
Abstract
Narrow therapeutic index (NTI) drugs are those drugs where small differences in dose or blood concentration may lead to serious therapeutic failures and/or adverse drug reactions that are life-threatening or result in persistent or significant disability or incapacity. This chapter describes various public perspectives on the interchangeability of NTI drugs, discusses the characteristics of NTI drugs, provides an overview of regulatory definitions and bioequivalence approaches taken by major regulatory bodies on NTI drugs, as well as presents case studies to illustrate an example process of classifying drugs with NTI. The FDA’s approach recommends a four-way, fully replicated, crossover design in healthy subjects or patients to permit simultaneous equivalence comparison of the mean and within-subject variability of the test and reference drug products, which offers greater assurance of therapeutic equivalence of NTI drug products. The challenges and future opportunities regarding NTI drugs are also discussed.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others


Keywords
- International Normalize Ratio
- Drug Product
- Reference Product
- Therapeutic Failure
- Narrow Therapeutic Index
These keywords were added by machine and not by the authors. This process is experimental and the keywords may be updated as the learning algorithm improves.
8.1 Introduction
Bioequivalence (BE) is defined as the absence of a significant difference in the rate and extent to which the active ingredient or active moiety in pharmaceutical equivalents or pharmaceutical alternatives becomes available at the site of drug action when administered at the same molar dose under similar conditions in an appropriately designed study. BE studies of systemically absorbed drug products are generally conducted by determining pharmacokinetic endpoints to compare the in vivo rate and extent of drug absorption of a test and a reference drug product in healthy subjects. A test product is considered bioequivalent to a reference product if the 90 % confidence intervals for the geometric mean test/reference ratios of the area under the drug’s plasma concentration versus time curve (AUC) and peak plasma concentration (C max) both fall within the predefined BE limits of 80.00–125.00 %.
Although this BE limit has been successfully used to approve thousands of generic drugs, questions persist about whether it is appropriate for narrow therapeutic index (NTI) drugs, for which small changes in blood concentration could potentially cause serious therapeutic failures and/or serious adverse drug reactions in patients. While health care professionals, pharmaceutical scientists, regulatory agencies, and consumer advocates agree that more stringent criteria for BE should be considered for regulatory approval of NTI drugs, they disagree about how much assurance is needed about the similarity of a generic and its original innovator (reference) product for NTI drugs to be considered therapeutically equivalent.
FDA recently reconsidered the BE approach for NTI drugs and recommends a new approach to demonstrate bioequivalence of NTI drugs (US Food and Drug Administration 2012). This chapter discusses various public perspectives on the interchangeability of NTI drugs, reviews definitions and regulatory BE approaches by various international regulatory bodies, and examines the key characteristics of NTI drugs. The discussion will focus on the FDA’s approach for NTI drugs with illustration of case studies.
8.2 Public Perspectives
There exist numerous anecdotal post-market reports that claim therapeutic failure or increased adverse events when switching from reference to generic drug products. There is, however, no well-controlled clinical study that demonstrates these events are related to switching between generic and reference drug products. Current spontaneous adverse event reporting systems are limited in their ability to compare safety signal between one drug product and another. As a result, the bulk of clinical evidence related to interchangeability of generic drugs is found in case reports and observational studies, which are difficult to prove causality.
Surveys of pharmacists and other health care professionals show that some believe that generic versions of certain drugs should not be dispensed (Kirking et al. 2001; Vasquez and Min 1999). Medical associations have issued various official positions on this issue. A 2006 joint position statement from the American Association of Clinical Endocrinologists, the Endocrine Society, and the American Thyroid Association raised concerns about FDA’s approach for evaluating BE of generic levothyroxine products and recommended that physicians not prescribe generic levothyroxine drug products. The American Medical Association (AMA) issued a report in 2007 (American Medical Association 2007; https://www.aace.com/files/position-statements/aace-tes-ata-thyroxineproducts.pdf) generally backing the use of generic drugs, but recommending continued research into the best approach to determine individual product BE and specifically advocating FDA to reexamine its BE criteria for levothyroxine. The 2006 position statement from the American Academy of Neurology opposed generic substitution of anticonvulsant drugs for the treatment of epilepsy without the attending physician’s approval (Liow et al. 2007). The American Society of Transplantation Conference report indicated that physicians supported the use of generic immunosuppressive agents in low-risk transplant recipients (Alloway 2003), however maintained that data is inadequate to make recommendations on the use of generic immunosuppressant medications in potentially at-risk patient populations (e.g., African Americans and pediatrics). The report recommended that demonstration of BE in at-risk patient populations be incorporated into the generic drug approval process. Although not all drugs in these categories are necessarily NTI drugs, the medical associations’ concerns about interchangeability point to areas for investigation.
Finally, in the United States, policies related to NTI drug substitution differ among states. Currently 13 states list specific NTI drugs that are considered nonsubstitutable (National Association of Boards of Pharmacy 2006). The pharmacy laws of North Carolina require that a prescription for an NTI drug be refilled “using only the same drug product by the same manufacturer that the pharmacist last dispensed under the prescription, unless the prescriber is notified by the pharmacist prior to the dispensing of another manufacturer’s product and the prescriber and the patient give documented consent to the dispensing of the other manufacturer’s product” (Pope 2009). Many states currently have mandatory generic substitution laws, although these laws may vary significantly. In Oklahoma, a pharmacist must obtain approval from the patient or prescriber before substituting with a generic product while in Vermont a physician must provide a statement of generic ineffectiveness to prevent generic substitution. The different approach states take to the regulation on generic substitution of NTI drugs underscores the continued uncertainties in the community.
8.3 Regulatory Definition of Narrow Therapeutic Index Drugs
Several terms are used to describe the drugs in which comparatively small differences in dose or concentration may lead to serious therapeutic failures and/or serious adverse drug reactions in patients, including narrow therapeutic index, narrow therapeutic range, narrow therapeutic ratio, narrow therapeutic window, and critical-dose drugs. Table 8.1 summarizes the terms for this type of drugs used by different regulatory bodies, as well as the drug list in regulatory guidance if provided.
Health Canada has long documented this category of drugs that required greater degree of assurance in bioequivalence studies and named them critical-dose drugs. Critical-dose drugs are defined as those drugs where comparatively small differences in dose or concentration lead to dose- and concentration-dependent, serious therapeutic failures and/or serious adverse drug reactions which may be persistent, irreversible, slowly reversible, or life-threatening, which could result in inpatient hospitalization or prolongation of existing hospitalization, persistent or significant disability or incapacity, or death (Health Canada 2012). Critical-dose drugs apply to products including, but not limited to, those containing cyclosporine, digoxin, flecainide, lithium, phenytoin, sirolimus, tacrolimus, theophylline, and warfarin.
European Medicines Agency (EMA) does not define a set of criteria to categorize drugs as NTI drugs and they decide it case-by-case by Committee for Human Medicinal Products (CHMP) whether an active substance is an NTI drug based on clinical considerations. For instance, in the “Questions & Answers: Positions on specific questions addressed to the pharmacokinetics working party” document, they specify that tacrolimus and cyclosporine are NTI drugs in their individual product bioequivalence guidance (European Medicines Agency).
Japan Pharmaceutical and Food Safety Bureau uses the term narrow therapeutic range drug but has no definition on it in relevant guidelines. Nonetheless, a long list of narrow therapeutic range drug is provided including mostly antiepileptic drugs, antidiabetic compounds, immunosuppressants, and others (Japan Pharmaceutical and Food Safety Bureau 2012a).
For United States Food and Drug Administration, the Code of Federal Regulations (21CFR320.33) uses narrow therapeutic ratio and defines it as follows:
-
(a)
There is less than a twofold difference in median lethal dose (LD50) and median effective dose (ED50) values
-
(b)
There is less than a twofold difference in the minimum toxic concentrations (MTC) and minimum effective concentrations (MEC) in the blood
-
(c)
Safe and effective use of the drug products requires careful titration and patient monitoring
The CFR definition about narrow therapeutic ratio highlights the importance of careful dosage titration and patient monitoring. However, it may not be clinically practical to assess it as the values of LD50, ED50, MTC, or MEC are frequently not available during drug development or even after approval. In its guidances to industry, FDA also identified narrow therapeutic range drug products as those containing certain drug substances that are “subject to therapeutic drug concentration or pharmacodynamic monitoring, and/or where product labeling indicates a narrow therapeutic range designation” (US Food and Drug Administration 2003, 2000).
The 2010 and 2011 FDA advisory committee meeting discussed the definitions of NTI drugs (US Food and Drug Administration 2010b, 2011). Following discussions in conjunction with the committee’s recommendations, FDA is using the term NTI and defining NTI drugs as those drugs where small differences in dose or blood concentration may lead to serious therapeutic failures and/or adverse drug reactions that are life-threatening or result in persistent or significant disability or incapacity (US Food and Drug Administration 2012).
8.4 Characteristic of Narrow Therapeutic Index Drugs
For NTI drugs, small differences in dose or concentration may lead to serious therapeutic failures and/or serious adverse drug reactions in patients. This section describes the general characteristics of NTI drugs, which can be used to classify certain drugs as NTI drugs.
First, we need to determine what are considered serious therapeutic failure or serious adverse drug reactions. If drug concentrations are below therapeutic concentrations for these indications, e.g., epilepsy, depression, schizophrenia, immunosuppression, cardiovascular disease, heart failure and atrial fibrillation, asthma and bronchospasm, anticoagulation, the therapeutic failures may be rated severe. Drug product black box warnings are generally considered as suggestion of severe toxicities. The severe toxicities can be hematological, cardiovascular, and neurological related such as bleeding, QT prolongation, arrhythmia, tachycardia, bradycardia, heart palpitations, hypertension, strokes, coma, seizures, and others. However, only severe toxicities relevant to drug substance are included to support the determination of NTI. For example, the cremophor vehicle for Taxol is thought to be responsible for most of the hypersensitive reactions seen with paclitaxel (Liebmann et al. 1993). The toxicities induced by cremophor in the drug product should not be considered in the determination of the drug substance as NTI. Further, the degree of adverse events or toxic effects should be evaluated based on the relative severity of the disease under investigation. For example, most clinicians will not treat a mild disease at the risk of serious side effects. Yet, one may tolerate more serious side effects to treat a life-threatening disease. Severe allergic reactions such as anaphylaxis are not considered in NTI determination since they are only pertaining to a small specific patient population and are not dose-/concentration-dependent.
Second, NTI drugs often have close therapeutic and toxic doses (or the associated blood/plasma concentrations). Adverse events can either possess their own dose-/concentration-response relationships or reflect extensions of therapeutic effects. Due to limitations in clinical studies, complete dose-/concentration-response curves are seldom developed. Therefore, therapeutic range data, blood concentration data associated with serious toxicity, and/or drug–drug interaction data can be used to estimate the ratio of toxic concentration to effective concentration. Table 8.2 lists some drugs’ therapeutic ranges and estimated toxic/effective concentration ratios. The estimated toxic/effective concentration ratios provide quantitative information about how close the effective and toxic concentrations are. It should be noted that not every drug would have therapeutic range data available. Further, the therapeutic range data are usually the mean estimates for a population, which may not reflect therapeutic range in an individual patient. In addition, the drug concentrations associated with serious toxicities are often not available, which adds challenges to define a clear range between effective and toxic dose/concentration.
Third, as small variations in drug exposure can have significant clinical impact, many NTI drugs are subject to therapeutic drug monitoring based on pharmacokinetic or pharmacodynamics measures. Nevertheless, not all drugs subject to therapeutic monitoring are NTI drugs. For example, clinicians may conduct therapeutic monitoring because patients have potential compliance problems or clinical observation alone could not optimize the drug dose.
Fourth, NTI drugs generally have small to medium within-subject variability (WSV). WSV is estimated via root mean square error (RMSE) values of the bioequivalence parameters C max and AUC0-t (Davit et al. 2008). Here, WSV refers to a measure of variability in blood concentration within the same subject, when the subject is administered two doses of the same formulation on two different occasions (Van Peer 2010). This variability may be intrinsic to the drug substance and/or the formulation, but also includes analytical variability, drug product quality variability, and unexplained random variation. WSV is of particular importance for NTI drugs because variations in plasma concentrations may have severe consequences. Approved drugs with narrow therapeutic indices should have exhibited small WSV. Otherwise, patients would routinely experience cycles of toxicity and lack of efficacy, and therapeutic monitoring would be useless (Benet 2006). A drug is considered highly variable if its WSV for C max and/or area under the curve (AUC) is greater than 30 % (Haidar et al. 2008). Table 8.3 summarizes the residual variability of PK parameters of six drugs from single-dose, two-way, crossover BE studies with mean residual coefficient of variation (CV) ranging from 5.7 to 21.7 %. The mean residual CV includes WSV as well as variations caused by differences between test and references formulations. Therefore, the actual WSV would be even smaller. All drugs in Table 8.3 possess low-to-moderate WSV. In some cases, the clinical use of NTI drug often involves small dose adjustments of less than 20 % (Parks 2006). There is the implicit assumption that product variation and specifically variation introduced by product substitution be less than the size of dose adjustments.
In summary, the following characteristics generally apply to NTI drugs: (a) sub-therapeutic concentrations may lead to serious therapeutic failure; (b) there is little separation between therapeutic and toxic doses (or the associated blood/plasma concentrations); (c) they are subject to therapeutic monitoring based on pharmacokinetic (PK) or pharmacodynamic (PD) measures; (d) they possess low-to-moderate (i.e., no more than 30 %) WSV; and (e) in clinical practice, doses are often adjusted in very small increments (less than 20 %). These characteristics can help the classification of drugs as NTI drugs.
8.5 Bioequivalence Approaches for Narrow Therapeutic Index Drugs
Bioequivalence (BE) studies are an integral component of the drug development and approval process. BE studies are designed to determine if there is a significant difference in the rate and extent to which the active drug ingredient, or active moiety, becomes available at the site of drug action. The conventional bioequivalence study is usually conducted in healthy subjects with pharmacokinetic (PK) endpoints using a single-dose, two-way, crossover study design. Samples of an accessible biologic fluid such as blood or urine are analyzed for drug concentrations, and pharmacokinetic measures such as AUC and peak concentration (C max), are obtained from the resulting concentration-time profiles. The BE parameters, AUC and C max, are statistically analyzed using a two one-sided test procedure to determine whether the average values for the measures estimated after administration of the test and reference products are comparable. Two products are generally judged bioequivalent if the 90 % confidence interval of the geometric mean ratio (GMR) of AUC and C max fall within 80–125 % (US Food and Drug Administration 2003).
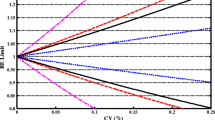
The BE limit of 80.00–125.00 % is based on the premise that a 20 % difference between test and reference product is not clinically significant. The two one-sided test procedure for evaluating BE simultaneously controls the average difference between the test and reference product and the precision with which the population averages are estimated. The precision is determined by the WSV of BE measures and the number of subjects in the study. A drug product with large WSV may need a large number of subjects to pass bioequivalence standards while a product with very low variability may pass with a larger difference in mean response, as shown in Fig. 8.1. The assumption that 20 % difference between test and reference product is not clinically significant may not hold for NTI drugs. Thus, the large difference in mean C max or AUC of two generic NTI products may potentially cause large plasma drug concentration fluctuation when patients switch between two products, potentially resulting in therapeutic failure or serious adverse events. As such, the conventional BE limits may be appropriate for most systemically available drug products, but not necessarily sufficient for NTI drugs.

Effect of variability on BE studies, where T is the test product and R is the reference product
Over the years various regulatory bodies have taken different bioequivalence approaches for NTI drugs (Table 8.4). Essentially, there are two approaches: Direct tightening of average bioequivalence limits and scaled average bioequivalence based on the WSV of the RLD.
8.5.1 Direct Tightening of Average Bioequivalence Limits
Considering that the bioequivalence limits of 80.00–125.00 % for the standard 90 % confidence interval may be too relaxed, some regulatory agencies take the approach of direct tightening of bioequivalence limits to a narrower range.
Health Canada requires the applicant to conduct a single-dose, two-way crossover or parallel study in healthy subjects or patients to demonstrate bioequivalence of NTI drugs. The criterion for the 90 % confidence interval of the relative mean AUC of the test to reference formulation is tightened to 90.0–112.0 % inclusive, whereas the criterion for the 90 % confidence interval of the relative mean C max of the test to reference formulation remains to be 80.0 and 125.0 % (Health Canada 2012). These requirements are to be met in both the fasted and fed states. Steady-state studies are not required for critical-dose drugs unless warranted by exceptional circumstances. If a steady-state study is required, the 90 % confidence interval of the relative mean C min of the test to reference formulation should also be between 80.0 and 125.0 % inclusive. If the bioequivalence study is conducted in patients who are already receiving the drug as part of treatment, Health Canada highly recommends that the study group be as homogeneous as possible with respect to predictable sources of variation in drug disposition.
EMA recommends the acceptance interval for AUC of NTI drugs be tightened to 90.00–111.11 % (European Medicines Agency 2010). Where C max is of particular importance for safety, efficacy, or drug level monitoring, the 90.00–111.11 % acceptance interval should also be applied for C max. Therapeutic Goods Administration (TGA) of Australia follows the EMA guideline about NTI drugs (Therapeutic Goods Administration of Australia). Certain countries within the European Union have more specific policies and guidance related to NTI drugs. For example, the Danish Medicines Agency requires that the 90 % confidence interval for the ratio of the test and reference products for AUC and C max must be within the 90.00–111.11 % (Danish Medicines Agency). Furthermore, the confidence interval must include 100 %.
As of South Africa Medicines Control Council, for general products, the 90 % confidence interval for the test/reference ratio of AUC and C max should lie within the acceptance interval of 80–125 % and 75–133 %, respectively (South Africa Medicines Control Council 2011). For NTI products, the 90 % confidence interval for the test/reference ratio of C max is tightened to 80–125 %.
In Japan, if the 90 % confidence interval of the difference in the average values of logarithmic C max and AUC between test and reference products is within log(0.80)–log(1.25), products are considered to be bioequivalent. In some cases the confidence interval is not within the above range, however the test products can still be accepted as bioequivalent if (1) the total sample size of the initial bioequivalence study is not less than 20 (n = 10/group) or pooled sample size of the initial and add-on subject studies is not less than 30, (2) the differences in average values of logarithmic C max and AUC between two products are within log(0.90)–log(1.11), and (3) dissolution rates of test and reference products are evaluated to be similar. These bioequivalence criteria apply to both conventional drug products and narrow therapeutic range products in Japan (Japan Pharmaceutical and Food Safety Bureau 2012b). However, for NTI drugs, a stricter requirement is used when applying biowaiver among different product strengths. For example, in the case of immediate release (IR) and enteric coated products containing NTI drugs, the test and reference are considered equivalent only if their dissolution profiles meet equivalence criteria and their average dissolution at 30 min are not less than 85 % under multiple testing conditions (Japan Pharmaceutical and Food Safety Bureau 2012a). For conventional drug products, they only need to meet the former dissolution criteria.
8.5.2 Reference-Scaled Average Bioequivalence Approach for NTI Drugs
The long time debate in the United States whether the BE limits of 80.00–125.00 % for the 90 % confidence interval is sufficient for NTI drugs was intensely discussed at the April 2010, FDA advisory committee meeting on NTI drugs (US Food and Drug Administration 2010a). The committee voted 11–2 that the BE criterion for the 90 % confidence interval to be within 80.00–125.00 % is insufficient for NTI drugs. Based on the input from the advisory committee, FDA conducted simulations to investigate the application of different BE approaches for NTI drugs, including the use of (1) direct tightening of BE limits and (2) tightening BE limits based on reference variability (the reference-scaled average BE approach). Variables evaluated in the simulations included WSV, sample size, and point estimate limit. The powers of a given study design using the reference-scaled average BE versus average BE approach were compared. Given the variation of WSV in NTI drugs (Yu 2011), the fixed average BE limits of 90–111 % can be too strict for truly equivalent generic drugs (i.e. GMR = 0.95–1.05) with medium WSV. The simulation results indicated that an approach that tightens BE limits based on reference variability is the preferred approach for evaluating BE of NTI drugs, i.e., the reference-scaled average bioequivalence approach. Based on these efforts, FDA is now recommending a four-way, fully replicated, crossover study design to demonstrate bioequivalence for NTI drugs. This study design permits not only the comparison of the mean of the test and reference drug products, but also the WSV of the test and reference drug products.
8.5.2.1 Mean Comparison
Because both test and reference drug products are given twice in each subject, the four-way, crossover, fully replicated study design enables the scaling of the acceptance BE limits to the WSV of the reference product. Scaled average BE for both AUC and C max is evaluated by testing the following null hypothesis (US Food and Drug Administration 2012):
(given θ > 0) versus the alternative hypothesis:
where μ T and μ R are the averages of the log-transformed measure (C max, AUC) for the test and reference drug products, respectively; usually testing is done at level α = 0.05; and θ is the scaled average BE limits. Furthermore,
where Δ is 1/0.9, the upper BE limit for Test/Reference ratio of geometric means, and σ W0 = 0.10. Note that rejection of the null hypothesis, H0, supports the conclusion of equivalence.
The baseline BE limits for NTI drugs are set at 90.00–111.11 % using the reference WSV (CV) of 10 %, but these limits would be scaled, based on the observed WSV of the reference product in the study. If reference WSV is less than or equal to 10%, then the reference-scaled BE limits are narrower than 90–111.11 %. If reference WSV is greater than 10 %, then the reference-scaled BE limits are wider than 90–111.11 %. These limits expand as the variability increases. However, since it is considered not desirable clinically to have these limits exceeded 80.00–125.00 %, FDA recommends that all BE studies on NTI drugs must pass both the reference-scaled approach and the unscaled average bioequivalence limits of 80.00–125.00 %. Because of these two criteria, the BE limits for these drug products would be tightened as shown in Fig. 8.2.

Implied BE limits of geometric mean ratios for NTI drugs
8.5.2.2 Within-Subject Variability Comparison
WSV is of particular importance for NTI drugs because variations in plasma concentrations may have serious consequences. If an NTI test drug product has much higher WSV than the reference drug product in a BE study, the larger variation in blood concentration may result in higher likelihood of serious therapeutic failures and/or adverse reactions. Therefore, the test/reference ratio of within-subject standard deviation is evaluated. WSV comparison of the test and reference drug products is carried out by a one-sided F test. The null hypothesis for this test is the following.
And the alternative hypothesis is:
where δ is the regulatory limit to declare the WSV of the test drug product is not greater than that of the reference drug product. The 90 % confidence interval of the ratio of the within-subject standard deviation of the test to reference drug product, σ WT/σ WR, is given by \( \left(\frac{s_{\mathrm{WT}}/{s}_{\mathrm{WR}}}{\sqrt{F_{\alpha /2}\left({v}_1,{v}_2\right)}},\frac{s_{\mathrm{WT}}/{s}_{\mathrm{WR}}}{\sqrt{F_{1-\alpha /2}\left({v}_1,{v}_2\right)}}\right), \) where s WT is the estimate of σ WT with v 1 as the degrees of freedom, s WR is the estimate of σ WR with v 2 as the degrees of freedom, F α/2(v 1, v 2) is the value of the F-distribution with v 1 (numerator) and v 2 (denominator) degrees of freedom that has a probability of α/2 to its right, and F 1 − α/2(v 1, v 2) is the value of the F-distribution with v 1 (numerator) and v 2 (denominator) degrees of freedom that has a probability of 1 − α/2 to its right. Here α is equal to 0.1. Equivalent WSV is declared when the upper limit of the 90 % confidence interval for σ WT/σ WR is less than or equal to 2.5—i.e. the test statistic is based on the upper limit of the 90 % confidence interval (Jiang et al. 2012; US Food and Drug Administration 2012).
The reference-scaled average BE approach has been used successfully to demonstrate the BE of highly variable drugs and drug products (Davit et al. 2012). Highly variable drugs and drug products are those having greater than 30 % of WSV in pharmacokinetic measures (AUC and/or C max), and generally exhibit a wide therapeutic window. Using the reference-scaled approach for highly variable drugs and drug products, the sample size required for a BE study is significantly reduced while avoiding the risk of allowing therapeutically inequivalent products to reach the market. Application of the reference-scaled approach for NTI drugs will tighten the BE limits of these drug products and circumvent the possibility of approving a generic product with a large mean difference from its reference drug product. Additional variability comparison will further reduce the risk of approving a generic drug product with a large variability difference from its reference drug product.
8.6 Case Studies
8.6.1 Warfarin (FDA) (http://www.clinicalpharmacology-ip.com/Forms/Monograph/monograph.aspx?cpnum=650&sec=moninte; http://www.thomsonhc.com/micromedex2/librarian/ND_T/evidencexpert/ND_PR/evidencexpert/CS/D343B0/ND_AppProduct/evidencexpert/DUPLICATIONSHIELDSYNC/175679/ND_PG/evidencexpert/ND_B/evidencexpert/ND_P/evidencexpert/PFActionId/evidencexpert.DisplayDrugpointDocument?docId=671285&contentSetId=100&title=Warfarin+Sodium&servicesTitle=Warfarin+Sodium&topicId=administrationMonitoringSection&subtopicId=null)
Warfarin is generally recognized as a NTI drug. Warfarin was first selected as a model drug to undergo a stepwise analysis to determine whether it satisfies all four general characteristics of NTI drugs: (1) sub-therapeutic concentrations may lead to serious therapeutic failure; (2) there is little separation between therapeutic and toxic doses (or the associated blood/plasma concentrations); (3) they are subject to therapeutic monitoring based on pharmacokinetic (PK) or pharmacodynamic (PD) measures; (4) they possess low-to-moderate WSV.
Warfarin is used for the following indication including (1) prophylaxis and/or treatment of venous thrombosis and its extension, and pulmonary embolism; (2) prophylaxis and/or treatment of the thromboembolic complications associated with atrial fibrillation and/or cardiac valve replacement; and (3) reducing the risk of death, recurrent myocardial infarction, and thromboembolic events such as stroke or systemic embolization after myocardial infarction (FDA). The dosage and administration of warfarin must be individualized for each patient according to the particular patient’s prothrombin (PT)/international normalized ratio (INR) response to the drug. If underdosed, failed treatment for the above indications may result in acute or recurrent thromboembolic episodes which are considered severe therapeutic failure. There is a black box warning in the warfarin label. If overdosed warfarin sodium can cause major or fatal bleeding, which are considered serious toxicity.
Warfarin’s dose–response relationship in an individual patient is unpredictable based on population data and therefore a new patient’s maintenance dose is difficult to predict. The label states that “It cannot be emphasized too strongly that treatment of each patient is a highly individualized matter. COUMADIN (Warfarin Sodium), a narrow therapeutic range (index) drug, may be affected by factors such as other drugs and dietary vitamin K. Dosage should be controlled by periodic determinations of prothrombin time (PT)/International Normalized Ratio (INR).” The relationship between warfarin dose and INR response is steep, which may lead to serious therapeutic failures and/or adverse drug reactions and make the selection of a maintenance dose challenging (Dalere et al. 1999) Based on the dose–response curve, one can estimate the toxic and effective doses for patients. INR < 2 or INR > 4 is considered likely to cause therapeutic failure or serious toxicity respectively (US Food and Drug Administration), the corresponding effective and toxic doses would be about 5 and 7 mg, respectively. Therefore, the ratio of toxic dose/effective dose is 1.4, which is very tight. In addition, some drug–drug interaction data also suggest that there is little separation between effective and toxic warfarin doses in patients. For example, studies have shown that rifampin increased the clearance of R-warfarin and S-warfarin 3.5-fold and 2-fold, respectively. Clinicians may need to increase warfarin’s daily dose by two- to threefolds within the 1st week of starting rifampin. Upon discontinuation of rifampin, warfarin doses need to be reduced by half (FDA).
Warfarin undergoes pharmacodynamic monitoring. The biomarker that is used to measure warfarin’s efficacy is the international normalized ratio (INR) and prothrombin time (PT). The INR is a good indicator of effectiveness and risk of bleeding during warfarin therapy. It is recommended to monitor INR levels in warfarin naïve patients starting after the initial two or three doses and at least once per month in patients receiving a stable dose regimen of warfarin (Ansell et al. 2008). Dose adjustment should be individualized to patient’s INR to ensure efficacy and prevent adverse reactions (e.g., excessive bleeding). Patients at a higher risk of bleeding may benefit from more frequent INR monitoring, careful dose adjustment to desired INR, and a shorter duration of therapy.
In addition, the ANOVA RMSE was calculated based on the results of 2-period 2-sequence crossover bioequivalence studies in healthy subjects. Table 8.3 provides a summary of RMSE from approved warfarin ANDAs reviewed between 1996 and 2008. The analysis suggested that warfarin has mean within-subject CV of 5.7 % and 12.7 % for AUC and C max, respectively. RMSE includes the variability between generic and reference drug products. Therefore, actual WSV would be even smaller.
In summary, warfarin therapeutic failure has serious consequences and overdose will cause severe toxicity. The dose that is minimally effective is relatively close to the minimum dose that leads to serious toxicity. Warfarin is subject to regular therapeutic monitoring based on INR, and has a small to medium (<30 %) WSV. Therefore, warfarin is classified as an NTI drug.
Since FDA has concluded that Warfarin sodium is a NTI drug, a fully replicated crossover design was recommended to demonstrate bioequivalence of generic warfarin sodium tablet in both fasting and fed states. The detailed statistical procedure and SAS code were provided in FDA individual bioequivalence guidance database (US Food and Drug Administration 2012) (see “Appendix”).
8.6.2 Tacrolimus (FDA)
Tacrolimus capsule is a calcineurin-inhibitor immunosuppressant indicated for the prophylaxis of organ rejection in patients receiving allogeneic kidney transplants, allogeneic liver transplants, or allogeneic heart transplants. It is often used concomitantly with azathioprine or mycophenolate mofetil (MMF) and adrenal corticosteroids. The consequences of underdosing including morbidity/mortality associated with graft rejection are of major clinical importance and can substantially affect clinical outcome.
Tacrolimus can cause serious toxicities including malignancies, infection, nephrotoxicity, neurotoxicity, and hypertension. The black box warnings in the tacrolimus label include malignancies and serious infections. Patients receiving immunosuppressants, including Prograf, are at increased risk of developing lymphomas and other malignancies, particularly of the skin. The risk appears to be related to the intensity and duration of immunosuppression rather than to the use of any specific agent. Patients receiving immunosuppressants, including Prograf, are at increased risk of developing bacterial, viral, fungal, and protozoal infections, including opportunistic infections. These infections may lead to serious, including fatal outcomes. Tacrolimus can cause acute or chronic nephrotoxicity, particularly when used in high doses. Acute nephrotoxicity is most often related to vasoconstriction of the afferent renal arteriole, which is characterized by increasing serum creatinine, hyperkalemia, and/or a decrease in urine output, and is typically reversible. Chronic CNI nephrotoxicity is associated with mostly irreversible histologic damage to all compartments of the kidneys, including glomeruli, arterioles, and tubulo-interstitium.
The toxic tacrolimus concentration is not well defined. Acute oral overdose has been associated with tacrolimus levels of 19–97 ng/ml. The initial oral dosage recommendations for adult patients with kidney, liver, or heart transplants along with recommendations for whole blood trough concentrations in the package insert are shown in Table 8.5. In the case of heart transplantation, the observed whole blood trough concentrations ranged from 10 to 20 ng/mL. The observed whole blood trough concentration range suggested that tacrolimus has a close effective trough concentration and trough concentration associated with serious toxicity. Masuda and Inui et al. reported that surveys of tacrolimus trough concentrations at the steady-state and clinical events revealed that patients with a trough concentration of between 10 and 20 ng/mL avoided acute cellular rejection, infections, and side effects and most of the adverse effects occurred at a blood concentration higher than 20 ng/mL. Since surveyed patients were safely discharged from hospital without complications, the trough blood concentration of tacrolimus ranging between 10 and 20 (ng/mL) was suggested to be the therapeutic range (Masuda and Inui 2006).
In addition, available drug–drug interaction data also suggest that tacrolimus has a close effective drug concentration and drug concentration associated with serious toxicity. Tacrolimus is metabolized mainly by CYP3A enzymes, drug substances known to inhibit these enzymes may increase tacrolimus whole blood concentrations. Drugs known to induce CYP3A enzymes may decrease tacrolimus whole blood concentration. For example, sirolimus (2–5 mg/day) decreases tacrolimus blood concentrations (mean AUC0-12 and C min by 30 %) vs tacrolimus alone. Sirolimus (1 mg/day) led to decrease in mean AUC0-12 and C min by ~3 % and 11 %, respectively. This extent of tacrolimus pharmacokinetic parameter changes was considered major. Thus, use of sirolimus, in combination with tacrolimus, for prevention of graft rejection is not recommended. However, if concurrent use is deemed necessary, monitoring patients closely for loss of tacrolimus efficacy is required. In summary, therapeutic range data and drug–drug interaction data provide quantitative estimate about the closeness of effective tacrolimus concentration and concentration associated with serious toxicity.
Monitoring of tacrolimus blood concentrations in conjunction with other laboratory and clinical parameters is considered an essential aid to patient management for the evaluation of rejection, toxicity, dose adjustments, and compliance. The relative risks of toxicity and efficacy failure are related to tacrolimus whole blood trough concentrations. Therefore, monitoring of whole blood trough concentrations is recommended to assist in the clinical evaluation of toxicity and efficacy failure. Factors influencing frequency of monitoring include but are not limited to hepatic or renal dysfunction, the addition or discontinuation of potentially interacting drugs and the post-transplant time.
In addition, the ANOVA RMSE from tacrolimus bioequivalence statistical analyses (Table 8.3) suggest that the WSV of tacrolimus is moderate.
In summary, for tacrolimus, therapeutic failure caused by underdose has serious consequences and overdose will cause severe toxicity. The minimum effective drug concentration is relatively close to the minimum drug concentration that leads to serious toxicity. Tacrolimus is subject to therapeutic drug monitoring based on trough whole blood concentration, and has medium (<30 %) WSV. Therefore, tacrolimus meets proposed NTI classification criteria and is an NTI drug.
Tacrolimus is also considered as a critical-dose drug by Health Canada based on the following (Health Canada 2012): (1) Tacrolimus may cause neurotoxicity and nephrotoxicity and the likelihood increases with higher blood levels; (2) Monitoring of tacrolimus blood levels in conjunction with other laboratory and clinical parameters is considered an essential aid to patient management. (3) In kidney transplant patients a significant correlation was found between tacrolimus levels and the incidence of both toxicity and rejection.
EMA also considers tacrolimus as a drug with a NTI (EMA 2012): (1) Tacrolimus is a drug that requires individual dose titration to achieve a satisfactory balance between maximizing efficacy and minimizing serious dose-related toxicity. Plasma level monitoring is routinely employed to facilitate dose titration; (2) Recommended Therapeutic Drug Monitoring schemes often set desirable levels close to the upper or lower limit of the therapeutic window (5 or 20 ng/ml); (3) The consequences of overdosing and of underdosing (including morbidity/mortality associated with graft rejection) are of major clinical importance and can substantially affect clinical outcome.
8.7 Future Perspectives
The adaptation of the BA/BE concept has enabled the approval of quality generic drug products. To provide enhanced assurance of the therapeutic equivalence of NTI drugs, FDA and other regulatory agencies have tightened their bioequivalence limits. As of Oct 2013, FDA has updated two product-specific bioequivalence recommendations and recommended reference-scaled bioequivalence approach for NTI drugs including warfarin sodium tablet and tacrolimus capsule. Broad implementation of this new bioequivalence approach is challenging because some drugs do not have an established NTI classification. It is imperative to establish a systematic process to identify and classify drugs as an NTI. Dose adjustment and therapeutic monitoring data in clinical practice may provide insight about the drug dose/concentration and response relationship. In 2013, FDA has initiated research projects to integrate clinical practice data with statistical tools to characterize the drug dose/concentration–response relationship and classify drugs with NTI (US Food and Drug Administration 2013).
Further, differences do exist in the determination and approval standards for NTI drugs among major regulatory bodies. Generic applicants have to conduct different types of bioequivalence studies for marketing the same generic NTI products in different regions of the world. A global NTI drug list and harmonized bioequivalence criteria are essential for creating lasting standards and will speed up the development and approval processes of generic NTI drugs.
References
21CFR320.33.
American Association of Clinical Endocrinologists (AACE), The Endocrine Society (TES), and American Thyroid Association (ATA). Joint position statement on the use and interchangeability of thyroxine products [Online]. https://www.aace.com/files/position-statements/aace-tes-ata-thyroxineproducts.pdf. Accessed
Digoxin [Online]. http://www.clinicalpharmacology-ip.com/Forms/Monograph/monograph.aspx?cpnum=190&sec=monmp. Accessed
Lithium [Online]. http://www.clinicalpharmacology-ip.com/Forms/Monograph/monograph.aspx?cpnum=351&sec=monmp. Accessed
Phenytoin [Online]. http://www.clinicalpharmacology-ip.com/Forms/drugoptions.aspx?cpnum=484&n=Phenytoin. Accessed
Theophylline [Online]. http://www.clinicalpharmacology-ip.com/Forms/Monograph/monograph.aspx?cpnum=599&sec=monmp. Accessed
Therapeutic Drug Levels [Online]. http://www.nlm.nih.gov/medlineplus/ency/article/003430.htm. Accessed
Warfarin [Online]. http://www.clinicalpharmacology-ip.com/Forms/Monograph/monograph.aspx?cpnum=650&sec=moninte. Accessed
Alloway RREA (2003) Report of the American Society of Transplantation conference on immunosuppressive drugs and the use of generic immunosuppressants. Am J Transplant 3:1211–1215
American Medical Association (2007) Generic substitution of narrow therapeutic index drugs. Report 2 of the council on science and public health [Online]. http://www.ama-assn.org/resources/doc/csaph/csaph2a07-fulltext.pdf. Accessed
Ansell J, Hirsh J, Hylek E, Jacobson A, Crowther M, Palareti G (2008) Pharmacology and management of the vitamin K antagonists: American College of Chest Physicians Evidence-Based Clinical Practice Guidelines (8th edition). Chest 133:160S–198S
Benet LZ (2006) Why highly variable drugs are safer? www.fda.gov/ohrms/dockets/ac/06/slides/2006-4241s2_2.ppt. Accessed
Dalere GM, Coleman RW, Lum BL (1999) A graphic nomogram for warfarin dosage adjustment. Pharmacotherapy 19:461–467
Danish Medicines Agency. http://laegemiddelstyrelsen.dk/en/topics/authorisation-and-supervision/licensing-of-medicines/marketing-authorisation/application-for-marketing-authorisation/bioequivalence-and-labelling-of-medicine--bstitution.aspx. Accessed
Davit BM, Conner DP, Fabian-Fritsch B, Haidar SH, Jiang X, Patel DT, Seo PR, Suh K, Thompson CL, Yu LX (2008) Highly variable drugs: observations from bioequivalence data submitted to the FDA for new generic drug applications. AAPS J 10:148–156
Davit BM, Chen ML, Conner DP, Haidar SH, Kim S, Lee CH, Lionberger RA, Makhlouf FT, Nwakama PE, Patel DT, Schuirmann DJ, Yu LX (2012) Implementation of a reference-scaled average bioequivalence approach for highly variable generic drug products by the US Food and Drug Administration. AAPS J 14:915–924
US Food and Drug Administration (2000) Guidance for industry: waiver of in vivo bioavailability and bioequivalence studies for immediate-release solid oral dosage forms based on a biopharmaceutics classification system [Online]. http://www.fda.gov/downloads/Drugs/…/Guidances/ucm070246.pdf. Accessed 16 June 2013
US Food and Drug Administration (2003) Guidance for industry: bioavailability and bioequivalence studies for orally administered drug products—general considerations [Online]. http://www.fda.gov/downloads/Drugs/…/Guidances/ucm070124.pdf. Accessed 16 June 2013
US Food and Drug Administration (2010a) April 13, 2010 Meeting of the Pharmaceutical Science and Clinical Pharmacology Advisory Committee: topic 1, revising the BE approaches for critical dose drugs [Online]. http://www.fda.gov/AdvisoryCommittees/Calendar/ucm203405.htm. Accessed 16 June 2013
US Food and Drug Administration (2010b) US FDA Pharmaceutical Science and Clinical Pharmacology Advisory Committee Meeting, April 13, 2010 [Online]. http://www.fda.gov/AdvisoryCommittees/Calendar/ucm203405.htm. Accessed
US Food and Drug Administration (2011) US FDA Pharmaceutical Science and Clinical Pharmacology Advisory Committee Meeting, July 26, 2011 [Online]. http://www.fda.gov/AdvisoryCommittees/Calendar/ucm261780.htm. Accessed
US Food and Drug Administration (2012) Draft guidance on warfarin sodium [Online]. http://www.fda.gov/downloads/Drugs/GuidanceComplianceRegulatoryInformation/Guidances/UCM201283.pdf. Accessed
US Food and Drug Administration (2013) Collection of dose adjustment and therapeutic monitoring data to aid narrow therapeutic index drug classification (U01) [Online]. http://grants.nih.gov/grants/guide/rfa-files/RFA-FD-13-020.html. Accessed
EMA (2014) Committee for Human Medicinal Products (CHMP) questions & answers: positions on specific questions addressed to the Pharmacokinetics Working Party. EMA/618604/2008 Rev. 9
European Medicines Agency (2010) Guideline on the investigation of bioequivalence [Online]. http://www.ema.europa.eu/docs/en_GB/document_library/Scientific_guideline/2010/01/WC500070039.pdf. Accessed
European Medicines Agency. Clinical efficacy and safety: clinical pharmacology and pharmacokinetics [Online]. http://www.ema.europa.eu/ema/index.jsp?curl=pages/regulation/general/general_content_000370.jsp&mid=WC0b01ac0580032ec5. Accessed
FDA. Warfarin sodium label [Online]. http://www.accessdata.fda.gov/scripts/cder/drugsatfda/index.cfm?fuseaction=Search.Overview&DrugName=WARFARIN%20SODIUM. Accessed
Japan Pharmaceutical and Food Safety Bureau (2012a) Guideline for bioequivalence studies for different strengths of oral solid dosage forms [Online]. http://www.nihs.go.jp/drug/be-guide(e)/strength/GL-E_120229_ganryo.pdf. Accessed
Japan Pharmaceutical and Food Safety Bureau (2012b) Guideline for bioequivalence studies of generic products [Online]. http://www.nihs.go.jp/drug/be-guide(e)/Generic/GL-E_120229_BE.pdf. Accessed
Haidar SH, Davit B, Chen ML, Conner D, Lee L, Li QH, Lionberger R, Makhlouf F, Patel D, Schuirmann DJ, Yu LX (2008) Bioequivalence approaches for highly variable drugs and drug products. Pharm Res 25:237–241
Health Canada (2012) Comparative bioavailability standards: formulations used for systemic effects [Online]. http://www.hc-sc.gc.ca/dhp-mps/alt_formats/pdf/prodpharma/applic-demande/guide-ld/bio/gd_standards_ld_normes-eng.pdf. Accessed
Jiang W, Schiurman D, Makhlouf F, Lionberger R, Zhang X, Patel D, Subramaniam S, Connor D, Davit B, Grosser S, Yu L (2012) Within-subject variability comparison of narrow therapeutic index drug products. In: American association of pharmaceutical scientist, Chicago, IL
Kirking DM, Gaither CA, Ascione FJ, Welage LS (2001) Pharmacists’ individual and organizational views on generic medications. J Am Pharm Assoc 41:723–728
Liebmann J, Cook JA, Mitchell JB (1993) Cremophor EL, solvent for paclitaxel, and toxicity. Lancet 342:1428
Liow K, Barkley GL, Pollard JR, Harden CL, Bazil CW (2007) Position statement on the coverage of anticonvulsant drugs for the treatment of epilepsy. Neurology 68:1249–1250
Masuda S, Inui K (2006) An up-date review on individualized dosage adjustment of calcineurin inhibitors in organ transplant patients. Pharmacol Ther 112:184–198
National Association of Boards of Pharmacy (2006) Survey of pharmacy laws. XIX: drug product selection laws http://www.nabp.net/publications/survey-of-pharmacy-law/
Parks MH (2006) Clinical perspectives on levothyroxine sodium products [Online]. http://www.fda.gov/ohrms/dockets/ac/06/slides/2006-4228S1-01-03-Parks%20Clinical.pdf. Accessed
Pope ND (2009) Generic substitution of narrow therapeutic index drugs. US Pharm (Generic Drug Review Suppl) 34:12–19
South Africa Medicines Control Council (2011) Biostudies [Online]. http://www.mccza.com/dynamism/default_dynamic.asp?grpID=30&doc=dynamic_generated_page.asp&categID=177&groupID=30. Accessed
Therapeutic Goods Administration of Australia. http://www.tga.gov.au/pdf/euguide/ewp140198rev1.pdf. Accessed
US Food and Drug Administration. Warfarin sodium label [Online]. http://www.accessdata.fda.gov/drugsatfda_docs/label/2011/009218s107lbl.pdf. Accessed
Van Peer A (2010) Variability and impact on design of bioequivalence studies. Basic Clin Pharmacol Toxicol 106:146–153
Vasquez EM, Min DI (1999) Transplant pharmacists’ opinions on generic product selection of critical-dose drugs. Am J Health Syst Pharm 56:615–621
Yu LX (2011) Quality and bioequivalence standards for narrow therapeutic index drugs [Online]. http://www.fda.gov/downloads/Drugs/DevelopmentApprovalProcess/HowDrugsareDevelopedandApproved/ApprovalApplications/AbbreviatedNewDrugApplicationANDAGenerics/UCM292676.pdf. Accessed Dec 2013
Disclaimer
The views presented in this article by the authors do not necessarily reflect those of the US FDA.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Appendix: Method for Statistical Analysis Using the Reference-Scaled Average Bioequivalence Approach for Narrow Therapeutic Index Drugs
Appendix: Method for Statistical Analysis Using the Reference-Scaled Average Bioequivalence Approach for Narrow Therapeutic Index Drugs
Step 1. Determine s WR, the estimate of within-subject standard deviation (SD) of the reference product, for the pharmacokinetic (PK) parameters AUC and C max. Calculation for s WR can be conducted as follows:
where: i = number of sequences m used in the study; [m = 2 for fully replicated design: TRTR and RTRT]; j = number of subjects within each sequence; T = Test product; R = Reference product
D ij = R ij1 − R ij2 (where 1 and 2 represent replicate reference treatments)
\( n={\displaystyle \sum_{i=1}^m{n}_j} \) (i.e. total number of subjects used in the study, while n i is the number of subjects used in sequence i)
Step 2. Use the reference-scaled procedure to determine BE for individual PK parameter(s).
Determine the 95 % upper confidence bound for:
Where:
-
\( \overline{Y_{\mathrm{T}}} \) and \( \overline{Y_{\mathrm{R}}} \) are the means of the ln-transformed PK endpoint (AUC and/or C max) obtained from the BE study for the test and reference products, respectively
-
\( \theta \equiv {\left(\frac{ \ln \left(\varDelta \right)}{\sigma_{\mathrm{W}0}}\right)}^2 \) (scaled average BE limit)
-
and σ W0 = 0.10 (regulatory constant), Δ = 1.11111 (=1/0.9, the upper BE limit)
The method of obtaining the upper confidence bound is based on Howe’s Approximation I, which is described in the following paper:
W.G. Howe (1974), Approximate Confidence Limits on the Mean of X + Y Where X and Y are Two Tabled Independent Random Variables, Journal of the American Statistical Association, 69 (347): 789–794.
Step 3. Use the unscaled average bioequivalence procedure to determine BE for individual PK parameter(s). Every study should pass the scaled average bioequivalence limits and also unscaled average bioequivalence limits of 80.00–125.00 %.
Step 4. Calculate the 90 % confidence interval of the ratio of the within-subject standard deviation of test product to reference product σ WT/σ WR. The upper limit of the 90 % confidence interval for σ WT/σ WR will be evaluated to determine if σ WT and σ WR are comparable. The proposed acceptance criteria for the upper limit of the 90 % equal-tails confidence interval for σ WT/σ WR is less than or equal to 2.5.
The (1 − α)100 % CI for \( \frac{\sigma_{\mathrm{WT}}}{\sigma_{\mathrm{WR}}} \) is given by
where
-
s WT is the estimate of σ WT with v 1 as the degree of freedom
-
s WR is the estimate of σ WR with v 2 as the degree of freedom
-
\( {F}_{\alpha /2,{\nu}_1,{\nu}_2} \) is the value of the F-distribution with ν 1 (numerator) and ν 2 (denominator) degrees of freedom that has probability of α/2 to its right.
-
\( {F}_{1-\alpha /2,{\nu}_1,{\nu}_2} \) is the value of the F-distribution with ν 1 (numerator) and ν 2 (denominator) degrees of freedom that has probability of 1 − α/2 to its right.
-
here α = 0.1.
-
If SAS® is used for statistical analysis*
-
PROC MIXED should be used for fully replicated (4-period, 2-sequence replicated crossover 4-way) BE studies
-
*not necessary to use SAS® if other software accomplishes same objectives
8.1.1 Example SAS Codes: 4-Period, 2-Sequence Replicated Crossover Study
For a bioequivalence study with the following sequence assignments in a fully replicated 4-way crossover design:
Period 1 | Period 2 | Period 3 | Period 4 | |
|---|---|---|---|---|
Sequence 1 | T | R | T | R |
Sequence 2 | R | T | R | T |
The following codes are an example of the determination of reference-scaled average bioequivalence for LAUCT. Assume that the datasets TEST and REF, have already been created, with TEST having all of the test observations and REF having all of the reference observations.
Dataset containing TEST 1 observations:

Dataset containing TEST 2 observations:

Dataset containing REFERENCE 1 observations:

Dataset containing REFERENCE 2 observations:

The number of subjects in each sequence is n1 and n2 for sequences 1 and 2, respectively.
Define the following quantities:
T ijk = kth observation (k = 1 or 2) on T for subject j within sequence i
R ijk = kth observation (k = 1 or 2) on R for subject j within sequence i
and
I ij is the difference between the mean of a subject’s (specifically subject j within sequence i) two observations on T and the mean of the subject’s two observations on R, while D ij is the difference between a subject’s two observations on R.
Determine I ij and D ij

Intermediate analysis—ilat

From the dataset IOUT2, calculate the following:
IOUT2:

Intermediate analysis—dlat

From the dataset DOUT1, calculate the following:
DOUT1: s2wr=estimate/ 2;
From the dataset DOUT2, calculate the following:
DOUT2: dfd=df;
From the above parameters, calculate the final 95 % upper confidence bound:

Calculate the unscaled average bioequivalence limits:
Calculation of unscaled 90 % bioequivalence confidence intervals:

Rights and permissions
Copyright information
© 2014 The United States Government
About this chapter
Cite this chapter
Jiang, W., Yu, L.X. (2014). Bioequivalence for Narrow Therapeutic Index Drugs. In: Yu, L., Li, B. (eds) FDA Bioequivalence Standards. AAPS Advances in the Pharmaceutical Sciences Series, vol 13. Springer, New York, NY. https://doi.org/10.1007/978-1-4939-1252-0_8
Download citation
DOI: https://doi.org/10.1007/978-1-4939-1252-0_8
Published:
Publisher Name: Springer, New York, NY
Print ISBN: 978-1-4939-1251-3
Online ISBN: 978-1-4939-1252-0
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)