
Abstract
Myocardial ischemia is universally accepted to be the result of an imbalance between oxygen supply and requirements to the myocardium. The presence of flow limiting coronary stenosis is the main recognized pathological mechanism underlying this condition. While revascularization procedures are performed with the aim to remove the flow limiting stenosis, traditional medical therapy with hemodynamic agents aim at reducing oxygen demand of the myocardium. However, although effective, none of these treatment strategies or their combination confers symptomatic relief in all patients, in this way underlying the need for further research in this area.
Metabolic derangement is critical in patients who presents with ischemic heart disease (IHD). Under normal conditions the heart derives most of its energy from β-oxidation of free fatty acids (FA). However, the healthy heart is able to easily switch from one substrate to another according to substrate availability, nutritional status, and exercise level. Paradoxically, during prolonged and severe ischemia the myocardium continues to derive most of its energy (50–70 %) from β-oxidation, despite a high rate of lactate production. At this stage it is believed that FA oxidation can turn to be detrimental in that, while requiring more oxygen, it produces less ATP. Given such metabolic derangements, pharmacological approaches aimed at rebalancing myocardial metabolism may play a key role in treatment of patients with IHD. In this scenario, therapeutic interventions aiming at a shift of myocardial substrate utilization towards glucose metabolism would particularly benefit cardiac efficiency and IHD symptoms. In the next session principal metabolic agents will be discussed to further address their role in IHD.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others

Keywords
1 Introduction
In the last thirty years mortality from cardiovascular disease has shown a global reduction. Such results have been mainly attributed to improved pharmacological and mechanical treatments, as well as efficacious educational programs. However, ischemic heart disease (IHD) remains the most important cause of death and morbidity in the western countries [1, 2]. Myocardial ischemia is commonly attributed to obstructive coronary artery disease (CAD) that, by limiting coronary blood flow, cause an unbalance between myocardial metabolic demands and blood supply. Based on this concept, anti-ischemic therapy has been mainly focused on the mechanical removal of the coronary obstructions, and/or pharmacological modulation of cardiac work and coronary blood flow, or both. These strategies are expected to restore an adequate supply/demand balance, improve symptoms and prolong survival. Unfortunately, an objective evaluation of available evidence does not confirm such expectations. Actually, several trials have reported that over 30 % of patients still experience angina despite “successful” coronary revascularization, by percutaneous coronary intervention (PCI) or by coronary artery bypass grafting (CABG) [3, 4]. Although with a certain degree of inertia, factors other than epicardial stenosis, such as microvascular dysfunction and metabolic derangement have been suggested as the underlying physiopathological mechanisms for persistent symptoms. Given the above mentioned incomplete success with current treatment, therapeutic strategies that target these “alternative physiopathological mechanisms” (i.e. metabolic modulation) have gained augmented interest.
Indeed, studies have shown that metabolic modulation therapy plays a critical role in the acute phase of ischemic events, impacting on the results of acute interventions, in the subsequent development of heart failure (HF)-stunned and hybernated myocardium- as well as for those who experience chronic stable angina [5]. A better understanding of the metabolic changes occurring at the time of ischemic events and following reperfusion are translating into new therapeutic opportunities.
2 Energy Metabolism and Myocardial Ischemia
As described in details in the previous chapters, the elevated energy demands of the heart are met by the hydrolysis of adenosine triphosphate (ATP), which is derived from a number of different metabolic pathways, including glycolysis and the mitochondrial oxidation of glucose, lactate, fatty acids (FA), and ketones. Under normal, aerobic conditions, 50–70 % of ATP requirements are derived from fatty acid ß-oxidation, with the remainder being mainly derived from carbohydrates oxidation, primarily glucose and lactate [6–8].
Importantly, the healthy heart is able to easily switch from one substrate to another according to substrate availability, nutritional status, and exercise level. For example, during increased workload (i.e. inotropic challenge) energy demands are primarily met with increase in the glycolytic rates and carbohydrate oxidation, whereas exogenous fatty acid oxidation largely remains unchanged [9, 10].
On the other hand, in conditions of chronic neuro-hormonal activation (i.e. heart failure, HF) there is an enhanced catecholamine induced activation of lipolysis and up-regulation of genes associated with FA use via peroxisome proliferator-activated receptor (PPAR) which ultimately lead to increased FA delivery to the heart. In addition, FA promote their own uptake and oxidation and antagonize the uptake of glucose, lactate, and pyruvate, in part through direct inhibition of pyruvate dehydrogenase. Mitochondrial effects of FA include uncoupling of cellular respiration, resulting in decreased ATP production and oxygen wasting. Thus, elevated blood levels of FA augment lactate and proton accumulation, decrease cellular pH, and disrupt cellular function. Other consequences of excessive FA concentration in the blood stream include impaired calcium handling, oxidative stress, reduced activity of the glucose transporter (GLUT)-4 and myocytes apoptosis [11].
Similarly, energy substrate utilization is a determinant factor of the capability of the heart to sustain ischemic insults [6, 12]. During mild to moderate ischemia, myocardial cells enhance glucose uptake in order to generate sufficient ATP for maintaining appropriate ionic gradients and calcium homeostasis. Paradoxically, during prolonged and severe ischemia, myocardium continues to derive most of its energy from FA β-oxidation, despite a high rate of lactate production. In this conditions, acute allosteric and/or covalent modulation of cardiac energy metabolism occur and high rates of FA oxidation further inhibit glucose oxidation due to competitive interaction (Randle phenomenon). Although the complete oxidation of FA yields more adenosine triphosphate (ATP) than does complete oxidation of glucose, it requires a greater amount of oxygen. Therefore, for a given amount of oxygen consumed, metabolism of glucose is more “energy efficient,” producing about 15 % more ATP. During sustained ischemia, when O2 availability is very low and metabolic supply is impeded, FA oxidation can turn to be detrimental. By requiring more oxygen, it produces less ATP and more reactive oxygen species (ROS), all events that concur in depressing mitochondrial respiratory efficiency. However, absolute FA and carbohydrates oxidation is limited by the lack of an adequate supply of oxygen to the muscle during ischemia. In the post-ischemic period, rates of FA oxidation rapidly recover in the face of depressed contractile function, whereas rates of glucose oxidation remain low [6, 13, 14].
In conclusion, alterations in the availability of oxygen and circulating energy substrates, as well as alterations in the mechanisms regulating substrate metabolism contribute to the metabolic phenotype during ischemia, and can be responsible for disruption of cell homeostasis, alterations in cellular and mitochondrial membrane structure, and altered cardiac contractility and efficiency.
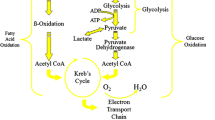
Based on this rationale, pharmacological agents and strategies that shift the balance of oxidative ATP production from FA β-oxidation towards glucose oxidation [15–21] increase cardiac post-ischemic recovery function and efficiency during chronic ischemia [22, 23]. Figures 1 and 2 encompass normal energy generation in the healthy heart and metabolic derangement in the ischemic heart, respectively.

Schematic representation of cardiac metabolism in normal condition. The heart derives most of its energy by fatty acids oxidation, but nutritional status (fasting, substrate availability) as well as exercise and hormones can modify fuel selection (see text for details)

Metabolic alterations during sustained period of ischemia. Glycolysis is the main catabolic pathway during ischemia, whereas fatty acid oxidation is enhanced in this period. As consequence less ATP is generated at the expenses of more protons production, which are ultimately responsible for myocardial acidosis and ions (mainly Calcium) accumulation (see text for more details). Red arrows: enhanced pathway; white arrows: almost abolished pathways
3 Metabolic Modulation Therapy
Given the interdependence between FA and glucose oxidation, metabolic modulation therapy with optimization of energy substrate utilization can be achieved by either inhibiting FA oxidation or stimulating glucose oxidation. This purpose can be achieved through three major strategies: (1) directly enhancing glucose oxidation; (2) decreasing the circulating levels of FA and/or their uptake by cardiac myocytes or mitochondrion; (3) directly inhibiting the enzymes that participate in FA oxidation.
3.1 Strategies to Enhance Glucose Oxidation
3.1.1 Dicholoroacetate (DCA)
The rate-limiting step for glucose oxidation is catalyzed by the pyruvate dehydrogenase (PDH) complex, which consists of PDH, PDH kinase (PDK), and PDH phosphatase (PDHP) enzymes [24]. Flux through PDH is regulated both by substrate/product ratios and by covalent modification. PDH flux is increased in response to increases in glycolysis and hence an increased generation of pyruvate, while PDH flux is decreased by increased ratios of mitochondrial NADH/NAD+ and acetyl-CoA/CoA [25]. With regards to covalent modification, PDHP dephosphorylates and activates PDH, whereas PDK, in response to acetyl-CoA and NADH (produced primarily from FA oxidation) phosphorylates and inhibits PDH; and thus restricts the oxidation of glycolytically derived pyruvate [24]. Dichloroacetate (DCA) inhibits PDK activity, thus stimulating the mitochondrial PDH with improved glycolysis and glucose oxidation coupling and decreased proton production [26].
Despite the promising experimental evidence, a contemporary PubMed search displays only two small studies of DCA, conducted on a total of less than 50 ischemic heart disease patients. No definitive conclusions can therefore be derived for clinical purposes. In addition, when used in other pathological conditions, DCA treatment has been associated with relevant neurotoxicity, thus further compromising the expectations for any future clinical utilization.
3.2 Strategies to Reduce Cellular/Mitochondrial FA Uptake
Many years ago, Oliver and coworkers developed the concept that suppression of circulating plasma non-esterified FA, and thus myocardial FA uptake and oxidation, reduced ischemic damage and ventricular arrhythmias during acute myocardial infarction (AMI) or exercise-induced angina. This concept was further developed in the 1980s with the demonstration that direct inhibition of FA transport into the mitochondria with oxfenicine increased glucose oxidation and decreases lactate production, which resulted in symptom relief in patients with stable angina.
3.2.1 Glucose–Insulin–Potassium (GIK) Solution
The rationale for this pharmacological approach originates from the early nineteenth century, when it was observed that chest pain could be relieved by administration of sugar. Later on, it was found that patients with AMI present high plasma levels of FA, within 30 minutes of the onset of chest pain, and such levels could be reduced by infusion of glucose, insulin and potassium (GIK) in experimental models of AMI [27]. Such a property becomes particularly relevant in patients with diabetes, who have blunted baseline glucose utilization capability due to insulin resistance. Indeed, GIK solutions have been shown to augment glucose utilization at the expense of FA uptake through increased GLUT-1 and GLUT-4 translocation to the sarcolemmal membrane. In animal models of AMI, early GIK administration was associated with reduced myocardial oxygen extraction, malignant arrhythmias, and myocardial infarct size and ultrastructural damage and improved systolic function. Despite these promising results, over the years there has been only one clinical study, the Immediate Myocardial Metabolic Enhancement During Initial Assessment and Treatment in Emergency care (IMMEDIATE) Trial, confirming the benefits with administration of GIK solution in the first critical hours after the onset of symptoms of AMI, when FA levels are elevated. This study showed that pre-hospital administration of intravenous GIK, compared with glucose placebo, was associated with lower rates of the composite outcome of cardiac arrest or in-hospital mortality [28].
However, subsequent trials designed to support the beneficial effects of GIK solution in preserving cardiac tissue during AMI did not confirm these results. In this regard, a meta-analysis, including a total of 16 randomized trials which investigated the role of GIK solution in 28,374 AMI patients from 1966 to 2008, was recently conducted. There were a total of 1,367 deaths (9.6 %) in the GIK group and 1,351 deaths (9.6 %) in the control group. The lack of benefit was also confirmed in the subgroup analysis of patients given high-dose GIK and in those whom reperfusion was not performed [29].
Overall, there is no definitive evidence to support the use of the GIK solution in clinical practice. Such controversies have been attributed to the possible opposed effects of hyperglycemia with glucose toxicity and insulin infusion, with hyperglycemia neutralizing the benefits of insulin [30]. Indeed the beneficial effects, if any, have been related to the various properties of insulin (i.e. vasodilator [31], anti-inflammatory [32], antioxidative [33], positive inotropic [34], prosurvival [35] and antifibrinolytic [36]). Therefore, studies designed to separate out the effects of hyperglycemia and glucose toxicity are needed in order to confirm these broad spectrum beneficial effects of insulin and its utility in the AMI setting.
3.2.2 Carnitine Palmitoyl Transferase Inhibitors
A strategy to inhibit mitochondrial uptake of FA is to suppress rate-limiting enzyme for the mitochondrial uptake of fatty acids like carnitine palmitoyl transferase (CPT) I or II. Perhexiline, a reversible CPT-1, and to a lesser extent, CPT-2 inhibitor has been shown to ameliorate angina [37], and, more recently to attenuate the increase in diastolic tension associated with myocardial ischemia, thereby improving myocardial efficiency [38]. Inhibition of CPT-1/CPT-2 by perhexiline increases myocardial oxygen utilization efficiency by at least 13 %. However, following perhexiline administration, cardiac efficiency increases by approximately 30 %, therefore suggesting additional mechanisms of effect [39]. However, interest in the long term administration of perhexiline has been diminished due to association with infrequent but serious hepatotoxicity and neuropathy that necessitates regular monitoring of plasma levels and makes perhexiline relatively contraindicated in patients with hepatic or renal dysfunction [40].
3.2.3 Malonyl-CoA Decarboxylase (MCD) Inhibitors
Malonyl-CoA is another potent, endogenous inhibitor of CPT-1 which decreases the uptake of FA into the mitochondria, thereby reducing mitochondrial FA β-oxidation. Malonyl-CoA decarboxylase (MCD) degrades malonyl-CoA and this effects leads to an increased fatty acid oxidation. Inhibition of MCD significantly increases malonyl-CoA levels, therefore causing a significant decrease in FA oxidation rates and a subsequent increase in glucose oxidation rates. In line with such pharmacodynamic properties, animal models of inhibition of MCD have shown a significant improvement in cardiac functional recovery of aerobically reperfused ischaemic hearts [15]. Inhibition of MCD in the heart appears to be a safe and a very promising therapeutic target for IHD but unfortunately has not yet been introduced for testing in a clinical setting.
3.3 Strategies to Reduce FA Oxidation
The concept of metabolic protection of the ischemic myocardium is in constant evolution and has recently been supported by clinical studies. The beneficial effect of coupling glycolysis to glucose oxidation explains the efficacy of anti-ischemic FA oxidation inhibitors such as trimetazidine and ranolazine. This is supported by the sub-cellular linkage between key glycolytic enzymes and the activity of two survival-promoting membrane-bound pumps, namely the sodium–potassium ATPase, and the calcium uptake pump of the sarcoplasmic reticulum (SERCA). Indeed, it has been demonstrated that ischemia induced disruption of cardiac metabolism can be minimized by metabolic agents that decrease oxidation of FA and increase the rate of combustion of glucose and lactate. In line with these considerations, the greatest progress in the use of metabolic therapy came with the advent of the direct inhibitors of myocardial FA oxidation, specifically TMZ and ranolazine [41].
3.3.1 Trimetazine
The anti-anginal efficacy of TMZ was established prior to the discovery that the drug acts via partial inhibition of myocardial FA oxidation (Fig. 3) [41, 42]. Initial preclinical studies demonstrated that it was cytoprotective in several models of myocardial ischaemia and reperfusion [43]. More recently, Kantor et al. showed that TMZ specifically inhibits the long-chain activity of the enzyme 3-ketoacyl-CoA thiolase [17]. This enzyme, commonly referred to as ‘3-KAT’, catalyzes the terminal reaction of FA β-oxidation, using long-chain 3-ketoacyl-CoA as a substrate, to generate acetyl-CoA. At clinically relevant concentrations TMZ does not inhibit the short or medium chain activity of 3-KAT. These results suggest that TMZ acts via inhibition of 3-KAT to reduce the NADH/NAD+ and acetyl-CoA/free CoA ratios in the mitochondrial matrix. This would remove inhibition on PDH, and increase the rate of glucose oxidation. Indeed, in the working rat heart, TMZ significantly increased the rate of glucose oxidation despite only modestly reducing the rate of FA oxidation [12, 17]. A recent meta-analysis proved that TMZ was as effective as Ca-channel blockers, long-acting nitrates, micorandil or ranolazine to improve exercise tolerance and/or clinical parameters, whether used as monotherapy or in combination therapy [44]. Clinical trials have also proved the efficacy of this metabolic agent in angina refractory to Ca2+-channel blockers, and have supported the superior benefit associated to the addition of this metabolic agent to classic hemodynamic agent, such as β-blockers or nitrates [45]. Furthermore, other studies have supported the use of TMZ to improve clinical manifestation in patients with stable IHD. Indeed, following chronic administration of TMZ a decrease of average number of attacks per week, reduction of mean weekly consumption of short acting nitrates, improvement of quality of life, lessening of severity of main clinical manifestations of chronic HF, and lowering of its functional class have been observed [44, 46–48]. Moreover, similar efficacy of TMZ has been demonstrated both in men and women, which allows recommendation of this metabolic myocardial cytoprotector to patients with IHD irrespective of gender [49, 50]. TMZ has been provided to confer cardioprotection also in patients undergoing CABG and PCI [44, 51].

TMZ inhibits LC-3KAT, one of the enzymes of fatty acid oxidation. In this way there is a reduced production of acetyl-CoA originating from fatty acid oxidation and therefore a relief of PDH activity. ATP adenosine triphosphate, TMZ trimetazidine
Beneficial effects of TMZ with reduction in left ventricular end-systolic volume, improved NYHA class, increased exercise duration and, more importantly, reduced all-cause mortality and cardiovascular events have been documented also in HF patients [52]. Unfortunately, although its clinical benefits have been documented since early eighties, trimetazidine still lacks a widespread clinical use and has only lately been included in the European guidelines (with a class IIb recommendation) for the management of chronic stable angina patients [53].
3.3.2 Ranolazine
In 2006, ranolazine (RNZ), a piperazine structurally related to TMZ was approved in the U.S. for the relief of angina in patients who remained symptomatic on BBs, CCB, or nitrates [54]. RNZ was shown to display antischemic properties through promotion of glucose oxidation at the expense of FA oxidation since early nineties [19]. However, additional properties such as reduction in intracellular calcium overload through inhibition of the late sodium channels have recently gained more attention [55]. These effects have been associated with a preserved mitochondrial structure, decreased intracellular calcium content and, finally, decreased post-ischemic ventricular fibrillation, myocardial stunning and infarct size. For this reason RNZ is currently considered a first generation of a new drug category (i.e. inhibitor of late sodium currents). Nonetheless, it is important to note that therapeutic concentrations at which a reduction in calcium overload is observed are similar to those at which an increase in glucose oxidation has been documented [19]. However, regardless of the action mechanism, RNZ has been shown to confer significant clinical benefit in angina patients. Its use is associated with a significant prolongation in exercise duration to angina and to ST-segment depression (1 mm), either in comparison or on top of defined anti-angina drugs [56]. Nonetheless, its effects on the morbidity of angina patients remain still to be determined. Despite QTc-prolonging action, clinical data have not shown a predisposition to torsades de pointes, and the medication has shown a reasonable safety profile even in those with structural heart disease. Other common side effects include dizziness and constipation.
Despite the “younger” age, as compared to TMZ, RNZ has evoked a major success and has gained a class IIa recommendation for angina relief in both European and American guidelines on management of stable ischemic heart disease [57].
4 Conclusions
Metabolic agents improve effectiveness of energy production, decrease the oxygen debt, and protect myocardial cells from the effects of ischemia. In this way they provide a valid alternative action mechanism as compared to classical hemodynamic agents which induce changes such as reduction in systemic vascular resistance, coronary vasodilatation, or negative inotropism. These effects offer particular advantage in patients in whom conventional treatment has proven insufficient or in those patients in whom conventional hemodynamic agents induce symptomatic hypotension, inappropriate bradycardia, or worsening HF. Unfortunately, a consistent part of the enthusiasm achieved from bench side research gets lost in translation when findings are applied into the clinical setting. For this reason, research efforts should be centered into the careful selection of the treatment agent as well as proper designation and support of clinical trials.
References
Go AS, Mozaffarian D, Roger VL et al (2013) Heart disease and stroke statistics–2013 update: a report from the American Heart Association. Circulation 127:e6–e245
Fox K, Garcia MA, Ardissino D et al (2006) Guidelines on the management of stable angina pectoris: executive summary: the task force on the management of stable angina pectoris of the European Society of Cardiology. Eur Heart J 27:1341–1381
Boden WE, O’Rourke RA, Teo KK et al (2007) Optimal medical therapy with or without PCI for stable coronary disease. N Engl J Med 356:1503–1516
(1997) Five-year clinical and functional outcome comparing bypass surgery and angioplasty in patients with multivessel coronary disease. A multicenter randomized trial. Writing Group for the Bypass Angioplasty Revascularization Investigation (BARI) Investigators. JAMA 277:715–721
Wolff AA, Rotmensch HH, Stanley WC et al (2002) Metabolic approaches to the treatment of ischemic heart disease: the clinicians’ perspective. Heart Fail Rev 7:187–203
Lopaschuk GD, Belke DD, Gamble J et al (1994) Regulation of fatty acid oxidation in the mammalian heart in health and disease. Biochim Biophys Acta 1213:263–276
Stanley WC, Recchia FA, Lopaschuk GD (2005) Myocardial substrate metabolism in the normal and failing heart. Physiol Rev 85:1093–1129
Lopaschuk GD, Ussher JR, Folmes CDL et al (2010) Myocardial fatty acid metabolism in health and disease. Physiol Rev 90(1):207–58
Goodwin GW, Taylor CS, Taegtmeyer H (1998) Regulation of energy metabolism of the heart during acute increase in heart work. J Biol Chem 273:29530–29539
Collins-Nakai RL, Noseworthy D, Lopaschuk GD (1994) Epinephrine increases ATP production in hearts by preferentially increasing glucose metabolism. Am J Physiol 267:H1862–H1871
Ashrafian H, Frenneaux MP, Opie LH (2007) Metabolic mechanisms in heart failure. Circulation 116:434–448
Lopaschuk GD (2001) Optimizing cardiac energy metabolism: how can fatty acid and carbohydrate metabolism be manipulated? Coron Artery Dis 12(Suppl 1):S8–S11
Lopaschuk GD, Stanley WC (1997) Glucose metabolism in the ischemic heart. Circulation 95:313–315
Stanley WC, Lopaschuk GD, McCormack JG (1997) Regulation of energy substrate metabolism in the diabetic heart. Cardiovasc Res 34:25–33
Dyck JR, Cheng JF, Stanley WC et al (2004) Malonyl coenzyme a decarboxylase inhibition protects the ischemic heart by inhibiting fatty acid oxidation and stimulating glucose oxidation. Circ Res 94:e78–e84
Dyck JR, Hopkins TA, Bonnet S et al (2006) Absence of malonyl coenzyme A decarboxylase in mice increases cardiac glucose oxidation and protects the heart from ischemic injury. Circulation 114:1721–1728
Kantor PF, Lucien A, Kozak R et al (2000) The antianginal drug trimetazidine shifts cardiac energy metabolism from fatty acid oxidation to glucose oxidation by inhibiting mitochondrial long-chain 3-ketoacyl coenzyme A thiolase. Circ Res 86:580–588
Lopaschuk GD, Barr R, Thomas PD et al (2003) Beneficial effects of trimetazidine in ex vivo working ischemic hearts are due to a stimulation of glucose oxidation secondary to inhibition of long-chain 3-ketoacyl coenzyme a thiolase. Circ Res 93:e33–e37
McCormack JG, Barr RL, Wolff AA et al (1996) Ranolazine stimulates glucose oxidation in normoxic, ischemic, and reperfused ischemic rat hearts. Circulation 93:135–142
Stanley WC, Morgan EE, Huang H et al (2005) Malonyl-CoA decarboxylase inhibition suppresses fatty acid oxidation and reduces lactate production during demand-induced ischemia. Am J Physiol Heart Circ Physiol 289:H2304–H2309
Taniguchi M, Wilson C, Hunter CA et al (2001) Dichloroacetate improves cardiac efficiency after ischemia independent of changes in mitochondrial proton leak. Am J Physiol Heart Circ Physiol 280:H1762–H1769
Liu B, Clanachan AS, Schulz R et al (1996) Cardiac efficiency is improved after ischemia by altering both the source and fate of protons. Circ Res 79:940–948
Liu Q, Docherty JC, Rendell JC et al (2002) High levels of fatty acids delay the recovery of intracellular pH and cardiac efficiency in post-ischemic hearts by inhibiting glucose oxidation. J Am Coll Cardiol 39:718–725
Sugden MC, Holness MJ (2003) Recent advances in mechanisms regulating glucose oxidation at the level of the pyruvate dehydrogenase complex by PDKs. Am J Physiol Endocrinol Metab 284:E855–E862
Spriet LL, Heigenhauser GJ (2002) Regulation of pyruvate dehydrogenase (PDH) activity in human skeletal muscle during exercise. Exerc Sport Sci Rev 30:91–95
McVeigh JJ, Lopaschuk GD (1990) Dichloroacetate stimulation of glucose oxidation improves recovery of ischemic rat hearts. Am J Physiol 259:H1079–H1085
Opie LH, Owen P (1976) Effect of glucose-insulin-potassium infusions on arteriovenous differences of glucose of free fatty acids and on tissue metabolic changes in dogs with developing myocardial infarction. Am J Cardiol 38:310–321
Selker HP, Beshansky JR, Sheehan PR et al (2012) Out-of-hospital administration of intravenous glucose-insulin-potassium in patients with suspected acute coronary syndromes: the IMMEDIATE randomized controlled trial. JAMA 307:1925–1933
Mamas MA, Neyses L, Fath-Ordoubadi F (2010) A meta-analysis of glucose-insulin-potassium therapy for treatment of acute myocardial infarction. Exp Clin Cardiol 15:e20–e24
Dandona P, Chaudhuri A, Ghanim H et al (2006) Anti-inflammatory effects of insulin and pro-inflammatory effects of glucose: relevance to the management of acute myocardial infarction and other acute coronary syndromes. Rev Cardiovasc Med 7(Suppl 2):S25–S34
Yki-Jarvinen H, Utriainen T (1998) Insulin-induced vasodilatation: physiology or pharmacology? Diabetologia 41:369–379
Albacker T, Carvalho G, Schricker T et al (2008) High-dose insulin therapy attenuates systemic inflammatory response in coronary artery bypass grafting patients. Ann Thorac Surg 86:20–27
Dandona P, Chaudhuri A, Ghanim H et al (2008) Use of insulin to improve glycemic control in diabetes mellitus. Cardiovasc Drugs Ther 22:241–251
Cottin Y, Lhuillier I, Gilson L et al (2002) Glucose insulin potassium infusion improves systolic function in patients with chronic ischemic cardiomyopathy. Eur J Heart Fail 4:181–184
Matsui T, Tao J, del Monte F et al (2001) Akt activation preserves cardiac function and prevents injury after transient cardiac ischemia in vivo. Circulation 104:330–335
Stegenga ME, van der Crabben SN, Levi M et al (2006) Hyperglycemia stimulates coagulation, whereas hyperinsulinemia impairs fibrinolysis in healthy humans. Diabetes 55:1807–1812
Cole PL, Beamer AD, McGowan N et al (1990) Efficacy and safety of perhexiline maleate in refractory angina. A double-blind placebo-controlled clinical trial of a novel antianginal agent. Circulation 81:1260–1270
Klassen GA, Zborowska-Sluis DT, Wright GJ (1980) Effects of oral perhexiline on canine myocardial flow distribution. Can J Physiol Pharmacol 58:543–549
Unger SA, Kennedy JA, McFadden-Lewis K et al (2005) Dissociation between metabolic and efficiency effects of perhexiline in normoxic rat myocardium. J Cardiovasc Pharmacol 46:849–855
Barclay ML, Sawyers SM, Begg EJ et al (2003) Correlation of CYP2D6 genotype with perhexiline phenotypic metabolizer status. Pharmacogenetics 13:627–632
Stanley WC (2002) Partial fatty acid oxidation inhibitors for stable angina. Expert Opin Investig Drugs 11:615–629
McClellan KJ, Plosker GL (1999) Trimetazidine. A review of its use in stable angina pectoris and other coronary conditions. Drugs 58:143–157
Vaillant F, Tsibiribi P, Bricca G et al (2008) Trimetazidine protective effect against ischemia-induced susceptibility to ventricular fibrillation in pigs. Cardiovasc Drugs Ther 22:29–36
Danchin N, Marzilli M, Parkhomenko A et al (2011) Efficacy comparison of trimetazidine with therapeutic alternatives in stable angina pectoris: a network meta-analysis. Cardiology 120:59–72
Chazov EI, Lepakchin VK, Zharova EA et al (2005) Trimetazidine in Angina Combination Therapy–the TACT study: trimetazidine versus conventional treatment in patients with stable angina pectoris in a randomized, placebo-controlled, multicenter study. Am J Ther J12:35–42
Grabczewska Z, Bialoszynski T, Szymanski P et al (2008) The effect of trimetazidine added to maximal anti-ischemic therapy in patients with advanced coronary artery disease. Cardiol J 15:344–350
Marzilli M (2003) Cardioprotective effects of trimetazidine: a review. Curr Med Res Opin 19:661–672
Marzilli M (2008) Does trimetazidine prevent myocardial injury after percutaneous coronary intervention? Nat Clin Pract Cardiovasc Med 5:16–17
Vasiuk Iu A, Shal’nova SA, Shkol’nik EL et al (2011) The (PRIMA) Study. Comparison of clinical effect of trimetazidine MR in men and women. Kardiologiia 51:11–15
Danchin N (2006) Clinical benefits of a metabolic approach with trimetazidine in revascularized patients with angina. Am J Cardiol 98:8J–13J
Martins GF, Siqueira Filho AG, Santos JB et al (2011) Trimetazidine on ischemic injury and reperfusion in coronary artery bypass grafting. Arq Bras Cardiol 97:209–216
Gao D, Ning N, Niu X et al (2011) Trimetazidine: a meta-analysis of randomised controlled trials in heart failure. Heart 97:278–286
Montalescot G, Sechtem U, Achenbach S et al (2013) 2013 ESC guidelines on the management of stable coronary artery disease: the Task Force on the management of stable coronary artery disease of the European Society of Cardiology. Eur Heart J 34:2949–3003
Chaitman BR (2006) Ranolazine for the treatment of chronic angina and potential use in other cardiovascular conditions. Circulation 113:2462–2472
Wasserstrom JA, Sharma R, O’Toole MJ et al (2009) Ranolazine antagonizes the effects of increased late sodium current on intracellular calcium cycling in rat isolated intact heart. J Pharmacol Exp Ther 331:382–391
Rousseau MF, Pouleur H, Cocco G et al (2005) Comparative efficacy of ranolazine versus atenolol for chronic angina pectoris. Am J Cardiol 95:311–316
Fihn SD, Gardin JM, Abrams J et al (2012) 2012 ACCF/AHA/ACP/AATS/PCNA/SCAI/STS guideline for the diagnosis and management of patients with stable ischemic heart disease: a report of the American College of Cardiology Foundation/American Heart Association task force on practice guidelines, and the American College of Physicians, American Association for Thoracic Surgery, Preventive Cardiovascular Nurses Association, Society for Cardiovascular Angiography and Interventions, and Society of Thoracic Surgeons. Circulation 126:e354–e471
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2014 Springer Science+Business Media New York
About this chapter
Cite this chapter
Guarini, G., Huqi, A., Marzilli, M. (2014). Metabolic Therapy for the Ischemic Heart. In: Lopaschuk, G., Dhalla, N. (eds) Cardiac Energy Metabolism in Health and Disease. Advances in Biochemistry in Health and Disease, vol 11. Springer, New York, NY. https://doi.org/10.1007/978-1-4939-1227-8_15
Download citation
DOI: https://doi.org/10.1007/978-1-4939-1227-8_15
Published:
Publisher Name: Springer, New York, NY
Print ISBN: 978-1-4939-1226-1
Online ISBN: 978-1-4939-1227-8
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)