
Abstract
Subependymal giant cell astrocytomas (SEGAs) are benign tumors (WHO grade I) that occur almost exclusively in the setting of tuberous sclerosis (TS), a well-defined, multi-system genetic syndrome. Most commonly originating from the region of the caudate nucleus, these tumors may cause obstruction of cerebrospinal fluid circulation leading to hydrocephalus. Less frequently, they may hemorrhage spontaneously, causing precipitous neurological impairment [1]. Mutations of the TSC-1 and TSC-2 genes, both effectors of the mTOR pathway (originally mammalian Target of Rapamycin, now formally mechanistic Target of Rapamycin), lead to the variably expressed systemic manifestations of TS; cardiac rhabdomyoma, renal angiolipomas, facial adenoma sebaceum, cortical tubers of the brain, and SEGAs. The standard treatment of symptomatic or enlarging SEGAs is surgical excision. Pharmacological effectors of the mTOR pathway, rapamycin (aka sirolimus) and its analogs have recently been shown to induce rapid involution of SEGAs; however, the optimal timing, dosage, safety, and duration of treatment remain areas of active clinical research. SEGAs in the context of TS represent an example of an emerging paradigm: targeted molecular-oncologic therapy.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others


Keywords
These keywords were added by machine and not by the authors. This process is experimental and the keywords may be updated as the learning algorithm improves.
Subependymal giant cell astrocytomas (SEGAs) are benign tumors (WHO grade I) that occur almost exclusively in the setting of tuberous sclerosis (TS), a well-defined, multi-system genetic syndrome (Table 10.1). Most commonly originating from the region of the caudate nucleus, these tumors may cause obstruction of cerebrospinal fluid circulation leading to hydrocephalus. Less frequently, they may hemorrhage spontaneously, causing precipitous neurological impairment [1]. Mutations of the TSC-1 and TSC-2 genes, both effectors of the mTOR pathway (originally mammalian Target of Rapamycin, now formally mechanistic Target of Rapamycin), lead to the variably expressed systemic manifestations of TS; cardiac rhabdomyoma, renal angiolipomas, facial adenoma sebaceum, cortical tubers of the brain, and SEGAs. The standard treatment of symptomatic or enlarging SEGAs is surgical excision. Pharmacological effectors of the mTOR pathway, rapamycin (aka sirolimus) and its analogs have recently been shown to induce rapid involution of SEGAs; however, the optimal timing, dosage, safety, and duration of treatment remain areas of active clinical research. SEGAs in the context of TS represent an example of an emerging paradigm: targeted molecular-oncologic therapy.
Incidence and Prevalence
SEGAs (subependymal giant cell tumors) typically occur in the first or second decade of life, predominately, yet not exclusively in patients with TS (tuberous sclerosis). Reports of neo- and prenatal diagnoses illustrate the developmental nature of these tumors [2–5]. Most SEGAs are related to tuberous sclerosis, occurring in approximately 1 per 5,000–10,000 births [6]. Although most SEGAs are associated with TS, the incidence of SEGA is only 5–10 % among patients with TS [7]. A small portion of SEGAs occur without clinical or genetic evidence of TS, or as “forme fruste” of the disorder displaying some characteristics [8]. Tuberous sclerosis occurs across all ethnicities and in both male and female, worldwide estimates are of 1–2 million affected individuals [6].
Genetics and Oncogenesis
Tuberous sclerosis is an autosomal dominant genetic disorder with high penetrance and variable expressivity. The majority of cases are due to de novo mutations, although inherited somatic mutations and gonadal mosaicism may also occur [9]. Somatic mosaicism may result in limited expression of TS. In cases of both spontaneous mutation and gonadal or somatic mosaicism, parental genetic testing may be normal. In cases of gonadal mosaicism, the possibility of transmission to future offspring remains, albeit at an unquantifiable rate. A variety of mutations of within two genes have been identified, TSC1 (chromosome 9) and TSC2 (chromosome 16), both effectors of the mTOR (mechanistic Target of Rapamycin) pathway. Identified aberrations, including mutation and deletion, lead to loss or attenuation of function. Sporadic SEGAs occurring without clinical or genetic evidence of TSC (tuberous sclerosis complex) may be due to dual somatic mutations of TSC1 or TSC2 [10, 11].
The TOR complexes influence many aspects of eukaryote physiology—largely via growth regulation, cell growth, proliferation, and survival (Fig. 10.1) [12]. The mTOR signaling pathway detects and integrates a variety of environmental conditions to regulate growth and homeostasis. Aberrations of the mTOR pathway have been implicated in a wide array of pathological processes including oncogenesis, obesity, type II diabetes, and neurodegenerative conditions. mTOR has been identified as an atypical serinine/threonine protein kinase belonging to the phosphoinositide 3-kinase (PI3K)-related kinase family. Interacting with other proteins, mTOR forms two complexes: mTOR complex 1 (mTORC1) and mTOR complex 2 (mTORC2). These complexes each have independent effectors and effects, as well as differing sensitivities to rapamycin and its analogs [13].

Overview of mTOR1 and mTOR2 interactions and effectors.
TSC1 (hamartin) and TSC2 (tuberin) form a heterodimer that is a key upstream regulator of mTORC1, functioning as a guanosine triphosphate (GTPase)-activating protein (GAP) for Ras homolog enriched in the brain (Rheb) (Fig. 10.2). The GTP-bound form of Rheb interacts directly with mTORC1, significantly enhancing its kinase activity [13]. TSC1/2 as a Rheb GTPase-activating protein negatively regulates mTORC1 by converting Rheb to its inactive GDP-bound state [14]. TSC1/2 integrates multiple upstream signals that attenuate mTORC1 including growth factors via PI3k and Ras pathways. The effector kinases of these pathways (Akt/PKB, ERK1/2, RSK1) directly phosphorylate the TSC1/2 dimer to inactivate it, resultantly activating mTORC1 [15]. Cytokines, such as TNFα, may also activate TORC1 by phosphorylation of TSC1/2 via Iκβ kinase β (IKKβ) [16]. The Wnt pathway, a regulator of diverse cellular processes including differentiation, proliferation, and polarity, also modulates mTOR. By inhibiting glycogen synthase kinase 3b, phosphorylation of TSC2 is reduced leading to activation of mTORC1 [17].

TSC1/2 complex interactions via a Rheb GTPase mediator leading to mTORC1 activation.
Hypoxia, mediated via transcriptional regulation of DNA damage response 1 (REDD1), activates TSC2 function [18]. mTORC1 is activated by DNA damage through a p53-mediated mechanism. The induction of TSC2 and Pten results in downregulation of PI3K-mTORC1 [19], and also, through induction of Sestrin1/2, activates AMPK [20]. Phosphatidic acid also activates mTORC1 [21].
mTORC1 may also be activated by amino acids (leucine and arginine), which are also required for activation of mTORC1 by some growth factors [22]. The mechanism of mTORC1 activation remains poorly understood, although it has been shown to involve the Rag GTPases and translocation of mTORC1 to the lysosomal surface [23].
Cellular processes regulated by mTORC1 include protein synthesis, lipid synthesis, energy metabolism, cell fate determination, autophagy, and cytoskeletal organization [13]. The role of the mTOR pathway in oncogenesis is evinced by mutations identified in human cancers and cancer syndromes. The loss of p53, a common observation in human cancers, promotes mTORC1 activation. Upstream from mTORC1 and mTORC2, components of the PI3K pathway are also often mutated in human tumors. Several human cancer syndromes, including TS and neurofibromatosis type I, are defined by mutations in upstream signaling components of mTOR complexes. Dysregulation of translation and protein synthesis downstream of mTORC1 by interaction with initiation factor 4E-binding proteins (4E-BP1/eIF4E) likely plays a significant role in tumorigenesis by promoting cell cycle progression [24]. Another hallmark of proliferating cancer cells, lipid synthesis is regulated by mTOR-PI3K activation of the lipogenic factor SREBP1, which requires mTORC1 signaling [25].
A complex, TSC1-TSC2 (hamartin-tuberin), via GTPase-activating protein acts as a negative regulator of mTORC1, a controller of anabolic processes. Multiple factors and cellular signaling pathways are integrated, leading to phosphorylation events, and resultantly mTORC1 activity [26]. Dysregulated mTOR activity subsequently results in abnormal cellular division and differentiation across tissue types and abnormal cellular enlargement is seen, as is the case in SEGAs.
Clinical Presentation
SEGAs usually present with signs and symptoms of cerebrospinal fluid obstruction due to the encroachment of the foramen of Monro either uni- or bilaterally. The onset of symptoms is usually insidious, with progressive headache, cognitive impairment, lethargy and finally, if unrecognized, coma and death. Occasionally, precipitous neurological decline or death due to acute hydrocephalus or intratumoral hemorrhage may occur [27–31]. Clinical history and findings suggestive of tuberous sclerosis may be present; epilepsy and other systemic manifestations may also lead to the diagnosis. SEGAs usually become symptomatic within the first two decades of life.
The diagnosis of TSC is based on clinical examination and confirmed with genetic testing. Cutaneous findings include hypomelanotic macules, facial angiofibromas, and shagreen patches. Oral lesions may include ungula or gingival fibromas. The three hallmark pathologies of TSC in the central nervous system (CNS) are cortical tubers, subependymal nodules, and SEGAs. Functional impairment of effected individuals may be due to seizures, intellectual disability, and/or developmental delay. Renal manifestation may include angiomyolipomas (AML), cysts, and renal cell carcinoma. Cardiac conditions, including rhabdomyoma and arrhythmias may be present. Pulmonary involvement is restricted to lymphangioleiomyomatosis (LAM). Consensus clinical diagnostic criteria (Table 10.1) were developed prior to reliable genetic testing and allow for stratification as definite, probable, or suspect TSC [32]. Patients with somatic TSC2 mutations, as a group, are most severely affected. Somatic mutations of TSC1 are less affected [33]. Patients with genetic mosaicism may have localized, minimal, or no clinical evidence of TSC.
Radiographic Characteristics
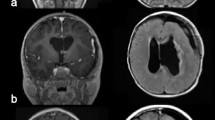
Location is of primary consideration in the radiologic suspicion of SEGA. Given that the vast majority of these tumors arise within the lateral ventricle in the caudothalamic groove, medial to the posterior caudate nucleus, SEGA should be strongly considered in the differential diagnosis of tumors in this region [34]. Growth on serial neuroimaging differentiates SEGAs from subependymal nodules. The radiologic identification of SEGA may be made on ultrasound (in neonates, and rarely prenates), computed tomography (CT), and magnetic resonance imaging (MRI) [4, 35] (Figs. 10.3 and 10.4).

Noncontrast CT scan, 3-year-old with TSC2 mutation. A SEGA is seen on the right, note calcification at the thalamocaudate groove.

Pre- and postgadolinium T1-weighted axial MRI, 9-year-old with TSC1 mutation. A SEGA is seen on the right, dense contrast enhancement is seen.
CT may show uni- or bilateral hyper-dense foci of calcification medial to the genu of the internal capsule (Fig. 10.3). In cases associated with TS, multiple calcifications and subependymal nodules (candle guttering) may be seen along the caudothalamic groove [36]. Ventriculomegaly may be identified unilaterally or bilaterally [37].
MRI characteristics mirror the heterogeneous pathology of SEGAs, with mixed signal intensities on T1- and T2-weighted imaging. SEGAs are usually hypo- and isointense on T1-weighted imaging, and iso- to hyperintense on T2-weighted imaging. Dense contrast enhancement is usually present, although it may occur in a heterogeneous pattern [38]. Calcified portions of the tumor, usually near the base of the tumor, typically appear hypodense on T2-weighted imaging (Fig. 10.4).
Pathology
The origin of almost all SEGAs is the wall of the lateral ventricle, from the region of the posterior caudate/basal ganglia, just medial to the genu of the internal capsule with projection into the frontal horn or body of the lateral ventricle. A focus of dense calcification is often present at the base of the tumor. They are well circumscribed, lobulated, angiomatous, and slow growing. Tumor-related cysts may be present. Malignant transformation is uncommon [39]. SEGAs in other locations have been reported, including the cerebral cortex [40], pineal region [41], and retina [42–44].
Histologically, SEGAs may display a wide range of astrocytic, glial, and neuronal differentiation. Three cell types predominate: small spindle cells, gemistocytic astrocytes, and giant cell with ganglionic features (Fig. 10.5). Mitotic index is usually low and necrosis is an uncommon finding. Nucleoli are usually distinct in all of the cell types and a finely granular chromatin pattern is common. SEGAs may display features associated with malignant potential, pleomorphism, mitotic figures, necrosis, and vascularity; however, true malignant behavior is exceedingly rare [39, 45]. (Table 10.2).

Subependymal giant cell astrocytoma shows large mostly polygonal cells with abundant cytoplasm and often vesicular eccentric nucleus with prominent nucleolus (a). The tumor cells share features of glial cells and are immunoreactive for glial fibrillary acidic protein (b, c) but also have neuronal features and are immunopositive for synaptophysin (inset).
Immunohistochemical staining is variably reactive for S-100 and GFAP—a reflection of the mixed astrocytic/glial composition and heterogeneity of the tumor. Neuronal markers including cytoskeletal components (neurofilaments, MAP2, class III Beta tubulin) and neurosecretory substances (serotonin, Beta endorphin, somatostatin) may also be positive [46]. The presence of both glial and neuronal markers within tumor cells supports the possibility that the originating cells of SEGAs have the potential to differentiate along glioneuronal in addition to neuroendocrine lineage, and to a greater degree than other mixed glioneuronal neoplasms [46]. Reported occurrence of SEGAs in the retina [42–44], with Mueller cell origin capable of dedifferentiation into pluripotent progenitor cell as their putative source, illustrates the potential mechanism of a common progenitor producing multiple cell types.
Treatment Options
The optimal treatment of SEGAs and other TSC-related conditions is an area of intense basic, translational, and clinical research. Recognition of the benign nature of these tumors, along with the potential for long life-expectancy mandates that treatment strategies not only result in long-term disease-free or progression-free survival, but also consider potential long-term complications and cost [47].
Observation
Serial clinic examination and radiologic surveillance are appropriate for incidentally discovered or small, stable, or slowly growing SEGAs. Rapid enlargement is unusual and clinical symptoms that are typically insidious allow for treatment on an elective basis [48–50].
Surgery
Various approaches for resection of SEGAs including craniotomy by transcallosal and transcortical approaches have been the reported. Early operative case series noted significant morbidity and mortality [51–53]. Contemporary series, however, with the aid of microdissection, stereotactic techniques, and modern pediatric neuroanesthetic techniques have significantly improved upon the results of these historical benchmarks [54]. A high rate of gross total resection, with little or no permanent neurological morbidity, can be expected at high-volume surgery centers [55–57]. Tumor recurrence after radiographically confirmed gross or radical subtotal resection is infrequent.
The preferred surgical approach depends upon a number of factors, ventricular size, prior surgery, and surgeon experience. Generally, smaller ventricular size favors a transcallosal approach. Significant ventriculomegally and the presence of an existing frontal resection cavity (i.e., from cortical tuber resection) may favor a transcortical approach. Additionally, success of a purely endoscopic approach via a single frontal burr hole has been reported and may be appropriate for some SEGAs [58].
Medical Therapy
SEGAs are slow-growing tumors with low, if any, potential for malignant transformation [39, 59, 60]. Conventional cytotoxic compounds do not have a role in their treatment. However, targeted medical therapy directed specifically at the implicated signal transduction pathways has emerged as a potentially effective and safe strategy to control SEGAs and other manifestations of TS. Progress in this area was initiated literally with the unearthing of rapamycin.
The discovery of the macrolide compound rapamycin began with a “bioprospecting” expedition to Easter Island (“Rapa Nui” in the native language). A soil sample obtained from the site included the bacterium Streptomyces hygroscopicus, from which a secondary metabolite with strong antiproliferative properties was obtained—rapamycin [61]. Eventually, the antifungal properties of rapamycin led to the discovery of its molecular targets—TOR1 and TOR2. Acting to suppress T-function, rapamycin was used in post-transplant patients as an immunosuppressant.
The mechanism of rapamycin and related compounds, known as rapalogs, upon the mTOR pathway is complicated; however, it is known to form a gain-of-function complex with FKBP12, a 12-kDa intracellular protein. This rapamycin-FKBP12 complex inhibits mTOR as component of mTORC1—although the molecular mechanism of this inhibition has not been elucidated. Current theories include impaired structural integrity of mTORC1 [62] and allosteric reduction of the complex’s kinase domain activity [63].
The first rapalog approved in the USA was Temsirolimus for advanced renal cell carcinoma. In 2012 Everolimus was approved by the Food and Drug Administration for the treatment of pediatric and adult patients with TSC who have SEGA that requires therapeutic intervention but cannot be curatively resected [12]. Case reports, clinical series and clinical trials, including a multicenter, placebo-controlled trial [64–68], have demonstrated ≥50 % volumetric reduction of SEGAs among treated patients. Notably, some trials have also demonstrated a meaningful reduction in seizure frequency during treatment [67, 69]. Common side effects include stomatitis, oral ulceration, and impaired wound healing [66, 70]. Metabolic side effects include hypercholesterolemia, hyperlipidemia, and hyperglycemia [71].
In addition to rapamycin and related compounds, the development of small molecule inhibitors of mTOR kinase activity has been investigated [72, 73]. As adenosine triphosphate (ATP)-competitive inhibitors of mTOR, these molecules block all phosphorylation targets downstream of mTORC1 and mTORC2. As a result, these compounds impair cell growth, proliferation, and tumor formation to a much greater extent than rapamycin, which solely inhibits mTORC1.
Radiosurgery
Stereotactic radiosurgery (SRS) has been used as a primary treatment of SEGAs and for tumor recurrence after initial surgical treatment in a small number of cases. Treatment doses of 13–14 Gee to the 50 % iso dose line have been used [74, 75]. A high rate of local control has been reported; however, instances of tumor progression have been noted, some retreated successfully with SRS, while others required surgical excision [74–76]. The risk of radiation-induced secondary tumors is a concern, especially in young patients, and development of glioblastoma has been reported after radiotherapy for SEGA in TSC [77].
Outcomes
CNS involvement is the leading cause of morbidity and mortality in patients with TSC and is usually related to status epilepticus or SEGAs [78]. Renal disease is the second leading cause of early death [9]. Cardiac or pulmonary involvement is also a potential cause of mortality in TSC [78]. Functionally, poorer cognitive outcomes have been shown for patients with bilateral cortical tubers and early age (<6 months) at the onset of seizures [79]. Tuber count or burden has not been shown to correlate with developmental outcome [80]. Treatment of disorders related to TSC, particularly SEGAs, is likely to undergo a tectonic shift. Rather than palliative and piecemeal strategies, targeted molecular therapies (rapalogs, multi kinase inhibitors, and others) are emerging. These agents may not only control tumor growth, but may also prevent CNS developmental malformations, control or prevent seizures, and ultimately lead to improved quality of life and outcomes.
References
Sterman H, Furlan AB, Matushita H, Teixeira MJ. Subependymal giant cell astrocytoma associated with tuberous sclerosis presenting with intratumoral bleeding. Case report and review of literature. Childs Nerv Syst. 2013;29:335–9.
Hahn JS, Bejar R, Gladson CL. Neonatal subependymal giant cell astrocytoma associated with tuberous sclerosis: MRI, CT, and ultrasound correlation. Neurology. 1991;41:124–8.
Oikawa S, Sakamoto K, Kobayashi N. A neonatal huge subependymal giant cell astrocytoma: case report. Neurosurgery. 1994;35:748–50.
Mirkin LD, Ey EH, Chaparro M. Congenital subependymal giant-cell astrocytoma: case report with prenatal ultrasonogram. Pediatr Radiol. 1999;29:776–80.
Nabbout R, Santos M, Rolland Y, Delalande O, Dulac O, Chiron C. Early diagnosis of subependymal giant cell astrocytoma in children with tuberous sclerosis. J Neurol Neurosurg Psychiatry. 1999;66:370–5.
Osborne JP, Fryer A, Webb D. Epidemiology of tuberous sclerosis. Ann N Y Acad Sci. 1991;615:125–7.
Braffman BH, Bilaniuk LT, Naidich TP, et al. MR imaging of tuberous sclerosis: pathogenesis of this phakomatosis, use of gadopentetate dimeglumine, and literature review. Radiology. 1992;183:227–38.
Kawahara I, Tsutsumi K, Hirose M, Matsuo Y, Yokoyama H. [Solitary subependymal giant cell astrocytoma: a forme fruste of tuberous sclerosis complex?]. No To Shinkei. 2004;56:585–91.
Northrup H, Koenig MK, Au KS. Tuberous sclerosis complex. In: Pagon RA, Adam MP, Ardinger HH, et al., editors. Gene reviews. Seattle: University of Washington; 1993. http://www.washington.edu/.
Ichikawa T, Wakisaka A, Daido S, et al. A case of solitary subependymal giant cell astrocytoma: two somatic hits of TSC2 in the tumor, without evidence of somatic mosaicism. J Mol Diagn. 2005;7:544–9.
Kashiwagi N, Yoshihara W, Shimada N, et al. Solitary subependymal giant cell astrocytoma: case report. Eur J Radiol. 2000;33:55–8.
Wander SA, Hennessy BT, Slingerland JM. Next-generation mTOR inhibitors in clinical oncology: how pathway complexity informs therapeutic strategy. J Clin Invest. 2011;121:1231–41.
Laplante M, Sabatini DM. mTOR signaling in growth control and disease. Cell. 2012;149:274–93.
Inoki K, Li Y, Xu T, Guan KL. Rheb GTPase is a direct target of TSC2 GAP activity and regulates mTOR signaling. Genes Dev. 2003;17:1829–34.
Ma L, Chen Z, Erdjument-Bromage H, Tempst P, Pandolfi PP. Phosphorylation and functional inactivation of TSC2 by Erk implications for tuberous sclerosis and cancer pathogenesis. Cell. 2005;121:179–93.
Lee DF, Kuo HP, Chen CT, et al. IKK beta suppression of TSC1 links inflammation and tumor angiogenesis via the mTOR pathway. Cell. 2007;130:440–55.
Inoki K, Ouyang H, Zhu T, et al. TSC2 integrates Wnt and energy signals via a coordinated phosphorylation by AMPK and GSK3 to regulate cell growth. Cell. 2006;126:955–68.
Brugarolas J, Lei K, Hurley RL, et al. Regulation of mTOR function in response to hypoxia by REDD1 and the TSC1/TSC2 tumor suppressor complex. Genes Dev. 2004;18:2893–904.
Stambolic V, MacPherson D, Sas D, et al. Regulation of PTEN transcription by p53. Mol Cell. 2001;8:317–25.
Budanov AV, Karin M. p53 target genes sestrin1 and sestrin2 connect genotoxic stress and mTOR signaling. Cell. 2008;134:451–60.
Foster DA. Phosphatidic acid signaling to mTOR: signals for the survival of human cancer cells. Biochim Biophys Acta. 2009;1791:949–55.
Hara K, Yonezawa K, Weng QP, Kozlowski MT, Belham C, Avruch J. Amino acid sufficiency and mTOR regulate p70 S6 kinase and eIF-4E BP1 through a common effector mechanism. J Biol Chem. 1998;273:14484–94.
Sancak Y, Bar-Peled L, Zoncu R, Markhard AL, Nada S, Sabatini DM. Ragulator-Rag complex targets mTORC1 to the lysosomal surface and is necessary for its activation by amino acids. Cell. 2010;141:290–303.
Dowling RJ, Topisirovic I, Alain T, et al. mTORC1-mediated cell proliferation, but not cell growth, controlled by the 4E-BPs. Science. 2010;328:1172–6.
Duvel K, Yecies JL, Menon S, et al. Activation of a metabolic gene regulatory network downstream of mTOR complex 1. Mol Cell. 2010;39:171–83.
Huang J, Manning BD. The TSC1-TSC2 complex: a molecular switchboard controlling cell growth. Biochem J. 2008;412:179–90.
Kalina P, Drehobl KE, Greenberg RW, Black KS, Hyman RA. Hemorrhagic subependymal giant cell astrocytoma. Pediatr Radiol. 1995;25:66–7.
Waga S, Yamamoto Y, Kojima T, Sakakura M. Massive hemorrhage in tumor of tuberous sclerosis. Surg Neurol. 1977;8:99–101.
Stavrinou P, Spiliotopoulos A, Patsalas I, et al. Subependymal giant cell astrocytoma with intratumoral hemorrhage in the absence of tuberous sclerosis. J Clin Neurosci. 2008;15:704–6.
Ogiwara H, Morota N. Subependymal giant cell astrocytoma with intratumoral hemorrhage. J Neurosurg Pediatr. 2013;11:469–72.
Mork SJ, Morild I, Giertsen JC. Subependymoma and unexpected death. Forensic Sci Int. 1986;30:275–80.
Roach ES, Smith M, Huttenlocher P, Bhat M, Alcorn D, Hawley L. Diagnostic criteria: tuberous sclerosis complex. Report of the Diagnostic Criteria Committee of the National Tuberous Sclerosis Association. J Child Neurol. 1992;7:221–4.
Dabora SL, Jozwiak S, Franz DN, et al. Mutational analysis in a cohort of 224 tuberous sclerosis patients indicates increased severity of TSC2, compared with TSC1, disease in multiple organs. Am J Hum Genet. 2001;68:64–80.
Barkovich AJ. Pediatric neuroimaging. 3rd ed. Philadelphia: Lippincott, Williams & Wilkins; 2000.
Hussain N, Curran A, Pilling D, et al. Congenital subependymal giant cell astrocytoma diagnosed on fetal MRI. Arch Dis Child. 2006;91:520.
Katz JS, Milla SS, Wiggins GC, Devinsky O, Weiner HL, Roth J. Intraventricular lesions in tuberous sclerosis complex: a possible association with the caudate nucleus. J Neurosurg Pediatr. 2012;9:406–13.
Di Rocco C, Iannelli A, Marchese E. On the treatment of subependymal giant cell astrocytomas and associated hydrocephalus in tuberous sclerosis. Pediatr Neurosurg. 1995;23:115–21.
Osborn AG. Diagnostic neuroradiology. 1st ed. St. Louis: Mosby-Year Book; 1994.
Grajkowska W, Kotulska K, Jurkiewicz E, et al. Subependymal giant cell astrocytomas with atypical histological features mimicking malignant gliomas. Folia Neuropathol. 2011;49:39–46.
Bollo RJ, Berliner JL, Fischer I, et al. Extraventricular subependymal giant cell tumor in a child with tuberous sclerosis complex. J Neurosurg Pediatr. 2009;4:85–90.
Dashti SR, Robinson S, Rodgers M, Cohen AR. Pineal region giant cell astrocytoma associated with tuberous sclerosis: case report. J Neurosurg. 2005;102:322–5.
Jakobiec FA, Brodie SE, Haik B, Iwamoto T. Giant cell astrocytoma of the retina. A tumor of possible Mueller cell origin. Ophthalmology. 1983;90:1565–76.
Margo CE, Barletta JP, Staman JA. Giant cell astrocytoma of the retina in tuberous sclerosis. Retina. 1993;13:155–9.
Jung CS, Hubbard II GB, Grossniklaus HE. Giant cell astrocytoma of the retina in a 1-month-old infant. J Pediatr Ophthalmol Strabismus. 2009. doi:10.3928/01913913-20091019-05. Epub 2009 Nov 2.
Telfeian AE, Judkins A, Younkin D, Pollock AN, Crino P. Subependymal giant cell astrocytoma with cranial and spinal metastases in a patient with tuberous sclerosis. Case report. J Neurosurg. 2004;100:498–500.
Lopes MB, Altermatt HJ, Scheithauer BW, Shepherd CW, VandenBerg SR. Immunohistochemical characterization of subependymal giant cell astrocytomas. Acta Neuropathol. 1996;91:368–75.
Roth J, Roach ES, Bartels U, et al. Subependymal giant cell astrocytoma: diagnosis, screening, and treatment. Recommendations from the International Tuberous Sclerosis Complex Consensus Conference 2012. Pediatr Neurol. 2013;49:439–44.
Ekici MA, Kumandas S, Per H, et al. Surgical timing of the subependymal giant cell astrocytoma (SEGA) with the patients of tuberous sclerosis complex. Turk Neurosurg. 2011;21:315–24.
de Ribaupierre S, Dorfmuller G, Bulteau C, et al. Subependymal giant-cell astrocytomas in pediatric tuberous sclerosis disease: when should we operate? Neurosurgery. 2007;60:83–9; discussion 89–90.
Clarke MJ, Foy AB, Wetjen N, Raffel C. Imaging characteristics and growth of subependymal giant cell astrocytomas. Neurosurg Focus. 2006;20:E5.
Ibrahim I, Young CA, Larner AJ. Fornix damage from solitary subependymal giant cell astrocytoma causing postoperative amnesic syndrome. Br J Hosp Med (Lond). 2009;70:478–9.
Jiang T, Jia G, Ma Z, Luo S, Zhang Y. The diagnosis and treatment of subependymal giant cell astrocytoma combined with tuberous sclerosis. Childs Nerv Syst. 2011;27:55–62.
Kotulska K, Borkowska J, Roszkowski M, et al. Surgical treatment of subependymal giant cell astrocytoma in tuberous sclerosis complex patients. Pediatr Neurol. 2014;50:307–12.
Harter DH, Bassani L, Rodgers SD, et al. A management strategy for intraventricular subependymal giant cell astrocytomas in tuberous sclerosis complex. J Neurosurg Pediatr. 2014;13:21–8.
Amin S, Carter M, Edwards RJ, et al. The outcome of surgical management of subependymal giant cell astrocytoma in tuberous sclerosis complex. Eur J Paediatr Neurol. 2013;17:36–44.
Lawton MT, Golfinos JG, Spetzler RF. The contralateral transcallosal approach: experience with 32 patients. Neurosurgery. 1996;39:729–34; discussion 734–5.
Moavero R, Pinci M, Bombardieri R, Curatolo P. The management of subependymal giant cell tumors in tuberous sclerosis: a clinician’s perspective. Childs Nerv Syst. 2011;27:1203–10.
Rodgers SD, Bassani L, Weiner HL, Harter DH. Stereotactic endoscopic resection and surgical management of a subependymal giant cell astrocytoma: case report. J Neurosurg Pediatr. 2012;9:417–20.
Sharma MC, Ralte AM, Gaekwad S, Santosh V, Shankar SK, Sarkar C. Subependymal giant cell astrocytoma–a clinicopathological study of 23 cases with special emphasis on histogenesis. Pathol Oncol Res. 2004;10:219–24.
Nagib MG, Haines SJ, Erickson DL, Mastri AR. Tuberous sclerosis: a review for the neurosurgeon. Neurosurgery. 1984;14:93–8.
Loewith R. A brief history of TOR. Biochem Soc Trans. 2011;39:437–42.
Kim DH, Sarbassov DD, Ali SM, et al. mTOR interacts with raptor to form a nutrient-sensitive complex that signals to the cell growth machinery. Cell. 2002;110:163–75.
Brunn GJ, Fadden P, Haystead TA, Lawrence Jr JC. The mammalian target of rapamycin phosphorylates sites having a (Ser/Thr)-Pro motif and is activated by antibodies to a region near its COOH terminus. J Biol Chem. 1997;272:32547–50.
Birca A, Mercier C, Major P. Rapamycin as an alternative to surgical treatment of subependymal giant cell astrocytomas in a patient with tuberous sclerosis complex. J Neurosurg Pediatr. 2010;6:381–4.
Franz DN, Leonard J, Tudor C, et al. Rapamycin causes regression of astrocytomas in tuberous sclerosis complex. Ann Neurol. 2006;59:490–8.
Franz DN, Belousova E, Sparagana S, et al. Efficacy and safety of everolimus for subependymal giant cell astrocytomas associated with tuberous sclerosis complex (EXIST-1): a multicentre, randomised, placebo-controlled phase 3 trial. Lancet. 2013;381:125–32.
Krueger DA, Care MM, Holland K, et al. Everolimus for subependymal giant-cell astrocytomas in tuberous sclerosis. N Engl J Med. 2010;363:1801–11.
Franz DN, Agricola KD, Tudor CA, Krueger DA. Everolimus for tumor recurrence after surgical resection for subependymal giant cell astrocytoma associated with tuberous sclerosis complex. J Child Neurol. 2013;28(5):602–7.
Kotulska K, Chmielewski D, Borkowska J, et al. Long-term effect of everolimus on epilepsy and growth in children under 3 years of age treated for subependymal giant cell astrocytoma associated with tuberous sclerosis complex. Eur J Paediatr Neurol. 2013;17(5):479–85.
Pengel LH, Liu LQ, Morris PJ. Do wound complications or lymphoceles occur more often in solid organ transplant recipients on mTOR inhibitors? A systematic review of randomized controlled trials. Transpl Int. 2011;24:1216–30.
Sivendran S, Agarwal N, Gartrell B, et al. Metabolic complications with the use of mTOR inhibitors for cancer therapy. Cancer Treat Rev. 2014;40(1):190–6.
Sini P, James D, Chresta C, Guichard S. Simultaneous inhibition of mTORC1 and mTORC2 by mTOR kinase inhibitor AZD8055 induces autophagy and cell death in cancer cells. Autophagy. 2010;6:553–4.
Liu Q, Thoreen C, Wang J, Sabatini D, Gray NS. mTOR mediated anti-cancer drug discovery. Drug Discov Today Ther Strateg. 2009;6:47–55.
Park KJ, Kano H, Kondziolka D, Niranjan A, Flickinger JC, Lunsford LD. Gamma Knife surgery for subependymal giant cell astrocytomas. Clinical article. J Neurosurg. 2011;114:808–13.
Henderson MA, Fakiris AJ, Timmerman RD, Worth RM, Lo SS, Witt TC. Gamma knife stereotactic radiosurgery for low-grade astrocytomas. Stereotact Funct Neurosurg. 2009;87:161–7.
Park YG, Kim EY, Chang JW, Chung SS. Volume changes following gamma knife radiosurgery of intracranial tumors. Surg Neurol. 1997;48:488–93.
Matsumura H, Takimoto H, Shimada N, Hirata M, Ohnishi T, Hayakawa T. Glioblastoma following radiotherapy in a patient with tuberous sclerosis. Neurol Med Chir (Tokyo). 1998;38:287–91.
Shepherd CW, Gomez MR. Mortality in the Mayo Clinic Tuberous Sclerosis Complex Study. Ann N Y Acad Sci. 1991;615:375–7.
Zaroff CM, Barr WB, Carlson C, et al. Mental retardation and relation to seizure and tuber burden in tuberous sclerosis complex. Seizure. 2006;15:558–62.
Kaczorowska M, Jurkiewicz E, Domanska-Pakiela D, et al. Cerebral tuber count and its impact on mental outcome of patients with tuberous sclerosis complex. Epilepsia. 2011;52:22–7.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2015 Springer Science+Business Media New York
About this chapter
Cite this chapter
Harter, D.H., Weiner, H.L., Zagzag, D. (2015). Subependymal Giant Cell Astrocytoma. In: Karajannis, M., Zagzag, D. (eds) Molecular Pathology of Nervous System Tumors. Molecular Pathology Library, vol 8. Springer, New York, NY. https://doi.org/10.1007/978-1-4939-1830-0_10
Download citation
DOI: https://doi.org/10.1007/978-1-4939-1830-0_10
Published:
Publisher Name: Springer, New York, NY
Print ISBN: 978-1-4939-1829-4
Online ISBN: 978-1-4939-1830-0
eBook Packages: MedicineMedicine (R0)