-identical sib
-
2.
Outcome dependent on pretransplantation clinical conditions, specifically the presence of hepatomegaly, extent of liver fibrosis, and magnitude of iron accumulation
-
3.
Risk of chronic graft-versus-host disease (GVHD) of variable severity: 5–8%
-
1.
-
7.
Cord blood transplantation (Kelly et al. 1997; Locatelli et al. 2003).
-
1.
From a related donor offers a good probability of a successful cure and is associated with a low risk of GVHD.
-
2.
Possibility of using cord blood obtained from unrelated donors with a decrease in the incidence of graft-versus-host disease.
-
3.
Human umbilical cord blood contains hematopoietic stem cells capable of reconstituting bone marrow.
-
1.
-
8.
Recent trends in the gene therapy of β-thalassemia (Finotti et al. 2015).
-
1.
A strong emphasis on the most recent findings, for β-thalassemia model systems
-
2.
For β-, γ-, and anti-sickling β-globin gene addition and combinatorial approaches including the latest results of clinical trials
-
3.
For novel approaches, such as transgene-mediated activation of γ-globin and genome editing using designer nucleases
-
1.
-
1.
-
1.

Peripheral blood smear from a 58-year-old woman with microcytic anemia and frequent target cells (codocytes). Hemoglobin electrophoresis showed an AA pattern with an increased hemoglobin A2 (5.8% by HPLC) consistent with beta+- thalassemia trait

Peripheral blood smear from a patient with alpha-thalassemia minor shows hypochromia, target cells (arrows), and anisopoikilocytosis (Wright-Giemsa stain, ×1,000)
References
Cao, A., & Galantello, R. (2010). Beta-thalassemia. Genetics in Medicine, 12, 61–76.
Cao, A., & Kan, Y. W. (2013). The prevention of thalassemia. Cold Spring Harbor Perspectives in Medicine, 3, 1–15.
Cao, A., Rosatelli, M. C., Monni, G., et al. (2002). Screening for thalassemia: A model of success. Obstetrics and Gynecology Clinics of North America, 29, 305–328.
Carr, S., Rubin, L., et al. (1995). Intrauterine therapy for homozygous alpha-thalassemia. Obstetrics and Gynecology, 85, 876.
Chen, H. (1992). Genetic testing & counseling for hemoglobinopathies. In H. Chen (Ed.), Ohio Department of Health the Resource Manual for hemoglobinopathies. An essential guide for health professionals (pp. 97–107). Columbus: Advisory Council on Newborn Screening for Hemoglobinopathies.
Cheung, M.-C., Goldberg, J., & Kan, Y. (1996). Prenatal diagnosis of sickle cell anaemia and thalassemia by analysis of fetal cells in maternal blood. Nature Genetics, 14, 264.
Dozy, A. M., Forman, E. N., Abuelo, D. N., et al. (1979). Prenatal diagnosis of homozygous alpha thalassemia. Journal of the American Medical Association, 241, 1610–1612.
Dumars, K. W., Boehm, G., Eckman, J. R., et al. (1996). Practical guide to the diagnosis of thalassemia. American Journal of Medical Genetics, 62, 29–37.
Finotti, A., Breda, L., Lederer, C. W., et al. (2015). Recent trends in the gene therapy of β-thalassemia. Journal of Blood Medicine, 6, 69–85.
Galanello, R., & Cao, A. (2011). Alpha-thalassemia. Genetics in Medicine, 13, 83–88.
Galanello, R., & Origa, R. (2010). Beta-thalassemia. Orphanet Journal of Rare Diseases, 6, 11–40.
Ghosh, A., Tang, M. H. Y., Liang, S. T., et al. (1987). Ultrasound evaluation of pregnancies at risk for homozygous α-thalassaemia-1. Prenatal Diagnosis, 7, 307–313.
Ghosh, A., Tang, M., Leung, M. P., et al. (1994). Cardiac blood flow studies in fetuses with haemoglobin Bart’s disease. Prenatal Diagnosis, 14, 627–632.
Giardini, C., & Lucarelli, G. (1999). Bone marrow transplantation for beta-thalassemia. Hematology/Oncology Clinics of North America, 13, 1059–1064.
Haddad, A., Tyan, P., Radwan, A., et al. (2014). β-thalassemia intermedia: a bird’s-eye view. Turkish Journal of Hematology, 31, 5–16.
Harteveld, C. L., & Higgs, D. R. (2010). Alpha-thalassaemia. Orphanet Journal of Rare Diseases, 5, 13–65.
Javid, B., & Said-Al-Naief, N. (2015). Craniofacial manifestations of b-thalassemia major. Oral and Maxillofacial Pathology, 119, e33–e40.
Kelly, N. (2012). Thalassemia. Pediatrics in Review, 33, 434–435.
Kelly, P., Kurtzberg, J., Vichinsky, E., et al. (1997). Umbilical cord blood stem cells: Application for the treatment of patients with hemoblobinopathies. Journal of Pediatrics, 130, 695–703.
Kokkali, G., Traeger-Synodinos, J., Vrettou, C., et al. (2007). Blastocyst biopsy versus cleavage stage biopsy and blastocyst transfer for preimplantation genetic diagnosis of β-thalassaemia: A pilot study. Human Reproduction, 22, 1443–1449.
Kukreja, A., Wadhwa, N., & Tiwari, A. (2013). Therapeutic role of natural agents in beta-thalassemia: A review. Journal of Pharmacy Research, 6, 954–959.
Kuliev, A., Rechitsky, S., Verlinsky, O., et al. (1998). Preimplantation diagnosis of thalassemias. Journal of Assisted Reproduction and Genetics, 15, 219–225.
Kuliev, A., Pakhalchuk, T., Verlinsky, O., et al. (2011). Preimplantation genetic diagnosis for hemoglobinopathies. Hemoglobin, 31, 273–277.
Lam, Y. H., Ghosh, A., Tang, M. H., et al. (1997). Second-trimester hydrops fetalis in pregnancies affected by homozygous alpha-thalassaemia-1. Prenatal Diagnosis, 17, 267–269.
Lam, Y. H., Tang, M. H. Y., Lee, C. P., et al. (1999). Cardiac blood flow studies in fetuses with homozygous α-thalassemia-1 at 12–13 weeks of gestation. Ultrasound in Obstetrics and Gynecology, 13, 48–51.
Li, X., Zhou, Q., Zhang, M., et al. (2015). Sonographic markers of fetal α-thalassemia major. Journal of Ultrasound in Medicine, 34, 197–206.
Locatelli, F., Rocha, V., Reed, W., et al. (2003). Related umbilical cord blood transplantation in patients with thalassemia and sickle cell disease. Blood, 101, 2137–2143.
Lucarelli, G., Giardini, C., & Baronciani, D. (1995). Bone marrow transplantation in thalassemia. Seminars in Hematology, 32, 297–303.
Nemtsas, P., Arnaotoglou, M., Perifanis, V., et al. (2015). Neurological complications of beta-thalassemia. Annals of Hematology, 94, 1261–1265.
Origa, R. (2015). Beta-thalassemia. GeneReviews. Updated May 14, 2015. Available at: http://www.ncbi.nlm.nih.gov/books/NBK1426/
Origa, R., Moi, P., Galanello, R., et al. (2013). Alpha-thalassemia. GeneReviews. Updated November 21, 2013. Available at: http://www.ncbi.nlm.nih.gov/books/NBK1435/
Picklesimer, A. H., Oepkes, D., Moise, K. J. Jr, et al. (2007). Determinants of the middle cerebral artery peak systolic velocity in the human fetus. American Journal or Obstetrics and Gynecology, 197, 526.e1–526.e4.
Rucknagel, D. L. (1992). Microcytosis and the thalassemias. In H. Chen (Ed.), Ohio Department of Health the Resource Manual for hemoglobinopathies. An essential guide for health professionals (pp. 15–18). Columbus: Advisory Council on Newborn Screening for Hemoglobinopathies.
Segel, G. B., Hirsh, M. G., & Feig, S. A. (2002). Managing anemia in pediatric office practice: Part 1. Pediatrics in Review, 23, 75–84.
Singer, S. T., Styles, L., Bojanowski, J., et al. (2000). Changing outcome of homozygous alpha-thalassemia: Cautious optimism. Journal of Pediatric Hematology/Oncology, 22, 539–542.
Taher, A. T., Musallam, K. M., Karimi, M., et al. (2012). Contemporary approaches to treatment of beta-thalassemia intermedia. Blood Reviews, 26S, S24–S27.
Thein, S. L. (2013). The molecular basis of β-thalassemia. Cold Spring Harbor Perspectives in Medicine, 3, 1–24.
Tongsong, T., Wanapirak, C., Srisomboon, J., et al. (1996). Antenatal sonographic features of 100 alpha-thalassemia hydrops fetalis fetuses. Journal of Clinical Ultrasound, 24, 73–77.
Verlinsky, Y., Ginsberg, N., Lifchez, A., et al. (1990). Analysis of the first polar body: Preconception genetic diagnosis. Human Reproduction, 5, 826–829.
Vichinsky, E. P. (2009). Alpha thalassemia major-new mutations, intrauterine management, and outcomes. American Society of Hematology Education Program, 1, 35–41.
Vichinsky, E. (2010). Complexity of alpha thalassemia: Growing health problem with new approaches to screening, diagnosis, and therapy. Annals of the New York Academy of Sciences, 1202, 180–187.
Vichinsky, E. P. (2013). Clinical manifestations of α-thalassemia. Cold Spring Harbor Perspectives in Medicine, 3, 1–10.
Weisz, B., Rosenbaum, O., Chayen, B., et al. (2009). Outcome of severely anaemic fetuses treated by intrauterine transfusions. Archives of Disease in Childhood. Fetal and Neonatal Edition, 94, F201–F204.
Yaish, H. M. (2010). Medscape reference. Updated April 30, 2010. Available at: http://emedicine.medscape.com/article/958850-overview
Zachaki, S., Vrettou, C., Destouni, A., et al. (2011). Novel and known microsatellite markers within the β-globin cluster to support robust preimplantation genetic diagnosis of β-thalassemia and sickle cell syndromes. Hemoglobin, 35, 56–66.
Author information
Authors and Affiliations
Rights and permissions
Copyright information
© 2017 Springer Science+Business Media LLC
About this entry
Cite this entry
Chen, H. (2017). Thalassemia. In: Atlas of Genetic Diagnosis and Counseling. Springer, New York, NY. https://doi.org/10.1007/978-1-4939-2401-1_227
Download citation
DOI: https://doi.org/10.1007/978-1-4939-2401-1_227
Published:
Publisher Name: Springer, New York, NY
Print ISBN: 978-1-4939-2400-4
Online ISBN: 978-1-4939-2401-1
eBook Packages: MedicineReference Module Medicine