
Abstract
Oxytocin (OT) and arginine vasopressin (AVP) are two small, related neuropeptides found in many mammalian species, including humans. These neuropeptides are associated with a range of social behaviors and their dysregulation has been associated with deficits in social behavior. In particular, the OT neuropeptide system has been investigated in Autism Spectrum Disorder (ASD), as well as in Prader-Willi Syndrome (PWS), Williams Syndrome (WS) and Fragile X Syndrome (FXS), all of which are characterized by marked social deficits. PWS, WS and FXS are caused by identified genetic mutations and provide insight into the developmental influences of the OT system. In particular, FXS is caused by a mutation in a single gene and up to 47 % of patients with FXS are diagnosed with ASD or also have autism related behaviors. Animal models of genetic neurodevelopmental disorders (NDD) are becoming a valuable tool to examine the role and relatedness of OT and AVP in the developing brain. We provide an example of how OT and AVP systems are altered with a mutation in the mouse Fragile X mental retardation 1 (Fmr1) gene which leads to FXS-like symptoms in Fmr1 knockout (KO) mice. By studying the OT and AVP systems in these rare disorders, we may further understand their mechanisms of action in ASD and in typical development. This chapter will summarize the current data pertaining to these NDD and the systems of OT and AVP.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others


Keywords
1 Introduction to OT & AVP Neuropeptide Hormones
Oxytocin (OT) and arginine vasopressin (AVP) are small mammalian neuropeptides nine amino acids in length, differing from each other by only two amino acids. OT is produced primarily in hypothalamic nuclei, including the supraoptic (SON) and paraventricular nuclei (PVN). AVP is also synthesized in the PVN and SON. However in males, additional brain regions including the amygdala and the bed nucleus of the stria terminalis (BNST) also produce AVP. OT and AVP of hypothalamic origins are transported from the SON and PVN to the mammalian posterior pituitary by neurosecretion where they are released into the blood stream, acting as hormones on target tissues. In addition, both OT and AVP are capable of moving throughout the central nervous system (CNS) via diffusion in the cerebral spinal fluid (CSF; Neumann and Landgraf 2012). The peptide producing oxytocin gene (OXT) is homologous with its evolutionarily related, vasopressin (AVP) gene. The human OXT and AVP genes are linked on chromosome 20p13 and are positioned in opposite transcriptional orientations, while separated by only 12 kb of DNA. Both have specific receptors, but their close evolutionary relationship permits cross-talk and interacting molecular systems. These neuropeptide hormones have receptors in various brain regions and throughout the body, including areas that are important for regulating social behavior and reactivity to stressors.
In both, the human and mouse genomes OT and AVP peptide genes are located adjacently on the same chromosome. Often, the blood levels of both peptides are highly correlated (Dai et al. 2012) suggesting a coordinated release. The receptors for both peptides are localized in specific areas of the nervous system, particularly in the brainstem and areas that play a role in social, adaptive behaviors, or in the regulation of the hypothalamic-pituitary-adrenal axis (HPA) and autonomic nervous system (ANS; Lim et al. 2004). Because OT and AVP are closely related and have the ability to act on the other’s receptors, it has been proposed that they evolved to interact and sometimes have opposing physiological effects. For example, both hormones have been shown to affect the control of the autonomic nervous system (ANS), with OT having primarily parasympathetic actions, and AVP serving as both a central and peripheral regulatory component of the sympathetic nervous system and HPA axis (Sawchenko and Swanson 1985; Kenkel et al. 2012). However, high levels of neuropeptides can be partial agonists for their homologous receptors and may result in AVP and OT pathway interactions (Chini et al. 1996).
Of particular importance in neurodevelopmental disorders (NDD) is the fact that OT and AVP can modulate social and repetitive behavior, as well as other manifestations of anxiety and state regulation (Carter 2007). Animal research has generally associated OT release or exposure with positive sociality, reduced anxiety and lower levels of reactivity to stressors (Carter 1998; Neumann and Landgraf 2012). AVP can influence anxiety and the regulation of the HPA axis and stress responses. In general, central AVP is described as anxiogenic (Landgraf and Wigger 2003). However, there also is evidence in rats that the effects of AVP are brain region specific and dose-dependent. For example, AVP may be anxiolytic if given in low doses (Appenrodt et al. 1998).
Mouse knockout (KO) studies of the OT receptor (OXTR) or OT regulators have reported decreased social memory or recognition (Jin et al. 2007; Ferguson et al. 2000; Takayanagi et al. 2005). Oxtr KO mice also showed decreased cognitive flexibility and a resistance to change learned pattern of behavior, comparable to restricted/repetitive interests (Sala et al. 2011). Both social deficits and behavioral rigidity were ameliorated by OT administration (Sala et al. 2011). The finding that OT continues to have effects in Oxtr KO mice supports the hypothesis that OT can influence behavior through other receptors, particularly the AVP receptors (e.g. AVPR1A and/or AVPR1B). Given the influence of these neuropeptides on brain regions affecting both social and repetitive behaviors, modulation of OT and AVP pathways are being explored as treatment targets for Fragile X Syndrome (FXS) and Autism Spectrum Disorders (ASD).
These studies set the stage for a series of recent studies of the effects of exogenous OT treatments in humans (Ebstein et al. 2012; Macdonald and Feifel 2013). For example, intranasal OT (IN-OT) administration in healthy human males increased prosocial behaviors and trust, especially as measured by computerized economic games (Baumgartner et al. 2008; Kirsch et al. 2005; Kosfeld et al. 2005). IN-OT may also increase gaze toward the eye region of faces (Guastella et al. 2008), and has been associated with improved facial memory (Rimmele et al. 2009), enhanced salience of social cues (Shamay-Tsoory et al. 2009), and improved performance on the reading the mind in the eyes task (RMET; Domes et al. 2007).
As previously reviewed, OT has been found to have anxiolytic effects, improve social interactions, reduce fear, and improve the ability of healthy volunteers to interpret subtle social cues (Macdonald and Macdonald 2010). In addition, OT dysfunction has been associated with neuropsychiatric disorders such as ASD in human studies (Ishak et al. 2011; Domes et al. 2007; Winslow and Insel 2004). By 2010, there were over 20 OT administration studies, which included ASD, schizophrenia, postpartum depression, posttraumatic stress disorder (PTSD), and irritable bowel syndrome (Macdonald and Macdonald 2010). As IN-OT has been associated with alterations in social decision-making, processing of social stimuli, certain social behaviors such as eye contact, and social memory, there has been a growing number of studies investigating its abilities to treat a range of neurobehavioral disorders.
2 Autism Spectrum Disorder and OT/AVP
In 1943, Leo Kanner described a male patient as having “stereotyped movements [and] … repetitions carried out in exactly the same way in which they had been performed originally” and also noted his social communication such that “he always seemed to be parroting what he had heard said to him at one time or another ….Words to him had a specifically literal, inflexible meaning. He seemed unable to generalize, to transfer an expression to another similar object or situation” (Kanner 1943). This group of symptoms later extended and described in detail is currently known as ASD. As described in the DSM-5 (American Psychiatric Association 2013), it is characterized by persistent deficits in social communication and social interaction across multiple contexts, and the diagnosis requires the presence of restricted, repetitive patterns of behaviors, interests, or activities. ASD is a heritable (Bailey et al. 1995) and highly heterogeneous disorder, caused by familial genetic risks in addition to possible gene-environment interactions during early development (Chaste and Leboyer 2012). Individuals with ASD may also have comorbid anxiety disorders, irritability and aggression, and come to clinical attention due to difficulties at home and school related to their communication deficits and restricted interests.
It was suggested by a number of researchers, including Waterhouse et al. as early as 1996, that dysfunction in the OT and AVP systems might contribute to the atypical social behaviors in ASD (Waterhouse et al. 1996). One of the first examinations of the association of OT specifically and ASD showed that children with ASD have low levels of plasma OT (Modahl et al. 1998). In particular, a subgroup of this sample identified as aloof using Wing’s diagnostic topology had the lowest levels, suggesting that those with the most severe socially aloof symptoms had more OT dysfunction. Building on these results with the same sample, it was also shown that there was an increase in OT-Gly, OT-Gly-Lys, and OT-Gly-Lys-Arg peptides, collectively known as OT-X precursors for OT, as well as an increase in the ratio of OT-X/OT, associated with the reduction in OT seen in the patients with ASD (Green et al. 2001). There was also a positive correlation between OT-X and checklist items associated with ASD, including stereotypies, while OT-X correlated negatively with an item describing atypical comfort-giving within the ASD group. Consequently, changes in OT processing, specifically a failure to completely process the prohormone OT-X, might lead to a deficiency in OT and thus exacerbate some of the symptoms of ASD. To our knowledge this study has not been replicated. However, other studies done in older patients have failed to report an OT deficiency, or have reported higher than expected OT levels in their ASD samples (Jansen et al. 2006; Miller et al. 2013). Future research will need to determine if differences in results of OT levels in individuals with ASD compared to controls reflect differences in the study populations and/or methods for assaying OT. It should be noted that these studies often prepared samples differently with varying plasma processing/extraction methods, and used different assay techniques. Additionally, ASD population differences included varying ages and recent data has suggested that some blood biomarkers like OT may change after puberty (Hammock et al. 2012). In addition, ASD is a very heterogeneous disorder and OT level differences may be specific to clinical and etiological subgroups within the broader ASD population.
2.1 Intranasal OT Studies in ASD
Currently medications for ASD concentrate on alleviating certain symptoms. Risperidone and aripiprazole may be used for irritability, whereas guanfacine and clonidine are used off label for aggression, and selective serotonin reuptake inhibitors (SSRI; i.e. escitalopram, fluoxetine, and sertraline) are used to treat anxiety or depression (Owley et al. 2010; Jaselskis et al. 1992). Recently, OT has been investigated to target the treatment of ASD’s core symptoms, social deficits and restrictive and repetitive behaviors (RRBs) .
Several studies using intravenous or IN-OT in patients with ASD have been conducted. It has been shown that nonapeptides, like AVP and OT, can be measured in CSF after intranasal administration (Born et al. 2002). Ease of giving intranasal drugs makes it preferred for most ASD studies, although more research needs to be conducted on how IN-OT reaches the brain and influences behavior. In addition to measuring OT responses to single dose challenges in ASD (Andari et al. 2010; Guastella et al. 2010), few studies have examined longer term treatment effects (Anagnostou et al. 2012). In addition to varying administration and duration study protocols, studies have often focused on symptom subdomains or defined social tasks such as RRBs (Hollander et al. 2003), emotion recognition (Guastella et al. 2010; Dadds et al. 2014), affective speech comprehension (Hollander et al. 2007), and facial recognition (Domes et al. 2013) .
As mentioned above, single dose studies or challenges have been utilized to study the acute and immediate effects of OT. An initial study in ASD examined the effects of a four-hour continuous dose of intravenous OT (Hollander et al. 2003). After 1 h of infusion there was a decrease in RRB (repeating and touching). This decrease lasted when measured after 4 h as well. More recently, a double-blind, randomized, placebo controlled study of IN-OT in 16 males with ASD (ages 12–19 years old) showed that a single IN-OT dose could improve the ability to recognize emotion, particularly in easy queries (Guastella et al. 2010). Single dose IN-OT studies have also been done to examine the effects of OT on trust behavior and visual scanning of faces (Andari et al. 2010). Individuals with ASD given OT had a significant preference for the “good player” (the computer player who tossed the ball back to you) that was similar to the control subjects also performing the task. This preference was further supported by the patients’ reporting of trust, towards the “good player” after OT administration. In the visual scanning task, ASD subjects specifically avoided the area of high expression, the eyes, when given placebo, but significantly increased their gaze fixation towards the eyes with OT .
Recently a multi-dose OT administration study was conducted by Dadds et al. (2014). Individuals with high functioning ASD received 12 or 24 IU (depending on the weight of the patient) IN placebo or OT once daily for 5 days. During these 5 days, the participant and their parents received daily parent-child interaction training and assessments of RRB, emotion recognition, social interaction skills, and general behavioral adjustment. While improvements over time were detected in both OT and placebo, there were no differences observed between the two groups. Several proposed possible explanations for these findings were: (1) emotion recognition was measured pre-post changes following multiple exposures versus while the patient was under the influence; (2) lower-order RRB, such as repeating, ordering and touching, respond to OT (Hollander et al. 2003), while higher-order RRB, such as ritualistic behavior and insistence on sameness, do not (Anagnostou et al. 2012); (3) increased eye gaze frequency is usually measured with artificial or computerized faces, while they utilized “real-life” interactions; (4) the OT receptor system disruptions in some patients with ASD may respond differently than in other ASD patients; and (5) differences between the studies regarding age and diagnostic characteristics of the sample. Positive results were also observed in a 6-week protocol, which showed that IN-OT is well tolerated when given daily and may improve social cognition and decrease RRB in adults (Anagnostou et al. 2012) . It may be important to note that reports of individual differences in the response to IN-OT are increasingly observed, although sample sizes in studies will need to be larger to explore individual response variation. While complex, the results from OT administration studies have provided a basis for understanding the role of OT. They aid in the development of study protocols that are focused on the population that will benefit the most from OT therapy .
2.2 OT and AVP Genetic Studies in ASD
Over the past decade, many studies have explored the role of genetics in ASD. It is theorized that the genetic heterogeneity of ASD could account for the complexity of its genetic etiology . When studying OT and AVP system genes, researchers often explore subphenotype scores such as social impairments in ASD. In a recent review, genetic polymorphisms of receptor and pathway regulators such as AVPR1a, OXTR, neurophysin I and II, and CD38 were discussed (Ebstein et al. 2009). Ebstein and colleagues presented preliminary data in their review regarding their findings about CD38, a transmembrane glycoprotein involved in OT secretion (Ebstein et al. 2009). Based on their studies of mice, they hypothesized a role for CD38 in ASD and its role in mediating OT, and in promoting nurturing behavior and social familiarity. (Jin et al. 2007) . They genotyped 12 tag single nucleotide polymorphisms (SNPs) across CD38 in 170 ASD trios and assessed IQ and social skills via the Vineland Adaptive Behavior Scales (VABS) in this sample. A significant association between categorical ASD measures (ADI-R; Lord et al. 1994 and ADOS-G; Lord et al. 2000) and CD38 SNPs was found, as well as between social skills in ASD (VABS) and CD38 (rs4634217, rs4516711, rs4508877 and rs3796867), haplotypes and VABS, and CD38 mRNA levels and VABS. Further supporting the role of CD38 in ASD, it has also been noted that there is reduced expression of CD38 in lymphoblastoid cells of patients with ASD (Lerer et al. 2010), and the rs3796863 CD38 SNP has been associated with high functioning ASD in some populations (Munesue et al. 2010). However, this CD38 SNP has also been shown to correlate with higher activation of fusiform brain regions in healthy males challenged with OT (Sauer et al. 2012) .
Candidate genes for ASD have also been culled from known genetic variants that are more broadly related to affiliative behavior (Yrigollen et al. 2008). In a study of 177 ASD probands from 151 families it was found that different OXTR SNPs were associated with stereotyped behavior, communication skills, an ADI-based diagnosis group and the ASD diagnosis acquired from multiple measurements. Specifically, there was a significant SNP in the OXT/AVP region (rs2740204; p= 0.016) associated with stereotyped behaviors (Yrigollen et al. 2008) .
In summary, many studies have focused on OXTR as a candidate gene for ASD. It has been shown that multiple SNPs are associated with stereotyped behaviors and communication skills (Yrigollen et al. 2008). Studies specifically examining identified populations such as Chinese Han have shown significant associations between ASD and two of its SNPs, rs2254298 and rs53576 (Wu et al. 2005). Also, in a study utilizing a Caucasian sample the rs2254298 SNP was associated with ASD diagnosis (Jacob et al. 2007). It should be noted that there was overtransmission of the G allele to the autistic Caucasian probands versus overtransmission of the A allele in the Chinese Han sample. More recently, the rs2268493, rs1042778 and rs7632287 SNPs have also been associated with ASD in a sample in which 95 % of individuals self identified as Caucasian (Campbell et al. 2011) .
2.3 OT Blood Levels in ASD
The limitations to direct access of the brain’s oxytocinergic pathways have constrained human research . Therefore, peripheral OT levels have been used as proxies for brain OT levels. A widely used measurement is plasma OT, but urine and salivary OT levels have also been examined. Several researchers have observed associations between peripheral, plasma OT and AVP levels and social stimuli (Kenkel et al. 2012; Schneiderman et al. 2012; Schradin et al. 2013; Seltzer et al. 2010; Wismer Fries et al. 2005). In particular, peripheral OT has been associated with human parental care, both maternal and paternal, such that parents display higher levels of OT than non-parents and low plasma OT levels in parents are associated with less parental touch, whereas higher levels correspond to longer durations of gaze synchrony and reporting of greater parental care during the parent’s childhood (Feldman et al. 2012) .
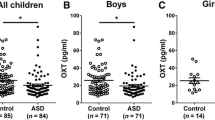
Studies of adolescents have also utilized peripheral measures of OT as well as AVP . In a recently published preliminary study of high functioning ASD individuals and typically developing controls, Miller et al. (2013) researched the direct connection between peripheral OT/AVP levels and ASD. Higher levels of OT were observed in all girls and were associated with greater anxiety. Across both sexes, higher OT levels were also associated with better pragmatic language. In addition, all boys had significantly higher levels of AVP. Gender differences were also noted within the ASD sample such that there was a positive association between AVP levels and RRB in ASD girls, but this was negatively (although non-significantly) associated with RRB in boys with ASD (Miller et al. 2013). Overall, these results suggest specific and sexually dimorphic mechanisms for OT and AVP with regard to anxiety and RRB .
Another important study that researched the connection between ASD and OT plasma levels was performed in 1998. Modahl et al. (1998), found plasma OT levels in children with ASD were lower than the control group. They also examined how social behaviors were associated with plasma OT levels. Modahl et al. found that oxytocin was positively associated with age for “normal” children but not for children with ASD. Additionally, physiological variables such as time of food intake were negatively associated with oxytocin level, and physical exercise and the presence of a respiratory condition were also associated with oxytocin level in the control group. The oxytocin levels of the children with ASD were not related to these physiological variables. Later, a study performed by Jansen et al. (2006) involved adults diagnosed with ASD, who were asked to perform a public speaking task. Interestingly, in this study, subjects showed normal cortisol responses and no change in response to the task in norepinephrine, epinephrine, OT or AVP. However both basal OT levels and heart rate were elevated in the ASD group compared to healthy controls. They hypothesized that the interplay between cortisol and OT may influence the effects of social interactions and support .
Peripheral measurement of serotonin (5-HT) has recently been investigated in conjunction with OT, as many studies have reported hyperserotonemia within a subgroup of individuals with ASD (Leventhal et al. 1990; Schain and Freedman 1961; Leboyer et al. 1999; Kuperman et al. 1985; Abramson et al. 1989; Chugani et al. 1999) and both 5-HT and OT are peripheral biomarkers that correspond to systems that interact in the brain. An analysis of whole blood 5-HT and plasma OT levels in children and adolescents with ASD showed a negative correlation, and this negative correlation was more prominent in younger children (Hammock et al. 2012). These results parallel findings in Oxtr KO mice which show that these mice had higher concentrations of whole-blood 5-HT, and the relationship between plasma OT levels and whole blood 5-HT levels are stronger in younger individuals (Hammock et al. 2012) .
Over the last few years studies measuring peripheral OT have increased. As discussed in McCullough et al. (2013) and Szeto et al. (2011), the methodologies can lead to vastly different results (increased values, decreased values or values differing by an order of magnitude). For example, Modahl et al. (1998) performed plasma extractions and then radioimmunoassays (RIA) whereas Miller et al. (2013) utilized an enzyme immunoassay (EIA) with different plasma preparation methods. Additionally, there is also specific laboratory generated RIA versus commercial EIA and RIA kits. When manufacturer instructions are followed, values obtained have a similar order of magnitude, but it has been noted that some of these kits may also be detecting closely related metabolites . Note that the studies often prepared samples differently with varying plasma processing/extraction methods, and use of different assay techniques. Future research will need to determine if differences in results about OT levels in ASD reflect differences in the study populations and/or methods for assaying OT. ASD population differences include varying ages and OT levels may change during development, especially after puberty. (Hammock et al. 2012). ASD is a very heterogeneous disorder and OT level differences may be specific to clinical and etiological subgroups within the broader ASD population. In addition, there is variability of OT plasma levels across typical and healthy populations. The inherent U-shaped distribution (Zhong et al. 2012) observed in normative populations may also add to variability in OT measurement in ASD studies .
3 Prader-Willi Syndrome and OT
Prader-Willi syndrome (PWS) is a complex disorder with multisystem effects and a distinct behavioral phenotype. It occurs in approximately 1/10,000 to 1/30,000 births, and is initially characterized by severe infantile hypotonia and difficulty feeding, although later in infancy and into adolescence individuals with PWS often eat excessively and develop morbid obesity. Other characteristics of PWS include hypogonadism, short stature, small hands and feet, and strabismus. The cognitive phenotype is marked by delayed motor and language development, and behavioral difficulties including compulsive behavior, stubbornness and temper tantrums (Bittel et al. 2007b; Cassidy et al. 2012). The many behavioral and psychiatric manifestations of PWS are evident in early childhood, and are characterized by hyperactivity, impulsivity, temper tantrums, emotional lability, anxiety and repetitive behavior (Borghgraef et al. 1990; Whitman and Accardo 1987; Gross-Tsur et al. 2001). Often this phenotype is suggestive of ASD as well as attention deficit hyperactivity disorder (ADHD; Cassidy et al. 2012). Face processing is also altered in individuals with PWS, as they have difficulty reading facial expressions (Whittington and Holland 2011) .
The cause of PWS is the lack of expression of specifically paternal genes located on chromosome 15q11.2-q13. Many of the genes expressed in this region come from the father, as those from the mother are normally inactivated. Consequently, either a lack of expression or absence of the paternal copy of the genes in this region leads to no expression (Saitoh et al. 1997). This may occur through microdeletions in the paternal chromosome, no copy of the paternal chromosome paired with two copies of the maternal chromosome or uniparental disomy (UPD) or imprinting defects due to epigenetic causes (Cassidy et al. 2012). The genes expressed in this region have been studied at length to develop models of PWS and to delineate their roles in the different aspects of the PWS phenotype. Such studies are complicated by differences in the behavioral phenotype between individuals with deletions and those with UPD, as those with UPD have a less severe phenotype (Bittel et al. 2007a) and higher verbal IQ scores (Dimitropoulos et al. 2000) .
While the deletion of no one individual gene has been found to cause PWS, research has shown that the lack of expression of multiple genes may be central to the syndrome’s expression. Specifically, five polypeptide coding genes, namely MKRN3, MAGEL2, MAGED1, NECDIN and SNURF-SNRPRN, have been shown to be centrally involved in PWS. Animal models lacking one of these genes have been developed for Magel2 (Boccaccio et al. 1999), Maged1 (Dombret et al. 2012), Necdin (Lavi-Itzkovitz et al. 2012; Muscatelli et al. 2000) and Snurf (Tsai et al. 1999), although none of these individual gene disruption models completely recapitulates the PWS phenotype.
Another line of approach to elucidate the physiological underpinnings of PWS has been to examine the OT system in individuals with PWS as well as in animal models. There is a deficit of OT producing neurons in the PVN in the brains of persons with PWS (Swaab et al. 1995), as well as lower levels of OT in CSF (Martin et al. 1998). IN-OT administration increases trust in others and decreases disruptive behavior in individuals with PWS (Tauber et al. 2011). In addition, administration of OT has also been shown to rescue behavior in a Maged1 deletion model of PWS in which there is a decrease in hypothalamic OT (Dombret et al. 2012) . Although rescue was not attempted in the Necdin model, this mutant also shows a reduction in OT producing neurons in the hypothalamus (Muscatelli et al. 2000). Consequently, there appears to be disruption of the OT system in individuals with PWS, which is recapitulated in different animal models. However, the exact mechanism of OT dysregulation is unclear .
4 Williams Syndrome and OT
Williams syndrome (WS) was first described over 50 years ago (Williams et al. 1961). The first reported cases were focused on infants with hypercalcemia, developmental delays, cardiac malformations and dysmorphic facial features (Morris 1993). However, better characterization of this syndrome has elucidated a distinct behavioral phenotype marked by an increased social drive paired with social fearlessness, poor judgment, difficulty forming peer relationships and high anxiety levels (Jarvinen et al. 2013). The cause of this disorder has been determined to be the deletion of 25–30 genes in the q11.23 region of either maternal or paternal chromosome 7 that span approximately 1.5 megabases (Ewart et al. 1993; Lowery et al. 1995; Korenberg et al. 2000; Schubert 2009) . ELN, the gene for elastin, was the first deleted gene identified and its absence is indicative of a diagnosis of WS. While ELN disruption affects connective tissue, particularly of the aorta (Lowery et al. 1995), other genes such as LIMK1, CYLN2, GTF2I and GTF2IRD1 are involved in the behavioral phenotype of WS (Jarvinen-Pasley et al. 2008). The deletion of GTF2I as well as GTF2IRD1 has been shown to be involved in the social phenotype specifically (Sakurai et al. 2011; Proulx et al. 2010) .
The social phenotype associated with WS is striking due to the hypersociability of the affected individuals, as well as the preference for novel social over non-social stimuli (Jarvinen-Pasley et al. 2008, 2010) and increased eye contact (Mervis et al. 2003). In addition, the speech of individuals with WS is marked by high levels of socially engaging language as compared to controls or individuals with other developmental disorders such as Down Syndrome (Jarvinen-Pasley et al. 2010; Jarvinen et al. 2013). However, this does not translate into the development of social relationships as individuals show difficulty with social adjustment (Gosch and Pankau 1994, 1997) and social judgment (Einfeld et al. 1997; Gosch and Pankau 1997). In addition, affected individuals show deficits in social understanding, as evidenced by difficulty identifying affect (Gagliardi et al. 2003; Plesa-Skwerer et al. 2006) or other’s mental states (Jarvinen-Pasley et al. 2008) .
The high sociability of individuals with WS positions this syndrome as a good mechanism through which to understand the biological underpinnings of social behavior. Mouse models of WS include GTF2I-deficient mice which display increased social interaction with novel mice and diminished social habituation (Sakurai et al. 2011) as well as Gtf2ird1 deletions, that also show increased sociability (Proulx et al. 2010). Recently, de novo duplications of regions of 7q11.23 have been shown to be associated with ASD, whereas deletions of the same region lead to WS (Sanders et al. 2011). Such opposite effects of gene expression leading to markedly contrasting phenotypes raises the issue of dosage effects, but it should be noted that both ASD and WS phenotypes include abnormal social relationships, although through different mechanisms. Whereas individuals with WS show prolonged face gaze, those with ASD display reduced face gaze (Riby and Hancock 2009). In addition, although children with WS and ASD display high levels of anxiety, individuals with ASD have higher levels of RRB as well as greater rates of social phobia and separation anxiety (Cascio et al. 2012) .
Dai et al. (2012) examined the possibility of dysregulation of OT in WS as it relates to the contrasting phenotypes of WS or ASD. This was done by examining deletions or increased expression, respectively, of genes in the region defining WS. They show increased baseline levels of OT in individuals with WS as compared to controls. Additionally, OT levels correlated positively with increased approach to strangers as well as decreased adaptive social behaviors. These results suggest that there may be a dose dependent effect of OT, as high levels may impair adaptive social behavior and may partly underlie the maladaptive social phenotype of WS .
5 Fragile X Syndrome
Named for the fragile site observed at Xq27.3, Fragile X Syndrome (FXS) is the most common inherited form of intellectual disability and the most common known single gene mutation associated with ASD (O’Donnell and Warren 2002) . World-wide prevalence estimates range from 1 case in 1000–4000 males and 1 case in 4000–6000 females (Brown 1990; Morton et al. 1997; Turner et al. 1996; Webb 2010). This rare genetic disorder is characterized by specific physical features, and cognitive and behavioral phenotypes (Berry-Kravis et al. 2002, 2011; McLennan et al. 2011). The physical features can include a long narrow face with large protruding ears, connective tissue abnormalities (i.e. hyperextensive joints), macroorchidism, macrocephaly, obesity (especially in young males), loose skin over the hands, a high arched palate, a vertical plantar crease and flat feet (Moy et al. 2009; Schapiro et al. 1995). The behavioral and social characteristics of FXS include hyperactivity, attention difficulties, mood lability, compulsive and perseverative behaviors, aggressive outbursts, learning deficits, developmental delays (including delayed speech development), social shyness, gaze avoidance, sensory hypersensitivity, withdrawal from touch and stereotypic movements and behaviors (i.e. hand flapping and rocking), poor motor coordination and echolalia (Hagerman et al. 2009; Hall 2009; Hall et al. 2009; Moy et al. 2009). Many of these behaviors are linked to the anxiety level of the individual, a meaningful link because physiological studies have noted increased sympathetic and decreased parasympathetic activity and poor coordination between the systems (Hall et al. 2009).
Cognitive tests have indicated a specific pattern of strengths and weaknesses. FXS individuals exhibit deficits in visuo-spatial tasks, quantitative skills, short-term and working memory, expressive language skills, sequential processing and executive function (Hall et al. 2012; Cornish et al. 1999; Kwon et al. 2001; Freund and Reiss 1991; Maes et al. 1994; Berry-Kravis et al. 2002) and relative strengths in receptive language skills, visual memory, acquisition of factual information, imitation skills and gestalt processing (Berry-Kravis et al. 2002). This population also has susceptibility to certain other neuropsychiatric disabilities including ASD, ADHD and anxiety disorders, as well as neurological disorders such as epilepsy (Pretorius et al. 1998; Hessl et al. 2001). While the genetic cause of FXS has been found, the neurological basis of FXS symptoms continues to be unknown. MRI studies have found that individuals with FXS have enlarged lateral ventricles and increased caudate nucleus volumes relative to control subjects (Reiss et al. 1995). Anatomical studies of post-mortem brains have revealed that dendritic spines of neocortical pyramidal neurons of FXS subjects are longer and thinner than those of matched controls (Rudelli et al. 1985; Wisniewski et al. 1991; Irwin et al. 2001; Hinton et al. 1991), indicating that the spines fail to mature normally in FXS patients (Irwin et al. 2001).
The majority of individuals with FXS have social anxiety and almost a third have symptoms that overlap with ASD (Hagerman et al. 2010). Published studies have reported the prevalence rate of FXS and autistic behaviors/ASD diagnosis to range from 25–47 %, however sample sizes are often small (Hatton et al. 2006; Morton et al. 1997). Like the disorder itself, autistic symptoms are more common in males than females. Individuals with both FXS and ASD often have poorer developmental outcomes, lower cognitive abilities, lower levels of adaptive behavior and more problem behaviors than FXS individuals with fewer ASD related comorbidities. Of the individuals with FXS and autistic behaviors, 15–40 % of males and a few females meet the diagnostic criteria for ASD (Berry-Kravis et al. 2002) and present with more severe communication deficits, stereotyped behaviors, and social anxiety versus social disinterest. In addition, males present with more severe developmental delays. Overlapping behaviors between ASD and FXS, such as eye gaze avoidance (Hall et al. 2009), have led many scientists to study FXS as a way to understand and possibly target treatment for ASD.
5.1 Molecular Biology of Fragile X Syndrome
Diagnosis is based on DNA analysis that identifies the number of CGG repeats in the fragile X mental retardation 1 (FMR1) gene at the Xq27.3 site (Turner et al. 1996) . In most affected individuals, this genetic disorder is caused by a trinucleotide (CGG) repeat expansion in the 5′ untranslated (promoter) region of the FMR1 gene. FMR1 encodes the fragile X mental retardation protein (FMRP) , a 69 kDa protein found in most adult and fetal tissues, with high concentrations in the brain and testes. The expression of FMRP in neural tissue seems to be experience-dependent. It is produced in the soma and near the synapse of neurons (Berry-Kravis et al. 2002, 2011), and is essential to the shaping of dendritic spines (Davidovic et al. 2011). The protein and network of mRNA targets and interacting proteins contribute to several forms of synaptic plasticity involving learning and memory processes, notably those induced by activation of type I metabotropic glutamate receptor (mGluR; Davidovic et al. 2011). It is hypothesized that a decrease in Fmr1 functionally affects the protein interaction network with direct consequences on signaling cascade and cellular metabolism (Davidovic et al. 2011).
There are two different FMR1 mutations, namely full mutation and premutation (Goodrich-Hunsaker et al. 2011a, b). Premutations make up approximately 10 % of male and 2–3 % of female ASD cases (Wang et al. 2010). These mutations are associated with Fragile X-associated tremor/ataxia syndrome (FXTAS; Wang et al. 2010), have a repeat length of 50–200 and do not usually cause mental deficits, although shyness and anxiety have been known to occur. Premutations influence translation of FMR1 mRNA (Feng et al. 1995) such that individuals with permutations produce excess FMR1 mRNA, yet synthesize lower than normal levels of FMRP (Tassone et al. 2000a, b). Upon female transmission the premutation can become a full mutation. FXS is caused by full mutation which is > 200 trinucleotide repeats, and results in hypermethylation of the gene and transcriptional silencing (Tassone et al. 2000a). This creates an FMRP deficiency in the brain which leads to FXS presentation (Tassone et al. 2000a; McLennan et al. 2011).
Very rarely have other mutations in the FMR1 gene involving deletions (Gedeon et al. 1992; Wohrle et al. 1992) or a point mutation (De Boulle et al. 1993) resulted in symptoms identical or even more severe than typical FXS. FMRP is an RNA-binding protein with three RNA-interacting motifs, namely two KH domains and one RGG box (Ashley et al. 1993; Siomi et al. 1993). Findings that a point mutation in one of the KH domains is sufficient to produce severe FXS (Siomi et al. 1994; De Boulle et al. 1993) points to the conclusion that this aspect of the protein is closely related to the clinical symptoms. While mainly found in the cytoplasm (Verheij et al. 1993; Devys et al. 1993), FMRP appears to be able to shuttle in and out of the nucleus (Feng et al. 1997) possibly as a carrier for specific mRNAs.
In the brain, FMRP appears in the cytoplasm of both the soma and dendrites of neurons (Devys et al. 1993; Feng et al. 1997), forms complexes with other proteins including the fragile X-related proteins 1 and 2 (FXR1P and FXR2P), along with mRNA in mRNA-proteins (mRNPs) in association with ribosomes (Feng et al. 1997; Corbin et al. 1997; Ceman et al. 2000; Eberhart et al. 1996; Khandjian et al. 1996). As mentioned earlier, levels of FMRP have been shown to be regulated by sensory experience (Irwin et al. 2005; Todd and Mack 2000; Todd et al. 2003a, b). Supporting this theory is mounting evidence which implicates FMRP in synaptic plasticity and findings showing that mice lacking FMRP have impaired long-term potentiation in somatosensory cortex (Li et al. 2002), visual cortex (Wilson and Cox 2007), olfactory cortex (Larson et al. 2005), cingulate cortex, and amygdala (Zhao et al. 2005), as well as enhanced long-term depression in hippocampus (Huber et al. 2002). In synaptosomal preparations, stimulation of mGluR results in an FMRP-dependent increase in protein synthesis (Weiler et al. 1997, 2004).
5.2 Fragile X Syndrome and OT
There are several reasons why treatment with OT has been investigated in FXS. There is evidence that some of the features of ASD and FXS may be reduced by OT (Bartz and Hollander 2006; Hall et al. 2012; Hollander et al. 2007). As described above, OT released endogenously or given exogenously has been associated with positive social behaviors, as well as reductions in anxiety, obsessiveness and stress reactivity. OT may also serve to counter the defensive behavioral strategies associated with stressful experiences, and the central release of AVP and other peptides, such as corticotropin-releasing factor (Carter 2007). IN-OT in males has increased “trust” in a computer game task (Kosfeld et al. 2005) and may reduce endocrine responses to psychosocial stressors in men and women (Heinrichs and Domes 2008).
Similarly to ASD, available treatments for FXS focus on managing symptoms. Stimulants are prescribed for ADHD symptoms, whereas SSRIs and antipsychotics treat aggression associated with anxiety and carbamazepine is used for treatment of seizures (Hampson et al. 2011). Currently, there are no treatments on the market targeting the molecular abnormalities of FXS (Gurkan and Hagerman 2012). However, recent studies have begun to investigate IN-OT due to the autistic-like behaviors observed in FXS and the social and anxiolytic effects of OT.
A 2012 study conducted by Hall and colleagues researched the effects of IN-OT on males with FXS. They hypothesized that the prosocial and anxiolytic effects of OT would reduce, if not alleviate, socially inappropriate behaviors and social anxiety in males with FXS. Of the ten low functioning males between the ages of 13 and 28 years (mean age = 21.3 years) recruited, eight subjects completed the study. All subjects were confirmed by standard Southern blot analysis to have a fully methylated full mutation. The study was set-up as a randomized double-blind placebo-controlled single-dose trial performed with intranasal administration of placebo, 24 IU OT and 48 IU OT.
As previously mentioned, extreme eye gaze avoidance and hyperarousal are exhibited by FXS individuals when experiencing stressful social situations. Hall and colleagues collected eye gaze frequency, heart rate (HR), respiratory sinus arrhythmia (RSA), heart rate variability (HRV) and salivary cortisol during the social challenge (10 min in length), which was conducted 50 min after OT administration. The first 5 min was a social proximity task, where the subjects sat quietly while a female researcher sat opposite with knees almost touching and read a magazine or book. The second five minute section was a social interaction task, here the experimenter while seated had a conversation with the subject, asking questions such as “tell me what movies you like to watch”. Before the conversation started, the subjects were instructed to look the experimenter in the eyes as much as possible while talking, they were additionally prompted by the researcher during the conversation when needed, similar to previous studies (Hall et al. 2012).
Confirmation of the hypothesis that OT would have beneficial consequences in FXS would be observed in increased eye gaze frequency, a reduction in physiological arousal, and a decrease in salivary cortisol. The researchers observed a significant main effect with OT. As compared to a placebo, 24 IU OT led to an increase in eye gaze frequency (p= 0.042). No differences were noticed between the two tasks of the social challenge. There was also a significant main effect of OT observed in the salivary cortisol levels. Salivary cortisol levels were looked at pre and post challenge, and showed a significant decrease for the 48 IU dose as compared to placebo (p= 0.05). However, no effects were observed in the physiological measurements (HR, HRV, and RSA). Hall and colleagues hypothesized that the administration of OT may dampen amygdala reactivity towards social stimuli that causes anxiety (Kirsch et al. 2005; Petrovic et al. 2008), decrease HPA axis activation, and increase social motivation (Witt et al. 1992; Witt and Insel 1992). At present, neither OT nor AVP are known to be targets of FMRP . Two points of future investigation that could reveal the interplay between OT and FMRP as proposed by Hall et al. (2012) were: (1) an experiment to determine the localization of mRNAs encoding FMRP and OT precursor in dendritic domains (Smith 2004), and (2) research into the non-coding BC1, a neuron-specific RNA polymerase III transcript and OT in hypothalamo-neurohypophyseal neurons (Tiedge et al. 1993) should be performed.
5.3 Animal Models of Fragile X Syndrome Provide Insight on Moderators of Neurodevelopmental Pathways
Animal models have proven indispensible in the understanding of diseases and disorders, and in the development of pharmaceuticals used to treat them. The quality of an animal model is ascertained based on how well it can meet certain criteria of validity, namely construct, face and predictive validity (Bernardet and Crusio 2006). How well the model’s behavioral traits resemble the core traits of the disorder is face validity. Predictive validity is established when a drug reduces or improves symptoms in both the model and human. Construct validity is the “quality” of the model, its ability to accurately measure or represent what it claims to be measuring (Cronbach and Meehl 1955). There currently are several FXS animal models, three mice and one drosophila model that meet multiple criteria.
Two homologous genes to Fmr1 in vertebrates are Fxr1 (fragile X related gene) and Fxr2. Their proteins, FXR1P and FXR2P are both expressed in the neural tissue, specifically in cell bodies, but they are also found in the dendrites near the synapse. Both Fxr1 and Fxr2 KO mice have been produced. FXR1P homozygous mice die within 24 hours of birth, while heterozygous mice exhibit abnormal limb musculature. FXR2P deficient mice have a normal lifespan and have learning deficits similar to Fmr1 KO with some differences and circadian rhythm deficits (Berry-Kravis et al. 2011). The fruit fly model, a mutant lacking dFmr1 (also known as dFxr) protein, exhibits overextension of neurites during development of mushroom bodies (brain region linked with memory) and have a behavioral phenotype that includes circadian rhythm abnormalities and altered courtship behavior (Berry-Kravis et al. 2011; Gatto and Broadie 2009).
One of the most well characterized animal models of FXS was developed in 1994 by the Dutch-Belgian Fragile X Consortium (1994), and is a mouse model with an identical molecular endpoint of the experimental model and the human disease (i.e., lack of FMRP throughout the lifespan). This mouse was created by inserting a neomycin cassette into exon 5 of the murine Fmr1 gene. The insert disrupts the transcription of Fmr1 mRNA causing an absence of FMRP . Even though the cause may not be identical, this mouse model exhibits behavioral similarities to FXS. Fmr1 KO mice are described as having lower than normal levels of initial social interaction (Mineur et al. 2006), lacking preference for social novelty, and displaying inappropriate social responses (Pietropaolo et al. 2011) versus wild-type (WT). In contrast, OTKO or AVPKO mice display high levels of social contact that does not diminish over time, thus failing to show familiarity (Crawley et al. 2007).
While macroorchidism is observed in the Fmr1 KO animals (Bakker et al. 1994) as well as in FXS patients, the behavioral patterns differ between the patients and the KO. By most accounts Fmr1 KO mice appear to have relatively normal behavior, but research has shown that the behavioral and cognitive deficits of the KO are actually quite subtle and parallel FXS patients (Paradee et al. 1999; D’Hooge et al. 1997; Peier et al. 2000; Berry-Kravis et al. 2002). Some behavioral phenotypes displayed in Fmr1 KO are deficits in object recognition memory (including a failure to habituate to objects), and impairment of spatial memory (Mineur et al. 2002).
Several studies indicate that Fmr1 KO mice are hyperactive and show indications of increased anxiety (Mineur et al. 2002; Spencer et al. 2005; Bakker et al. 1994), and overreact to sensory stimuli (Chen and Toth 2001; Frankland et al. 2004; Nielsen et al. 2002). Fmr1 KO mice also exhibit abnormal social interactions, including a general reduction in social contact and a failure to show social recognition (Bernardet and Crusio 2006; Mineur et al. 2006; Spencer et al. 2005; Yan et al. 2004). Also, similar to FXS patients, these mice exhibit sensory hyperresponsiveness, especially to auditory stimuli. Loud tones may induce audiogenic seizures. In some tasks there is variability in the results (i.e. complex visual and auditory discriminant tasks and activity level in an open field). This variability may come from different mouse strains reflecting the effect of different genetic backgrounds on the the expression of the symptoms. This is also hypothesized to be the basis for the variability observed in the symptoms of FXS patients. In work by Pietropaolo et al. (2011), the validity of the Fmr1 KO mouse on the C57BL/6 (B6) background was tested against WT and Fmr1 KO on the FVB background. They found the Fmr1 KO on the B6 background to be a good model for FXS and a suitable model for ASD (Pietropaolo et al. 2011; Yan et al. 2004).
These mice also have neuropathologic phenotypes that are similar to those of FXS patients, including density of dendritic spines of pyramidal neurons in the visual and somatosensory cortices that are greater in adult Fmr1 KO than WT. In some brain areas, in both mice Fmr1 KO and FXS patients, the appearance of the spines are more similar to developing versus mature spines (Berry-Kravis et al. 2002). Absence of FMRP in both humans and mice results in improper development of dendritic spines on cortical pyramidal neurons (Comery et al. 1997; Irwin et al. 2000, 2001, 2002). The use of the Fmr1 KO mouse has also provided some insight into the normal cellular function of FMRP. The subtle cognitive deficits of Fmr1 KO mice present difficulties for preclinical testing of potential treatments, and highlight how complex the relationship between the mouse and human phenotypes are. One possibility is that cognitive processes in which FMRP plays a vital role in humans are poorly developed in mice; thus mice lacking FMRP are not particularly disabled, at least compared to severely-affected patients. A second possibility is that other proteins can compensate for FMRP in mice but not in humans. Third, the behavioral paradigms thus far applied to the mouse model may not efficiently assay cognitive domains most affected in FXS.
5.4 Examining OT in Neurodevelopmental Animal Models: A Way to Examine Early Effects?
Below we present preliminary data measuring the OT and AVP systems in a mouse model in order to learn more about FXS pathway interactions during development. The animal model was generated with WT and Fmr1 KO mice from a colony founded with stock obtained from the Jackson Laboratory (Bar Harbor, ME, USA) that was backcrossed onto a B6 background > 10 generations. Mice were genotyped using primers described previously (The Dutch-Belgian Fragile-X Consortium 1994). Cells were stained using the immunocytochemical (ICC) staining procedures, following protocols described in early work on OT and AVP in voles (Yamamoto et al. 2004). All sections were double-stained for NeuN (a marker that stains cell nuclei only in neurons), which allowed precise localization of cytoarchitechtonic boundaries. Stained sections were mounted on subbed slides and examined with OT and AVP antibodies (OT antibodies were generously provided by M. Morris and AVP antibodies were obtained from MP Biomedical #647171, formerly ICN; Solon, OH, USA). Slices of tissue for each animal were categorized as described in Paxinos and Franklin (2004) and carefully matched across subjects to allow comparable sections. The imaged slides captured at 10X, were coded and scored by an experimentally blind scorer using Image J (NIH, Bethesda, MD) software. Cells in the PVN of the hypothalamus regions were stained separately for OT and AVP (N= 6–7 mice per group). Boxed sampling areas were: 125 × 125 μm2 (PVN total staining density), 250 × 375 μm2 (PVN fibers), 93.75 × 93.75 μm2 for cell counts bilaterally in both the PVN and SON.
Preliminary results suggest a significant reduction in both OT-positive (Fig. 14.1) and AVP-positive (Fig. 14.2) cells in the PVN of Fmr1 KO as compared to WT (Table 14.1). While not significant, there is a trend for fewer OT-positive cells in the SON (Table 14.2). We also measured, by cell count, the abundance of OXTR positive cells in the hippocampus, and retrosplenial granular and piriform cortices. None of these areas showed a significant change in cell density as compared to WT mice (p> 0.05). Earlier work in voles has suggested that both OT and AVP may support a general tendency toward social contact (Cho et al. 1999). The absence of either OT or AVP in the presence of the other neuropeptide did not produce an “asocial” animal. However, selective social preferences, such as those necessary for pair bond formation, appear to require stimulation of both OT and AVP receptors.

Expression in the paraventricular nucleus (PVN) of OT, as measured by ICC, is reduced in Fmr1 KO mice, compared to the wild-type (WT). (Reprinted from Brain Research, Francis et al. 2014, Copyright (2014) with permission from Elsevier)

Expression in the paraventricular nucleus (PVN) of AVP. In Fmr1 KO mice, as compared to the wild-type (WT) AVP expression is reduced as measured by ICC. (Reprinted from Brain Research, Francis et al. 2014, Copyright (2014) with permission from Elsevier)
Although the preliminary data shown here for Fmr1 KO mice needs to be replicated in a larger sample size and in other animal models , we include these findings as an example of possible approaches to examining the role of peptides, including OT and AVP, in molecularly characterized genetic syndromes. Work across these models also could provide additional insight regarding the role of OT and AVP in early development, especially in syndromes in which atypical trajectories occur.
6 Conclusion and Next Steps
While each of the disorders described here (ASD, PWS, WS and FXS) is unique, each one is characterized by atypical social behaviors and in many cases a tendency toward high levels of anxiety. Given the importance of OT and AVP to mammalian social behaviors and anxiety, the neuropeptides’ investigative value in these syndromes is not unexpected. This review summarized the possible role of OT in these NDD through experiments conducted by others and ourselves (Table 14.3). Each of these early developmental disorders displayed alterations in the OT system and may represent many molecular pathways that lead to a commonly disrupted neuropeptide hormone system. Our preliminary data suggests decreased numbers of OT-positive and AVP-positive cells in the PVN of Fmr1 KO mice, a mouse model for FXS. Individuals with PWS have shown lower levels of OT in CSF and fewer OT producing cells in the PVN. Lower plasma OT levels have also been detected in some children or a subgroup of ASD affected children. In contrast with WS, which is characterized by hypersociability, a positive correlation was found between OT levels and increased stranger approach and decreased adaptive social behavior. Knowledge of the functionality of the OXTR in WS remains to be studied. Given the rarity of these disorders and the complex animal models needed to research these disorders, many of these studies have small sample sizes. These significant studies, however, can motivate future research on these disorders and other NDD, especially those disorders with dysfunctional social behaviors as a symptom.
ASD, as described above, is the primary NDD presumed to be associated with dysregulation of the OT system. It is striking, however, that other disorders with phenotypes marked by abnormal social behavior as well as anxiety (manifested in RRB) have abnormalities in OT production as measured in blood and CSF (Table 14.3). For example, individuals with PWS, like those in ASD, have difficulty with social competence (Dimitropoulos et al. 2013), are aloof and avoid eye contact (Dimitropoulos et al. 2009). Furthermore, RRB is also evidenced in PWS (Greaves et al. 2006), although to a lesser degree than in ASD as measured using the Repetitive Behavior Scale-Revised (RBS-R; Flores et al. 2011). A subset of the chromosomal region associated with PWS is also associated with an increased risk for ASD, as maternally inherited duplications of the 15q11-13 region are associated with 1–3 % of ASD cases (Bolton et al. 2001; Cook et al. 1997; Vorstman et al. 2006).
Williams syndrome and ASD also share commonalities, namely abnormal social phenotypes and anxiety. Individuals with WS, unlike those with PWS, show a phenotype that is markedly different from ASD. Although both groups are at risk for anxiety, individuals with ASD show higher levels of social phobia and separation anxiety, as well as higher rates of RRB. However, children with WS have higher scores on measures of generalized anxiety (Rodgers et al. 2012). WS is characterized by an increase in OT levels (Dai et al., 2012), as well as a deletion of the 7q11.23 region as opposed to a de novo duplication which leads to ASD (Sanders et al., 2011). Thus, it is likely that some ASD and WS symptoms are related to genetic dosage effects. Studies of FX and ASD mechanism may also inform each other, as mutations in mGluR5 can contribute to the diagnosis of FXS or ASD, and mGluR5 antagonists have shown promise in alleviating ASD symptoms in mouse models (Silverman et al. 2012) as well as Fragile X pathology.
As summarized in this review, animal and human research to date has shown that dysregulation of the OT system is associated with marked deficits in social behavior as well as anxiety. This commonality across multiple NDD may indicate a shared OT pathway that is affected during development. The use of animal models , particularly those developed for FXS, WS and PWS, will provide insight into such a pathway, as these disorders have well characterized genetics, whereas there are over 103 disease genes and 44 genomic loci reported to be involved in ASD (Betancur 2011). However, unlike in ASD, there is a lack of human data on the pathophysiology of FXS, WS and PWS, as well as pharmacological interventions. Ideally, scientists want to identify specific molecular pathways to target distinct syndromes and disorders for treatment. However, many effective treatments modulate common neurochemical or hormone pathways that are downstream from etiologically contributing factors (e.g. drugs for hypertension). Combining the strengths of human and animal model studies across these NDD may provide important clues into the role of OT in development, in addition to elucidating the complex neurophysiology and treatment targets for FXS, PWS, WS and ASD.
References
Abramson RK, Wright HH, Carpenter R, Brennan W, Lumpuy O, Cole E, Young SR (1989) Elevated blood serotonin in autistic probands and their first-degree relatives. J Autism Dev Disord 19:397–407
American Psychiatric Association (2013) Diagnostic and statistical manual of mental disorders, 5th edn. American Psychiatric Publishing, Arlington
Anagnostou E, Soorya L, Chaplin W, Bartz J, Halpern D, Wasserman S, Wang AT, Pepa L, Tanel N, Kushki A, Hollander E (2012) Intranasal oxytocin versus placebo in the treatment of adults with autism spectrum disorders: a randomized controlled trial. Mol Autism 3:16
Andari E, Duhamel JR, Zalla T, Herbrecht E, Leboyer M, Sirigu A (2010) Promoting social behavior with oxytocin in high-functioning autism spectrum disorders. Proc Natl Acad Sci U S A 107:4389–4394
Appenrodt E, Schnabel R, Schwarzberg H (1998) Vasopressin administration modulates anxiety-related behavior in rats. Physiol Behav 64:543–547
Ashley CT Jr, Wilkinson KD, Reines D, Warren ST (1993) FMR1 protein: conserved RNP family domains and selective RNA binding. Science 262:563–566
Bailey A, Le Couteur A, Gottesman I, Bolton P, Simonoff E, Yuzda E, Rutter M (1995) Autism as a strongly genetic disorder: evidence from a British twin study. Psychol Med 25:63–77
Bakker CE, Verheij C, Willemsen R, Vanderhelm R, Oerlemans F, Vermey M, Bygrave A, Hoogeveen AT, Oostra BA, Reyniers E, Deboulle K, Dhooge R, Cras P, Vanvelzen D, Nagels G, Martin JJ, Dedeyn PP, Darby JK, Willems PJ (1994) Fmr1 knockout mice—a model to study fragile-X mental-retardation. Cell 78:23–33
Bartz JA, Hollander E (2006) The neuroscience of affiliation: forging links between basic and clinical research on neuropeptides and social behavior. Horm Behav 50:518–528
Baumgartner T, Heinrichs M, Vonlanthen A, Fischbacher U, Fehr E (2008) Oxytocin shapes the neural circuitry of trust and trust adaptation in humans. Neuron 58:639–650
Bernardet M, Crusio WE (2006) Fmr1 KO mice as a possible model of autistic features. Scientific World Journal 6:1164–1176
Berry-Kravis E, Grossman AW, Crnicz LS, Greenough WT (2002) Understanding fragile X syndrome. Curr Paediatr 12:316–324
Berry-Kravis E, Knox A, Hervey C (2011) Targeted treatments for fragile X syndrome. J Neurodev Disord 3:193–210
Betancur C (2011) Etiological heterogeneity in autism spectrum disorders: more than 100 genetic and genomic disorders and still counting. Brain Res 1380:42–77
Bittel DC, Kibiryeva N, McNulty SG, Driscoll DJ, Butler MG, White RA (2007a) Whole genome microarray analysis of gene expression in an imprinting center deletion mouse model of Prader-Willi syndrome. Am J Med Genet A 143:422–429
Bittel DC, Kibiryeva N, Sell SM, Strong TV, Butler MG (2007b) Whole genome microarray analysis of gene expression in Prader-Willi syndrome. Am J Med Genet A 143:430–442
Boccaccio I, Glatt-Deeley H, Watrin F, Roeckel N, Lalande M, Muscatelli F (1999) The human MAGEL2 gene and its mouse homologue are paternally expressed and mapped to the Prader-Willi region. Hum Mol Genet 8:2497–2505
Bolton PF, Dennis NR, Browne CE, Thomas NS, Veltman MW, Thompson RJ, Jacobs P (2001) The phenotypic manifestations of interstitial duplications of proximal 15q with special reference to the autistic spectrum disorders. Am J Med Genet 105:675–685
Borghgraef M, Fryns JP, Van Den Berghe H (1990) Psychological profile and behavioural characteristics in 12 patients with Prader-Willi syndrome. Genet Counsel 1:141–150
Born J, Lange T, Kern W, McGregor GP, Bickel U, Fehm HL (2002) Sniffing neuropeptides: a transnasal approach to the human brain. Nat Neurosci 5:514–516
Brown WT (1990) The fragile X: progress toward solving the puzzle. Am J Hum Genet 47:175–180
Campbell DB, Datta D, Jones ST, Batey Lee E, Sutcliffe JS, Hammock EA, Levitt P (2011) Association of oxytocin receptor (OXTR) gene variants with multiple phenotype domains of autism spectrum disorder. J Neurodev Disord 3:101–112
Carter CS (1998) Neuroendocrine perspectives on social attachment and love. Psychoneuroendocrinology 23:779–818
Carter CS (2007) Sex differences in oxytocin and vasopressin: implications for autism spectrum disorders? Behav Brain Res 176:170–186
Cascio CJ, Foss-Feig JH, Heacock JL, Newsom CR, Cowan RL, Benningfield MM, Rogers BP, Cao A (2012) Response of neural reward regions to food cues in autism spectrum disorders. J Neurodev Disord 4:9
Cassidy SB, Schwartz S, Miller JL, Driscoll DJ (2012) Prader-Willi syndrome. Genet Med 14:10–26
Ceman S, Nelson R, Warren ST (2000) Identification of mouse YB1/p50 as a component of the FMRP-associated mRNP particle. Biochem Biophys Res Comm 279:904–908
Chaste P, Leboyer M (2012) Autism risk factors: genes, environment, and gene-environment interactions. Dialogue Clin Neurosci 14:281–292
Chen L, Toth M (2001) Fragile X mice develop sensory hyperreactivity to auditory stimuli. Neurosci 103:1043–1050
Chini B, Mouillac B, Balestre MN, Trumpp-Kallmeyer S, Hoflack J, Hibert M, Andriolo M, Pupier S, Jard S, Barberis C (1996) Two aromatic residues regulate the response of the human oxytocin receptor to the partial agonist arginine vasopressin. FEBS Lett 397:201–206
Cho MM, DeVries AC, Williams JR, Carter CS (1999) The effects of oxytocin and vasopressin on partner preferences in male and female prairie voles (Microtus ochrogaster). Behav Neurosci 113:1071–1079
Chugani DC, Muzik O, Behen M, Rothermel R, Janisse JJ, Lee J, Chugani HT (1999) Developmental changes in brain serotonin synthesis capacity in autistic and nonautistic children. Ann Neurol 45:287–295
Comery TA, Harris JB, Willems PJ, Oostra BA, Irwin SA, Weiler IJ, Greenough WT (1997) Abnormal dendritic spines in fragile X knockout mice: maturation and pruning deficits. Proc Natl Acad Sci U S A 94:5401–5404
Cook EH Jr, Lindgren V, Leventhal BL, Courchesne R, Lincoln A, Shulman C, Lord C, Courchesne E (1997) Autism or atypical autism in maternally but not paternally derived proximal 15q duplication. Am J Hum Genet 60:928–934
Corbin F, Bouillon M, Fortin A, Morin S, Rousseau F, Khandjian EW (1997) The fragile X mental retardation protein is associated with poly(A)+mRNA in actively translating polyribosomes. Hum Mol Genet 6:1465–1472
Cornish KM, Munir F, Cross G (1999) Spatial cognition in males with Fragile-X syndrome: evidence for a neuropsychological phenotype. Cortex 35:263–271
Crawley JN, Chen T, Puri A, Washburn R, Sullivan TL, Hill JM, Young NB, Nadler JJ, Moy SS, Young LJ, Caldwell HK, Young WS (2007) Social approach behaviors in oxytocin knockout mice: comparison of two independent lines tested in different laboratory environments. Neuropeptides 41:145–163
Cronbach LJ, Meehl PE (1955) Construct validity in psychological tests. Psychol Bull 52:281–302
D’Hooge R, Nagels G, Franck F, Bakker CE, Reyniers E, Storm K, Kooy RF, Oostra BA, Willems PJ, De Deyn PP (1997) Mildly impaired water maze performance in male Fmr1 knockout mice. Neuroscience 76:367–376
Dadds MR, Macdonald E, Cauchi A, Williams K, Levy F, Brennan J (2014) Nasal oxytocin for social deficits in childhood autism: a randomized controlled trial. J Autism Dev Disord 44:521–531
Dai L, Carter CS, Ying J, Bellugi U, Pournajafi-Nazarloo H, Korenberg JR (2012) Oxytocin and vasopressin are dysregulated in Williams Syndrome, a genetic disorder affecting social behavior. PloS One 7:e38513
Davidovic L, Navratil V, Bonaccorso CM, Catania MV, Bardoni B, Dumas ME (2011) A metabolomic and systems biology perspective on the brain of the fragile X syndrome mouse model. Genome Res 21:2190–2202
De Boulle K, Verkerk AJ, Reyniers E, Vits L, Hendrickx J, Van Roy B, Van den Bos F, de Graaff E, Oostra BA, Willems PJ (1993) A point mutation in the FMR-1 gene associated with fragile X mental retardation. Nat Genet 3:31–35
Devys D, Lutz Y, Rouyer N, Bellocq JP, Mandel JL (1993) The FMR-1 protein is cytoplasmic, most abundant in neurons and appears normal in carriers of a fragile X premutation. Nat Genet 4:335–340
Dimitropoulos A, Feurer ID, Roof E, Stone W, Butler MG, Sutcliffe J, Thompson T (2000) Appetitive behavior, compulsivity, and neurochemistry in Prader-Willi syndrome. Ment Retard Dev Disabil Res Rev 6:125–130
Dimitropoulos A, Ho AY, Klaiman C, Koenig K, Schultz RT (2009) A comparison of behavioral and emotional characteristics in children with autism, Prader-Willi syndrome, and Williams syndrome. J Ment Health Res Intellect Disabil 2:220–243
Dimitropoulos A, Ho A, Feldman B (2013) Social responsiveness and competence in Prader-Willi syndrome: direct comparison to autism spectrum disorder. J Autism Dev Disord 43:103–113
Dombret C, Nguyen T, Schakman O, Michaud JL, Hardin-Pouzet H, Bertrand MJ, De Backer O (2012) Loss of Maged1 results in obesity, deficits of social interactions, impaired sexual behavior and severe alteration of mature oxytocin production in the hypothalamus. Hum Mol Genet 21:4703–4717
Domes G, Heinrichs M, Michel A, Berger C, Herpertz S (2007) Oxytocin improves “mind-reading” in humans. Biol Psychiatry 61:731–733
Domes G, Sibold M, Schulze L, Lischke A, Herpertz SC, Heinrichs M (2013) Intranasal oxytocin increases covert attention to positive social cues. Psychol Med 43:1747–1753
Eberhart DE, Malter HE, Feng Y, Warren ST (1996) The fragile X mental retardation protein is a ribonucleoprotein containing both nuclear localization and nuclear export signals. Hum Mol Genet 5:1083–1091
Ebstein RP, Israel S, Lerer E, Uzefovsky F, Shalev I, Gritsenko I, Riebold M, Salomon S, Yirmiya N (2009) Arginine vasopressin and oxytocin modulate human social behavior. Ann N Y Acad Sci 1167:87–102
Ebstein RP, Knafo A, Mankuta D, Chew SH, Lai PS (2012) The contributions of oxytocin and vasopressin pathway genes to human behavior. Horm Behav 61:359–379
Einfeld SL, Tonge BJ, Florio T (1997) Behavioral and emotional disturbance in individuals with Williams syndrome. Am J Ment Retard 102:45–53
Ewart AK, Morris CA, Atkinson D, Jin W, Sternes K, Spallone P, Stock AD, Leppert M, Keating MT (1993) Hemizygosity at the elastin locus in a developmental disorder, Williams syndrome. Nat Genet 5:11–16
Feldman R, Zagoory-Sharon O, Weisman O, Schneiderman I, Gordon I, Maoz R, Shalev I, Ebstein RP (2012) Sensitive parenting is associated with plasma oxytocin and polymorphisms in the OXTR and CD38 genes. Biol Psychiatry 72:175–181
Feng Y, Zhang F, Lokey LK, Chastain JL, Lakkis L, Eberhart D, Warren ST (1995) Translational suppression by trinucleotide repeat expansion at FMR1. Science 268:731–734
Feng Y, Absher D, Eberhart DE, Brown V, Malter HE, Warren ST (1997) FMRP associates with polyribosomes as an mRNP, and the I304N mutation of severe fragile X syndrome abolishes this association. Mol Cell 1:109–118
Ferguson JN, Young LJ, Hearn EF, Matzuk MM, Insel TR, Winslow JT (2000) Social amnesia in mice lacking the oxytocin gene. Nat Genet 25:284–288
Flores CG, Valcante G, Guter S, Zaytoun A, Wray E, Bell L, Jacob S, Lewis MH, Driscoll DJ, Cook EH Jr, Kim SJ (2011) Repetitive behavior profiles: consistency across autism spectrum disorder cohorts and divergence from Prader-Willi syndrome. J Neurodev Disord 3:316–324
Francis SM, Sagar A, Levin-Decanini T, Liu W, Carter CS, Jacob S. (2014) Brain Res. 11;1580:199–218
Frankland PW, Wang Y, Rosner B, Shimizu T, Balleine BW, Dykens EM, Ornitz EM, Silva AJ (2004) Sensorimotor gating abnormalities in young males with fragile X syndrome and Fmr1-knockout mice. Mol Psychiatry 9:417–425
Freund LS, Reiss AL (1991) Cognitive profiles associated with the fra(X) syndrome in males and females. Am J Med Genet 38:542–547
Gagliardi C, Frigerio E, Burt DM, Cazzaniga I, Perrett DI, Borgatti R (2003) Facial expression recognition in Williams syndrome. Neuropsychologia 41:733–738
Gatto CL, Broadie K (2009) Temporal requirements of the fragile X mental retardation protein in modulating circadian clock circuit synaptic architecture. Front Neural Circuits 3:8
Gedeon AK, Baker E, Robinson H, Partington MW, Gross B, Manca A, Korn B, Poustka A, Yu S, Sutherland GR, Mulley JC (1992) Fragile X syndrome without CCG amplification has an FMR1 deletion. Nat Genet 1:341–344
Goodrich-Hunsaker NJ, Wong LM, McLennan Y, Srivastava S, Tassone F, Harvey D, Rivera SM, Simon TJ (2011a) Young adult female fragile X premutation carriers show age- and genetically-modulated cognitive impairments. Brain Cogn 75:255–260
Goodrich-Hunsaker NJ, Wong LM, McLennan Y, Tassone F, Harvey D, Rivera SM, Simon TJ (2011b) Adult female fragile X premutation carriers exhibit age- and CGG repeat length-related impairments on an attentionally based enumeration task. Front Hum Neurosci 5:63
Gosch A, Pankau R (1994) Social-emotional and behavioral adjustment in children with Williams-Beuren syndrome. Am J Med Genet 53:335–339
Gosch A, Pankau R (1997) Personality characteristics and behaviour problems in individuals of different ages with Williams syndrome. Dev Med Child Neurol 39:527–533
Greaves N, Prince E, Evans DW, Charman T (2006) Repetitive and ritualistic behaviour in children with Prader-Willi syndrome and children with autism. J Intellect Disabil Res 50:92–100
Green L, Fein D, Modahl C, Feinstein C, Waterhouse L, Morris M (2001) Oxytocin and autistic disorder: alterations in peptide forms. Biol Psychiatry 50:609–613
Gross-Tsur V, Landau YE, Benarroch F, Wertman-Elad R, Shalev RS (2001) Cognition, attention, and behavior in Prader-Willi syndrome. J Child Neurol 16:288–290
Guastella AJ, Mitchell PB, Dadds MR (2008) Oxytocin increases gaze to the eye region of human faces. Biol Psychiatry 63:3–5
Guastella AJ, Einfeld SL, Gray KM, Rinehart NJ, Tonge BJ, Lambert TJ, Hickie IB (2010) Intranasal oxytocin improves emotion recognition for youth with autism spectrum disorders. Biol Psychiatry 67:692–694
Gurkan CK, Hagerman RJ (2012) Targeted treatments in autism and fragile X syndrome. Res Autism Spectr Disord 6:1311–1320
Hagerman RJ, Berry-Kravis E, Kaufmann WE, Ono MY, Tartaglia N, Lachiewicz A, Kronk R, Delahunty C, Hessl D, Visootsak J, Picker J, Gane L, Tranfaglia M (2009) Advances in the treatment of fragile X syndrome. Pediatrics 123:378–390
Hagerman R, Hoem G, Hagerman P (2010) Fragile X and autism: intertwined at the molecular level leading to targeted treatments. Mol Autism 1:12
Hall SS (2009) Treatments for fragile X syndrome: a closer look at the data. Dev Disabil Res Rev 15:353–360
Hall SS, Lightbody AA, Huffman LC, Lazzeroni LC, Reiss AL (2009) Physiological correlates of social avoidance behavior in children and adolescents with fragile x syndrome. J Am Acad Child Adolesc Psychiatry 48:320–329
Hall SS, Lightbody AA, McCarthy BE, Parker KJ, Reiss AL (2012) Effects of intranasal oxytocin on social anxiety in males with fragile X syndrome. Psychoneuroendocrinology 37:509–518
Hammock E, Veenstra-VanderWeele J, Yan Z, Kerr TM, Morris M, Anderson GM, Carter CS, Cook EH, Jacob S (2012) Examining autism spectrum disorders by biomarkers: example from the oxytocin and serotonin systems. J Am Acad Child Adolesc Psychiatry 51:712–721
Hampson DR, Adusei DC, Pacey LK (2011) The neurochemical basis for the treatment of autism spectrum disorders and Fragile X Syndrome. Biochem Pharmacol 81:1078–1086
Hatton DD, Sideris J, Skinner M, Mankowski J, Bailey DB Jr, Roberts J, Mirrett P (2006) Autistic behavior in children with fragile X syndrome: prevalence, stability, and the impact of FMRP. Am J Med Genet A 140A:1804–1813
Heinrichs M, Domes G (2008) Neuropeptides and social behaviour: effects of oxytocin and vasopressin in humans. Prog Brain Res 170:337–350
Hessl D, Dyer-Friedman J, Glaser B, Wisbeck J, Barajas RG, Taylor A, Reiss AL (2001) The influence of environmental and genetic factors on behavior problems and autistic symptoms in boys and girls with fragile X syndrome. Pediatrics 108:E88
Hinton VJ, Brown WT, Wisniewski K, Rudelli RD (1991) Analysis of neocortex in three males with the fragile X syndrome. Am J Med Genet 41:289–294
Hollander E, Novotny S, Hanratty M, Yaffe R, DeCaria CM, Aronowitz BR, Mosovich S (2003) Oxytocin infusion reduces repetitive behaviors in adults with autistic and Asperger’s disorders. Neuropsychopharmacology 28:193–198
Hollander E, Bartz J, Chaplin W, Phillips A, Sumner J, Soorya L, Anagnostou E, Wasserman S (2007) Oxytocin increases retention of social cognition in autism. Biol Psychiatry 61:498–503
Huber KM, Gallagher SM, Warren ST, Bear MF (2002) Altered synaptic plasticity in a mouse model of fragile X mental retardation. Proc Natl Acad Sci U S A 99:7746–7750
Irwin SA, Galvez R, Greenough WT (2000) Dendritic spine structural anomalies in fragile-X mental retardation syndrome. Cereb Cortex 10:1038–1044
Irwin SA, Patel B, Idupulapati M, Harris JB, Crisostomo RA, Larsen BP, Kooy F, Willems PJ, Cras P, Kozlowski PB, Swain RA, Weiler IJ, Greenough WT (2001) Abnormal dendritic spine characteristics in the temporal and visual cortices of patients with fragile-X syndrome: a quantitative examination. Am J Med Genet 98:161–167
Irwin SA, Idupulapati M, Gilbert ME, Harris JB, Chakravarti AB, Rogers EJ, Crisostomo RA, Larsen BP, Mehta A, Alcantara CJ, Patel B, Swain RA, Weiler IJ, Oostra BA, Greenough WT (2002) Dendritic spine and dendritic field characteristics of layer V pyramidal neurons in the visual cortex of fragile-X knockout mice. Am J Med Genet 111:140–146
Irwin SA, Christmon CA, Grossman AW, Galvez R, Kim SH, DeGrush BJ, Weiler IJ, Greenough WT (2005) Fragile X mental retardation protein levels increase following complex environment exposure in rat brain regions undergoing active synaptogenesis. Neurobiol Learn Mem 83:180–187
Ishak WW, Kahloon M, Fakhry H (2011) Oxytocin role in enhancing well-being: a literature review. J Affect Disord 130:1–9
Jacob S, Brune CW, Carter CS, Leventhal BL, Lord C, Cook EH Jr (2007) Association of the oxytocin receptor gene (OXTR) in Caucasian children and adolescents with autism. Neurosci Lett 417:6–9
Jansen LM, Gispen-de Wied CC, Wiegant VM, Westenberg HG, Lahuis BE, van Engeland H (2006) Autonomic and neuroendocrine responses to a psychosocial stressor in adults with autistic spectrum disorder. J Autism Dev Disord 36:891–899
Jarvinen-Pasley A, Bellugi U, Reilly J, Mills DL, Galaburda A, Reiss AL, Korenberg JR (2008) Defining the social phenotype in Williams syndrome: a model for linking gene, the brain, and behavior. Dev Psychopathol 20:1–35
Jarvinen-Pasley A, Adolphs R, Yam A, Hill KJ, Grichanik M, Reilly J, Mills D, Reiss AL, Korenberg JR, Bellugi U (2010) Affiliative behavior in Williams syndrome: social perception and real-life social behavior. Neuropsychologia 48:2110–2119
Jarvinen A, Korenberg JR, Bellugi U (2013) The social phenotype of Williams syndrome. Curr Opin Neurobiol 23:414–422
Jaselskis CA, Cook EH Jr, Fletcher KE, Leventhal BL (1992) Clonidine treatment of hyperactive and impulsive children with autistic disorder. J Clin Psychopharmacol 12:322–327
Jin D, Liu HX, Hirai H, Torashima T, Nagai T, Lopatina O, Shnayder NA, Yamada K, Noda M, Seike T, Fujita K, Takasawa S, Yokoyama S, Koizumi K, Shiraishi Y, Tanaka S, Hashii M, Yoshihara T, Higashida K, Islam MS, Yamada N, Hayashi K, Noguchi N, Kato I, Okamoto H, Matsushima A, Salmina A, Munesue T, Shimizu N, Mochida S, Asano M, Higashida H (2007) CD38 is critical for social behaviour by regulating oxytocin secretion. Nature 446:41–45
Kanner L (1943) Autistic disturbances of affective contact. Nerv Child 2:217–250
Kenkel WM, Paredes J, Yee JR, Pournajafi-Nazarloo H, Bales KL, Carter CS (2012) Neuroendocrine and behavioural responses to exposure to an infant in male prairie voles. J Neuroendocrinol 24:874–886
Khandjian EW, Corbin F, Woerly S, Rousseau F (1996) The fragile X mental retardation protein is associated with ribosomes. Nat Genet 12:91–93
Kirsch P, Esslinger C, Chen Q, Mier D, Lis S, Siddhanti S, Gruppe H, Mattay VS, Gallhofer B, Meyer-Lindenberg A (2005) Oxytocin modulates neural circuitry for social cognition and fear in humans. J Neurosci 25:11489–11493
Korenberg JR, Chen XN, Hirota H, Lai Z, Bellugi U, Burian D, Roe B, Matsuoka R (2000) VI. Genome structure and cognitive map of Williams syndrome. J Cogn Neurosci 12(Suppl 1):89–107
Kosfeld M, Heinrichs M, Zak PJ, Fischbacher U, Fehr E (2005) Oxytocin increases trust in humans. Nature 435:673–676
Kuperman S, Beeghly JH, Burns TL, Tsai LY (1985) Serotonin relationships of autistic probands and their first-degree relatives. J Am Acad Child Psychiatry 24:186–190
Kwon H, Menon V, Eliez S, Warsofsky IS, White CD, Dyer-Friedman J, Taylor AK, Glover GH, Reiss AL (2001) Functional neuroanatomy of visuospatial working memory in fragile X syndrome: relation to behavioral and molecular measures. Am J Psychiatry 158:1040–1051
Landgraf R, Wigger A (2003) Born to be anxious: neuroendocrine and genetic correlates of trait anxiety in HAB rats. Stress 6:111–119
Larson J, Jessen RE, Kim D, Fine AK, du Hoffmann J (2005) Age-dependent and selective impairment of long-term potentiation in the anterior piriform cortex of mice lacking the fragile X mental retardation protein. J Neurosci 25:9460–9469
Lavi-Itzkovitz A, Tcherpakov M, Levy Z, Itzkovitz S, Muscatelli F, Fainzilber M (2012) Functional consequences of necdin nucleocytoplasmic localization. PLoS One 7:e33786
Leboyer M, Philippe A, Bouvard M, Guilloud-Bataille M, Bondoux D, Tabuteau F, Feingold J, Mouren-Simeoni MC, Launay JM (1999) Whole blood serotonin and plasma beta-endorphin in autistic probands and their first-degree relatives. Biol Psychiatry 45:158–163
Lerer E, Levi S, Israel S, Yaari M, Nemanov L, Mankuta D, Nurit Y, Ebstein RP (2010) Low CD38 expression in lymphoblastoid cells and haplotypes are both associated with autism in a family-based study. Autism Res 3:293–302
Leventhal BL, Cook EH Jr, Morford M, Ravitz A, Freedman DX (1990) Relationships of whole blood serotonin and plasma norepinephrine within families. J Autism Dev Disord 20:499–511
Li J, Pelletier MR, Perez Velazquez JL, Carlen PL (2002) Reduced cortical synaptic plasticity and GluR1 expression associated with fragile X mental retardation protein deficiency. Mol Cell Neurosci 19:138–151
Lim MM, Wang Z, Olazabal DE, Ren X, Terwilliger EF, Young LJ (2004) Enhanced partner preference in a promiscuous species by manipulating the expression of a single gene. Nature 429:754–757
Lord C, Rutter M, Le Couteur A (1994) Autism Diagnostic Interview-Revised: a revised version of a diagnostic interview for caregivers of individuals with possible pervasive developmental disorders. J Autism Dev Disord 24:659–685
Lord C, Risi S, Lambrecht L, Cook EH Jr, Leventhal BL, DiLavore PC, Pickles A, Rutter M (2000) The autism diagnostic observation schedule-generic: a standard measure of social and communication deficits associated with the spectrum of autism. J Autism Dev Disord 30:205–223
Lowery MC, Morris CA, Ewart A, Brothman LJ, Zhu XL, Leonard CO, Carey JC, Keating M, Brothman AR (1995) Strong correlation of elastin deletions, detected by FISH, with Williams syndrome: evaluation of 235 patients. Am J Hum Genet 57:49–53
Macdonald K, Feifel D (2013) Helping oxytocin deliver: considerations in the development of oxytocin-based therapeutics for brain disorders. Front Neurosci 7:35
Macdonald K, Macdonald TM (2010) The peptide that binds: a systematic review of oxytocin and its prosocial effects in humans. Harv Rev Psychiatry 18:1–21
Maes B, Fryns JP, Van Walleghem M, Van den Berghe H (1994) Cognitive functioning and information processing of adult mentally retarded men with fragile-X syndrome. Am J Med Genet 50:190–200
Martin A, State M, Anderson GM, Kaye WM, Hanchett JM, McConaha CW, North WG, Leckman JF (1998) Cerebrospinal fluid levels of oxytocin in Prader-Willi syndrome: a preliminary report. Biol Psychiatry 44:1349–1352
McCullough, ME, Churchland, PS, Mendez, AJ (2013). Problems with measuring peripheral oxytocin: can the data on oxytocin and human behavior be trusted? Neurosci Biobehav Rev 37:1485–1492
McLennan Y, Polussa J, Tassone F, Hagerman R (2011) Fragile X syndrome. Curr Genomics 12:216–224
Mervis CB, Morris CA, Klein-Tasman BP, Bertrand J, Kwitny S, Appelbaum LG, Rice CE (2003) Attentional characteristics of infants and toddlers with Williams syndrome during triadic interactions. Dev Neuropsychol 23:243–268
Miller M, Bales KL, Taylor SL, Yoon J, Hostetler CM, Carter CS, Solomon M (2013) Oxytocin and vasopressin in children and adolescents with autism spectrum disorders: sex differences and associations with symptoms. Autism Res 6:91–102
Mineur YS, Sluyter F, de Wit S, Oostra BA, Crusio WE (2002) Behavioral and neuroanatomical characterization of the Fmr1 knockout mouse. Hippocampus 12:39–46
Mineur YS, Huynh LX, Crusio WE (2006) Social behavior deficits in the Fmr1 mutant mouse. Behavioural Brain Research 168:172–175
Modahl C, Green L, Fein D, Morris M, Waterhouse L, Feinstein C, Levin H (1998) Plasma oxytocin levels in autistic children. Biol Psychiatry 43:270–277
Morris CA (1993) Williams syndrome. In: Pagon RA, Adam MP, Bird TD, Dolan CR, Fong CT, Stephens K (eds) Gene reviews. University of Washington-Seattle, Seattle
Morton JE, Bundey S, Webb TP, MacDonald F, Rindl PM, Bullock S (1997) Fragile X syndrome is less common than previously estimated. J Med Genet 34:1–5
Moy SS, Nadler JJ, Young NB, Nonneman RJ, Grossman AW, Murphy DL, D’Ercole AJ, Crawley JN, Magnuson TR, Lauder JM (2009) Social approach in genetically engineered mouse lines relevant to autism. Genes Brain Behav 8:129–142
Munesue T, Yokoyama S, Nakamura K, Anitha A, Yamada K, Hayashi K, Asaka T, Liu HX, Jin D, Koizumi K, Islam MS, Huang JJ, Ma WJ, Kim UH, Kim SJ, Park K, Kim D, Kikuchi M, Ono Y, Nakatani H, Suda S, Miyachi T, Hirai H, Salmina A, Pichugina YA, Soumarokov AA, Takei N, Mori N, Tsujii M, Sugiyama T, Yagi K, Yamagishi M, Sasaki T, Yamasue H, Kato N, Hashimoto R, Taniike M, Hayashi Y, Hamada J, Suzuki S, Ooi A, Noda M, Kamiyama Y, Kido MA, Lopatina O, Hashii M, Amina S, Malavasi F, Huang EJ, Zhang J, Shimizu N, Yoshikawa T, Matsushima A, Minabe Y, Higashida H (2010) Two genetic variants of CD38 in subjects with autism spectrum disorder and controls. Neurosci Res 67:181–191
Muscatelli F, Abrous DN, Massacrier A, Boccaccio I, Le Moal M, Cau P, Cremer H (2000) Disruption of the mouse Necdin gene results in hypothalamic and behavioral alterations reminiscent of the human Prader-Willi syndrome. Hum Mol Genet 9:3101–3110
Neumann ID, Landgraf R (2012) Balance of brain oxytocin and vasopressin: implications for anxiety, depression, and social behaviors. Trends Neurosci 35:649–659
Nielsen DM, Derber WJ, McClellan DA, Crnic LS (2002) Alterations in the auditory startle response in Fmr1 targeted mutant mouse models of fragile X syndrome. Brain Res 927:8–17
O’Donnell WT, Warren ST (2002) A decade of molecular studies of fragile X syndrome. Annu Rev Neurosci 25:315–338
Owley T, Brune CW, Salt J, Walton L, Guter S, Ayuyao N, Gibbons RD, Leventhal BL, Cook EH (2010) A pharmacogenetic study of escitalopram in autism spectrum disorders. Autism Res 3:1–7
Paradee W, Melikian HE, Rasmussen DL, Kenneson A, Conn PJ, Warren ST (1999) Fragile X mouse: strain effects of knockout phenotype and evidence suggesting deficient amygdala function. Neuroscience 94:185–192
Paxinos G, Franklin KBJ (2004) The mouse brain in stereotaxic coordinates, compact 2nd edn. Elsevier Academic Press, Amsterdam
Peier AM, McIlwain KL, Kenneson A, Warren ST, Paylor R, Nelson DL (2000) (Over)correction of FMR1 deficiency with YAC transgenics: behavioral and physical features. Hum Mol Genet 9:1145–1159
Petrovic P, Kalisch R, Singer T, Dolan RJ (2008) Oxytocin attenuates affective evaluations of conditioned faces and amygdala activity. J Neurosci 28:6607–6615
Pietropaolo S, Guilleminot A, Martin B, D’Amato FR, Crusio WE (2011) Genetic-Background Modulation of Core and Variable Autistic-Like Symptoms in Fmr1 Knock-Out Mice. PloS One 6:e17073
Plesa-Skwerer D, Faja S, Schofield C, Verbalis A, Tager-Flusberg H (2006) Perceiving facial and vocal expressions of emotion in individuals with Williams syndrome. Am J Ment Retard 111:15–26
Pretorius PH, King MA, Pan TS, de Vries DJ, Glick SJ, Byrne CL (1998) Reducing the influence of the partial volume effect on SPECT activity quantitation with 3D modelling of spatial resolution in iterative reconstruction. Phys Med Biol 43:407–420
Proulx E, Young EJ, Osborne LR, Lambe EK (2010) Enhanced prefrontal serotonin 5-HT(1A) currents in a mouse model of Williams-Beuren syndrome with low innate anxiety. J Neurodev Disord 2:99–108
Reiss AL, Abrams MT, Greenlaw R, Freund L, Denckla MB (1995) Neurodevelopmental effects of the FMR-1 full mutation in humans. Nat Med 1:159–167
Riby D, Hancock PJ (2009) Looking at movies and cartoons: eye-tracking evidence from Williams syndrome and autism. J Intellect Disabil Res 53:169–181
Rimmele U, Hediger K, Heinrichs M, Klaver P (2009) Oxytocin makes a face in memory familiar. J Neurosci 29:38–42
Rodgers J, Riby DM, Janes E, Connolly B, McConachie H (2012) Anxiety and repetitive behaviours in autism spectrum disorders and Williams syndrome: a cross-syndrome comparison. J Autism Dev Disord 42:175–180
Rudelli RD, Brown WT, Wisniewski K, Jenkins EC, Laure-Kamionowska M, Connell F, Wisniewski HM (1985) Adult fragile X syndrome. Clinico-neuropathologic findings. Acta neuropathologica 67:289–295
Saitoh S, Buiting K, Cassidy SB, Conroy JM, Driscoll DJ, Gabriel JM, Gillessen-Kaesbach G, Glenn CC, Greenswag LR, Horsthemke B, Kondo I, Kuwajima K, Niikawa N, Rogan PK, Schwartz S, Seip J, Williams CA, Nicholls RD (1997) Clinical spectrum and molecular diagnosis of Angelman and Prader-Willi syndrome patients with an imprinting mutation. Am J Med Genet 68:195–206
Sakurai T, Dorr NP, Takahashi N, McInnes LA, Elder GA, Buxbaum JD (2011) Haploinsufficiency of Gtf2i, a gene deleted in Williams Syndrome, leads to increases in social interactions. Autism Res 4:28–39
Sala M, Braida D, Lentini D, Busnelli M, Bulgheroni E, Capurro V, Finardi A, Donzelli A, Pattini L, Rubino T, Parolaro D, Nishimori K, Parenti M, Chini B (2011) Pharmacologic rescue of impaired cognitive flexibility, social deficits, increased aggression, and seizure susceptibility in oxytocin receptor null mice: a neurobehavioral model of autism. Biol Psychiatry 69:875–882
Sanders SJ, Ercan-Sencicek AG, Hus V, Luo R, Murtha MT, Moreno-De-Luca D, Chu SH, Moreau MP, Gupta AR, Thomson SA, Mason CE, Bilguvar K, Celestino-Soper PB, Choi M, Crawford EL, Davis L, Wright NR, Dhodapkar RM, DiCola M, DiLullo NM, Fernandez TV, Fielding-Singh V, Fishman DO, Frahm S, Garagaloyan R, Goh GS, Kammela S, Klei L, Lowe JK, Lund SC, McGrew AD, Meyer KA, Moffat WJ, Murdoch JD, O’Roak BJ, Ober GT, Pottenger RS, Raubeson MJ, Song Y, Wang Q, Yaspan BL, Yu TW, Yurkiewicz IR, Beaudet AL, Cantor RM, Curland M, Grice DE, Gunel M, Lifton RP, Mane SM, Martin DM, Shaw CA, Sheldon M, Tischfield JA, Walsh CA, Morrow EM, Ledbetter DH, Fombonne E, Lord C, Martin CL, Brooks AI, Sutcliffe JS, Cook EH Jr, Geschwind D, Roeder K, Devlin B, State MW (2011) Multiple recurrent de novo CNVs, including duplications of the 7q11.23 Williams syndrome region, are strongly associated with autism. Neuron 70:863–885
Sauer C, Montag C, Worner C, Kirsch P, Reuter M (2012) Effects of a common variant in the CD38 gene on social processing in an oxytocin challenge study: possible links to autism. Neuropsychopharmacology 37:1474–1482
Sawchenko PE, Swanson LW (1985) Localization, colocalization, and plasticity of corticotropin-releasing factor immunoreactivity in rat brain. Fed Proc 44:221–227
Schain RJ, Freedman DX (1961) Studies on 5-hydroxyindole metabolism in autistic and other mentally retarded children. J Pediatr 58:315–320
Schapiro MB, Murphy DG, Hagerman RJ, Azari NP, Alexander GE, Miezejeski CM, Hinton VJ, Horwitz B, Haxby JV, Kumar A, White B, Grady CL (1995) Adult fragile X syndrome: neuropsychology, brain anatomy, and metabolism. Am J Med Genet 60:480–493
Schneiderman I, Zagoory-Sharon O, Leckman JF, Feldman R (2012) Oxytocin during the initial stages of romantic attachment: relations to couples’ interactive reciprocity. Psychoneuroendocrinology 37:1277–1285
Schradin C, Kenkel W, Krackow S, Carter CS (2013) Staying put or leaving home: endocrine, neuroendocrine and behavioral consequences in male African striped mice. Horm Behav 63:136–143
Schubert C (2009) The genomic basis of the Williams-Beuren syndrome. Cell Mol Life Sci 66:1178–1197
Seltzer LJ, Ziegler TE, Pollak SD (2010) Social vocalizations can release oxytocin in humans. Proc Biol Sci 277:2661–2666
Shamay-Tsoory SG, Fischer M, Dvash J, Harari H, Perach-Bloom N, Levkovitz Y (2009) Intranasal administration of oxytocin increases envy and schadenfreude (gloating). Biol Psychiatry 66:864–870
Silverman JL, Smith DG, Rizzo SJ, Karras MN, Turner SM, Tolu SS, Bryce DK, Smith DL, Fonseca K, Ring RH, Crawley JN (2012) Negative allosteric modulation of the mGluR5 receptor reduces repetitive behaviors and rescues social deficits in mouse models of autism. Sci Trans Med 4:131ra151
Siomi H, Siomi MC, Nussbaum RL, Dreyfuss G (1993) The protein product of the fragile X gene, FMR1, has characteristics of an RNA-binding protein. Cell 74:291–298
Siomi H, Choi M, Siomi MC, Nussbaum RL, Dreyfuss G (1994) Essential role for KH domains in RNA binding: impaired RNA binding by a mutation in the KH domain of FMR1 that causes fragile X syndrome. Cell 77:33–39
Smith R (2004) Moving molecules: mRNA trafficking in Mammalian oligodendrocytes and neurons. Neuroscientist 10:495–500
Spencer CM, Alekseyenko O, Serysheva E, Yuva-Paylor LA, Paylor R (2005) Altered anxiety-related and social behaviors in the Fmr1 knockout mouse model of fragile X syndrome. Genes Brain Behav 4:420–430
Swaab DF, Purba JS, Hofman MA (1995) Alterations in the hypothalamic paraventricular nucleus and its oxytocin neurons (putative satiety cells) in Prader-Willi syndrome: a study of five cases. J Clin Endocrinol Metab 80:573–579
Szeto, A, McCabe, PM, Nationmm DA, Tabak, BA, Rossetti, MA, McCullough, ME, Schneiderman, N, Mendez, AJ. (2011) Evaluation of enzyme immunoassay and radioimmunoassay methods for the measurement of plasma oxytocin. Psychosom Med 73:393–400
Takayanagi Y, Yoshida M, Bielsky IF, Ross HE, Kawamata M, Onaka T, Yanagisawa T, Kimura T, Matzuk MM, Young LJ, Nishimori K (2005) Pervasive social deficits, but normal parturition, in oxytocin receptor-deficient mice. Proc Natl Acad Sci U S A 102:16096–16101
Tassone F, Hagerman RJ, Chamberlain WD, Hagerman PJ (2000a) Transcription of the FMR1 gene in individuals with fragile X syndrome. Am J Med Genet 97:195–203
Tassone F, Hagerman RJ, Taylor AK, Gane LW, Godfrey TE, Hagerman PJ (2000b) Elevated levels of FMR1 mRNA in carrier males: a new mechanism of involvement in the fragile-X syndrome. Am J Hum Genet 66:6–15
Tauber M, Mantoulan C, Copet P, Jauregui J, Demeer G, Diene G, Roge B, Laurier V, Ehlinger V, Arnaud C, Molinas C, Thuilleaux D (2011) Oxytocin may be useful to increase trust in others and decrease disruptive behaviours in patients with Prader-Willi syndrome: a randomised placebo-controlled trial in 24 patients. Orphanet J Rare Dis 6:47
The Dutch-Belgian Fragile-X Consortium (1994) Fmr1 knockout mice: a model to study fragile X mental retardation. The Dutch-Belgian Fragile X Consortium. Cell 78:23–33
Tiedge H, Zhou A, Thorn NA, Brosius J (1993) Transport of BC1 RNA in hypothalamo-neurohypophyseal axons. J Neurosci 13:4214–4219
Todd PK, Mack KJ (2000) Sensory stimulation increases cortical expression of the fragile X mental retardation protein in vivo. Brain Res Mol Brain Res 80:17–25
Todd PK, Mack KJ, Malter JS (2003a) The fragile X mental retardation protein is required for type-I metabotropic glutamate receptor-dependent translation of PSD-95. Proc Natl Acad Sci U S A 100:14374–14378
Todd PK, Malter JS, Mack KJ (2003b) Whisker stimulation-dependent translation of FMRP in the barrel cortex requires activation of type I metabotropic glutamate receptors. Brain Res Mol Brain Res 110:267–278
Tsai TF, Jiang YH, Bressler J, Armstrong D, Beaudet AL (1999) Paternal deletion from Snrpn to Ube3a in the mouse causes hypotonia, growth retardation and partial lethality and provides evidence for a gene contributing to Prader-Willi syndrome. Hum Mol Genet 8:1357–1364
Turner G, Webb T, Wake S, Robinson H (1996) Prevalence of fragile X syndrome. Am J Med Genet 64:196–197
Verheij C, Bakker CE, de Graaff E, Keulemans J, Willemsen R, Verkerk AJ, Galjaard H, Reuser AJ, Hoogeveen AT, Oostra BA (1993) Characterization and localization of the FMR-1 gene product associated with fragile X syndrome. Nature 363:722–724
Vorstman JA, Staal WG, van Daalen E, van Engeland H, Hochstenbach PF, Franke L (2006) Identification of novel autism candidate regions through analysis of reported cytogenetic abnormalities associated with autism. Mol Psychiatry 11:1, 18–28
Wang LW, Berry-Kravis E, Hagerman RJ (2010) Fragile X: leading the way for targeted treatments in autism. Neurotherapeutics 7:264–274
Waterhouse L, Fein D, Modahl C (1996) Neurofunctional mechanisms in autism. Psychol Rev 103:457–489
Webb S (2010) Drugmakers dance with autism. Nat Biotechnol 28:772–774
Weiler IJ, Irwin SA, Klintsova AY, Spencer CM, Brazelton AD, Miyashiro K, Comery TA, Patel B, Eberwine J, Greenough WT (1997) Fragile X mental retardation protein is translated near synapses in response to neurotransmitter activation. Proc Natl Acad Sci U S A 94:5395–5400
Weiler IJ, Spangler CC, Klintsova AY, Grossman AW, Kim SH, Bertaina-Anglade V, Khaliq H, de Vries FE, Lambers FA, Hatia F, Base CK, Greenough WT (2004) Fragile X mental retardation protein is necessary for neurotransmitter-activated protein translation at synapses. Proc Natl Acad Sci U S A 101:17504–17509
Whitman BY, Accardo P (1987) Emotional symptoms in Prader-Willi syndrome adolescents. Am J Med Genet 28:897–905
Whittington J, Holland T (2011) Recognition of emotion in facial expression by people with Prader-Willi syndrome. J Intellect Disabil Res 55:75–84
Williams JC, Barratt-Boyes BG, Lowe JB (1961) Supravalvular aortic stenosis. Circulation 24:1311–1318
Wilson BM, Cox CL (2007) Absence of metabotropic glutamate receptor-mediated plasticity in the neocortex of fragile X mice. Proc Natl Acad Sci U S A 104:2454–2459
Winslow JT, Insel TR (2004) Neuroendocrine basis of social recognition. Curr Opin Neurobiol 14:248–253
Wismer Fries AB, Ziegler TE, Kurian JR, Jacoris S, Pollak SD (2005) Early experience in humans is associated with changes in neuropeptides critical for regulating social behavior. Proc Natl Acad Sci U S A 102:17237–17240
Wisniewski KE, Segan SM, Miezejeski CM, Sersen EA, Rudelli RD (1991) The Fra(X) syndrome: neurological, electrophysiological, and neuropathological abnormalities. Am J Med Genet 38:476–480
Witt DM, Insel TR (1992) Central oxytocin antagonism decreases female reproductive behavior. Ann N Y Acad Sci 652:445–447
Witt DM, Winslow JT, Insel TR (1992) Enhanced social interactions in rats following chronic, centrally infused oxytocin. Pharmacol Biochem Behav 43:855–861
Wohrle D, Kotzot D, Hirst MC, Manca A, Korn B, Schmidt A, Barbi G, Rott HD, Poustka A, Davies KE et al (1992) A microdeletion of less than 250 kb, including the proximal part of the FMR-I gene and the fragile-X site, in a male with the clinical phenotype of fragile-X syndrome. Am J Hum Genet 51:299–306
Wu S, Jia M, Ruan Y, Liu J, Guo Y, Shuang M, Gong X, Zhang Y, Yang X, Zhang D (2005) Positive association of the oxytocin receptor gene (OXTR) with autism in the Chinese Han population. Biol Psychiatry 58:74–77
Yamamoto Y, Cushing BS, Kramer KM, Epperson PD, Hoffman GE, Carter CS (2004) Neonatal manipulations of oxytocin alter expression of oxytocin and vasopressin immunoreactive cells in the paraventricular nucleus of the hypothalamus in a gender-specific manner. Neuroscience 125:947–955
Yan QJ, Asafo-Adjei PK, Arnold HM, Brown RE, Bauchwitz RP (2004) A phenotypic and molecular characterization of the fmr1-tm1Cgr fragile X mouse. Genes Brain Behav 3:337–359
Yrigollen CM, Han SS, Kochetkova A, Babitz T, Chang JT, Volkmar FR, Leckman JF, Grigorenko EL (2008) Genes controlling affiliative behavior as candidate genes for autism. Biol Psychiatry 63:911–916
Zhao MG, Toyoda H, Ko SW, Ding HK, Wu LJ, Zhuo M (2005) Deficits in trace fear memory and long-term potentiation in a mouse model for fragile X syndrome. J Neurosci 25:7385–7392
Acknowledgements
This work was supported in part by NIH K23MH082121 (SJ). The authors would also like to thank Jeanine Leary and Jennifer Speak for their assistance in preparing the text.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2015 Springer Science+Business Media New York
About this chapter
Cite this chapter
Levin-Decanini, T., Francis, S., Sagar, A., Liu, W., Carter, C., Jacob, S. (2015). Oxytocin and Vasopressin in Autism and Genetic Syndromes. In: Fatemi, S. (eds) The Molecular Basis of Autism. Contemporary Clinical Neuroscience. Springer, New York, NY. https://doi.org/10.1007/978-1-4939-2190-4_14
Download citation
DOI: https://doi.org/10.1007/978-1-4939-2190-4_14
Published:
Publisher Name: Springer, New York, NY
Print ISBN: 978-1-4939-2189-8
Online ISBN: 978-1-4939-2190-4
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)