
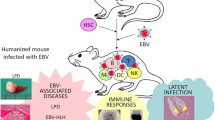
Abstract
Epstein–Barr virus (EBV) is a lymphotropic herpes virus associated with a variety of diseases including infectious mononucleosis, lymphoproliferative diseases, and malignant diseases such as Burkitt lymphoma and Hodgkin lymphoma. EBV is also implicated in various autoimmune diseases. Humans are the only natural host of EBV and small animal models of EBV infection have not been available, although a few new-world monkeys can be infected with the virus experimentally. Humanized mice harboring functioning human lymphocytes can be readily infected with EBV and have reproduced cardinal features associated with human EBV infection, including lymphoproliferative disease, hemophagocytic lymphohistiocytosis, erosive arthritis resembling rheumatoid arthritis, and asymptomatic persistent infection. EBV-specific T-cell responses are induced in humanized mice that protect them from uncontrolled proliferation of EBV-infected cells. Chronic active EBV infection, an EBV-associated T/NK lymphoproliferative disease, that could not be reproduced in humanized mice, has been recapitulated by xenogeneic transplantation of patient’s peripheral blood mononuclear cells to NOG mice. This chapter summarizes features of human EBV infection that were reproduced in humanized mouse models and mouse xenograft models, and shows how they have been utilized to analyze EBV pathogenesis and normal and aberrant human immune responses to the virus.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others

Keywords
- Epstein–Barr virus
- Humanized mouse
- Lymphoproliferative disease
- Hemophagocytic lymphohistiocytosis
- Rheumatoid arthritis
- Chronic active EBV infection
- Latent infection
- Immune responses
- Xenograft model
1 Introduction
Epstein–Barr virus (EBV) , a lymphotropic γ herpesvirus, was first identified in a primary culture of Burkitt lymphoma [1]. Although subsequent studies indicated that EBV is etiologically involved in a number of human malignancies, the virus was found to be a ubiquitous virus latently infecting more than 90 % of the adult population worldwide [1]. Primary EBV infection occurs most often in childhood and is usually asymptomatic or accompanied by only nonspecific flu-like symptoms. Between 25 and 75 % of primarily EBV infection in adolescence and young adulthood results in the development of infectious mononucleosis (IM) [1]. In a restricted fraction of hosts, EBV is involved in the pathogenesis of a wide variety of diseases, including lymphomas, carcinomas, B-cell lymphoproliferative diseases (LPDs) in immunocompromised hosts, and T- and NK-cell LPDs endemic mainly in east Asia [1]. In addition, EBV has been implicated in various autoimmune diseases including rheumatoid arthritis (RA) [2]. EBV has a unique biological activity to transform B lymphocytes into continuously proliferating lymphoblastoid cell lines (LCLs). EBV-transformed lymphoblastoid cells are normally removed by the virus-specific cytotoxic T lymphocytes (CTL) [3], but in immunocompromised hosts such as transplant recipients and AIDS patients, they may proliferate unlimitedly to cause B-cell LPDs such as posttransplant lymphoproliferative disease (PTLD) and AIDS-associated lymphomas.
EBV has a genome of approximately 170 kbp encoding more than 80 genes. EBV-transformed LCLs express six nuclear proteins (EBV nuclear antigens (EBNAs) 1, 2, 3A, 3B, 3C, and LP), three membrane proteins (latent membrane proteins (LMPs) 1, 2A, and 2B), and two sets of untranslated RNAs (EBV-encoded small RNAs (EBERs) 1 and 2, and BamHI-A rightward transcripts (BARTs)) [4]. EBV-infected cells observed in the virus-associated malignancies and LPDs exhibit various patterns of viral gene expression that are characteristic of the respective diseases. These patterns can be categorized into three groups: latency I, characterized by the expression of EBNA1, EBERs, and BARTs, is seen in Burkitt lymphoma and gastric carcinoma; latency II, characterized by the expression of EBNA1, LMP1, LMP2A, EBERs, and BARTs, is found in CAEBV, Hodgkin lymphoma, and nasopharyngeal carcinoma; latency III, characterized by the expression of all EBV-encoded proteins and RNAs identified in EBV-transformed LCLs, is observed in PTLD and AIDS-associated lymphomas [1].
Humans are the only natural host of EBV, but several new-world monkeys and rabbits can be infected with the virus experimentally [5–8]. Cotton-top tamarins that develop B-cell lymphomas following EBV infection were used for the evaluation of a candidate vaccine against the virus [9]. They are, however, endangered species and cannot be used in large numbers. A rhesus monkey lymphocryptovirus homologous to EBV was shown to reproduce key features of human EBV infection, including oral transmission, atypical lymphocytosis, and lymphadenopathy [10, 11]. Rhesus monkeys are thus a promising animal model, although they have disadvantages common to primate models, including limited accessibility and high costs. Small animal models for EBV infection were not available until the development of humanized mice.
2 EBV Pathogenesis in Humanized Mice
2.1 B-Cell Lymphoproliferative Disease
EBV studies with humanized mice started with scid-hu PBL mice. In 1988, Mosier and others demonstrated that intraperitoneal transplantation of peripheral blood mononuclear cells (PBMC) isolated from EBV-infected healthy carriers induced EBV-positive B-cell lymphomas in C.B-17 scid mice [12]. Similar transplantation of PBMC derived from EBV-uninfected donors did not cause lymphomas but subsequent infection with EBV resulted in B-cell lymphomas [13]. Pathological and virological studies demonstrated that these B-cell lymphomas are similar to EBV-associated B-cell LPD in immunocompromised hosts [14, 15]. This early humanized mouse model of EBV-associated B-cell LPD provided various insights into EBV-induced lymphomagenesis. For example, a critical role for CD4+ T cells in in vivo proliferation of EBV-infected B cells was revealed in this model [16, 17]. The scid-hu PBL mouse model also revealed the involvement of human IL-10 and CXCL12/CXCR4 signaling in lymphomagenesis by EBV [18, 19]. Analysis on the IFN-γ gene polymorphism indicated that the A/A genotype for the base + 874 was more prevalent in donors of PBMC that generated aggressive lymphoproliferation in scid-hu PBL mice [20]. Biological studies showed that the IFN-γ allele with adenosine at + 874 was associated with inefficient CTL restimulation, probably explaining the above finding [20]. A study with NOD/scid mice transplanted with human PBMC revealed an important role of plasmacytoid dendritic cells in cellular immune responses against EBV [21].
Although scid-hu PBL mice were the remarkable first small animal model of EBV-associated B-cell LPD, they had disadvantages including inability to mount primary immune responses and a tendency to develop graft-versus-host disease. These disadvantages were removed in new-generation humanized mouse models that were prepared by transplanting human hematopoietic stem cells (HSC) to immunodeficient mice of various strains (e.g. NOD/Shi-scid Il2rg null (NOG), Balb/c Rag2 -/- Il2rg -/- (BRG), and NOD/LtSz-scid Il2rg -/- (NSG)) [22–25]. Models for EBV infection based on new-generation humanized mice are summarized in Table 39.1. Traggiai and others reconstituted human T cells, B cells, and dendritic cells in BRG mice and demonstrated that these mice can be infected with EBV and develop B-cell lymphoproliferation [23]. Characterization of EBV-induced LPD in humanized mice and its comparison to the original human disease was performed by Yajima and others [26] (Figure 39.1). They showed that the development of LPD in EBV-infected humanized NOG (hu-NOG) mice were dependent on viral dose; mice inoculated with more than 102 50 % transforming dose (TD50) tended to develop LPD whereas those inoculated with less than 101 TD50 mostly remained asymptomatic. They demonstrated the latency III type EBV gene expression, expression of B-cell activation markers and germinal center markers, as well as the histology of diffuse large B-cell lymphoma (DLBCL), indicating that EBV-induced LPD closely resembling PTLD and AIDS-associated lymphomas was reproduced (Figure 39.1) [26]. It is interesting that EBV-infected B cells morphologically similar to Hodgkin cells and Reed–Sternberg cells were occasionally seen in EBV-induced LPD in hu-NOG mice (Figure 39.1c). EBV-induced B-cell LPD with similar characteristics was also reproduced in humanized NSG (hu-NSG) mice and BLT NSG mice [27–30]. One of the earliest new-generation humanized mice was prepared by transplanting NOD/scid mice with human HSC, resulting in reconstitution of B cells and myeloid cells but not T cells [31]. Note that B-cell LPD generated in these mice following EBV infection exhibited the latency II type EBV gene expression [31].

EBV-induced lymphoproliferative disease in humanized NOG mice. a Photograph of an EBV-infected mouse showing tumors in the cervical area. b Photographs of spleens, liver, lymph node, and kidney from EBV-infected mice with lymphoproliferative disorder. The upper-left panel shows the spleen from an uninfected mouse. c Photomicrographs of HE-stained tissues of lymphoproliferative disorder. An arrow indicates a Reed–Sternberg-like cell and arrowheads Hodgkin-like cells. d Immunohistochemical staining for lymphocyte surface markers (CD3, CD20, CD23, and Mum1) and EBV-encoded proteins (LMP1 and EBNA2), as well as in situ hybridization for EBER in a lymph node of a mouse bearing lymphoproliferative disorder. The bottom-right panel represents double staining for EBER and CD20. e and f RT-PCR detection of latent-cycle (e) and lytic-cycle (f) EBV gene expression in tumors from EBV-infected hNOG mice. Spleen tumors from three different mice were examined for the expression of EBNAs 1 and 2, LMPs 1, 2A and 2B, EBER1, BZLF1, BMRF1, and BLLF1. RNA samples from LCL e and anti-IgG-treated Akata cells f were used as positive controls, and those of EBV-negative Akata cells (e and f) were used as negative controls. Assays were done with (+) or without (−) reverse transcriptase (RT) in f. Expression of GAPDH was examined as reference. g Double staining of EBER and CD20 in the spleen of a hu-NOG mouse persistently infected with EBV without developing lymphoproliferative disorder. EBER is stained navy in the nucleus and CD20 is stained brown in the membrane. (Reproduced from [26], by permission of Oxford University Press)
Preclinical studies of experimental therapies for EBV-associated LPD have been carried out so far mainly in scid-hu PBL mice. The list of tested regimens includes anti-CTLA-4 antibody [32], rituximab and IL-2 in combination [33], GM-CSF and IL-2 in combination [34], low-dose IL-2 [35], and the combination of CD13/CD19-bispecific antibody, CD28 specific antibody, and autologous T cells [36]. Ganciclovir induced complete regression of B-cell tumors in C.B-17 scid mice generated by transplantation of EBV-transformed LCL cells harboring a thymidine kinase gene driven by EBNA2 [37]. Gurer and others prepared a fusion protein of EBNA1 and the heavy chain of antibody against DEC-205, an endocytic receptor on dendritic cells, and showed that vaccination of hu-NSG mice with this fusion protein primed EBNA1-specific T cells and induced anti-EBNA1 antibodies [38].
2.2 EBV-HLH
Primary EBV infection can be complicated, although in rare occasions, by hemophagocytic lymphohistiocytosis (HLH) , a serious hyperinflammatory condition associated with highly activated but ineffective immune responses [39]. Monoclonal proliferation of EBV-infected T or NK (most often CD8+ T) cells have been consistently observed in EBV-HLH [40, 41]. Overproduction of cytokines by EBV-infected T or NK cells as well as by activated macrophages and T cells reacting to the virus is thought to play a central role in the pathogenesis [42]. Sato and others described systemic organ infiltration of activated CD8+ T cells, IFN-γ cytokinemia, normocytic anemia, thrombocytopenia, and hemophagocytosis in EBV-infected hu-NOG mice, where EBV was found restrictedly in B cells [43]. Based on these findings, they proposed that EBV-infected hu-NOG mice may be a useful model of EBV-HLH. EBV-HLH associated with proliferation of the virus-infected B cells has been recently described in patients with X-linked lymphoproliferative disease 1 (XLP-1) and XLP-2 [44] . Using the same EBV-infected hu-NOG mice, Yajima and others observed predominantly B-cell LPD [26]. Because there were a number of differences in the protocols of Sato et al. [43] and Yajima et al. [26], including the sex of NOG mice, age at transplantation of HSC, and the route of transplantation, these differences might explain the different results. As described later, xenogenic transplantation of PBMC obtained from patients with EBV-HLH also reproduced cardinal features of this disease including proliferation of EBV-infected T cells and hypercytokinemia [45].
2.3 Rheumatoid Arthritis
Rheumatoid arthritis (RA) is a common autoimmune disease associated with progressive disability and systemic complications [46]. Evidence for the involvement of EBV in the RA pathogenesis includes increased EBV-infected B cells in patients’ peripheral blood, infiltration of EBV-specific activated CD8+ T cells in affected joints, and impaired functions of patients’ T cells to suppress the outgrowth of EBV-transformed B cells [47]. Furthermore, cynovial EBV infection with the expression of viral proteins including LMP1 has been repeatedly observed in RA lesions [48, 49]. These lines of evidence are, however, rather circumstantial and direct evidence for the causal relationship has been missing, primarily because of the lack of an appropriate animal model. Recently, Kuwana and others reported that EBV-infected hu-NOG mice developed erosive arthritis with massive synovial cell proliferation and infiltration of human CD4+ T cells, CD8+ T cells, B cells, and macrophages [50]. A histological structure termed pannus, particularly characteristic to RA and involved in the destruction of bone tissues, was found in the arthritis lesions of hu-NOG mice. These results, although restricted to histopathological findings, indicate that EBV triggers the development of erosive arthritis morphologically resembling RA in humanized mice and suggest that the virus is responsible for the development of RA in humans as a trigger. Further studies are required to clarify whether this arthritis in mice is generated by a similar mechanism as that for the original human disease. Effects of antibodies specific to cytokines such as TNF-α, IL-1, and IL-6, that are known to be involved in the pathogenesis of RA, can be readily tested in this model. Inasmuch as there is a strong association between certain HLA types and RA, it will be an interesting experiment to test whether humanized mice prepared with HSC with high-risk HLA types have an increased frequency of arthritis following infection with EBV. The search for signs of other autoimmune diseases such as multiple sclerosis, Sjögren syndrome, and systemic lupus erythematosus in EBV-infected hu-NOG mice is underway.
3 Immune Responses to EBV in Humanized Mice
New-generation humanized mice mount a primary immune response to EBV [22, 23]. The spleen of humanized BRG (hu-BRG) mice contained an increased number of T cells after infection with EBV and CD8+ T cells isolated from them proliferated vigorously upon stimulation with autologous EBV-transformed LCL [23]. In BLT mice developed by Melkus et al. [22], transplanted human thymus tissues enabled restriction of T cells by human MHC and hence efficient EBV-specific T-cell responses. These pioneering works were followed by more detailed descriptions of EBV-specific T-cell responses in humanized mice of various strains [26, 29, 43, 51]. EBV-specific CD4+ and CD8+ T-cell clones were established from EBV-infected hu-NSG mice and found to lyse HLA-matched LCLs [29]. Antibodies specific to the human MHC class-I inhibited the production of IFN-γ by CD8+ T cells that were isolated from EBV-infected BLT or hu-NOG mice and stimulated with autologous LCL, indicating that these T cells recognized EBV epitopes presented by the human MHC class I [22, 26]. In accordance with these results, human MHC class I tetramers presenting EBV epitopes identified EBV-specific CD8+ T cells in hu-NOG mice [43]. Depletion of either CD4+ or CD8+ T cells by administration of anti-CD4 or anti-CD8 antibody, respectively, caused more aggressive proliferation of EBV-infected cells and reduced the life span of infected mice, indicating that T-cell responses induced in humanized mice play a protective role [29, 51]. More directly, CD8+ T cells isolated from EBV-infected hu-NOG mice suppressed transformation of autologous B cells by EBV [51]. EBV infection of hu-NOG and hu-NSG mice induced marked proliferation of CD8+ T cells, but only a minor fraction of them appeared to be EBV-specific [26, 29, 43, 51]; the nature of the remaining part of proliferating CD8+ T cells is not clear. One study with NOD/scid mice transplanted with human fetal thymic cells demonstrated an important role for EBV-induced CD8+ NKT cells in the suppression of tumorigenesis by EBV-associated Hodgkin lymphoma and nasopharyngeal carcinoma cells [52].
It is a puzzling question how EBV-specific T-cell responses restricted by human MHC were induced in humanized mice that did not have human thymus tissues transplanted [26, 29, 43, 51]. T-cell responses specific to other viruses including HIV-1 [53], an adenovirus vector expressing HCG glycoproteins [54], HSV-2 [55], and influenza virus [56] have also been induced in humanized mice without human thymic tissues. Regarding this issue, experiments with humanized NOG I-A-/- mice performed by Watanabe and others suggested that although murine MHC plays a major role in the positive selection of T cells in humanized mice, human HSC-derived cells, conceivably B cells or dendritic cells, are also involved in the positive selection , possibly explaining T-cell restriction by human MHC observed in humanized mice [57]. This problem of T-cell education was averted in humanized mice expressing human MHC; NSG-derived mouse strains with a human HLA-A2 transgene that were reconstituted with HSC having the same HLA allele mounted efficient EBV-specific T-cell responses restricted by the particular HLA type [29, 58].
Antibody responses to EBV were analyzed in hu-NOG mice and found much less efficient than T-cell responses [26]. Only 3 out of 40 mice produced IgM antibody to P18BFRF3, a major component of the viral capsid antigen (VCA). No IgG antibody to EBV-encoded proteins was detected. This inefficiency in antibody responses, especially that in the IgG response, appears common to humanized mouse models of other viruses including HIV-1 [53, 59, 60]. The finding that transfer of functional human T cells expressing TCR specific to hemagglutinin (HA) improved HA-specific IgG responses in hu-NOG mice suggests that suboptimal interactions between T and B cells underlie the inefficient production of antigen-specific IgG antibodies in these mice [57]. Better antibody responses in BLT mice support this notion [61, 62].
4 Persistent EBV Infection in Humanized Mice
EBV establishes asymptomatic persistent infection in human hosts that is dependent on T-cell immunosurveillance. In immunocompetent hosts, EBV-transformed cells with expression of highly immunogenic viral proteins are efficiently removed by CTLs and the virus is found persisting in memory B cells where all viral protein expressions are shut down. Yajima and others reported that the majority of hu-NOG mice inoculated with low doses of EBV (< 101 TD50) did not develop LPD and survived up to 6 months without any apparent signs of diseases [26]. EBV DNA was detected in the peripheral blood only for the first several weeks following inoculation and thereafter remained undetectable throughout the course of observation. Upon autopsy at 6 months postinfection, no macroscopic pathological changes were observed, but occasional EBER-positive cells were found in their spleen and liver, indicating that EBV persisted in the mice. These EBER-positive cells were CD20-positive B cells, but their morphology did not resemble that of resting memory B cells (Figure 39.1g) [26]. mRNAs coding for EBNA1, EBNA2, LMP1, and LMP2A were detected by RT-PCR analyses of RNA obtained from the spleen or liver of these persistently infected mice, indicating the presence of latency III cells (Yajima et al. unpublished results). Persistent EBV infection in hu-NOG mice therefore does not completely reproduce EBV latency in humans. Because proper differentiation of memory B cells requires intricate interactions of B and T cells that have not been reproduced in current humanized mice [57], reproduction of bona fide EBV latency in memory B cells may require more sophisticated humanized mice. Nevertheless, it is an interesting question how immune responses are involved in the induction and maintenance of persistent EBV infection in hu-NOG mice. It is interesting that the blood EBV DNA level fluctuated in a few persistently infected mice and there the rise in the EBV DNA level was immediately followed by the increase in CD8+ T cells and subsequent decline of the EBV DNA level, suggesting an effective T-cell control of EBV-infected cells [51]. If this persistent infection can be disrupted and EBV-positive LPD is induced by certain immunosuppressive regimens, it will be an excellent model of EBV-associated LPD in immunocompromised hosts.
The exact mechanism by which EBV establishes latent infection in memory B cells is not clear. Cocco and others examined EBV gene expression, surface marker expression, and hypermutation of the IG gene variable region in a single cell level in lymphoid tissues of EBV-infected hu-BRG mice [63]. Although they found mainly EBV-infected naïve B cells in the mantle zone, they identified infected cells in latency II that carried mutations in Ig genes in germinal centers. They proposed that these results support the previously presented hypothesis that EBV infects naïve B cells and induces their differentiation into memory B cells via germinal center reactions.
5 EBV Reverse Genetics in Humanized Mice
A number of EBV mutants with their particular genes knocked-out by homologous recombination have been prepared and revealed specific functions of these genes [4]. Mutations of certain EBV genes, however, did not generate any new phenotype in in vitro experiments, although strong conservation of these genes among natural EBV isolates suggests that they have important functions. EBNA3B is a representative of such genes; the EBNA3B knock-out virus retained its capacity to transform B cells and its role in the EBV life cycle is not known. Recent work by White and others demonstrated that an EBNA3B-KO EBV mutant induced more aggressive LPD in hu-NSG mice as compared with wild-type EBV, suggesting a tumor suppressor-like function of this gene [30]. B cells transformed by this mutant virus secreted less T-cell chemoattractant CXCL10 and thereby escaped T-cell–mediated killing. EBNA3B might thus function as a safety guard so that the virus should not be a life-threatening harm to the host.
BZLF1 is an immediate-early gene of EBV and acts as a switch from the latent to lytic cycle of EBV infection. Knocking out the BZLF gene did not affect the in vitro transforming activity of EBV and its involvement in lymphomagenesis was first elucidated in experiments with humanized mice. Ma and others generated EBV recombinants with the BZLF1 gene knocked out or with enhanced BZLF1 expression and demonstrated that BZLF1 enhanced EBV-induced lymphomagenesis in NSG-BLT mice, although the exact mechanism was not clear [27, 28].
There are a number of EBV genes such as BHRF1 (encoding an Bcl-2-like antiapoptotic protein) [64], BXLF1 (encoding EBV thymidine kinase) [65], and BCRF1 (encoding a cytokine highly homologous to human IL-10) [66], loss-of-function mutants of which exhibit no or only minor phenotype alteration in in vitro studies. Examination of these EBV mutants in humanized mice might reveal their critical roles in the EBV life cycle and pathogenesis.
6 Mouse Xenograft Models of EBV-Associated T/NK-Cell LPD
Chronic active EBV infection (CAEBV) is a disease with high morbidity and mortality characterized by prolonged IM-like symptoms and elevated EBV DNA load in the peripheral blood [67–69] . Similar to EBV-HLH, monoclonal or oligoclonal proliferation of EBV-infected T or NK cells is consistently observed in this disease. In EBV-infected hu-NOG mice, however, EBV infection of neither T nor NK cells was recognized. To recapitulate CAEBV and EBV-HLH, Imadome and others transplanted PBMC isolated from patients to NOG mice and recapitulated major features of the two diseases, including systemic monoclonal proliferation of EBV-infected T or NK cells and hypercytokinemia [45] (Figure 39.2). Experiments with these models showed that EBV-infected T and NK cells did not engraft if CD4+ T cells (whether or not infected with EBV) were removed from PBMC. When CD4+ T cells were depleted in vivo just following transplantation of PBMC by administrating the OKT-4 antibody specific to CD4, engraftment of EBV-infected cells was consistently prevented [45]. Furthermore, administration of the antibody after engraftment of EBV-infected cells was also effective and reduced peripheral blood EBV DNA load to an undetectable level, suggesting that therapeutic approaches targeting CD4+ T cells might be possible (Imadome and others, unpublished results).

A xenograft mouse model of chronic active EBV infection. a Photographs of a model mouse showing splenomegaly and of the excised spleen. This mouse was transplanted with PBMC from a CAEBV patient of the CD8 type. Spleen from a control NOG mouse is also shown. b Photomicrographs of various tissues of the mouse shown in a. Upper panels: liver tissue was stained with hematoxylin-eosin (HE), antibodies specific to human CD3 or CD20, or by ISH with an EBER probe; the rightmost panel is a double staining with EBER and human CD45RO. Bottom panels: EBER ISH in the spleen, kidney, lung, and small intestine. c Photomicrographs of the spleen and liver tissues obtained from NOG mice transplanted with PBMC from CAEBV patients of the CD4, γδT, and NK types, respectively. Tissues were stained by EBER-ISH or by double staining with EBER-ISH and human CD45RO. (From [45]. Reproduced under Creative Commons Attribution (CC BY) license.)
Abbreviations
- EBV:
-
Epstein–Barr virus
- IM:
-
Infectious mononucleosis
- LPD:
-
Lymphoproliferative disease
- RA:
-
Rheumatoid arthritis
- LCL:
-
Lymphoblastoid cell line
- CTL:
-
Cytotoxic T lymphocyte
- EBNA:
-
EBV nuclear antigen
- LMP:
-
Latent membrane protein
- EBER:
-
EBV-encoded small RNA
- BART:
-
BamHI-A rightward transcript
- CAEBV:
-
Chronic active EBV infection
- PBMC:
-
Peripheral blood mononuclear cells
- HSC:
-
Hematopoietic stem cell
- HLH:
-
Hemophagocytic lymphohistiocytosis
- XLP:
-
X-linked lymphoproliferative disease
- HA:
-
Hemagglutinin
- NOG:
-
NOD/Shi-scid Il2rg null
- BRG:
-
Balb/c Rag2 -/- Il2rg -/-
- NSG:
-
NOD/LtSz-scid Il2rg -/-
References
Rickinson AB, Kieff ED. Epstein–Barr virus. In: Knipe DM, Howley PM, editors. Fields virology. 5. edn. Philadelphia: Lippincott Williams and Wilkins; 2007. pp. 2655–700.
Niller HH, Wolf H, Ay E, Minarovits J. Epigenetic dysregulation of Epstein–Barr virus latency and development of autoimmune disease. Adv Exp Med Biol. 2011;711:82–102.
Hislop AD, Taylor GS, Sauce D, Rickinson AB. Cellular responses to viral infection in humans: lessons from Epstein–Barr virus. Annu Rev Immunol. 2007;25:587–617.
Kieff ED, Rickinson AB. Epstein–Barr virus and its replication. In: Knipe DM, Howley PM, editors. Fields virology. Philadelphia: Lippincott Williams and Wilkins; 2007. pp. 2603–54.
Shope T, Dechairo D, Miller G. Malignant lymphoma in cotton top marmosets after inoculation with Epstein–Barr virus. Proc Natl Acad Sci U S A. 1973;70(9):2487–91.
Johannessen I, Crawford DH. In vivo models for Epstein–Barr virus (EBV)-associated B cell lymphoproliferative disease (BLPD). Rev Med Virol. 1999;9(4):263–77.
Epstein MA. zur Hausen H, Ball G, Rabin H. Pilot experiments with EB virus in owl monkeys (Aotus trivirgatus). III. Serological and biochemical findings in an animal with reticuloproliferative disease. Int J Cancer. 1975;15(1):17–22.
Takashima K, Ohashi M, Kitamura Y, Ando K, Nagashima K, Sugihara H, et al. A new animal model for primary and persistent Epstein–Barr virus infection: human EBV-infected rabbit characteristics determined using sequential imaging and pathological analysis. J Med Virol. 2008;80(3):455–66.
Epstein MA, Morgan AJ, Finerty S, Randle BJ, Kirkwood JK. Protection of cottontop tamarins against Epstein–Barr virus-induced malignant lymphoma by a prototype subunit vaccine. Nature. 1985;318(6043):287–9.
Moghaddam A, Rosenzweig M, Lee-Parritz D, Annis B, Johnson RP, Wang F. An animal model for acute and persistent Epstein–Barr virus infection. Science. 1997;276(5321):2030–3.
Wang F. A new animal model for Epstein–Barr virus pathogenesis. Curr Top Microbiol Immunol. 2001;258:201–19.
Mosier DE, Gulizia RJ, Baird SM, Wilson DB. Transfer of a functional human immune system to mice with severe combined immunodeficiency. Nature. 1988;335(6187):256–9.
Cannon MJ, Pisa P, Fox RI, Cooper NR. Epstein-Barr virus induces aggressive lymphoproliferative disorders of human B cell origin in SCID/hu chimeric mice. J Clin Invest. 1990;85(4):1333–7.
Rowe M, Young LS, Crocker J, Stokes H, Henderson S, Rickinson AB. Epstein-Barr virus (EBV)-associated lymphoproliferative disease in the SCID mouse model: implications for the pathogenesis of EBV-positive lymphomas in man. J Exp Med. 1991;173(1):147–58.
Mosier DE, Gulizia RJ, Baird SM, Spector S, Spector D, Kipps TJ, et al. Studies of HIV infection and the development of Epstein-Barr virus-related B cell lymphomas following transfer of human lymphocytes to mice with severe combined immunodeficiency. Curr Top Microbiol Immunol. 1989;152:195–9.
Veronese ML, Veronesi A, D’Andrea E, Del Mistro A, Indraccolo S, Mazza MR, et al. Lymphoproliferative disease in human peripheral blood mononuclear cell-injected SCID mice. I. T lymphocyte requirement for B cell tumor generation. J Exp Med. 1992;176(6):1763–7.
Johannessen I, Asghar M, Crawford DH. Essential role for T cells in human B-cell lymphoproliferative disease development in severe combined immunodeficient mice. Br J Haematol. 2000;109(3):600–10.
Baiocchi RA, Ross ME, Tan JC, Chou CC, Sullivan L, Haldar S, et al. Lymphomagenesis in the SCID-hu mouse involves abundant production of human interleukin-10. Blood. 1995;85(4):1063–74.
Piovan E, Tosello V, Indraccolo S, Cabrelle A, Baesso I, Trentin L, et al. Chemokine receptor expression in EBV-associated lymphoproliferation in hu/SCID mice: implications for CXCL12/CXCR4 axis in lymphoma generation. Blood. 2005;105(3):931–9.
Dierksheide JE, Baiocchi RA, Ferketich AK, Roychowdhury S, Pelletier RP, Eisenbeis CF, et al. IFN-gamma gene polymorphisms associate with development of EBV + lymphoproliferative disease in hu PBL-SCID mice. Blood. 2005;105(4):1558–65.
Lim WH, Kireta S, Russ GR, Coates PT. Human plasmacytoid dendritic cells regulate immune responses to Epstein-Barr virus (EBV) infection and delay EBV-related mortality in humanized NOD-SCID mice. Blood. 2007;109(3):1043–50.
Melkus MW, Estes JD, Padgett-Thomas A, Gatlin J, Denton PW, Othieno FA, et al. Humanized mice mount specific adaptive and innate immune responses to EBV and TSST-1. Nat Med. 2006;12(11):1316–22.
Traggiai E, Chicha L, Mazzucchelli L, Bronz L, Piffaretti JC, Lanzavecchia A, et al. Development of a human adaptive immune system in cord blood cell-transplanted mice. Science. 2004;304(5667):104–7.
Ito M, Hiramatsu H, Kobayashi K, Suzue K, Kawahata M, Hioki K, et al. NOD/SCID/gamma(c)(null) mouse: an excellent recipient mouse model for engraftment of human cells. Blood. 2002;100(9):3175–82.
Shultz LD, Lyons BL, Burzenski LM, Gott B, Chen X, Chaleff S, et al. Human lymphoid and myeloid cell development in NOD/LtSz-scid IL2R gamma null mice engrafted with mobilized human hemopoietic stem cells. J Immunol. 2005;174(10):6477–89.
Yajima M, Imadome K, Nakagawa A, Watanabe S, Terashima K, Nakamura H, et al. A new humanized mouse model of Epstein-Barr virus infection that reproduces persistent infection, lymphoproliferative disorder, and cell-mediated and humoral immune responses. J Infect Dis. 2008;198(5):673–82.
Ma SD, Hegde S, Young KH, Sullivan R, Rajesh D, Zhou Y, et al. A new model of Epstein-Barr virus infection reveals an important role for early lytic viral protein expression in the development of lymphomas. J Virol. 2011;85(1):165–77.
Ma SD, Yu X, Mertz JE, Gumperz JE, Reinheim E, Zhou Y, et al. An Epstein-Barr Virus (EBV) mutant with enhanced BZLF1 expression causes lymphomas with abortive lytic EBV infection in a humanized mouse model. J Virol. 2012;86(15):7976–87.
Strowig T, Gurer C, Ploss A, Liu YF, Arrey F, Sashihara J, et al. Priming of protective T cell responses against virus-induced tumors in mice with human immune system components. J Exp Med. 2009;206(6):1423–34.
White RE, Ramer PC, Naresh KN, Meixlsperger S, Pinaud L, Rooney C, et al. EBNA3B-deficient EBV promotes B cell lymphomagenesis in humanized mice and is found in human tumors. J Clin Invest. 2012;122(4):1487–502.
Islas-Ohlmayer M, Padgett-Thomas A, Domiati-Saad R, Melkus MW, Cravens PD, Martin Mdel P, et al. Experimental infection of NOD/SCID mice reconstituted with human CD34+ cells with Epstein-Barr virus. J Virol. 2004;78(24):13891–900.
May KF Jr, Roychowdhury S, Bhatt D, Kocak E, Bai XF, Liu JQ, et al. Anti-human CTLA-4 monoclonal antibody promotes T-cell expansion and immunity in a hu-PBL-SCID model: a new method for preclinical screening of costimulatory monoclonal antibodies. Blood. 2005;105(3):1114–20.
Eisenbeis CF, Grainger A, Fischer B, Baiocchi RA, Carrodeguas L, Roychowdhury S, et al. Combination immunotherapy of B-cell non-Hodgkin's lymphoma with rituximab and interleukin-2: a preclinical and phase I study. Clin Cancer Res. 2004;10(18 Pt 1):6101–10.
Baiocchi RA, Ward JS, Carrodeguas L, Eisenbeis CF, Peng R, Roychowdhury S, et al. GM-CSF and IL-2 induce specific cellular immunity and provide protection against Epstein-Barr virus lymphoproliferative disorder. J Clin Invest. 2001;108(6):887–94.
Baiocchi RA, Caligiuri MA. Low-dose interleukin 2 prevents the development of Epstein-Barr virus (EBV)-associated lymphoproliferative disease in scid/scid mice reconstituted i.p. with EBV-seropositive human peripheral blood lymphocytes. Proc Natl Acad Sci U S A. 1994;91(12):5577–81.
Bohlen H, Manzke O, Titzer S, Lorenzen J, Kube D, Engert A, et al. Prevention of Epstein-Barr virus-induced human B-cell lymphoma in severe combined immunodeficient mice treated with CD3xCD19 bispecific antibodies, CD28 monospecific antibodies, and autologous T cells. Cancer Res. 1997;57(9):1704–9.
Franken M, Estabrooks A, Cavacini L, Sherburne B, Wang F, Scadden DT. Epstein-Barr virus-driven gene therapy for EBV-related lymphomas. Nat Med. 1996;2(12):1379–82.
Gurer C, Strowig T, Brilot F, Pack M, Trumpfheller C, Arrey F, et al. Targeting the nuclear antigen 1 of Epstein-Barr virus to the human endocytic receptor DEC-205 stimulates protective T-cell responses. Blood. 2008;112(4):1231–9.
Henter JI, Horne A, Arico M, Egeler RM, Filipovich AH, Imashuku S, et al. HLH-2004: diagnostic and therapeutic guidelines for hemophagocytic lymphohistiocytosis. Pediatr Blood Cancer. 2007;48(2):124–31.
Kikuta H, Sakiyama Y, Matsumoto S, Oh-Ishi T, Nakano T, Nagashima T, et al. Fatal Epstein-Barr virus-associated hemophagocytic syndrome. Blood. 1993;82(11):3259–64.
Kawaguchi H, Miyashita T, Herbst H, Niedobitek G, Asada M, Tsuchida M, et al. Epstein-Barr virus-infected T lymphocytes in Epstein-Barr virus-associated hemophagocytic syndrome. J Clin Invest. 1993;92(3):1444–50.
Lay JD, Tsao CJ, Chen JY, Kadin ME, Su IJ. Upregulation of tumor necrosis factor-alpha gene by Epstein-Barr virus and activation of macrophages in Epstein-Barr virus-infected T cells in the pathogenesis of hemophagocytic syndrome. J Clin Invest. 1997;100(8):1969–79.
Sato K, Misawa N, Nie C, Satou Y, Iwakiri D, Matsuoka M, et al. A novel animal model of Epstein-Barr virus-associated hemophagocytic lymphohistiocytosis in humanized mice. Blood. 2011;117(21):5663–73.
Yang X, Wada T, Imadome K, Nishida N, Mukai T, Fujiwara M, et al. Characterization of Epstein-Barr virus (EBV)-infected cells in EBV-associated hemophagocytic lymphohistiocytosis in two patients with X-linked lymphoproliferative syndrome type 1 and type 2. Herpesviridae. 2012;3(1):1.
Imadome K, Yajima M, Arai A, Nakazawa A, Kawano F, Ichikawa S, et al. Novel mouse xenograft models reveal a critical role of CD4 + T cells in the proliferation of EBV-infected T and NK cells. PLoS Pathog. 2011;7(10):e1002326.
McInnes IB, Schett G. The pathogenesis of rheumatoid arthritis. N Engl J Med. 2011;365(23):2205–19.
Toussirot E, Roudier J. Pathophysiological links between rheumatoid arthritis and the Epstein-Barr virus: an update. Joint Bone Spine. 2007;74(5):418–26.
Takei M, Mitamura K, Fujiwara S, Horie T, Ryu J, Osaka S, et al. Detection of Epstein-Barr virus-encoded small RNA 1 and latent membrane protein 1 in synovial lining cells from rheumatoid arthritis patients. Int Immunol. 1997;9(5):739–43.
Takeda T, Mizugaki Y, Matsubara L, Imai S, Koike T, Takada K. Lytic Epstein-Barr virus infection in the synovial tissue of patients with rheumatoid arthritis. Arthritis Rheum. 2000;43(6):1218–25.
Kuwana Y, Takei M, Yajima M, Imadome K, Inomata H, Shiozaki M, et al. Epstein-Barr virus induces erosive arthritis in humanized mice. PLoS ONE. 2011;6(10):e26630.
Yajima M, Imadome K, Nakagawa A, Watanabe S, Terashima K, Nakamura H, et al. T cell-mediated control of Epstein-Barr virus infection in humanized mice. J Infect Dis. 2009;200(10):1611–5.
Yuling H, Ruijing X, Li L, Xiang J, Rui Z, Yujuan W, et al. EBV-induced human CD8 + NKT cells suppress tumorigenesis by EBV-associated malignancies. Cancer Res. 2009;69(20):7935–44.
Gorantla S, Makarov E, Finke-Dwyer J, Gebhart CL, Domm W, Dewhurst S, et al. CD8 + cell depletion accelerates HIV-1 immunopathology in humanized mice. J Immunol. 2010;184(12):7082–91.
Marodon G, Desjardins D, Mercey L, Baillou C, Parent P, Manuel M, et al. High diversity of the immune repertoire in humanized NOD.SCID.gamma c-/- mice. Eur J Immunol. 2009;39(8):2136–45.
Kwant-Mitchell A, Ashkar AA, Rosenthal KL. Mucosal innate and adaptive immune responses against herpes simplex virus type 2 in a humanized mouse model. J Virol. 2009;83(20):10664–76.
Yu CI, Gallegos M, Marches F, Zurawski G, Ramilo O, Garcia-Sastre A, et al. Broad influenza-specific CD8+ T-cell responses in humanized mice vaccinated with influenza virus vaccines. Blood. 2008;112(9):3671–8.
Watanabe Y, Takahashi T, Okajima A, Shiokawa M, Ishii N, Katano I, et al. The analysis of the functions of human B and T cells in humanized NOD/shi-scid/gammac(null) (NOG) mice (hu-HSC NOG mice). Int Immunol. 2009;21(7):843–58.
Shultz LD, Saito Y, Najima Y, Tanaka S, Ochi T, Tomizawa M, et al. Generation of functional human T-cell subsets with HLA-restricted immune responses in HLA class I expressing NOD/SCID/IL2r gamma(null) humanized mice. Proc Natl Acad Sci U S A. 2010;107(29):13022–7.
Baenziger S, Tussiwand R, Schlaepfer E, Mazzucchelli L, Heikenwalder M, Kurrer MO, et al. Disseminated and sustained HIV infection in CD34+ cord blood cell-transplanted Rag2-/-gamma c-/- mice. Proc Natl Acad Sci U S A. 2006;103(43):15951–6.
Watanabe S, Terashima K, Ohta S, Horibata S, Yajima M, Shiozawa Y, et al. Hematopoietic stem cell-engrafted NOD/SCID/IL2Rgamma null mice develop human lymphoid systems and induce long-lasting HIV-1 infection with specific humoral immune responses. Blood. 2007;109(1):212–8.
Brainard DM, Seung E, Frahm N, Cariappa A, Bailey CC, Hart WK, et al. Induction of robust cellular and humoral virus-specific adaptive immune responses in human immunodeficiency virus-infected humanized BLT mice. J Virol. 2009;83(14):7305–21.
Sun Z, Denton PW, Estes JD, Othieno FA, Wei BL, Wege AK, et al. Intrarectal transmission, systemic infection, and CD4+ T cell depletion in humanized mice infected with HIV-1. J Exp Med. 2007;204(4):705–14.
Cocco M, Bellan C, Tussiwand R, Corti D, Traggiai E, Lazzi S, et al. CD34+ cord blood cell-transplanted Rag2-/- gamma(c)-/- mice as a model for Epstein-Barr virus infection. Am J Pathol. 2008;173(5):1369–78.
Lee MA, Yates JL. BHRF1 of Epstein-Barr virus, which is homologous to human proto-oncogene bcl2, is not essential for transformation of B cells or for virus replication in vitro. J Virol. 1992;66(4):1899–906.
Shimizu N, Yoshiyama H, Takada K. Clonal propagation of Epstein-Barr virus (EBV) recombinants in EBV-negative Akata cells. J Virol. 1996;70(10):7260–3.
Swaminathan S, Hesselton R, Sullivan J, Kieff E. Epstein-Barr virus recombinants with specifically mutated BCRF1 genes. J Virol. 1993;67(12):7406–13.
Okano M. Overview and problematic standpoints of severe chronic active Epstein-Barr virus infection syndrome. Crit Rev Oncol Hematol. 2002;44(3):273–82.
Straus SE. Acute progressive Epstein-Barr virus infections. Annu Rev Med. 1992;43:437–49.
Kimura H. Pathogenesis of chronic active Epstein-Barr virus infection: is this an infectious disease, lymphoproliferative disorder, or immunodeficiency? Rev Med Virol. 2006;16(4):251–61.
Acknowledgments
This study was supported by grants from the Ministry of Health, Labour and Welfare of Japan (H24-Nanchi-046 and H22-AIDS-I-002), the Grant of National Center for Child Health and Development (25-9), a grant for the Research on Publicly Essential Drugs and Medical Devices from The Japan Health Sciences Foundation (KHD1221), and the Grant-in-Aid for Scientific Research (C) (H22-22590430).
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2014 Springer Science+Business Media New York
About this chapter
Cite this chapter
Fujiwara, S., Matsuda, G., Imadome, KI. (2014). Epstein–Barr Virus Infection in Humanized Mice. In: Poluektova, L., Garcia, J., Koyanagi, Y., Manz, M., Tager, A. (eds) Humanized Mice for HIV Research. Springer, New York, NY. https://doi.org/10.1007/978-1-4939-1655-9_39
Download citation
DOI: https://doi.org/10.1007/978-1-4939-1655-9_39
Published:
Publisher Name: Springer, New York, NY
Print ISBN: 978-1-4939-1654-2
Online ISBN: 978-1-4939-1655-9
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)