
Abstract
Complete spinal cord injury, characterized by complete loss of motor, sensory, and autonomous function below the level of injury, requires robust therapeutic approaches to replace damaged neural tissue, including astroglia, oligodendroglia, and neurons. Defined stem and progenitor cell populations, from virtually any developmental stage, can differentiate into glia and neurons, thereby having the capacity to replace degenerated spinal cord tissue in a phenotypically appropriate fashion. Numerous preclinical in vivo studies in various spinal cord injury models have demonstrated that stem cell transplantation strategies can indeed promote morphological, and in some cases functional, recovery, via different mechanisms, including remyelination, axonal growth, and regeneration or neuronal replacement. To date, two neural stem cell-based transplantation strategies have moved to phase I clinical trials. This review provides an overview on the current status of neural stem cell transplantation in spinal cord injury and discusses future perspectives in the field.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others

Keywords
- Neuroregeneration
- Spinal cord injury
- Stem cells
- Transplantation
- Functional recovery
- Remyelination
- Axon growth
- Neuron replacement
Introduction
Approximately 12,000 new spinal cord injury (SCI) cases are reported in the USA each year (https://www.nscisc.uab.edu/PublicDocuments/fact_figures_docs/Facts%202012%20Feb%20Final.pdf). The sudden impairment/loss of motor, sensory, and autonomic functions turns the life of mostly young patients instantly upside down. To date, effective restorative therapies that target molecular and cellular mechanisms known to prevent structural and functional recovery are not available.
Most spinal cord injuries are induced by blunt trauma with subsequent compression of the spinal cord, leading to the disruption of axonal tracts. The loss of spinal cord parenchyma, including astroglia, oligodendroglia, and neurons, as well as microglial activation, macrophage and leukocyte infiltration, and demyelination of preserved axons, represents key factors contributing to the poor intrinsic regenerative capacity of the adult spinal cord. Moreover, growth-inhibitory factors, such as myelin-based inhibitors [i.e., Nogo, MAG (myelin-associated protein), and OMgp (oligodendrocyte myelin glycoprotein)], inhibitory extracellular matrix components [i.e., CSPG (chondroitin sulfate proteoglycans)], diffusible chemorepellents [i.e., semaphorins, ephrins, and netrin (Busch and Silver 2007; Giger et al. 2010)], and the lack of growth-promoting molecules [i.e., neurotrophic factors], represent additional components preventing recovery from SCI (Lu and Tuszynski 2008; Thuret et al. 2006).
To achieve improvement in the most severe complete SCI cases, cell replacement strategies are likely needed to compensate for the loss of spinal cord parenchyma at the site of injury, as well as remote regions, with the ultimate goal to induce functional recovery in spinal cord-injured individuals (Barnabe-Heider and Frisen 2008; Hulsebosch 2002; Collins 1983). Solely enhancing the intrinsic capacity for cell replacement seems insufficient to promote beneficial structural repair after a devastating trauma with substantial tissue damage.
Ideally, grafted cells should serve as a substrate for axonal growth, axonal remyelination, and replacement of lost neurons or as neuronal relays to reconnect damaged axons and secrete growth factors to promote axonal outgrowth/remyelination. Studies have identified numerous cell types that meet some or all of these objectives. The most attractive cell types can differentiate into neural phenotypes of the adult spinal cord in a phenotypically appropriate fashion. Olfactory ensheathing cells and Schwann cells derived from the peripheral nervous system (PNS) and the transition zone between the PNS and central nervous system (CNS), respectively, cannot replace lost cells in a phenotypically appropriate fashion. These cells do not reside in the CNS (Field et al. 2003; Ramon-Cueto and Avila 1998), yet they can elicit substantial axonal regrowth and myelination in the injured adult CNS (Akiyama et al. 2004; Blakemore and Crang 1985; Honmou et al. 1996; Imaizumi et al. 2000; Kohama et al. 2001; Weidner et al. 1999; Li et al. 1997; Lu et al. 2002; Ramon-Cueto et al. 2000; Sasaki et al. 2004; Steward et al. 2006; Takami et al. 2002; Lu et al. 2006). Other cell types, such as fibroblasts, which are not cellular components of the CNS, cannot mimic glial or neuronal function and can only serve as cellular substrates for axonal growth and/or vehicles for the local delivery of growth-promoting factors after genetic modification (Grill et al. 1997; Tuszynski et al. 2003; Jin et al. 2002). The present review will focus on neural stem/progenitor cells, which have the potential to differentiate into astroglia, oligodendroglia, and neurons and are thus theoretically able to reconstitute the original CNS tissue composition as close as possible.
Stem Cell Nomenclature
The developmental stage, at which cells are isolated, defines stem cells as embryonic, fetal, or adult stem cells.
Embryonic Stem Cells
Embryonic stem (ES) cells are derived from the inner cell mass of the blastocyst and remain pluripotent, meaning that they retain the potential to give rise to all cell types of the three germ layers (ectoderm, endoderm, and mesoderm), with exception to extraembryonic tissues (Marcus and Woodbury 2008). The first ES cells were isolated and cultivated from mice, and a few years later, human-derived ES cells followed (Reubinoff et al. 2000; Thomson et al. 1998). ES cells have the capacity for unlimited proliferation in vitro (Evans and Kaufman 1981; Martin 1981; McDonald et al. 1999), representing a promising cell type for transplantation paradigms. However, major concerns regarding ethical issues, the necessity for immunosuppression, the risk for tumor formation, and possible chromosomal instability after long-term cultivation challenge the widespread application of this particular cell type (McDonald et al. 1999; Vats et al. 2005; Nussbaum et al. 2007).
Fetal Stem Cells
Fetal stem cells are postembryonic cells, which are multipotent. Thus, they can only differentiate into cell types within a specific lineage, e.g., neural stem cells or hematopoietic stem cells. Fetal stem cells are isolated from specific regions, similar to adult stem cells, but they lack the differentiation potential of ES cells. The proliferative capacity of fetal stem cells is still superior in comparison to adult stem cells (Rao 1999; Pojda et al. 2005; Johe et al. 1996; Wu et al. 2002a; O'Donoghue and Fisk 2004). However, the need for immunosuppression following transplantation, as well as ethical concerns, presents major drawbacks for this method.
Glial- or neuronal-restricted precursor cells (GRPs or NRPs) can be isolated from the fetal spinal cord (rat E12 through E14) and are capable of self-renewal (Rao et al. 1998; Lee et al. 2000). GRPs can be maintained in culture for a prolonged time without loosing their differentiation potential (Wu et al. 2002b; Davies et al. 2006; Rao and Mayer-Proschel 1997; Rao et al. 1998; Gregori et al. 2002). In addition, they can differentiate into two distinct astrocyte populations (type 1 and type 2 astrocytes), as well as oligodendrocytes, but not into neurons (Wu et al. 2002b; Han et al. 2004). NRPs, on the other hand, can differentiate into several neuronal phenotypes, but not into oligodendrocytes or astrocytes (Kalyani and Rao 1998; Mujtaba et al. 1999; Rao 1999).
Adult Neural Stem/Progenitor Cells
Adult neural stem/progenitor cells (NSCs) are multipotent and can be isolated from two neurogenic regions: 1) the subventricular zone of the lateral ventricles, which normally gives rise to neurons of the olfactory bulb, and 2) the subgranular zone of the hippocampal dentate gyrus. In addition, adult NSCs also exist in non-neurogenic regions of the CNS, such as the striatum, neocortex, and spinal cord (Palmer et al. 1999; Palmer et al. 1995; Yamamoto et al. 2001; Weiss et al. 1996). Besides their potential for self-renewal, adult NSCs proliferate and can be expanded in vitro in the presence of epidermal growth factor (EGF) and basic fibroblast growth factor (bFGF) (Roy et al. 2000; Wachs et al. 2003; Gritti et al. 1996). After withdrawal of growth factors and/or exposure to serum, adult NSCs differentiate into neurons, astrocytes, and oligodendrocytes in vitro (Arsenijevic et al. 2001; Gage et al. 1995; Johansson et al. 1999; Reynolds and Weiss 1992; Weiss et al. 1996; Shihabuddin et al. 1997; Cao et al. 2002).
Induced Pluripotent Stem Cells
Induced pluripotent stem cells (iPSCs) are obtained by reprogramming differentiated somatic cells to induce a pluripotent, embryonic-like state. By introducing four genes (Oct3/4, Sox2, c-Myc, and Klf4) to mouse and human somatic cells, such as fibroblasts, these cells have been reprogrammed to become iPSCs (Takahashi and Yamanaka 2006; Park et al. 2008; Takahashi et al. 2007; Yu et al. 2007; Nakagawa et al. 2008). Another method to generate iPSCs is to replace the oncogenes c-Myc and Klf4 with NANOG and LIN28 (Yu et al. 2007). Similar to ES cells, iPSCs give rise to cells of all three germ layers (Takahashi and Yamanaka 2006). In the context of potential clinical applications, iPSC isolation from patient skin allows for autologous transplantation, thus avoiding ethical concerns and immunological problems (Salewski et al. 2010; Fujimoto et al. 2012; Tsuji et al. 2011). However, the yield of iPSCs obtainable from adult skin biopsies remains rather low. Furthermore, uncontrolled proliferation of iPSC grafts is a major concern and has to be closely monitored (Yamanaka 2009; Takahashi and Yamanaka 2006; Maherali et al. 2007; Maherali and Hochedlinger 2008; Wernig et al. 2007). Due to potential epigenetic alterations, inappropriate reprogramming, and the possibility of chromosomal aberrations, iPSCs are thought to be more tumorigenic than ES cells (Ben-David and Benvenisty 2011; Wernig et al. 2008; Yamanaka 2007; Miura et al. 2009). Therefore, each iPSC line needs to be pre-evaluated to assess the risk of tumor formation, as well as to confirm their safety following cell grafting, before their use can be translated into clinical applications (Tsuji et al. 2010).
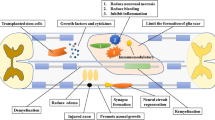
Therapeutic Targets of Neural Stem Cell Transplantation
Remyelination
Axonal demyelination and loss of nerve conduction resulting from oligodendrocyte loss contributes to functional impairment after SCI (Gledhill et al. 1973; Hulsebosch 2002; Basso et al. 1996). Post-mortem studies in spinal cord-injured individuals indicate a preserved rim of axons in motor complete SCI (Kakulas 1999; Sherwood et al. 1992). However, these spared axons are frequently demyelinated (Kakulas 1999). The intrinsic capacity for oligodendroglial replacement, and consecutive remyelination of demyelinated axons in the adult spinal cord, remains limited. The poor remyelination capacity is at least partially due to inadequate proliferation and terminal differentiation of oligodendroglial precursor cells into mature myelinating oligodendrocytes (Levine and Reynolds 1999). Therefore, approaches to enhance oligodendrocyte myelination represent a viable option for repairing the injured spinal cord.
In vitro, adult NSCs differentiate into neurons, astroglia, and oligodendroglia (Cao et al. 2002; Shihabuddin et al. 1997; Weiss et al. 1996). However, following transplantation into the injured spinal cord, adult NSCs differentiate predominantly into astroglia, with only few cells becoming oligodendroglia, while neuronal differentiation is essentially never found (Akiyama et al. 2001; Vroemen et al. 2003; Hofstetter et al. 2005; Cao et al. 2001; Pfeifer et al. 2006; Pfeifer et al. 2004; Mothe and Tator 2008; Vroemen et al. 2007; Karimi-Abdolrezaee et al. 2006).
Similar results have been reported for fetal-derived NSCs, which generate only a small proportion of cells (<10 %) that differentiate into oligodendrocytes up to 8 months post-transplantation in the injured spinal cord (Cao et al. 2001; Iwanami et al. 2005; Wu et al. 2002a). Other studies have shown that both ES cell and fetal NSC grafts contain significantly higher proportions of cells that express markers for mature oligodendrocytes (Salazar et al. 2010; Yasuda et al. 2011; Cummings et al. 2005; McDonald et al. 1999). The grafting of human brain-derived fetal NSCs into spinal cord-injured immunodeficient and myelin-deficient mice yielded up to 64 % adenomatous polyposis coli (APC)-positive oligodendrocytes, which were also associated with remyelination of axons 4 months after transplantation (Cummings et al. 2005).
Increased cell survival and oligodendroglial differentiation in vivo has been shown after infusion of growth factors (PDGF-AA, bFGF, and EGF), in combination with adult NSC grafts (Karimi-Abdolrezaee et al. 2006), grafting of ciliary neurotrophic factor (CNTF) expressing oligodendrocyte progenitor cells (OPCs) (Cao et al. 2010) or multi-neurotrophin-expressing GRPs (with BDNF and NT-3 activity) (Cao et al. 2005; Cao et al. 2010), and after grafting of neurogenin-2-expressing adult NSCs (Hofstetter et al. 2005). Survival/differentiation of graft-derived oligodendrocytes varied between 50 and 400 % in these studies. In contrast, the number of transplanted oligodendrocytes decreased, and neuronal differentiation increased following grafting of fetal NSCs in combination with valproic acid administration, a histone deacetylase inhibitor (Abematsu et al. 2010).
The pre-differentiation of NSCs into OPCs, which are already restricted toward an oligodendroglial lineage, is another method that has been utilized to increase absolute numbers of ES cell- and NSC-derived oligodendrocytes (Cao et al. 2010; Keirstead et al. 2005; Nistor et al. 2005). Depending on the maturation stage, OPC differentiation, survival, and migration differ significantly. Immature OPCs survive better after transplantation, migrate more extensively from the implantation site, and remyelinate more efficiently (Archer et al. 1997; Groves et al. 1993; Warrington et al. 1993; Zhang et al. 1999; Franklin 2002). In addition, OPCs derived from fetal or adult tissue, which have been purified by fluorescence-activated cell sorting of A2B5+/PSA-NCAM− cells, result in different characteristics. Both OPC phenotypes mediate robust myelination; however, adult-derived OPCs myelinate more rapidly, generate a higher number of oligodendrocytes, and ensheath more host axons per donor cell. In addition, adult cells migrate shorter distances compared to fetal-derived OPCs (Windrem et al. 2004).
Graft-induced remyelination is mostly visualized by toluidine-blue- or Luxol fast blue-stained semi-thin sections and electron microscopy (Abematsu et al. 2010; Cao et al. 2010; Cummings et al. 2005; Keirstead et al. 2005; Yasuda et al. 2011; Zhang et al. 2007). The ratio between myelin sheath thickness and axon diameter can be used to discriminate remyelination by Schwann cells or oligodendrocytes from preserved myelin. Remyelinated axons typically exhibit thin myelin sheaths relative to axon diameter (Keirstead et al. 2005). These measurements indicate that oligodendrocyte-mediated remyelination, rather than prevention of oligodendroglial cell death, accounts for the observed behavioral recovery. Remyelination by grafted human fetal NSC-derived oligodendroglia has also been verified in shiverer mice, where engrafted human cells were selectively ablated with diphtheria toxin, resulting in secondary demyelination (Cummings et al. 2005).
In terms of locomotor recovery, minimal improvement of around 2 points on the Basso, Beattie, & Bresnahan (BBB) motor recovery scale (range from 0 to 21; 0 no observable movement vs. 21 normal locomotion) (Basso et al. 1996) was the most common finding in studies examining remyelination (Cao et al. 2005; Hofstetter et al. 2005; Karimi-Abdolrezaee et al. 2006; Keirstead et al. 2005; McDonald et al. 1999; Mitsui et al. 2005; Zhang et al. 2007). Two studies grafting human ES cell-derived OPCs and CNTF-overexpressing rat OPCs, respectively, reported an average improvement of around 4 points on the BBB locomotor rating scale (Cao et al. 2010; Keirstead et al. 2005), which was paralleled by restoration of neurophysiological parameters (transcranial magnetic motor-evoked potentials, magnetic inter-enlargement reflex responses) (Cao et al. 2010). In addition to oligodendroglial differentiation/remyelination, axonal sprouting around the injury site was reported as a structural correlate of transplanting CNTF-overexpressing OPCs. Transplantation of fetal NSCs into the contused spinal cord of non-obese diabetic (NOD)/severe combined immunodeficiency (SCID) mice elicited restoration of motor-evoked potentials and improved locomotor function (2 points in the Basso Mouse Scale; BMS). In control animals receiving grafts of fetal shiverer mice-derived NSCs, only thin, poorly formed, myelin sheaths were observed as the correlate of impaired functional outcome (Yasuda et al. 2011).
Axonal Regrowth
Axonal regrowth can contribute to structural repair via two different mechanisms: (1) transected axonal endings regrow across or around the lesion and reinnervate previous target neurons distal to the lesion; (2) remaining intact axons that bypass the lesion site extend collateral sprouts, which may partially compensate for transected axons. To accomplish true axonal regeneration across the lesion, a growth-permissive substrate that fills the cystic lesion cavity needs to be introduced. Cellular grafts not only provide a growth-permissive scaffold but also further recruit endogenous growth-promoting cells, such as Schwann cells, into the lesion site. Neurotrophic factors or factors that neutralize inhibitory molecules can also promote collateral sprouting of axons. Local delivery of such growth-promoting molecules can be achieved via cell grafts, which produce relevant growth factors either naturally or following genetic modification.
Overall, few studies have reported neural stem/progenitor cell graft-mediated axonal regeneration/sprouting. Enhanced regrowth of anterogradely labeled corticospinal axons for a short distance into the graft has been shown following grafting of adult spinal cord-derived NSCs into rat cervical dorsal column transections (Pfeifer et al. 2004). Of note, grafted adult NSCs alone are not able to fill the cystic lesion site. Co-grafting with fibroblasts or bone-marrow stromal cells (Sandner et al. 2012) represents an effective strategy to maintain NSCs within the lesion site. Regenerating axons were found to be co-localized with graft-derived glial-fibrillary-acidic-protein GFAP-expressing astroglia, suggesting that NSC-derived astrocytes provide a cellular scaffold for injured axons (Pfeifer et al. 2004). Similar mechanisms have been described following transplantation of GRP-derived astroglia into the injured rat spinal cord (Davies et al. 2006). Fetal spinal cord-derived GRPs were purified through A2B5 sorting, and consecutive exposure to bone morphogenetic protein 4 (BMP4) in vitro resulted in differentiation of A2B5-negative astroglia. These GFAP-expressing astrocytes formed a permissive substrate for regenerating the ascending sensory and descending rubrospinal axons through and across the graft after unilateral lesions of the dorsal or dorsolateral funiculi. This observed axonal regeneration was paralleled by improved locomotor recovery, as assessed by grid-walk analysis. Interestingly, fetal grafts without astroglial differentiation failed to promote functional improvement.
Unmodified spinal cord-derived fetal NSCs, grafted as cell suspensions into rat spinal cord dorsal column crush lesions, were not able to fill the cystic lesion site and failed to promote corticospinal and sensory axonal regeneration (Webber et al. 2007). Infusion of CNTF antibody, in combination with fetal NSC-soaked Gelfoam grafted into a thoracic over-hemisection lesion in adult rats, resulted in regrowth of anterogradely labeled corticospinal axons at and caudal to the lesion site (Ishii et al. 2006). This effect was attributed to CNTF deprivation with consecutive reduction of astroglial scar formation rather than direct or indirect graft-mediated mechanisms. Limited axonal sprouting has also been reported in studies using co-grafts of fetal/neonatal cortical or hippocampal NSCs, together with olfactory ensheathing or Schwann cells (Wang et al. 2010; Wang et al. 2011), although the underlying mechanisms remain unknown. Neuroprotection, rather than graft-mediated enhancement of axonal growth, might be one mechanism resulting in increased axonal density in animals with grafts of stem/progenitor cells. Increased density of serotonergic, noradrenergic, and corticotrophin-releasing factor-positive axons caudal to the lesion site has been demonstrated following transplantation of NRPs/GRPs in rat thoracic contusion SCI. However, grafted cells were not spatially associated with these axons. Functionally, locomotor and bladder functions (reduced detrusor hyperreflexia) were improved (Mitsui et al. 2005).
Several studies reporting axonal sprouting/regeneration also described partial functional recovery (Davies et al. 2006; Mitsui et al. 2005; Wang et al. 2010; Wang et al. 2011). An impressive improvement of locomotor function (BBB scores of 17 vs. 9.1 in the control group) within 12 weeks post-injury was demonstrated after co-grafting of fetal NSCs and olfactory ensheathing cells (Wang et al. 2010). Neonatal NSCs overexpressing TrkC, co-grafted with NT-3-overexpressing Schwann cells, also induced superior locomotor function (BBB score improvement from 0.5 in control animals to 7.6 in grafted animals). Moreover, motor- and sensory-evoked potentials recovered almost to the level of uninjured animals (Wang et al. 2011). Independent investigators need to replicate these findings before firm conclusions can be drawn. Taken together, at present there is only limited evidence for robust functional recovery by neural stem cell-mediated axonal regeneration.
Neuronal Replacement
Significant degeneration of lower motor neurons is observed in cervical and lumbar spinal cord injuries, which contributes to severe and irreversible paresis in upper and lower extremities. In these cases, therapeutic efforts should focus on neuronal replacement strategies, rather than aiming for long-distance axonal regeneration to promote functional recovery. In addition, transplanted neurons might serve as “relays” across the lesion in the injured spinal cord. Transected axons may form synapses with graft-derived neurons that can extend into the distal spinal cord to connect with appropriate target neurons.
A key prerequisite for neuronal replacement is that grafted stem/progenitor cells differentiate into appropriate mature neurons. However, unmodified adult NSCs do not differentiate into neurons after grafting to the injured spinal cord (Pfeifer et al. 2004; Pfeifer et al. 2006; Vroemen et al. 2003; Mothe and Tator 2008). In contrast, in the majority of studies, fetal NSCs have been shown to differentiate into neurons after spinal cord grafting (Yan et al. 2007; Okada et al. 2005; Cummings et al. 2005; Cummings et al. 2006; Ogawa et al. 2002), as demonstrated by immunohistochemistry (beta-III-tubulin, NeuN, and defined neurotransmitter phenotypes). Some of the fetal NSC graft-derived neurons also exhibited electron microscopic evidence of synapse formation with the host neurons (Cummings et al. 2005). Only limited evidence exists that relevant integration of graft-derived neurons into the spinal or supraspinal circuitry occurs. Robust neuronal differentiation into GABAergic and cholinergic neurons was demonstrated after grafting NSCs from human fetal spinal cord into the lumbar spinal cord of nude rats. Graft-derived neurons were innervated by host axons and subsequently established synapses with host neurons (Yan et al. 2007). However, these effects were investigated in a peripheral nerve lesion model (lumbar rhizotomy and excitotoxic root lesion), not in a SCI model. E13.5 rat spinal cord-derived NRPs/GRPs differentiated into GABAergic and glutamatergic neurons after grafting into a rat spinal cord lesion model (dorsal column transection) (Bonner et al. 2010). Axonal outgrowth over a limited distance into the host spinal cord from these neurons was promoted by injection of a lentiviral vector expressing the neurotrophic factor BDNF. In a subsequent study, the same cells grafted into a dorsal column lesion at cervical level C1, followed by BDNF-expressing lentiviral injections, elicited sensory axonal regeneration into the graft, as well as graft-derived axonal outgrowth into the dorsal column nuclei along the neurotrophin gradient. Synaptic connections between sensory axons and graft-derived neurons on one side, and graft-derived neurons and dorsal column nuclei on the other side, were confirmed by electron microscopy and electrophysiological analysis. These findings provide convincing evidence for a “relay” function of stem cell grafts (Bonner et al. 2011).
Transplanted fetal spinal cord tissue into the neonatal injured spinal cord supports survival of axotomized neurons and axonal regrowth, resulting in locomotor improvement (Bregman and Reier 1986; Bregman 1987; Diener and Bregman 1998; Bernstein-Goral and Bregman 1993, 1997; Houle and Reier 1988). Fetal spinal cord grafts within the adult uninjured spinal cord were shown to promote host-derived axonal growth into the transplant. However, axons failed to grow across the graft and back into the host spinal cord (Kunkel-Bagden et al. 1992; Bregman et al. 1997; Bernstein-Goral and Bregman 1993). Based on a more recent study, which demonstrated remarkable axonal growth after grafting green-fluorescent-protein GFP-expressing rat cortical E14 NSCs into the lesioned rat motor cortex (Gaillard et al. 2007), NSCs derived from E14 GFP-transgenic rat spinal cord were transplanted subacutely into the completely transected adult rat spinal cord (Lu et al. 2012). The NSC grafts differentiated into astrocytes, oligodendrocytes, and neurons. Graft-derived axons elongated robustly from the thoracic graft site both into the white matter of the lumbar spinal cord and cervical spinal cord in a highly oriented fashion. Frequently, axon collaterals terminated in the spinal cord gray matter, and GFP-expressing axon terminals formed synapses with host dendrites, which suggested that graft-derived neurons form functional relays in the injured spinal cord. Importantly, axonal growth occurred despite the presence of numerous growth-inhibitory molecules in the injured spinal cord, underlining the importance of a proper neuron-intrinsic axonal regrowth capacity. A remarkable locomotor improvement of 5 points in the BBB locomotor scale was paralleled by partial restoration of nerve conduction across the transection site. Both locomotor and electrophysiological recoveries were abolished by spinal cord re-transection. In subsequent experiments, grafts of human fetal-derived stem cell lines achieved comparable results after thoracic spinal cord transection (Lu et al. 2012), which highlights their relevance for a potential clinical translation.
Neuronal replacement was also proposed as an underlying mechanism of functional recovery after grafting of iPSCs (Fujimoto et al. 2012; Nori et al. 2011; Tsuji et al. 2010). In these studies, murine- or human-derived neural progenitors generated from iPSCs were grafted into NOD/SCID mice at 7 to 9 days following SCI. The grafted cells survived, integrated into the host tissue, and differentiated into all three CNS lineages (neurons, astrocytes, and oligodendrocytes). The majority of these cells differentiated into neurons (50–75 %) (Fujimoto et al. 2012; Nori et al. 2011), which formed synaptic connections with host neurons. Roughly one third of the grafted iPSCs differentiated into astroglia and oligodendroglia. The exact mechanisms underlying the observed functional recovery remain to be determined.
Stem Cell Transplantation: Clinical Application
Clinical trials are mandatory to assess the risks and benefits of a cell-based therapy before such an approach can be translated into a routine clinical application. The point in time when a promising experimental therapy should be investigated in a clinical trial is frequently the subject of major controversy. On one side, expectations and hopes of patients asking for effective therapies to cure severe disabilities have to be addressed. On the other side, careful scientists ask for caution in conducting premature clinical trials. To ensure that clinical trials in SCI are justified and conducted properly, a consensus for standards for the appropriate conduct of clinical trials has been established (Tuszynski et al. 2007; Steeves et al. 2007; Lammertse et al. 2007; Fawcett et al. 2007). A number of case series/clinical trials using cell-based therapies in SCI have been conducted, which indicated different degrees of compliance with these guidelines (Hug and Weidner 2012). The majority of studies have investigated bone-marrow-derived cell grafts (Pal et al. 2009; Callera and do Nascimento 2006; Chernykh et al. 2007; Deda et al. 2008; Geffner et al. 2008; Kumar et al. 2009; Park et al. 2005; Sykova et al. 2006; Yoon et al. 2007; Saito et al. 2008). In this chapter, only clinical trials investigating NSC-based therapies will be reviewed.
In December 2010, StemCells, Inc. initiated a phase I/II clinical trial based on preclinical evidence that fetal human brain-derived stem cells can differentiate into myelinating oligodendrocytes and synapse-forming neurons mediating functional recovery in rodent models of SCI (Cummings et al. 2006; Cummings et al. 2005; Hooshmand et al. 2009; Salazar et al. 2010; Uchida et al. 2000). The trial received final approval in July 2011 and is currently recruiting patients. The aim of the trial is to assess the safety and preliminary efficacy in 12 patients with AIS (American Spinal Injury Association impairment scale) grade A–C thoracic SCI at three to 12 months post-injury. Patients receive cell suspension grafts injected directly into the SCI lesion site, followed by transient immunosuppression. Subsequently, patients will be assessed over a 1-year period to monitor safety and to measure recovery of sensation, motor, and bowel/bladder function. An additional 4-year follow-up observational study will conclude the study. As of January 2013, four patients have been enrolled. No side effects have been reported to date (Dr. Armin Curt, Zürich, personal communication).
Based on preclinical evidence (Sharp et al. 2010; Keirstead et al. 2005) that human ES cell-derived oligodendrocyte progenitor cells enhance remyelination and neuroprotection after SCI, Geron Corp. initiated a multicenter trial (Mayor 2010). In 2009, the first clinical trial to evaluate safety and efficacy (recovery of neurological function) following human ES cell transplantation in acute SCI was approved. Patients with a complete thoracic SCI (AIS A) were included. Two million human ES cell-derived oligodendrocyte progenitor cells were injected into the spinal cord lesion site within 14 days after injury, accompanied by immunosuppression with tacrolimus. Surprisingly, in November 2011, the company discontinued the clinical trial for financial reasons after five patients underwent the cell transplantation procedure. Apparently, the establishment of proper cell purification protocols, problems with cyst formation after transplantation, and slow patient recruitment caused an unexpected rise in costs. Over the course of 12 years from bench to bedside, developmental costs for this therapeutic approach were estimated at $170 million (http://www.bbc.co.uk/news/health-11517680). According to the company, the therapy did not promote any improvement, but was well tolerated with no serious adverse events. (Kaiser 2011).
Summary and Conclusions
Preclinical studies investigating neural stem cell grafting in SCI reported partial structural and functional improvement. Based on this evidence, two clinical trials investigating neural stem cell grafts in SCI subjects were initiated. Unfortunately, the first phase I/II trial investigating ES cell-derived grafting in acute SCI was terminated. Despite the disappointment related to the premature termination of this clinical trial, the experience gathered from this pioneer study will significantly facilitate planning and execution of future clinical studies of this kind.
Current concepts employing neural stem cell grafts, as glial scaffolds for regenerating axons (Pfeifer et al. 2004; Davies et al. 2006) or as relays after neuronal differentiation (Bonner et al. 2011; Lu et al. 2012), hold great promise for promoting substantial structural restoration as a prerequisite for a successful regenerative therapy in SCI. Future research efforts need to focus on accessible stem cell sources, which do not raise ethical concerns. iPSCs – once issues of tumor formation and efficient cell propagation and differentiation have been addressed – may serve as a reliable and effective stem cell-based regenerative therapy in SCI. Moreover, relevant glial or neuronal phenotypes need to be enriched and further modified in their capacity to promote structural repair. Robust restoration of spinal neural circuits in animal models of chronic severe spinal cord contusion injury will be the most valuable predictor for successful clinical trials in SCI.
References
Abematsu, M., Tsujimura, K., Yamano, M., Saito, M., Kohno, K., Kohyama, J., et al. (2010). Neurons derived from transplanted neural stem cells restore disrupted neuronal circuitry in a mouse model of spinal cord injury. Journal of Clinical Investigation, 120(9), 3255–3266. doi:10.1172/JCI42957.
Akiyama, Y., Honmou, O., Kato, T., Uede, T., Hashi, K., & Kocsis, J. D. (2001). Transplantation of clonal neural precursor cells derived from adult human brain establishes functional peripheral myelin in the rat spinal cord. Experimental Neurology, 167(1), 27–39. doi:10.1006/exnr.2000.7539. S0014-4886(00)97539-3 [pii].
Akiyama, Y., Lankford, K., Radtke, C., Greer, C. A., & Kocsis, J. D. (2004). Remyelination of spinal cord axons by olfactory ensheathing cells and Schwann cells derived from a transgenic rat expressing alkaline phosphatase marker gene. Neuron Glia Biology, 1(1), 47–55.
Archer, D. R., Cuddon, P. A., Lipsitz, D., & Duncan, I. D. (1997). Myelination of the canine central nervous system by glial cell transplantation: A model for repair of human myelin disease. Nature Medicine, 3(1), 54–59.
Arsenijevic, Y., Villemure, J. G., Brunet, J. F., Bloch, J. J., Deglon, N., Kostic, C., et al. (2001). Isolation of multipotent neural precursors residing in the cortex of the adult human brain. Experimental Neurology, 170(1), 48–62. doi:10.1006/exnr.2001.7691.
Barnabe-Heider, F., & Frisen, J. (2008). Stem cells for spinal cord repair. Cell Stem Cell, 3(1), 16–24. doi:10.1016/j.stem.2008.06.011. S1934-5909(08)00290-7 [pii].
Basso, D. M., Beattie, M. S., & Bresnahan, J. C. (1996). Graded histological and locomotor outcomes after spinal cord contusion using the NYU weight-drop device versus transection. Experimental Neurology, 139(2), 244–256. doi:10.1006/exnr.1996.0098.
Ben-David, U., & Benvenisty, N. (2011). The tumorigenicity of human embryonic and induced pluripotent stem cells. Nature Reviews. Cancer, 11(4), 268–277. doi:10.1038/nrc3034.
Bernstein-Goral, H., & Bregman, B. S. (1993). Spinal cord transplants support the regeneration of axotomized neurons after spinal cord lesions at birth: A quantitative double-labeling study. Experimental Neurology, 123(1), 118–132. doi:10.1006/exnr.1993.1145.
Bernstein-Goral, H., & Bregman, B. S. (1997). Axotomized rubrospinal neurons rescued by fetal spinal cord transplants maintain axon collaterals to rostral CNS targets. Experimental Neurology, 148(1), 13–25. doi:10.1006/exnr.1997.6640.
Blakemore, W. F., & Crang, A. J. (1985). The use of cultured autologous Schwann cells to remyelinate areas of persistent demyelination in the central nervous system. Journal of Neurological Sciences, 70(2), 207–223.
Bonner, J. F., Blesch, A., Neuhuber, B., & Fischer, I. (2010). Promoting directional axon growth from neural progenitors grafted into the injured spinal cord. Journal of Neuroscience Research, 88(6), 1182–1192. doi:10.1002/jnr.22288.
Bonner, J. F., Connors, T. M., Silverman, W. F., Kowalski, D. P., Lemay, M. A., & Fischer, I. (2011). Grafted neural progenitors integrate and restore synaptic connectivity across the injured spinal cord. Journal of Neuroscience, 31(12), 4675–4686. doi:10.1523/JNEUROSCI.4130-10.2011.
Bregman, B. S. (1987). Spinal cord transplants permit the growth of serotonergic axons across the site of neonatal spinal cord transection. Brain Research, 431(2), 265–279.
Bregman, B. S., Diener, P. S., McAtee, M., Dai, H. N., & James, C. (1997). Intervention strategies to enhance anatomical plasticity and recovery of function after spinal cord injury. Advances in Neurology, 72, 257–275.
Bregman, B. S., & Reier, P. J. (1986). Neural tissue transplants rescue axotomized rubrospinal cells from retrograde death. Journal of Comparative Neurology, 244(1), 86–95. doi:10.1002/cne.902440107.
Busch, S. A., & Silver, J. (2007). The role of extracellular matrix in CNS regeneration. Current Opinion in Neurobiology, 17(1), 120–127. doi:10.1016/j.conb.2006.09.004. S0959-4388(07)00002-5 [pii].
Callera, F., & do Nascimento, R. X. (2006). Delivery of autologous bone marrow precursor cells into the spinal cord via lumbar puncture technique in patients with spinal cord injury: A preliminary safety study. Experimental Hematology, 34(2), 130–131. doi:10.1016/j.exphem.2005.11.006.
Cao, Q., He, Q., Wang, Y., Cheng, X., Howard, R. M., Zhang, Y., et al. (2010). Transplantation of ciliary neurotrophic factor-expressing adult oligodendrocyte precursor cells promotes remyelination and functional recovery after spinal cord injury. Journal of Neuroscience, 30(8), 2989–3001. doi:10.1523/JNEUROSCI.3174-09.2010.
Cao, Q. L., Howard, R. M., Dennison, J. B., & Whittemore, S. R. (2002). Differentiation of engrafted neuronal-restricted precursor cells is inhibited in the traumatically injured spinal cord. Experimental Neurology, 177(2), 349–359.
Cao, Q., Xu, X. M., Devries, W. H., Enzmann, G. U., Ping, P., Tsoulfas, P., et al. (2005). Functional recovery in traumatic spinal cord injury after transplantation of multineurotrophin-expressing glial-restricted precursor cells. Journal of Neuroscience, 25(30), 6947–6957. doi:10.1523/JNEUROSCI.1065-05.2005.
Cao, Q. L., Zhang, Y. P., Howard, R. M., Walters, W. M., Tsoulfas, P., & Whittemore, S. R. (2001). Pluripotent stem cells engrafted into the normal or lesioned adult rat spinal cord are restricted to a glial lineage. Experimental Neurology, 167(1), 48–58.
Chernykh, E. R., Stupak, V. V., Muradov, G. M., Sizikov, M. Y., Shevela, E. Y., Leplina, O. Y., et al. (2007). Application of autologous bone marrow stem cells in the therapy of spinal cord injury patients. Bulletin of Experimental Biology and Medicine, 143(4), 543–547.
Collins, W. F. (1983). A review and update of experiment and clinical studies of spinal cord injury. Paraplegia, 21(4), 204–219.
Cummings, B. J., Uchida, N., Tamaki, S. J., Salazar, D. L., Hooshmand, M., Summers, R., et al. (2005). Human neural stem cells differentiate and promote locomotor recovery in spinal cord-injured mice. Proceedings of the National Academy of Sciences of the United States of America, 102(39), 14069–14074. doi:10.1073/pnas.0507063102.
Cummings, B. J., Uchida, N., Tamaki, S. J., & Anderson, A. J. (2006). Human neural stem cell differentiation following transplantation into spinal cord injured mice: Association with recovery of locomotor function. Neurological Research, 28(5), 474–481. doi:10.1179/016164106X115116.
Davies, J. E., Huang, C., Proschel, C., Noble, M., Mayer-Proschel, M., & Davies, S. J. (2006). Astrocytes derived from glial-restricted precursors promote spinal cord repair. Journal of Biology, 5(3), 7. doi:10.1186/jbiol35.
Deda, H., Inci, M. C., Kurekci, A. E., Kayihan, K., Ozgun, E., Ustunsoy, G. E., et al. (2008). Treatment of chronic spinal cord injured patients with autologous bone marrow-derived hematopoietic stem cell transplantation: 1-year follow-up. Cytotherapy, 10(6), 565–574. doi:10.1080/14653240802241797.
Diener, P. S., & Bregman, B. S. (1998). Fetal spinal cord transplants support growth of supraspinal and segmental projections after cervical spinal cord hemisection in the neonatal rat. Journal of Neuroscience, 18(2), 779–793.
Evans, M. J., & Kaufman, M. H. (1981). Establishment in culture of pluripotential cells from mouse embryos. Nature, 292(5819), 154–156.
Fawcett, J. W., Curt, A., Steeves, J. D., Coleman, W. P., Tuszynski, M. H., Lammertse, D., et al. (2007). Guidelines for the conduct of clinical trials for spinal cord injury as developed by the ICCP panel: Spontaneous recovery after spinal cord injury and statistical power needed for therapeutic clinical trials. Spinal Cord, 45(3), 190–205. doi:10.1038/sj.sc.3102007.
Field, P., Li, Y., & Raisman, G. (2003). Ensheathment of the olfactory nerves in the adult rat. Journal of Neurocytology, 32(3), 317–324. doi:10.1023/B:NEUR.0000010089.37032.48.
Franklin, R. J. (2002). Remyelination of the demyelinated CNS: The case for and against transplantation of central, peripheral and olfactory glia. Brain Research Bulletin, 57(6), 827–832.
Fujimoto, Y., Abematsu, M., Falk, A., Tsujimura, K., Sanosaka, T., Juliandi, B., et al. (2012). Treatment of a mouse model of spinal cord injury by transplantation of human induced pluripotent stem cell-derived long-term self-renewing neuroepithelial-like stem cells. Stem Cells, 30(6), 1163–1173. doi:10.1002/stem.1083.
Gage, F. H., Coates, P. W., Palmer, T. D., Kuhn, H. G., Fisher, L. J., Suhonen, J. O., et al. (1995). Survival and differentiation of adult neuronal progenitor cells transplanted to the adult brain. Proceedings of the National Academy of Sciences of the United States of America, 92(25), 11879–11883.
Gaillard, A., Prestoz, L., Dumartin, B., Cantereau, A., Morel, F., Roger, M., et al. (2007). Reestablishment of damaged adult motor pathways by grafted embryonic cortical neurons. Nature Neuroscience, 10(10), 1294–1299. doi:10.1038/nn1970.
Geffner, L. F., Santacruz, P., Izurieta, M., Flor, L., Maldonado, B., Auad, A. H., et al. (2008). Administration of autologous bone marrow stem cells into spinal cord injury patients via multiple routes is safe and improves their quality of life: Comprehensive case studies. Cell Transplantation, 17(12), 1277–1293.
Giger, R. J., Hollis, E. R., 2nd, & Tuszynski, M. H. (2010). Guidance molecules in axon regeneration. Cold Spring Harbor Perspectives in Biology, 2(7), a001867. doi:10.1101/cshperspect.a001867.
Gledhill, R. F., Harrison, B. M., & McDonald, W. I. (1973). Demyelination and remyelination after acute spinal cord compression. Experimental Neurology, 38(3), 472–487.
Gregori, N., Proschel, C., Noble, M., & Mayer-Proschel, M. (2002). The tripotential glial-restricted precursor (GRP) cell and glial development in the spinal cord: Generation of bipotential oligodendrocyte-type-2 astrocyte progenitor cells and dorsal-ventral differences in GRP cell function. Journal of Neuroscience, 22(1), 248–256.
Grill, R., Murai, K., Blesch, A., Gage, F. H., & Tuszynski, M. H. (1997). Cellular delivery of neurotrophin-3 promotes corticospinal axonal growth and partial functional recovery after spinal cord injury. Journal of Neuroscience, 17(14), 5560–5572.
Gritti, A., Parati, E. A., Cova, L., Frolichsthal, P., Galli, R., Wanke, E., et al. (1996). Multipotential stem cells from the adult mouse brain proliferate and self-renew in response to basic fibroblast growth factor. Journal of Neuroscience, 16(3), 1091–1100.
Groves, A. K., Barnett, S. C., Franklin, R. J., Crang, A. J., Mayer, M., Blakemore, W. F., et al. (1993). Repair of demyelinated lesions by transplantation of purified O-2A progenitor cells. Nature, 362(6419), 453–455. doi:10.1038/362453a0.
Han, S. S., Liu, Y., Tyler-Polsz, C., Rao, M. S., & Fischer, I. (2004). Transplantation of glial-restricted precursor cells into the adult spinal cord: Survival, glial-specific differentiation, and preferential migration in white matter. Glia, 45(1), 1–16. doi:10.1002/glia.10282.
Hofstetter, C. P., Holmstrom, N. A., Lilja, J. A., Schweinhardt, P., Hao, J., Spenger, C., et al. (2005). Allodynia limits the usefulness of intraspinal neural stem cell grafts; directed differentiation improves outcome. Nature Neuroscience, 8(3), 346–353. doi:10.1038/nn1405.
Honmou, O., Felts, P. A., Waxman, S. G., & Kocsis, J. D. (1996). Restoration of normal conduction properties in demyelinated spinal cord axons in the adult rat by transplantation of exogenous Schwann cells. Journal of Neuroscience, 16(10), 3199–3208.
Hooshmand, M. J., Sontag, C. J., Uchida, N., Tamaki, S., Anderson, A. J., & Cummings, B. J. (2009). Analysis of host-mediated repair mechanisms after human CNS-stem cell transplantation for spinal cord injury: Correlation of engraftment with recovery. PloS One, 4(6), e5871. doi:10.1371/journal.pone.0005871.
Houle, J. D., & Reier, P. J. (1988). Transplantation of fetal spinal cord tissue into the chronically injured adult rat spinal cord. Journal of Comparative Neurology, 269(4), 535–547. doi:10.1002/cne.902690406.
Hug, A., & Weidner, N. (2012). From bench to beside to cure spinal cord injury: Lost in translation? International Review of Neurobiology, 106, 173–196. doi:10.1016/B978-0-12-407178-0.00008-9.
Hulsebosch, C. E. (2002). Recent advances in pathophysiology and treatment of spinal cord injury. Advances in Physiology Education, 26(1–4), 238–255.
Imaizumi, T., Lankford, K. L., & Kocsis, J. D. (2000). Transplantation of olfactory ensheathing cells or Schwann cells restores rapid and secure conduction across the transected spinal cord. Brain Research, 854(1–2), 70–78.
Ishii, K., Nakamura, M., Dai, H., Finn, T. P., Okano, H., Toyama, Y., et al. (2006). Neutralization of ciliary neurotrophic factor reduces astrocyte production from transplanted neural stem cells and promotes regeneration of corticospinal tract fibers in spinal cord injury. Journal of Neuroscience Research, 84(8), 1669–1681. doi:10.1002/jnr.21079.
Iwanami, A., Kaneko, S., Nakamura, M., Kanemura, Y., Mori, H., Kobayashi, S., et al. (2005). Transplantation of human neural stem cells for spinal cord injury in primates. Journal of Neuroscience Research, 80(2), 182–190. doi:10.1002/jnr.20436.
Jin, Y., Fischer, I., Tessler, A., & Houle, J. D. (2002). Transplants of fibroblasts genetically modified to express BDNF promote axonal regeneration from supraspinal neurons following chronic spinal cord injury. Experimental Neurology, 177(1), 265–275.
Johansson, C. B., Svensson, M., Wallstedt, L., Janson, A. M., & Frisen, J. (1999). Neural stem cells in the adult human brain. Experimental Cell Research, 253(2), 733–736. doi:10.1006/excr.1999.4678.
Johe, K. K., Hazel, T. G., Muller, T., Dugich-Djordjevic, M. M., & McKay, R. D. (1996). Single factors direct the differentiation of stem cells from the fetal and adult central nervous system. Genes and Development, 10(24), 3129–3140.
Kaiser, J. (2011). Embryonic stem cells. Researchers mull impact of Geron’s sudden exit from field. Science, 334(6059), 1043. doi:10.1126/science.334.6059.1043.
Kakulas, B. A. (1999). A review of the neuropathology of human spinal cord injury with emphasis on special features. Journal of Spinal Cord Medicine, 22(2), 119–124.
Kalyani, A. J., & Rao, M. S. (1998). Cell lineage in the developing neural tube. Biochemistry and Cell Biology, 76(6), 1051–1068.
Karimi-Abdolrezaee, S., Eftekharpour, E., Wang, J., Morshead, C. M., & Fehlings, M. G. (2006). Delayed transplantation of adult neural precursor cells promotes remyelination and functional neurological recovery after spinal cord injury. Journal of Neuroscience, 26(13), 3377–3389. doi:10.1523/JNEUROSCI.4184-05.2006.
Keirstead, H. S., Nistor, G., Bernal, G., Totoiu, M., Cloutier, F., Sharp, K., et al. (2005). Human embryonic stem cell-derived oligodendrocyte progenitor cell transplants remyelinate and restore locomotion after spinal cord injury. Journal of Neuroscience, 25(19), 4694–4705. doi:10.1523/JNEUROSCI.0311-05.2005.
Kohama, I., Lankford, K. L., Preiningerova, J., White, F. A., Vollmer, T. L., & Kocsis, J. D. (2001). Transplantation of cryopreserved adult human Schwann cells enhances axonal conduction in demyelinated spinal cord. Journal of Neuroscience, 21(3), 944–950.
Kumar, A. A., Kumar, S. R., Narayanan, R., Arul, K., & Baskaran, M. (2009). Autologous bone marrow derived mononuclear cell therapy for spinal cord injury: A phase I/II clinical safety and primary efficacy data. Experimental and Clinical Transplantation, 7(4), 241–248.
Kunkel-Bagden, E., Dai, H. N., & Bregman, B. S. (1992). Recovery of function after spinal cord hemisection in newborn and adult rats: Differential effects on reflex and locomotor function. Experimental Neurology, 116(1), 40–51.
Lammertse, D., Tuszynski, M. H., Steeves, J. D., Curt, A., Fawcett, J. W., Rask, C., et al. (2007). Guidelines for the conduct of clinical trials for spinal cord injury as developed by the ICCP panel: Clinical trial design. Spinal Cord, 45(3), 232–242. doi:10.1038/sj.sc.3102010.
Lee, J. C., Mayer-Proschel, M., & Rao, M. S. (2000). Gliogenesis in the central nervous system. Glia, 30(2), 105–121.
Levine, J. M., & Reynolds, R. (1999). Activation and proliferation of endogenous oligodendrocyte precursor cells during ethidium bromide-induced demyelination. Experimental Neurology, 160(2), 333–347. doi:10.1006/exnr.1999.7224.
Li, Y., Field, P. M., & Raisman, G. (1997). Repair of adult rat corticospinal tract by transplants of olfactory ensheathing cells. Science, 277(5334), 2000–2002.
Lu, J., Feron, F., Mackay-Sim, A., & Waite, P. M. (2002). Olfactory ensheathing cells promote locomotor recovery after delayed transplantation into transected spinal cord. Brain, 125(Pt 1), 14–21.
Lu, P., & Tuszynski, M. H. (2008). Growth factors and combinatorial therapies for CNS regeneration. Experimental Neurology, 209(2), 313–320. doi:10.1016/j.expneurol.2007.08.004. S0014-4886(07)00305-6 [pii].
Lu, P., Wang, Y., Graham, L., McHale, K., Gao, M., Wu, D., et al. (2012). Long-distance growth and connectivity of neural stem cells after severe spinal cord injury. Cell, 150(6), 1264–1273. doi:10.1016/j.cell.2012.08.020.
Lu, P., Yang, H., Culbertson, M., Graham, L., Roskams, A. J., & Tuszynski, M. H. (2006). Olfactory ensheathing cells do not exhibit unique migratory or axonal growth-promoting properties after spinal cord injury. Journal of Neuroscience, 26(43), 11120–11130. doi:10.1523/JNEUROSCI.3264-06.2006.
Maherali, N., & Hochedlinger, K. (2008). Guidelines and techniques for the generation of induced pluripotent stem cells. Cell Stem Cell, 3(6), 595–605. doi:10.1016/j.stem.2008.11.008.
Maherali, N., Sridharan, R., Xie, W., Utikal, J., Eminli, S., Arnold, K., et al. (2007). Directly reprogrammed fibroblasts show global epigenetic remodeling and widespread tissue contribution. Cell Stem Cell, 1(1), 55–70. doi:10.1016/j.stem.2007.05.014.
Marcus, A. J., & Woodbury, D. (2008). Fetal stem cells from extra-embryonic tissues: Do not discard. Journal of Cellular and Molecular Medicine, 12(3), 730–742. doi:10.1111/j.1582-4934.2008.00221.x.
Martin, G. R. (1981). Isolation of a pluripotent cell line from early mouse embryos cultured in medium conditioned by teratocarcinoma stem cells. Proceedings of the National Academy of Sciences of the United States of America, 78(12), 7634–7638.
Mayor, S. (2010). First patient enters trial to test safety of stem cells in spinal injury. BMJ, 341, c5724. doi:10.1136/bmj.c5724.
McDonald, J. W., Liu, X. Z., Qu, Y., Liu, S., Mickey, S. K., Turetsky, D., et al. (1999). Transplanted embryonic stem cells survive, differentiate and promote recovery in injured rat spinal cord. Nature Medicine, 5(12), 1410–1412. doi:10.1038/70986.
Mitsui, T., Shumsky, J. S., Lepore, A. C., Murray, M., & Fischer, I. (2005). Transplantation of neuronal and glial restricted precursors into contused spinal cord improves bladder and motor functions, decreases thermal hypersensitivity, and modifies intraspinal circuitry. Journal of Neuroscience, 25(42), 9624–9636. doi:10.1523/JNEUROSCI.2175-05.2005.
Miura, K., Okada, Y., Aoi, T., Okada, A., Takahashi, K., Okita, K., et al. (2009). Variation in the safety of induced pluripotent stem cell lines. Nature Biotechnology, 27(8), 743–745. doi:10.1038/nbt.1554.
Mothe, A. J., & Tator, C. H. (2008). Transplanted neural stem/progenitor cells generate myelinating oligodendrocytes and Schwann cells in spinal cord demyelination and dysmyelination. Experimental Neurology, 213(1), 176–190. doi:10.1016/j.expneurol.2008.05.024. S0014-4886(08)00244-6 [pii].
Mujtaba, T., Piper, D. R., Kalyani, A., Groves, A. K., Lucero, M. T., & Rao, M. S. (1999). Lineage-restricted neural precursors can be isolated from both the mouse neural tube and cultured ES cells. Developmental Biology, 214(1), 113–127. doi:10.1006/dbio.1999.9418.
Nakagawa, M., Koyanagi, M., Tanabe, K., Takahashi, K., Ichisaka, T., Aoi, T., et al. (2008). Generation of induced pluripotent stem cells without Myc from mouse and human fibroblasts. Nature Biotechnology, 26(1), 101–106. doi:10.1038/nbt1374.
Nistor, G. I., Totoiu, M. O., Haque, N., Carpenter, M. K., & Keirstead, H. S. (2005). Human embryonic stem cells differentiate into oligodendrocytes in high purity and myelinate after spinal cord transplantation. Glia, 49(3), 385–396. doi:10.1002/glia.20127.
Nori, S., Okada, Y., Yasuda, A., Tsuji, O., Takahashi, Y., Kobayashi, Y., et al. (2011). Grafted human-induced pluripotent stem-cell-derived neurospheres promote motor functional recovery after spinal cord injury in mice. Proceedings of the National Academy of Sciences of the United States of America, 108(40), 16825–16830. doi:10.1073/pnas.1108077108.
Nussbaum, J., Minami, E., Laflamme, M. A., Virag, J. A., Ware, C. B., Masino, A., et al. (2007). Transplantation of undifferentiated murine embryonic stem cells in the heart: Teratoma formation and immune response. The FASEB Journal, 21(7), 1345–1357. doi:10.1096/fj.06-6769com.
O'Donoghue, K., & Fisk, N. M. (2004). Fetal stem cells. Best Practice & Research. Clinical Obstetrics & Gynaecology, 18(6), 853–875. doi:10.1016/j.bpobgyn.2004.06.010.
Ogawa, Y., Sawamoto, K., Miyata, T., Miyao, S., Watanabe, M., Nakamura, M., et al. (2002). Transplantation of in vitro-expanded fetal neural progenitor cells results in neurogenesis and functional recovery after spinal cord contusion injury in adult rats. Journal of Neuroscience Research, 69(6), 925–933. doi:10.1002/jnr.10341.
Okada, S., Ishii, K., Yamane, J., Iwanami, A., Ikegami, T., Katoh, H., et al. (2005). In vivo imaging of engrafted neural stem cells: Its application in evaluating the optimal timing of transplantation for spinal cord injury. The FASEB Journal, 19(13), 1839–1841. doi:10.1096/fj.05-4082fje.
Pal, R., Venkataramana, N. K., Bansal, A., Balaraju, S., Jan, M., Chandra, R., et al. (2009). Ex vivo-expanded autologous bone marrow-derived mesenchymal stromal cells in human spinal cord injury/paraplegia: A pilot clinical study. Cytotherapy, 11(7), 897–911. doi:10.3109/14653240903253857. 10.3109/14653240903253857 [pii].
Palmer, T. D., Markakis, E. A., Willhoite, A. R., Safar, F., & Gage, F. H. (1999). Fibroblast growth factor-2 activates a latent neurogenic program in neural stem cells from diverse regions of the adult CNS. Journal of Neuroscience, 19(19), 8487–8497.
Palmer, T. D., Ray, J., & Gage, F. H. (1995). FGF-2-responsive neuronal progenitors reside in proliferative and quiescent regions of the adult rodent brain. Molecular and Cellular Neuroscience, 6(5), 474–486. doi:10.1006/mcne.1995.1035.
Park, H. C., Shim, Y. S., Ha, Y., Yoon, S. H., Park, S. R., Choi, B. H., et al. (2005). Treatment of complete spinal cord injury patients by autologous bone marrow cell transplantation and administration of granulocyte-macrophage colony stimulating factor. Tissue Engineering, 11(5–6), 913–922. doi:10.1089/ten.2005.11.913.
Park, I. H., Zhao, R., West, J. A., Yabuuchi, A., Huo, H., Ince, T. A., et al. (2008). Reprogramming of human somatic cells to pluripotency with defined factors. Nature, 451(7175), 141–146. doi:10.1038/nature06534.
Pfeifer, K., Vroemen, M., Blesch, A., & Weidner, N. (2004). Adult neural progenitor cells provide a permissive guiding substrate for corticospinal axon growth following spinal cord injury. European Journal of Neuroscience, 20(7), 1695–1704. doi:10.1111/j.1460-9568.2004.03657.x.
Pfeifer, K., Vroemen, M., Caioni, M., Aigner, L., Bogdahn, U., & Weidner, N. (2006). Autologous adult rodent neural progenitor cell transplantation represents a feasible strategy to promote structural repair in the chronically injured spinal cord. Regenerative Medicine, 1(2), 255–266. doi:10.2217/17460751.1.2.255.
Pojda, Z., Machaj, E. K., Oldak, T., Gajkowska, A., & Jastrzewska, M. (2005). Nonhematopoietic stem cells of fetal origin–how much of today’s enthusiasm will pass the time test? Folia histochemica et cytobiologica/Polish Academy of Sciences, Polish Histochemical and Cytochemical Society, 43(4), 209–212.
Ramon-Cueto, A., & Avila, J. (1998). Olfactory ensheathing glia: Properties and function. Brain Research Bulletin, 46(3), 175–187.
Ramon-Cueto, A., Cordero, M. I., Santos-Benito, F. F., & Avila, J. (2000). Functional recovery of paraplegic rats and motor axon regeneration in their spinal cords by olfactory ensheathing glia. Neuron, 25(2), 425–435. S0896-6273(00)80905-8 [pii].
Rao, M. S. (1999). Multipotent and restricted precursors in the central nervous system. The Anatomical Record, 257(4), 137–148.
Rao, M. S., & Mayer-Proschel, M. (1997). Glial-restricted precursors are derived from multipotent neuroepithelial stem cells. Developmental Biology, 188(1), 48–63. doi:10.1006/dbio.1997.8597.
Rao, M. S., Noble, M., & Mayer-Proschel, M. (1998). A tripotential glial precursor cell is present in the developing spinal cord. Proceedings of the National Academy of Sciences of the United States of America, 95(7), 3996–4001.
Reubinoff, B. E., Pera, M. F., Fong, C. Y., Trounson, A., & Bongso, A. (2000). Embryonic stem cell lines from human blastocysts: Somatic differentiation in vitro. Nature Biotechnology, 18(4), 399–404. doi:10.1038/74447.
Reynolds, B. A., & Weiss, S. (1992). Generation of neurons and astrocytes from isolated cells of the adult mammalian central nervous system. Science, 255(5052), 1707–1710.
Roy, N. S., Wang, S., Jiang, L., Kang, J., Benraiss, A., Harrison-Restelli, C., et al. (2000). In vitro neurogenesis by progenitor cells isolated from the adult human hippocampus. Nature Medicine, 6(3), 271–277. doi:10.1038/73119.
Saito, F., Nakatani, T., Iwase, M., Maeda, Y., Hirakawa, A., Murao, Y., et al. (2008). Spinal cord injury treatment with intrathecal autologous bone marrow stromal cell transplantation: The first clinical trial case report. Journal of Trauma, 64(1), 53–59. doi:10.1097/TA.0b013e31815b847d.
Salazar, D. L., Uchida, N., Hamers, F. P., Cummings, B. J., & Anderson, A. J. (2010). Human neural stem cells differentiate and promote locomotor recovery in an early chronic spinal cord injury NOD-scid mouse model. PloS One, 5(8), e12272. doi:10.1371/journal.pone.0012272.
Salewski, R. P., Eftekharpour, E., & Fehlings, M. G. (2010). Are induced pluripotent stem cells the future of cell-based regenerative therapies for spinal cord injury? Journal of Cellular Physiology, 222(3), 515–521. doi:10.1002/jcp.21995.
Sandner, B., Rivera, F.J., Aigner, L., Blesch, A. & Weidner, N.. (2012). Bone morphogenetic proteins prevent mesenchymal stromal cell-mediated oligodendroglial differentiation of transplanted adult neural progenitor cells in the injured spinal cord (Program No. 531.09). 2012 Neuroscience Meeting Planner. New Orleans, LA: Society for Neuroscience. Online.
Sasaki, M., Lankford, K. L., Zemedkun, M., & Kocsis, J. D. (2004). Identified olfactory ensheathing cells transplanted into the transected dorsal funiculus bridge the lesion and form myelin. Journal of Neuroscience, 24(39), 8485–8493. doi:10.1523/JNEUROSCI.1998-04.2004.
Sharp, J., Frame, J., Siegenthaler, M., Nistor, G., & Keirstead, H. S. (2010). Human embryonic stem cell-derived oligodendrocyte progenitor cell transplants improve recovery after cervical spinal cord injury. Stem Cells, 28(1), 152–163. doi:10.1002/stem.245.
Sherwood, A. M., Dimitrijevic, M. R., & McKay, W. B. (1992). Evidence of subclinical brain influence in clinically complete spinal cord injury: Discomplete SCI. Journal of Neurological Sciences, 110(1–2), 90–98.
Shihabuddin, L. S., Ray, J., & Gage, F. H. (1997). FGF-2 is sufficient to isolate progenitors found in the adult mammalian spinal cord. Experimental Neurology, 148(2), 577–586. doi:10.1006/exnr.1997.6697.
Steeves, J. D., Lammertse, D., Curt, A., Fawcett, J. W., Tuszynski, M. H., Ditunno, J. F., et al. (2007). Guidelines for the conduct of clinical trials for spinal cord injury (SCI) as developed by the ICCP panel: Clinical trial outcome measures. Spinal Cord, 45(3), 206–221. doi:10.1038/sj.sc.3102008.
Steward, O., Sharp, K., Selvan, G., Hadden, A., Hofstadter, M., Au, E., et al. (2006). A re-assessment of the consequences of delayed transplantation of olfactory lamina propria following complete spinal cord transection in rats. Experimental Neurology, 198(2), 483–499. doi:10.1016/j.expneurol.2005.12.034.
Sykova, E., Homola, A., Mazanec, R., Lachmann, H., Konradova, S. L., Kobylka, P., et al. (2006). Autologous bone marrow transplantation in patients with subacute and chronic spinal cord injury. Cell Transplantation, 15(8–9), 675–687.
Takahashi, K., Tanabe, K., Ohnuki, M., Narita, M., Ichisaka, T., Tomoda, K., et al. (2007). Induction of pluripotent stem cells from adult human fibroblasts by defined factors. Cell, 131(5), 861–872. doi:10.1016/j.cell.2007.11.019.
Takahashi, K., & Yamanaka, S. (2006). Induction of pluripotent stem cells from mouse embryonic and adult fibroblast cultures by defined factors. Cell, 126(4), 663–676. doi:10.1016/j.cell.2006.07.024.
Takami, T., Oudega, M., Bates, M. L., Wood, P. M., Kleitman, N., & Bunge, M. B. (2002). Schwann cell but not olfactory ensheathing glia transplants improve hindlimb locomotor performance in the moderately contused adult rat thoracic spinal cord. Journal of Neuroscience, 22(15), 6670–6681. 20026636.
Thomson, J. A., Itskovitz-Eldor, J., Shapiro, S. S., Waknitz, M. A., Swiergiel, J. J., Marshall, V. S., et al. (1998). Embryonic stem cell lines derived from human blastocysts. Science, 282(5391), 1145–1147.
Thuret, S., Moon, L. D., & Gage, F. H. (2006). Therapeutic interventions after spinal cord injury. Nature Reviews. Neuroscience, 7(8), 628–643. doi:10.1038/nrn1955. nrn1955 [pii].
Tsuji, O., Miura, K., Fujiyoshi, K., Momoshima, S., Nakamura, M., & Okano, H. (2011). Cell therapy for spinal cord injury by neural stem/progenitor cells derived from iPS/ES cells. Neurotherapeutics, 8(4), 668–676. doi:10.1007/s13311-011-0063-z.
Tsuji, O., Miura, K., Okada, Y., Fujiyoshi, K., Mukaino, M., Nagoshi, N., et al. (2010). Therapeutic potential of appropriately evaluated safe-induced pluripotent stem cells for spinal cord injury. Proceedings of the National Academy of Sciences of the United States of America, 107(28), 12704–12709. doi:10.1073/pnas.0910106107.
Tuszynski, M. H., Grill, R., Jones, L. L., Brant, A., Blesch, A., Low, K., et al. (2003). NT-3 gene delivery elicits growth of chronically injured corticospinal axons and modestly improves functional deficits after chronic scar resection. Experimental Neurology, 181(1), 47–56.
Tuszynski, M. H., Steeves, J. D., Fawcett, J. W., Lammertse, D., Kalichman, M., Rask, C., et al. (2007). Guidelines for the conduct of clinical trials for spinal cord injury as developed by the ICCP Panel: Clinical trial inclusion/exclusion criteria and ethics. Spinal Cord, 45(3), 222–231. doi:10.1038/sj.sc.3102009.
Uchida, N., Buck, D. W., He, D., Reitsma, M. J., Masek, M., Phan, T. V., et al. (2000). Direct isolation of human central nervous system stem cells. Proceedings of the National Academy of Sciences of the United States of America, 97(26), 14720–14725. doi:10.1073/pnas.97.26.14720.
Vats, A., Tolley, N. S., Bishop, A. E., & Polak, J. M. (2005). Embryonic stem cells and tissue engineering: Delivering stem cells to the clinic. Journal of the Royal Society of Medicine, 98(8), 346–350. doi:10.1258/jrsm.98.8.346.
Vroemen, M., Aigner, L., Winkler, J., & Weidner, N. (2003). Adult neural progenitor cell grafts survive after acute spinal cord injury and integrate along axonal pathways. European Journal of Neuroscience, 18(4), 743–751.
Vroemen, M., Caioni, M., Bogdahn, U., & Weidner, N. (2007). Failure of Schwann cells as supporting cells for adult neural progenitor cell grafts in the acutely injured spinal cord. Cell and Tissue Research, 327(1), 1–13.
Wachs, F. P., Couillard-Despres, S., Engelhardt, M., Wilhelm, D., Ploetz, S., Vroemen, M., et al. (2003). High efficacy of clonal growth and expansion of adult neural stem cells. Laboratory Investigation, 83(7), 949–962.
Wang, G., Ao, Q., Gong, K., Zuo, H., Gong, Y., & Zhang, X. (2010). Synergistic effect of neural stem cells and olfactory ensheathing cells on repair of adult rat spinal cord injury. Cell Transplantation, 19(10), 1325–1337. doi:10.3727/096368910X505855.
Wang, J. M., Zeng, Y. S., Wu, J. L., Li, Y., & Teng, Y. D. (2011). Cograft of neural stem cells and schwann cells overexpressing TrkC and neurotrophin-3 respectively after rat spinal cord transection. Biomaterials. doi:10.1016/j.biomaterials.2011.06.036.
Warrington, A. E., Barbarese, E., & Pfeiffer, S. E. (1993). Differential myelinogenic capacity of specific developmental stages of the oligodendrocyte lineage upon transplantation into hypomyelinating hosts. Journal of Neuroscience Research, 34(1), 1–13. doi:10.1002/jnr.490340102.
Webber, D. J., Bradbury, E. J., McMahon, S. B., & Minger, S. L. (2007). Transplanted neural progenitor cells survive and differentiate but achieve limited functional recovery in the lesioned adult rat spinal cord. Regenerative Medicine, 2(6), 929–945. doi:10.2217/17460751.2.6.929.
Weidner, N., Grill, R. J., & Tuszynski, M. H. (1999). Elimination of basal lamina and the collagen “scar” after spinal cord injury fails to augment corticospinal tract regeneration. Experimental Neurology, 160(1), 40–50.
Weiss, S., Dunne, C., Hewson, J., Wohl, C., Wheatley, M., Peterson, A. C., et al. (1996). Multipotent CNS stem cells are present in the adult mammalian spinal cord and ventricular neuroaxis. Journal of Neuroscience, 16(23), 7599–7609.
Wernig, M., Meissner, A., Foreman, R., Brambrink, T., Ku, M., Hochedlinger, K., et al. (2007). In vitro reprogramming of fibroblasts into a pluripotent ES-cell-like state. Nature, 448(7151), 318–324. doi:10.1038/nature05944.
Wernig, M., Zhao, J. P., Pruszak, J., Hedlund, E., Fu, D., Soldner, F., et al. (2008). Neurons derived from reprogrammed fibroblasts functionally integrate into the fetal brain and improve symptoms of rats with Parkinson's disease. Proceedings of the National Academy of Sciences of the United States of America, 105(15), 5856–5861. doi:10.1073/pnas.0801677105.
Windrem, M. S., Nunes, M. C., Rashbaum, W. K., Schwartz, T. H., Goodman, R. A., McKhann, G., 2nd, et al. (2004). Fetal and adult human oligodendrocyte progenitor cell isolates myelinate the congenitally dysmyelinated brain. Nature Medicine, 10(1), 93–97. doi:10.1038/nm974.
Wu, Y. Y., Mujtaba, T., Han, S. S., Fischer, I., & Rao, M. S. (2002a). Isolation of a glial-restricted tripotential cell line from embryonic spinal cord cultures. Glia, 38(1), 65–79.
Wu, P., Tarasenko, Y. I., Gu, Y., Huang, L. Y., Coggeshall, R. E., & Yu, Y. (2002b). Region-specific generation of cholinergic neurons from fetal human neural stem cells grafted in adult rat. Nature Neuroscience, 5(12), 1271–1278. doi:10.1038/nn974.
Yamamoto, S., Yamamoto, N., Kitamura, T., Nakamura, K., & Nakafuku, M. (2001). Proliferation of parenchymal neural progenitors in response to injury in the adult rat spinal cord. Experimental Neurology, 172(1), 115–127. doi:10.1006/exnr.2001.7798.
Yamanaka, S. (2007). Strategies and new developments in the generation of patient-specific pluripotent stem cells. Cell Stem Cell, 1(1), 39–49. doi:10.1016/j.stem.2007.05.012.
Yamanaka, S. (2009). A fresh look at iPS cells. Cell, 137(1), 13–17. doi:10.1016/j.cell.2009.03.034.
Yan, J., Xu, L., Welsh, A. M., Hatfield, G., Hazel, T., Johe, K., et al. (2007). Extensive neuronal differentiation of human neural stem cell grafts in adult rat spinal cord. PLoS Medicine, 4(2), e39. doi:10.1371/journal.pmed.0040039.
Yasuda, A., Tsuji, O., Shibata, S., Nori, S., Takano, M., Kobayashi, Y., et al. (2011). Significance of Remyelination by Neural Stem/Progenitor Cells Transplanted into the Injured Spinal Cord. Stem Cells, 29(12), 1983–1994. doi:10.1002/stem.767.
Yoon, S. H., Shim, Y. S., Park, Y. H., Chung, J. K., Nam, J. H., Kim, M. O., et al. (2007). Complete spinal cord injury treatment using autologous bone marrow cell transplantation and bone marrow stimulation with granulocyte macrophage-colony stimulating factor: Phase I/II clinical trial. Stem Cells, 25(8), 2066–2073. doi:10.1634/stemcells.2006-0807.
Yu, J., Vodyanik, M. A., Smuga-Otto, K., Antosiewicz-Bourget, J., Frane, J. L., Tian, S., et al. (2007). Induced pluripotent stem cell lines derived from human somatic cells. Science, 318(5858), 1917–1920. doi:10.1126/science.1151526.
Zhang, S. C., Ge, B., & Duncan, I. D. (1999). Adult brain retains the potential to generate oligodendroglial progenitors with extensive myelination capacity. Proceedings of the National Academy of Sciences of the United States of America, 96(7), 4089–4094.
Zhang, X., Zeng, Y., Zhang, W., Wang, J., Wu, J., & Li, J. (2007). Co-transplantation of neural stem cells and NT-3-overexpressing Schwann cells in transected spinal cord. Journal of Neurotrauma, 24(12), 1863–1877. doi:10.1089/neu.2007.0334.
Acknowledgments
This work was supported by Wings for Life, Spinal Cord Research Foundation (to B.S.), the International Foundation for Research in Paraplegia (P119 to A.B. and N.W.) and the EU (IRG268282 to A.B.).
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2015 Springer Science+Business Media New York
About this chapter
Cite this chapter
Sandner, B., Prang, P., Blesch, A., Weidner, N. (2015). Stem Cell-Based Therapies for Spinal Cord Regeneration. In: Kuhn, H., Eisch, A. (eds) Neural Stem Cells in Development, Adulthood and Disease. Stem Cell Biology and Regenerative Medicine. Humana Press, New York, NY. https://doi.org/10.1007/978-1-4939-1908-6_9
Download citation
DOI: https://doi.org/10.1007/978-1-4939-1908-6_9
Published:
Publisher Name: Humana Press, New York, NY
Print ISBN: 978-1-4939-1907-9
Online ISBN: 978-1-4939-1908-6
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)