
Abstract
RNA-binding proteins (RBPs) play pivotal roles in multiple cellular pathways from transcription to RNA turnover by interacting with RNA sequence and/or structural elements to form distinct RNA–protein complexes. Since these complexes are required for the normal regulation of gene expression, mutations that alter RBP functions may result in a cascade of deleterious events that lead to severe disease. Here, we focus on a group of hereditary disorders, the microsatellite expansion diseases, which alter RBP activities and result in abnormal neurological and neuromuscular phenotypes. While many of these diseases are classified as adult-onset disorders, mounting evidence indicates that disruption of normal RNA–protein interaction networks during embryogenesis modifies developmental pathways, which ultimately leads to disease manifestations later in life. Efforts to understand the molecular basis of these disorders has already uncovered novel pathogenic mechanisms, including RNA toxicity and repeat-associated non-ATG (RAN) translation, and current studies suggest that additional surprising insights into cellular regulatory pathways will emerge in the future.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others

Keywords
- Amyotrophic lateral sclerosis/frontotemporal dementia
- Microsatellite
- Myotonic dystrophy
- Neurological disease
- Oculopharyngeal muscular dystrophy
- RNA processing
- RNA toxicity
- Spinocerebellar ataxia
1 Introduction
1.1 Benefits and Problems with a Repetitive Genome
Nearly half of the human genome consists of repetitive DNA sequences composed mostly of interspersed and transposon-derived repeats but also tandem repeats (TRs) (Gemayel et al. 2010). Microsatellites, also known as TRs and simple sequence repeats (SSRs), are often defined as repeating units of ≤10 base pairs (bp) while larger repeats are referred to as minisatellites (>10 bp) and macrosatellites or megasatellites (>135 bp). Microsatellites, which account for 3–5 % of mammalian genomes, are highly polymorphic since the DNA replication, repair, and recombination machineries have intrinsic problems handling these unusual repetitive sequences due to their tendency to form imperfect hairpins, quadraplex-like and slipped-stranded structures (Lopez Castel et al. 2010; Mirkin 2007). Many microsatellites are bidirectionally transcribed (Batra et al. 2010; Budworth and McMurray 2013) and repeat length polymorphism is common with mutation rates 10–100,000 fold higher than other genomic regions (Jansen et al. 2012). In the overall population, repeat lengths for a given allele vary moderately and unaffected individuals may harbor alleles with a different number of repeats within the normal range. However, once an allele expands beyond a critical length threshold, instability is greatly amplified and the mutation manifests into a pathological state. Because expansions and contractions occur during cell division and error-prone DNA repair, affected patient tissues are composed of cells containing varying numbers of microsatellite repeats in the disease allele, a process termed somatic mosaicism (Lopez Castel et al. 2010; Mirkin 2007). Furthermore, continuous expansions of mutant repeat lengths often occur during aging, which might explain the progressive nature of many of these neurological diseases. Microsatellite expansions and contractions also occur in the germ line, which affects the repeat length passed on from one generation to the next. Comparison of average repeat lengths among patients within pedigrees shows that successive generations often have progressively larger repeats. Moreover, this increase in repeat length often correlates with an increase in disease severity and earlier age-of-onset of disease symptoms, hence providing a genetic explanation for the observation of intergenerational anticipation (Friedman 2011).
Although the remarkable abundance of repeats in the human genome hints at functionality, early reports classified these sequences as an evolutionary artifact or non-functional “junk” DNA (Doolittle and Sapienza 1980; Ohno 1972; Orgel and Crick 1980). More recent studies indicate that repetitive DNA might serve valuable cellular functions. Microsatellites occur in the protein-coding regions of ~17 % of human genes and the 10–20 % of eukaryotic genes that contain microsatellite repeats are often important for cellular regulatory pathways (Gemayel et al. 2010; Jansen et al. 2012). For example, TRs in budding yeast are primarily found within genes encoding cell-surface and important regulatory proteins, including chromatin modification and transcription factors. However, TRs are not restricted to eukaryotes. Indeed, TRs facilitate antigenic variation in pathogenic prokaryotes as a mechanism to evade host defense systems (Gemayel et al. 2010; Mrazek et al. 2007). Repeat unit variations in promoter regions may lead to changes in gene expression while TR variations in coding regions can result in frameshift mutations and the production of truncated proteins. Thus, simple sequence repeats serve regulatory functions and catalyze adaptations beneficial to pathogen survival. Despite these examples of TR functions, functional roles for microsatellite variability in human genes have not been well documented although TRs are enriched in vertebrate genes that control organ and/or body morphology (Gemayel et al. 2010). Due to their relatively high mutation rate, variability in microsatellite repeat number within a gene might offer evolutionary and regulatory advantages. For example, variations in GC-rich repeats within the normal range positioned within a promoter could have modest, but advantageous, effects on transcriptional activity while similar repeats in the 5′ untranslated region (5′ UTR) could modulate translation and the number of encoded proteins available during a particular developmental window (Fig. 10.1a). However, most studies have emphasized the detrimental effects of larger microsatellite expansions. How do expanded microsatellites cause disease? Research has revealed several distinct mechanisms, some of which are novel and unique, at least currently, to repeat disease. Relevant to this review, some of these mechanisms involve RNA-binding proteins and altered RNA processing.

Microsatellites in normal and mutant genes associated with hereditary diseases discussed in this chapter. (a) A normal five exon tandem repeat (TR) gene with 5′ and 3′ UTRs (open boxes), coding region (black boxes), introns (thick grey line), and microsatellites (green boxes) located in promoter, noncoding (UTRs, intron) and coding regions. Copy number variation, due to errors during DNA replication or repair, may result in a modest variation in TR number (shown here as 3–4 TRs) that might influence specific regulatory pathways depending on the TR location in the allele. (b) TR expansions result in the diseases discussed in this chapter. The position of each TR is shown (single green box) together with normal (N) allele and mutant (red triangle) length ranges
1.2 Microsatellites in Disease
Over two decades ago, the field of unstable microsatellite disease was initiated by seminal findings on the molecular etiology of fragile X syndrome (FXS), caused by a CGGexp in the fragile X mental retardation 1 (FMR1) gene, and spinal and bulbar muscular atrophy (SBMA), which is due to CAGexp mutations in the androgen receptor (AR) gene (La Spada et al. 1991; Verkerk et al. 1991) (Fig. 10.1b). Currently, dozens of neurological diseases have been traced back to microsatellite expansion mutations in additional genes with the most recent discovery that a GGGGCCexp in the C9ORF72 gene is responsible for the most common cause of familial amyotrophic lateral sclerosis (ALS) and frontotemporal dementia (FTD) (C9-linked ALS/FTD) (DeJesus-Hernandez et al. 2011; Renton et al. 2011).
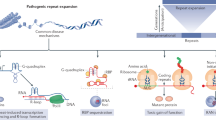
Immediately following the discovery of unstable microsatellites in FXS and SBMA, several perplexing questions emerged regarding how expanded microsatellites cause disease. Why are such unstable and disease-prone repetitive sequences so prevalent in the human genome, and in some cases, conserved during evolution (Buschiazzo and Gemmell 2010; Gemayel et al. 2010). Why are so many microsatellites located in noncoding regions? The discovery of microsatellite expansion disorders has been accompanied by global initiatives to characterize the associated disease pathologies and uncover the molecular mechanisms involved in this group of neurological disorders. Several common mechanistic themes have emerged (Fig. 10.2). Many of the repeat expansion diseases involve the nervous and musculoskeletal systems, follow a dominant inheritance pattern and show genetic anticipation. Despite widespread expression of certain disease genes, neurological and neuromuscular systems display a particular vulnerability to mutant microsatellite genes. Histological analysis of patient tissue reveals that many of these disorders are characterized by cellular aggregates of homopolymeric proteins, notably polyglutamine (polyQ) inclusions (Orr 2012b; Zoghbi and Orr 2009). In some diseases, patient cells contain nuclear foci harboring RNA expressed from the expanded genes along with specific proteins. Intriguingly, RNA-binding protein misregulation and compromised RNA metabolism have become recurrent disease themes (Echeverria and Cooper 2012; Poulos et al. 2011).

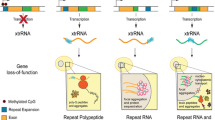
Proposed pathogenic mechanisms for microsatellite repeat diseases. Microsatellite expansions (red triangles) in noncoding (UTRs, small blue boxes; introns, black lines) regions may result in RNA (yellow boxes) gain-of-function (RNA GOF), protein (grey and colored circles) loss-of-function (protein LOF), or repeat-associated non-ATG (RAN) protein GOF (RAN GOF). In conventional coding regions (open reading frame, ORF), expansions might cause a protein GOF effect such as the CAG expansion in SCA3 which results in a polyGln (polyQ, brown circles) expansion that either accumulates in cellular inclusions or alters mutant ATXN3 (mATXN3) interactions. Note that RAN translation of the C9ORF72 GGGGCCexp repeat in ALS/FTD produces polyGlyAla (GA), polyGlyPro (GP), and polyGlyArg (GR) dipeptide repeat proteins due to the recognition an RNA secondary structure (red hairpin) by the ribosome (orange ellipsoids)
In this chapter, we focus on the molecular mechanisms underlying microsatellite expansion diseases which include: (1) RNA gain-of-function, in which the repeat-containing RNA transcript expressed from the mutant gene sequesters trans-acting factors; (2) protein loss-of-function due to microsatellite expansion mutations that render a protein non-functional or represses expression; (3) protein gain-of-function whereby a repeat expansion in a coding region contributes a deleterious function to the mutant protein; (4) repeat associated non-ATG (RAN) translation that results in homopolymeric or heteropolymeric peptides translated from the repeat region independent of a normal initiation codon. As an additional layer of complexity, bidirectional transcription through repeat DNA enables multiple pathogenic mechanisms from a single locus. Examples of microsatellite expansion diseases that illustrate each of these molecular mechanisms, and their effect on RNA-binding protein function, are the focus of this chapter.
2 RNA Toxicity and Protein Sequestration in Myotonic Dystrophy
2.1 Tipping the Balance Between Antagonistic RNA Processing Factors
The discovery that the transcription of some microsatellite expansions results in the synthesis of toxic RNAs originated from studies designed to elucidate the molecular etiology of the most frequent form of muscular dystrophy in adults, myotonic dystrophy (dystrophia myotonica, DM) (Echeverria and Cooper 2012; Poulos et al. 2011; Udd and Krahe 2012). Although DM is classified as a muscular dystrophy, multiple tissues are affected including skeletal muscle (hyperexcitability or myotonia, weakness/wasting), the heart (arrhythmias and conduction block), the visual system (dust-like cataracts), the reproductive system, (testicular atrophy), and the brain (hypersomnia, executive dysfunction, and cerebral atrophy). Interestingly, some disease manifestations, such as brain/muscle atrophy and alopecia (premature balding), resemble the normal aging process (Martin 2005). A distinguishing feature of DM is that microsatellite expansions in two unrelated genes cause disease. DM type 1 (DM1) is associated with a CTGexp in the dystrophia myotonica protein kinase (DMPK) gene, which encodes a serine–threonine kinase, while DM type 2 is caused by a CCTGexp in CNBP, the cellular nucleic acid binding protein gene that encodes a factor implicated in both transcription and translation (Brook et al. 1992; Fu et al. 1992; Jansen et al. 1992; Liquori et al. 2001; Mahadevan et al. 1992) (Fig. 10.1b). Unaffected individuals have 5–37 CTG repeats in the DMPK gene and 10–26 CCTG repeats in CNBP. For DM1, CTGexp lengths expand to 50- > 3,000 repeats while in DM2 the CCTGexp ranges from 75 to >11,000 repeats. In contrast to DM2, which does not have a congenital form, very large (>1,000 repeats) DMPK CTGexp mutations also cause congenital DM1 (CDM) characterized by neonatal hypotonia (floppy baby) and intellectual disability.
Several experimental findings argue that DM is an RNA-mediated, or RNA gain-of-function (GOF), disease (Fig. 10.2). The DMPK and CNBP expansions are located in noncoding regions and mutant DMPK and CNBP C(C)UGexp RNAs accumulate in RNA foci in the nucleus (Davis et al. 1997; Margolis et al. 2006; Poulos et al. 2011; Ranum and Cooper 2006; Taneja et al. 1995). Attempts to model DM in mouse transgenic models led to the observation that CTGexp mutations cause DM-relevant phenotypes irrespective of gene context and the degree of pathology correlates with transgene expression level (Mankodi et al. 2000; Sicot and Gomes-Pereira 2013). How does C(C)UGexp RNA expression lead to DM? C(C)UGexp RNAs could: (1) promote formation of nuclear RNA foci that impair normal nuclear functions such as RNA processing and nuclear export pathways; (2) fold into a structure that possesses an inherent dominant-negative function; (3) sequester specific proteins leading to loss-of-function of these factors. Interestingly, the current model for DM pathogenesis implicates all of these mechanisms. Transcription of expanded C(C)TG repeats leads to the synthesis of C(C)UGexp RNAs that gain toxic functions, including the recruitment and sequestration of the muscleblind-like (MBNL) proteins and hyperphosphorylation, and increased levels of CUGBP1 and ETR3-like factors (CELF) (Kuyumcu-Martinez et al. 2007; Miller et al. 2000). The MBNL and CELF proteins are antagonistic alternative splicing factors that function during postnatal development by promoting either adult (MBNL) or fetal (CELF) splicing patterns (Charizanis et al. 2012; Du et al. 2010; Ho et al. 2004; Kanadia et al. 2003a, b; Lin et al. 2006; Philips et al. 1998; Timchenko et al. 1996). Thus, DM pathogenesis involves RNA gain-of-function leading to protein loss, and gain, of function and the persistence of, or reversion to, fetal splicing patterns in adult tissues.
Besides MBNL and CELF, other RNA processing factors have been implicated in DM. HnRNP H binds in vitro to DMPK-derived CUGexp RNAs that also contain a splicing branch point distal to the repeat region and siRNA knockdown of this protein rescues nuclear retention of CUGexp-containing RNAs (Kim et al. 2005). HnRNP H protein levels are also elevated in DM1 myoblasts and hnRNP H or CELF1 overexpression leads to the formation of a repressor complex that inhibits splicing of insulin receptor (IR) exon 11 (Paul et al. 2006). HnRNP H exists in a complex with MBNL1 and 9 other proteins (hnRNP H2, H3, F, A2/B1, K, L, DDX5, DDX17, and DHX9) in normal myoblast extracts, but the stoichiometry of these complexes is altered in DM1 extracts (Paul et al. 2011). Staufen1 (STAU1) is a double-stranded (ds)RNA-binding protein that is also misregulated in DM1 skeletal muscle (Ravel-Chapuis et al. 2012). STAU1 levels increase in human DM1, and CUGexp mouse, skeletal muscle, but it is not sequestered in nuclear CUGexp RNA foci. STAU1 promotes nuclear export and translation of CUGexp mRNAs, and its overexpression rescues abnormal splicing of several pre-mRNAs misspliced in DM, so increased levels of STAU1 may be a compensatory response that ameliorates the DM phenotype.
An additional type of RNA-binding protein appears to modulate the interaction of MBNL proteins with pathogenic C(C)UGexp RNAs. MBNL proteins contain tandem zinc finger (ZnF) domains that bind preferentially to YGCY (Y = pyrimidine) RNA elements (Charizanis et al. 2012; Du et al. 2010; Goers et al. 2010; Wang et al. 2012a). Structural analysis of MBNL ZnFs indicates that they target GC steps and induce an antiparallel orientation on these bound elements due to inter-ZnF linker topology (Teplova and Patel 2008). MBNL proteins do not possess a high affinity for uninterrupted Watson–Crick RNA duplexes but instead show preferential binding to imperfect duplexes, particularly with U–U and C–U/U–C mismatches, and steady-state fluorescence quenching analysis confirms that MBNL1 binding alters helical RNA structures (Fu et al. 2012). Since CUGexp RNAs fold into RNA hairpin structures (Krzyzosiak et al. 2012), it is interesting that the RNA helicase p68/DDX5 acts to modulate MBNL1 binding activity (Laurent et al. 2012). The p68/DDX5 helicase colocalizes with nuclear RNA foci and has a stimulatory effect on MBNL1 binding to both CUGexp and MBNL1 splicing target RNA binding sites.
2.2 Pathogenic RNAs Disrupt Additional Regulatory Pathways Including Pre-miR Processing, mRNA Trafficking, and Translation
While the impact of C(C)UGexp RNA expression on the regulation of alternative splicing has received considerable attention, these toxic RNAs have also been reported to affect other regulatory pathways. In DM1 skeletal muscle biopsies, microRNA (miR) expression patterns are variable between studies with miR-206 overexpression compared to controls (Gambardella et al. 2010) while another study found that miR-1 and miR-335 were upregulated, miRs 29b,c and miR-33 were downregulated, and miR-1, miR133b and miR-206 were mislocalized (Perbellini et al. 2011). Mis-processing of miR-1 occurs in the DM1 heart and this has been linked to MBNL loss-of-function. MBNL1 normally recognizes a UGC motif in the pre-miR-1 loop to facilitate processing because it competes with LIN28, which blocks Dicer processing via ZCCHC11/TUT4-mediated pre-miR-1 uridylation (Rau et al. 2011).
Several studies have provided evidence that muscle weaknesses and wasting in DM may arise from impaired muscle differentiation due to alterations in CELF1 activity and the resulting effects on translation of specific target mRNAs. As mentioned previously, CELF1 protein levels increase in DM and CELF1 overexpression in transgenic mice inhibits myogenesis and causes MEF2A and p21 overexpression (Timchenko et al. 2004). Several studies have suggested that DM2 is caused by CNBP haploinsufficiency (Chen et al. 2007; Huichalaf et al. 2009; Raheem et al. 2010) although other groups report that CNBP protein levels are not altered in this disease (Botta et al. 2006; Margolis et al. 2006; Massa et al. 2010). Interestingly, CNBP binds to the 5′ UTRs of terminal oligopyrimidine (TOP) genes encoding a variety of proteins important for translational regulation, including PABPC1, eIF1a, and eIF2, so CCUGexp expression has been proposed to impact the rate of global protein synthesis (Huichalaf et al. 2009; Schneider-Gold and Timchenko 2010).
RNA trafficking is another critical regulatory pathway that is altered by the expansion mutations in DM. Early work provided evidence that MBNL2 regulates the localized expression of integrin α3 to adhesion complexes (Adereth et al. 2005). More recently, transcriptome analysis has indicated that MBNL proteins regulate mRNA localization in vertebrate and invertebrate (Drosophila) cells resulting in effects on both translation and protein secretion (Wang et al. 2012a). For future studies, it will be important to link distinct disease manifestations of DM to alterations in the localization of specific RNAs.
2.3 Are RNA Foci Important Features of RNA Toxicity?
Nuclear RNA foci are a hallmark pathological feature of DM as well as other neurological diseases that may be linked to RNA-mediated toxicity (e.g., C9-linked ALS/FTD). However, the exact role of RNA foci in the induction and/or maintenance of DM-associated pathology remains controversial (Junghans 2009; Mahadevan 2012; Wojciechowska and Krzyzosiak 2011). RNA foci might be pathological entities that interfere directly with normal nuclear functions or protective protein–RNA complexes designed to block toxic RNAs from reaching the cytoplasm. For DM1, RNA foci are composed of both MBNL proteins and CUGexp RNAs. Early studies in DM1 cells indicated that RNA foci are insoluble aggregates (Davis et al. 1997; Taneja et al. 1995) and thus formation of MBNL-C(C)UGexp complexes might effectively inhibit MBNL dissociation and enhance disease. An argument against this possibility comes from single-particle tracking, fluorescence recovery after photobleaching (FRAP) and fluorescence loss in photobleaching (FLIP) experiments, which indicate that (CUG)145 RNA transcripts expressed in C2C12 myoblasts undergo stochastic aggregation/disaggregation cycles. An early FRAP study provided evidence for rapid exchange between unbound GFP-Mbnl1 and GFP-Mbnl1 bound to aggregates (Ho et al. 2005). However, when GFP-MBNL1 was expressed at levels comparable to endogenous Mbnl1 levels, considerably less freely diffusing GFP-Mbnl1 was observed (Querido et al. 2011). Thus, RNA foci may be dynamic structures but MBNL proteins could be effectively sequestered in non-focal MBNL-C(C)UGexp complexes. In addition, the dynamics of RNA foci formation and MBNL-C(C)UGexp complex stability may be profoundly influenced by the larger repeats in DM1 and DM2 myofibers and neurons compared to other cell types. Irrespective of potential roles in pathogenesis, RNA foci and alternative splicing alternations are important biomarkers of RNA-mediated disease (Cardani et al. 2006, 2009; Nakamori et al. 2013).
3 Dual Disease Mechanisms from a Single Gene: Fragile X and FMR1
3.1 FMR1 Epigenetic Silencing and Misregulated Translation
Fragile X syndrome (FXS) is the most common form of inherited mental retardation with an incidence of approximately 1 in 5,000 individuals. The symptoms of FXS are variable but include intellectual disability, behavioral abnormalities such as attention deficit hyperactivity disorder (ADHD) and autistic behavior, childhood seizures, connective tissue defects, and macroorchidism (abnormally large testes) (Hernandez et al. 2009; McLennan et al. 2011; Nelson et al. 2013). Unlike many of the other microsatellite diseases, which tend to be inherited in an autosomal dominant pattern, FXS is an X-linked disorder and the mutation creates a fragile site, a locus that is prone to gaps or breaks. FXS arises when the FMR1 CGGexp repeat, which ranges between 6 and 52 repeats in unaffected individuals, expands beyond 230 repeats (Fig. 10.1b). These mutant repeats create an expanded CpG island in the DNA that becomes aberrantly methylated and coupled with histone deacetylation (Coffee et al. 1999; Sutcliffe et al. 1992) (Fig. 10.2). Thus, the primary molecular basis for this neurological disease is epigenetic silencing and loss of function (LOF) of the encoded protein, FMRP.
FMRP is an RNA-binding protein that interacts with polyribosomes to regulate local protein synthesis (Bhakar et al. 2012; Wang et al. 2012b). FMRP interacts with mRNA via two K homology (KH) domains, KH1 and KH2, as well as an arginine- and glycine-rich (RGG) domain (Siomi et al. 1993). While the RGG domain has been reported to bind to G quartets, FMRP has also been reported to bind to the coding regions of mRNA independent of these G-rich structures (Darnell et al. 2001, 2011). Tight regulation of synaptic protein synthesis in response to neuronal activity is thought to be essential for neuronal processes such as those involved in learning and memory formation. FMRP located in neurons at synapses binds to mRNA targets to fine-tune translation regulation in an activity-dependent manner (Akins et al. 2009; Bassell and Warren 2008). A combination of FMRP binding site analysis in mouse brain using high-throughput sequencing and cross-linking/immunoprecipitation (HITS-CLIP) with a polyribosome-programmed translation system revealed that FMRP binds to transcripts of presynaptic and postsynaptic proteins to stall ribosomes until the appropriate cellular signals are present (Darnell et al. 2011). The current molecular model of FXS suggests dysregulation of local protein synthesis as a consequence of FMRP loss leads to neuronal dysfunction and neurological manifestations of the disease. Consistent with this model, Fmr1 null mice have increased translation of Fmrp target mRNAs and have similar abnormal neuron morphology and dendritic abnormities as seen in FXS brains (Berman and Willemsen 2009). Fmr1 null mice also display phenotypes that mirror FXS symptoms (Bhogal and Jongens 2010). Similarly, point mutations in the human FMR1 gene that disrupt the FMRP RNA binding domain KH2 also result in FXS symptoms (Nelson et al. 2013). Cumulatively, these observations strongly suggest that loss of FMRP underlies the neuronal dysfunction seen in patients and is the primary molecular cause of FXS.
3.2 Fragile X-Associated Tremor/Ataxia Syndrome and RNA Toxicity
In contrast to FXS, FMR1 transcriptional activity does not decrease in patients with the related disease, fragile X-associated tremor/ataxia syndrome (FXTAS) (Peprah et al. 2010). Whereas FXS results from a full mutation, or (CGG)>230, more moderately sized premutation alleles of (CGG)55–200 cause FXTAS (Fig. 10.1b). Premutation carriers may suffer from a late-onset neurodegenerative disorder now known as FXTAS, with a typical age of onset in the early 60s, or fragile X-associated primary ovarian insufficiency (FXPOI) with infertility prior to age 40 (Hagerman and Hagerman 2004; Leehey and Hagerman 2012). Clinical features of FXTAS differ from those in FXS and include gait ataxia, progressive action tremor, autonomic dysfunction, and neurodegeneration. The distinct clinical outcomes of FXS and FXTAS result from different pathogenic mechanisms since CGGexp repeats in the premutation range do not repress FMR1 expression. On the contrary, FXTAS patients have up to eightfold higher FMR1 RNA levels with normal to slightly reduced levels of FMRP (Kenneson et al. 2001; Peprah et al. 2010; Tassone et al. 2000a, b, c). Current evidence points towards rCGGexp RNA toxicity in FXTAS. As in DM, FMR1 repeat-containing RNA accumulates in the nucleus (Tassone et al. 2004a, b). Furthermore, rCGGexp expression is sufficient to cause nuclear inclusions and neurodegeneration irrespective of its context in the FMR1 gene, as demonstrated by mouse models expressing a CGGexp reporter gene (Hashem et al. 2009). The presence of ribonuclear inclusions led researchers to ask whether FXTAS follows the RNA toxicity paradigm set by DM, in which cellular factors bind to the mutant RNA expansion and are sequestered away from their normal cellular functions (Fig. 10.2, RNA GOF). Indeed, several RNA-binding proteins have been proposed to be sequestered by rCGGexp repeats, including MBNL1, hnRNP A2/B1, hnRNP G, Sam68, and Pur α (Li and Jin 2012; Tassone and Hagerman 2012).
A combination of animal studies and in vitro assays identified hnRNP A2/B1 and Purα as candidate proteins that bind to rCGGexp RNAs and are titrated away from their normal functions (Jin et al. 2007; Muslimov et al. 2011; Sofola et al. 2007). HnRNP A2/B1 is an abundant nuclear RNA-binding protein containing a glycine-rich and two RRM domains and disruption of hnRNP A2/B1 function has been proposed to alter RNA processing in FXTAS. Furthermore, hnRNP A2/B1 tethers another RNA-binding protein to CGGexp RNA, CELF1, previously implicated in DM pathogenesis (Sofola et al. 2007). Overexpression of either hnRNP A2/B1 or CELF1 in a fly model of FXTAS alleviates the neurodegenerative phenotype observed in this model. Furthermore, CGGexp RNA induces mislocalization of hnRNP A2/B1 target RNAs from dendrites to the neuronal cell body, presumably via titration of hnRNP A2/B1 onto the repeats. Delivery of additional hnRNP A2/B1 to neurons restores dendritic localization of target RNA, supporting the proposed role of hnRNP A2/B1 in FXTAS (Muslimov et al. 2011). Purα is an RNA- and DNA-binding protein and has diverse roles in transcription activation, DNA replication and mRNA localization. In the brain, Purα is involved in neuronal cell proliferation and neurodevelopment and plays a role in regulating the dendritic and axonal localization of mRNA targets (Hokkanen et al. 2012; Johnson et al. 2006; Ohashi et al. 2000). Like hnRNP A2/B1, overexpression of Purα suppresses neurodegeneration in the fly model of FXTAS and this protein was identified in nuclear inclusions in FXTAS patient brains, suggesting that sequestration of Pur α may indeed play a role in the disease pathogenesis (Jin et al. 2007).
Another RNA-binding protein found to be present in FXTAS nuclear inclusions is Sam68 (Src-associated substrate during mitosis of 68 kDa) (Sellier et al. 2010). Sam68 is an alternative splicing regulator that has diverse roles in cellular signaling, apoptosis, and neuronal functions. Mice lacking Sam68 display motor coordination defects reminiscent of ataxia (Lukong and Richard 2008; Ramakrishnan and Baltimore 2011; Sellier et al. 2010). Sam68 localizes to nuclear inclusions following transfection of rCGGexp repeats in cell models and, in turn, recruits other RNA-binding proteins, including MBNL1 and hnRNP G (Sellier et al. 2010). Missplicing of pre-mRNA targets of Sam68 has been reported to occur in FXTAS patient brains, and overexpression of a Sam68 mutant lacking CGG binding function in cell culture is sufficient to rescue the splicing abnormalities.
FXTAS-associated rCGGexp nuclear inclusions also contain other proteins, including ubiquitin, heat-shock proteins such as αB-crystallin, lamin A/C, myelin basic protein, and DROSHA/DGCR8 (Greco et al. 2002; Iwahashi et al. 2006; Sellier et al. 2013). DGCR8 is a double-stranded RNA-binding protein and the DROSHA/DGCR8 complex is responsible for the miRNA processing step that converts pri-miRNA into pre-miRNA. DGCR8 binds to rCGGexp RNA and recruits its partner DROSHA into the inclusions. Levels of mature miRNA decrease in FXTAS patient brains, and overexpression of DGCR8 rescues rCGGexp-induced neuronal cell death, suggesting that miRNA dysregulation contributes to neuronal dysfunction in FXTAS (Sellier et al. 2013).
Cumulatively, these studies suggest that multiple RNA-binding proteins may be titrated by rCGGexp in FXTAS resulting in widespread dysregulation of RNA processing and neurodegeneration. Larger CGGexp mutations in FMR1 lead to FMRP loss-of-function and the subsequent loss of translational regulation and synaptic dysfunction seen in FXS. The observation that microsatellite expansions in a noncoding region of a single gene cause several distinct disease outcomes and disrupt multiple molecular pathways has also been observed in DM1/CDM and is likely to be repeated for other unstable microsatellite diseases.
4 Compound Threats: RNA and Protein Toxicity
Microsatellite expansion diseases have been traditionally classified as either or protein- or RNA-mediated. However, these expansions can also pose compound threats highlighted by the recent discoveries of bidirectional transcription through repeat regions and RAN translation (Batra et al. 2010; Zu et al. 2011). Indeed, in vitro studies suggest that CAG•CTG expansion mutations have the potential to produce nine toxic molecules including two pathogenic RNAs (CAGexp, CTGexp) as well as seven homopolymeric proteins produced by conventional and RAN translation (Pearson 2011; Zu et al. 2011). Below, we discuss expansion diseases, including HDL2 and several spinocerebellar ataxias, in which both RNA and protein toxicity have been implicated.
4.1 Huntington Disease-Like 2
Huntington disease-like 2 (HDL2) is a microsatellite expansion disease that is thought to trigger the production of a combination of toxic RNA and protein species (Margolis et al. 2004). HDL2 is a phenocopy of Huntington’s disease (HD), both of which are dominantly inherited diseases characterized by motor coordination defects, dementia, cortical and striatal neurodegeneration, and eventual death within decades of diagnosis (Rudnicki et al. 2008). HD is caused by a CAGexp in the coding region of the Huntingtin (HTT) gene that results in the expression of a HTT protein with an expanded polyglutamine tract (polyGln or polyQ) (Ha and Fung 2012). HD is a member of the CAGexp neurological disorders characterized by neuronal inclusions of ubiquitinated polyGln. PolyGln is thought to be neurotoxic due to several mechanisms including blockage of the ubiquitin proteasome system (UPS) and disruption of mitochondrial function (Finkbeiner 2011; Orr and Zoghbi 2007).
Although the striking clinical similarities between HD and HDL2 suggest that these diseases have a similar pathogenic mechanism, the mutation responsible for HDL2 was originally described as a CTGexp in the junctophilin-3 (JPH3) gene, which is primarily expressed in the brain (Holmes et al. 2001). Whereas unaffected individuals have 6–27 CTG repeats, HDL2 patients have expansions of 41–58 CTG repeats (Fig. 10.1b). The JPH3 protein is involved in formation of a structure that connects the plasma membrane with the endoplasmic reticulum to help control the release of calcium ions during neuronal activity. Loss of JPH3 expression may contribute to HDL2 pathogenesis (Seixas et al. 2012). While the CTGexp mutation is present in alternatively spliced JPH3 exon 2a, variations in transcription and alternative splicing cause the repeat to be located in different transcript regions. JPH3 transcripts that include exon 2a encode a truncated JPH3 isoform while the CTGexp may result in translation of polyleucine and polyalanine tracts in the truncated protein depending on which exon 2a 3′ splice site is utilized. A third exon 2a splice variant places the CTGexp in the 3′UTR suggesting that toxic CTGexp RNAs could be expressed and CTGexp nuclear RNA foci are detectable in HDL2 neurons (Rudnicki et al. 2007). These foci not only contain CTGexp RNA but also MBNL1, the splicing factor implicated in DM, and several MBNL1-dependent missplicing events seen in DM are also observed in HDL2 neurons.
Despite this data, the RNA-mediated model of HDL2 pathogenesis cannot explain the remarkable commonalities between HDL2 and HD. This discrepancy has now been addressed by the discovery of bidirectional transcription through the repeat that creates an antisense transcript containing a CAGexp encoding polyGln as seen in HD (Wilburn et al. 2011). Early histological analysis of HDL2 patient brains also revealed ubiquitin-positive polyGln nuclear inclusions, reminiscent of those observed in HD, independent of RNA foci (Rudnicki et al. 2008; Walker et al. 2002). Cumulatively, these results indicate that toxic RNA and protein (Fig. 10.2, RNA GOF and protein GOF) may interact synergistically to wreak havoc on neuronal pathways although polyGln toxicity appears to play the predominant role in HDL2.
4.2 Spinocerebellar Ataxia Types 3, 8 and 10
A similar interplay of protein and RNA toxicity has been observed in spinocerebellar ataxia types 3, 8, and 10 (SCA3, SCA8, SCA10). The SCAs are a large group of inherited neurological diseases in which neurological dysfunction in the cerebellum and brainstem causes motor coordination defects known as ataxias (Hersheson et al. 2012; Orr 2012a). SCAs can result from mutations in 37 genes (SCA1-37), but intriguingly several of the mutations are microsatellite repeat expansions including coding CAGexp mutations in SCA1, 2, 3, 6, 7, and 17 and noncoding CTGexp in SCA8, CAGexp in SCA12, ATTCTexp in SCA10, TGGAA in SCA31, and GGCCTGexp in SCA36 (Matilla-Duenas et al. 2012; Serrano-Munuera et al. 2013).
4.2.1 SCA3: Toxic ATXN3 Protein and CAGexp RNA
SCA3 (Machado–Joseph disease, MJD) is characterized by late-onset ataxia and neurodegeneration and is the most prevalent SCA worldwide (Orr 2012a). SCA3 is associated with CAG repeats that expand from the normal (12–37) to a pathogenic (61–84) range resulting in an extended polyGln tract in the C-terminus of the ataxin-3 (ATXN3) protein. ATXN3 is a deubiquitinating enzyme (DUB) that is involved in protein homeostasis and transcription and may regulate the expression of genes involved in stress response pathways (Orr 2012a). As with other polyGln diseases, SCA3 neurons contain nuclear inclusions and the extended polyGln tract triggers proteolytic cleavage of mutant ATXN3 protein. These cleavage products are detectable in patient brains and fragment accumulation in transgenic mice is neurotoxic (Goti et al. 2004; Haacke et al. 2006; Paulson et al. 1997; Warrick et al. 1998; Wellington et al. 1998) (Fig. 10.2, protein GOF).
While many studies indicate that SCA3 disease is predominantly caused by the mutant ATXN3 protein (Costa Mdo and Paulson 2012; Orr 2012a), SCA3 rCAGexp RNA has also been implicated in SCA3 pathogenesis. Similar to DM, a toxic RNA-induced protein sequestration hypothesis has been tested in a fly SCA3 model as well as CAGexp-expressing mouse and nematode models (Hsu et al. 2011; Li et al. 2008; Wang et al. 2011). To evaluate the toxicity of CAGexp independent of polyGln, interruption of CAGexp with CAA repeats, which still encode polyGln but do not cause RNA toxicity, mitigates the neurodegeneration observed in the fly SCA3 model (Li et al. 2008). Furthermore, expression of a CAGexp in the 3′UTR of a reporter gene in human cell lines is sufficient to elicit MBNL1-containing nuclear foci and MBNL1-dependent splicing changes previously reported in DM (Mykowska et al. 2011). The RNA-binding protein Orb2, also known as cytoplasmic polyadenylation element (CPE)-binding protein 1 (CPEB1), has been implicated in SCA3 since it is a modifier of neurotoxicity in the SCA3 fly model (Shieh and Bonini 2011). Orb2/CPEB1 contains two RRMs and a zinc finger domain, which it uses to bind the CPE of target mRNAs and regulate translation (Richter 2007). The mRNA targets of Orb2/CPEB1 are enriched for functions related to synaptic plasticity and neuronal growth, suggesting that this protein plays a role in learning and memory. Orb2 overexpression in the SCA3 fly model partially suppresses neurotoxicity hinting that the rCAGexp RNAs might interfere with normal Orb2 functions and lead to the loss of translation regulation in SCA3 neurons (Shieh and Bonini 2011). Similarly, Orb2 colocalizes with CGGexp nuclear foci and is a genetic modifier in a fly FXTAS model (Cziko et al. 2009).
4.2.2 SCA8: Bidirectional Transcription and the Discovery of RAN Translation
SCA8 is characterized by ataxia, slurred speech, and abnormal eye movements (nystagmus) and is associated with a CAG•CTG expansion that undergoes bidirectional transcription to produce a CAGexp from ATXN8 and a CUGexp RNA from the ataxin-8 opposite strand (AXN8OS) gene (Ikeda et al. 2008; Moseley et al. 2006). Unaffected individuals have 16–50 CAG•CTG repeats, whereas patients have expansions of 71–1,300 repeats (Fig. 10.1b). Originally, SCA8 pathogenesis was thought to arise primarily from RNA toxicity due to CUGexp expression from the noncoding AXN8OS gene (Koob et al. 1999). As predicted, CUGexp RNA foci colocalize with MBNL1 in SCA8 neurons and MBNL1 overexpression reverses SCA8-induced splicing errors (Chen et al. 2009; Daughters et al. 2009). However, ATXN8 also encodes a short ATXN8 protein that consists primarily of polyGln and SCA8 neurons contain polyGln and ubiquitin-positive inclusions in the nucleus similar to those observed in HD and other polyGln diseases (Moseley et al. 2006) (Fig. 10.3).

Bidirectional transcription across microsatellite expansions may generate both pathogenic RNAs and proteins. Transcription of a CTGexp mutation in the ATXN8OS gene (composed of noncoding exons A–D) produces toxic RNA hairpins (red) that either sequester the MBNL proteins or produce polyLeu (L, pink circles), polyCys (C, turquoise) or polyAla (A, blue) by RAN translation. Transcription of the CAGexp on the opposite strand (ATXN8) results in RAN-generated polyGln (Q, orange), polyAla (A, blue) or polySer (S, green) homopolymer polypeptides or a toxic RNA that might sequester an unknown rCAGexp binding protein (CAGBP). While all six RAN proteins are observed in transfected cells, only polyGln (asterisk), which is initiated with a conventional methionine codon (green) in ATXN8, and polyAla (asterisk) have been detected in vivo
Discovery of a third pathogenic mechanism in SCA8 has profound implications not only on the microsatellite expansion and neurological disease fields but also on our understanding of translational regulatory mechanisms. While investigating the role of polyGln in SCA8, ATXN8 expression constructs were generated to remove the ATG initiation codon upstream of the CAGexp encoding polyGln (Cleary and Ranum 2013; Zu et al. 2011). Surprisingly, removal of the start codon fails to ablate translation and polyGln is still expressed. More surprisingly, the translation of the CAGexp repeat occurs in all three open reading frames (ORFs) leading to co-expression of polyGln, polySer, and polyAla in the absence of frameshifting. Immunological evidence as well as direct protein sequencing supports the existence of this repeat-associated non-ATG, or RAN, translation. All three RAN proteins translated from CAGexp RNAs (polyGln, polyAla, polySer) accumulate in transfected cells and polyGln and polyAla proteins have been shown to accumulate in vivo in mouse and human SCA8 cerebellar Purkinje cells (Cleary and Ranum 2013; Zu et al. 2011) (Figs. 10.2 and 10.3).
Currently, RAN translation has been implicated in four microsatellite expansion diseases including ALS/FTD, DM1, FXTAS as well as SCA8 (Ash et al. 2013; Mori et al. 2013b; Todd et al. 2013; Zu et al. 2011). For FXTAS, considerable evidence supports the hypothesis that this disease is RNA-mediated, but patient neurons also contain large and ubiquitin-positive nuclear inclusions, a hallmark of protein-mediated disease. This paradox led to the recent discovery of RAN translation in FXTAS with in vitro and in vivo evidence for the accumulation of polyglycine (polyGly) containing aggregates (Todd et al. 2013). Additional studies show that polyAla can also be expressed across CGGexp constructs in transfected cells. Suppression of polyGly expression partially rescues CGGexp-induced toxicity and loss of cell viability in FXTAS models and polyGly accumulates in ubiquitinated intranuclear inclusions in patient brains. Thus, some microsatellite expansion diseases, previously labeled as either RNA mediated or protein mediated, involve multiple pathogenic mechanisms and toxic agents (Ling et al. 2013). Clearly, reexamination of patient brain sections from other microsatellite diseases for RAN protein aggregates should now be a priority.
4.2.3 SCA10: AUUCUexp RNA, hnRNP K and Apoptosis
Another RNA-binding protein, hnRNP K (HNRNPK), is implicated in spinocerebellar ataxia type 10 (SCA10). SCA10 is an autosomal dominant disorder characterized by ataxia, seizures, mild peripheral nerve and cognitive impairment. Disease results from an ATTCT repeat expansion in intron 9 of the ATXN10 gene (Teive et al. 2011). This pentanucleotide repeat is relatively short in the normal population (10–32) while SCA10 patients have very large expansions of 800–4,500 repeats (Fig. 10.1b). Although the ATTCTexp does not alter ATXN10 expression levels or splicing (Wakamiya et al. 2006), the AUUCUexp RNA accumulates in nuclear and cytoplasmic foci in SCA10 mouse models and patient fibroblasts (White et al. 2010, 2012). The presence of cytoplasmic foci is a somewhat surprising observation although these structures may be released from the nuclear compartment during mitosis. In vitro, hnRNP K binds to AUUCUexp RNA and colocalizes with AUUCUexp-containing foci (White et al. 2010). HnRNP K contains three KH domains that have a high affinity for C-rich clusters and SCA10 fibroblasts show aberrant splicing of the hnRNP K target β-tropomyosin, suggesting that loss of hnRNP K function contributes to missplicing in SCA10. HnRNPK also acts as a docking protein for multiple factors to modulate chromatin remodeling, transcription, and translation to facilitate cross-talk between multiple gene expression tiers (Bomsztyk et al. 2004). Another study suggests that loss of hnRNP K function triggers apoptosis in AUUCUexp-expressing cells (White et al. 2010). Sequestration of hnRNP K by AUUCUexp RNA blocks its binding to PKCδ, which could allow PKCδ to promote caspase 3-mediated apoptosis. In accordance with this model, overexpression of hnRNP K suppresses the AUUCUexp-induced apoptosis pathway and partially rescues cell viability. Nevertheless, additional cell and animal models must be developed to substantiate the hnRNP K LOF model for SCA10.
5 An Intrinsic Curse: Microsatellite Expansions in RNA-Binding Proteins
5.1 SCA2 and ATXN2
While some expansion mutations indirectly affect RNA-binding protein function via sequestration by repeat expansion RNAs, other diseases stem from expansion mutations in genes encoding RNA-binding proteins. One of the most common types of ataxia, spinocerebellar ataxia type 2 (SCA2), is caused by a CAGexp in ATNX2, the gene encoding the RNA-binding protein ataxin-2 (Orr 2012a; Rub et al. 2013). SCA2 patients suffer from cerebellar ataxia in addition to decreased reflexes and polyneuropathy, which affects peripheral nerves throughout the body. Cell death has been observed in both SCA2 motor neurons and Purkinje cells. The genetic basis of SCA2 is an expansion from 15–24 to 32–200 CAG repeats in exon 1 of ATXN2, which results in a polyGln expansion in the ATXN2 N-terminus (Fig. 10.1b).
ATXN2 has been implicated in various cellular processes including Golgi-mediated transport, calcium regulation, formation of stress granules and P-bodies, and regulation of RNA post-transcriptional modification and translation (Magana et al. 2013). Several lines of evidence suggest that ATXN2 participates directly and indirectly in RNA metabolism. The ATXN2 protein contains an Sm-like (Lsm) domain, also found in factors that function in pre-RNA processing and mRNA decay (Albrecht et al. 2004), as well as a PAM2 domain that mediates ATXN2 binding to PABPC1, the major cytoplasmic poly(A)-binding protein involved in regulating poly(A) tail length, mRNA stability, and translation (Ralser et al. 2005). ATXN2 and PABPC1 colocalize in cells, assemble onto polyribosomes and are recruited to stress granules, which suggests that ATXN2 and PABPC1 cooperate to sequester mRNAs in stress granules to downregulate their translation during cell stress (Magana et al. 2013; Satterfield and Pallanck 2006).
Another RNA-binding protein, originally identified as a novel protein that interacts with ATXN2, is ataxin-2-binding protein (A2BP1, now known as RBFOX1) (Shibata et al. 2000). RBFOX1 is predominantly expressed in the brain and skeletal muscle and belongs to the RBFOX family of alternative splicing factors (Fogel et al. 2012; Gehman et al. 2011; Underwood et al. 2005). Murine Rbfox1 binds directly to pre-mRNA to activate or repress exon inclusion in a position-dependent manner (Sun et al. 2012). Rbfox1 null mice have widespread splicing abnormalities in the brain, as well as neuronal excitability and seizures. A combination of splicing microarrays and binding site analysis using individual-nucleotide resolution cross-linking immunoprecipitation (iCLIP) reveals that Rbfox1 regulates the alternative splicing of gene transcripts involved in membrane excitation and synaptic transmission (Gehman et al. 2011). Similarly, knockdown of RBFOX1 in human neuronal cultures results in splicing abnormalities in genes involved in neuronal development (Fogel et al. 2012). The binding of RBFOX1 to ATXN2 suggests that the two proteins interact to regulate alternative splicing in the brain and that RBFOX1 may play a role in SCA2.
Several molecular mechanisms have been proposed for SCA2 pathogenesis, including formation of polyGln aggregates and deleterious gain-of-function of mutant ATXN2. Mutant ATXN2, but not the wild-type protein, has been observed to interact directly with the calcium channel InsP3R1 in neuronal cultures, which may lead to altered calcium signaling and excitotoxic cell death (Liu et al. 2009). Treatment of the cells with an inhibitor of another calcium channel, the ryanodine receptor, reduces cell death highlighting the calcium regulatory pathway as a potential therapeutic target for SCA2. The expanded polyGln tract in mutant ATXN2 also triggers ATXN2 protein misfolding and polyGln aggregation in ubiquinated intranuclear and cytoplasmic inclusions in SCA2 neurons (Huynh et al. 2000; Koyano et al. 1999). In addition, mutant ATXN2 undergoes proteolytic cleavage and the C-terminal fragment might have an altered function compared to the holoprotein while the N-terminal fragment, containing the polyGln expansion, accumulates in potentially toxic aggregates (Huynh et al. 2000). Interestingly, intermediate-sized ATXN2 expansions are associated with an increased risk for developing amyotrophic lateral sclerosis (ALS) and parkinsonism (Elden et al. 2010; Simon-Sanchez et al. 2005). Supporting a link between ATXN2 and ALS, ATXN2 accumulates in discrete foci, possibly stress granules, in degenerating motor neurons of ALS patients (Li et al. 2013). Furthermore, ATXN2 interacts in an RNA-dependent manner with TDP-43, which plays a key role in ALS pathology, and modifies TDP-43 toxicity in several ALS model systems (Elden et al. 2010). These results underscore the role of ATXN2 in RNA processing and turnover and provide genetic and mechanistic links between ATXN2 and ALS.
5.2 Oculopharyngeal Muscular Dystrophy and PABPN1
Another neuromuscular disorder that arises from a microsatellite expansion in the coding region of an RNA-binding protein is oculopharyngeal muscular dystrophy (OPMD) (Brais 2009; Messaed and Rouleau 2009). OPMD is a late-onset muscular dystrophy characterized by muscle weakness, ptosis (eyelid drooping), and dysphagia (difficulty in swallowing that may lead to aspiration pneumonia). OPMD is caused by a GCNexp in PABPN1, which encodes the major nuclear polyadenylate-binding protein that plays a vital role in pre-mRNA 3′ end processing and poly(A) tail formation (Banerjee et al. 2013).
Initially, OPMD was associated with GCGexp expansions (Brais et al. 1998), but additional mutations, including point mutations, have also been identified that result in a stretch of GCN codons encoding an expanded polyalanine (polyAla) tract in the N-terminus of PABPN1 (Nakamoto et al. 2002; Robinson et al. 2006). Whereas unaffected individuals typically have 10 GCN repeats, patients have 11–17 repeats, resulting in one to seven additional alanine residues (Fig. 10.1b). Similar to other neurological disorders, a central mystery is why a mutation in an essential and ubiquitously expressed protein causes a late-onset disease that primarily affects specific tissues, in this case facial, tongue, and extremity muscles. Histological analysis of patient skeletal muscle biopsies reveals hallmarks of muscular dystrophies, such as changes in muscle fiber size variability, centralized myonuclei, and an overall loss of myofibers with excessive fibrous and fatty tissue (Tome et al. 1997). Moreover, OPMD muscle fibers have mutant PABPN1 intranuclear inclusions that appear as zones of tubular filaments (Tome et al. 1997; Tome and Fardeau 1980).
Controversy exists over the toxicity of these inclusions and whether they induce toxicity or serve a protective role in confining the mutant protein. Several lines of evidence support the latter hypothesis. In an OPMD cell model, the most toxic form of mutant PABPN1 is soluble and not in insoluble inclusions while inclusion disruption increases cell death (Messaed et al. 2007). Using PABPN1 constructs with variable polyAla length, mutant PABPN1 is most toxic when the expansions are longer although these expanded proteins do not form inclusions (Klein et al. 2008). In a similar vein, an OPMD fly model recapitulates the muscle phenotype observed in the disease, despite the fact that it lacks inclusions (Chartier et al. 2006). PABPN1 forms oligomeric structures prior to inclusion formation so soluble and oligomeric forms of mutant PABPN1 may contribute to the cellular toxicity (Raz et al. 2011). In addition to inclusion-independent models of PABPN1 toxicity, several inclusion-dependent models have been proposed. PABPN1 expansion mutations may cause a dominant-negative effect leading to inclusions that, in turn, sequester other cellular factors and disrupts several cellular pathways. While PABPN1 is an integral constituent of the inclusions, other co-localizing components include transcription factors, polyadenylated RNA, RNA-binding proteins (including CELF1), and components of the UPS (Brais 2009; Corbeil-Girard et al. 2005). As noted previously, UPS dysfunction has been implicated in other neurodegenerative diseases (Ciechanover and Brundin 2003; Dennissen et al. 2012) and addition of a proteasome inhibitor in an OPMD cell model exacerbates inclusion formation in a dose-dependent manner while overexpression of heat shock proteins decreases inclusions (Abu-Baker et al. 2003). Furthermore, transcriptome analysis indicates that genes encoding UPS components are misregulated in OPMD (Anvar et al. 2011). The specificity of the UPS involvement in OPMD is highlighted by the observation that the UPS is consistently misregulated in OPMD while other protein degradation pathways are not. These results have led to a disease model in which an age-related decrease in UPS function causes accumulation of misfolded PABPN1 that aggregates, forms inclusions that, in turn, recruit UPS thus further blocking the UPS in OPMD cells (Raz et al. 2013). In addition to blocking the UPS, recruitment of other cellular factors to aggregates may disrupt additional cellular pathways. To support the toxicity of aggregate formation, oral treatment of anti-aggregation drugs attenuates pathology in an OPMD mouse model (Davies et al. 2005, 2006).
Inclusion-dependent models of OPMD propose that a reduction of soluble PABPN1 due to aggregation leads to PABPN1 loss-of-function. PABPN1 consists of an N-terminal region with the polyAla stretch, a central domain with a conserved RNA recognition motif (RRM) and a C-terminal domain that may also be involved in fibril formation (Winter et al. 2012). PABPN1 was originally characterized as a nuclear factor that increases processivity and efficiency of poly(A) addition by poly(A) polymerase. PABPN1 coats the poly(A) tail, possibly to maintain poly(A) tail length that impacts downstream events including nuclear RNA export, mRNA translation and stability (Banerjee et al. 2013). PABPN1 also appears to shuttle from nucleus to cytoplasm, potentially aiding in nuclear export and translation, and then cooperates with, or is replaced by, its cytoplasmic partner PABPC1 (Lemay et al. 2010). Knockdown of PABPN1 in primary mouse myoblasts prepared from extraocular, pharyngeal, and limb muscles causes shortening of poly(A) tails and accumulation of nuclear poly(A) RNA, supporting a role for PABPN1 in nuclear export (Apponi et al. 2010). Furthermore, loss of PABPN1 in myoblasts results in decreased proliferation and differentiation hinting that myogenesis might be compromised in OPMD. Recently, PABPN1 was found to regulate alternative cleavage and polyadenylation (APA) (de Klerk et al. 2012; Jenal et al. 2012). PABPN1 depletion induces selection of proximal cleavage sites and widespread 3′ UTR shortening. Analysis of OPMD patient cells, as well as an OPMD mouse model, also show 3′ UTR shortening suggesting that mutant PABPN1 and PABPN1 depletion are equivalent phenomena that result in APA dysregulation. These results support a model in which PABPN1 loss-of-function in OPMD leads to widespread molecular abnormalities, including shortening of 3′UTRs, decrease in poly(A) tail length, and blockage of poly(A)-mRNA nuclear export, which results in defects in myogenesis and muscle function. In addition to this mRNA-centric view, PABPN1 also promotes the turnover of a class of long noncoding (lnc)RNAs (Beaulieu et al. 2012). Further investigation into the normal functions of PABPN1 are required to clarify how disruption of these processes results in the distinct pathological phenotypes associated this neuromuscular disease.
6 The Horizon in Microsatellite Expansion Disorders
6.1 C9ORF72 Expansions in ALS/FTD
The microsatellite expansion field has received increased attention with the discovery that a G4C2 exp mutation in the C9ORF72 gene is the most common known cause of familial amyotrophic lateral sclerosis (ALS) and frontotemporal dementia (FTD) (DeJesus-Hernandez et al. 2011; Renton et al. 2011). Prior to this discovery, there were indications that these two diseases represent different manifestations of a spectrum disorder with common genetics, pathology, and clinical features. Patients diagnosed with ALS, also known as Lou Gehrig’s disease, suffer from muscle wasting and paralysis resulting from denervation caused by the death of upper and lower motor neurons (Van Damme and Robberecht 2013). In contrast, FTD is a condition characterized by loss of neurons in the frontal and temporal lobes of the brain, which results in behavioral changes, dementia, and speech abnormalities or progressive non-fluent aphasia. However, FTD patients may suffer from ALS-like symptoms, such as motor deficits caused by motor neuron dysfunction (Lomen-Hoerth et al. 2002). Conversely, up to 50 % of ALS patients also develop FTD-like symptoms, including dementia and cognitive changes, related to impairment of frontotemporal functions (Giordana et al. 2011).
A positive family history of disease or related symptoms occurs in approximately 10 % of ALS, and 25–50 % of FTD, patients (Graff-Radford and Woodruff 2007; Gros-Louis et al. 2006; Rohrer et al. 2009). These cases are referred to as familial ALS and FTD, while the remainder of cases are sporadic. FTD has been linked to mutations in several genes including microtubule-associated protein tau (MAPT), progranulin (GRN), valosin-containing protein (VCP), charged multivesicular body protein 2B (CHMP2B), and C9ORF72 (Rademakers et al. 2012). Similarly, ALS-linked mutations include superoxide dismutase 1 (SOD1), ataxin 2 (ATXN2), TATA-binding protein associated factor 15 (TAF15), Ewing’s sarcoma breakpoint region 1 (EWSR1), angiogenin (ANG), senataxin (SETX), fused in sarcoma (FUS), TDP-43 (TARDBP), and C9ORF72 (Robberecht and Philips 2013). Many of the genes have been linked to both ALS and FTD but are more commonly associated with one of these diseases (Al-Chalabi et al. 2012). While the overlapping genetics and clinical features support the view of ALS/FTD is a spectrum disorder, even more striking are the commonalities between FTD and ALS molecular pathology. Most FTD and ALS cases have characteristic neuronal cytoplasmic inclusions, which contain certain RNA-binding proteins. For example, mutations in FUS and TDP-43 are less common in FTD than ALS, yet both ALS and FTD patient neurons often contain cytoplasmic aggregates of FUS or TDP-43 even without a corresponding gene mutation (Kwiatkowski et al. 2009; Neumann et al. 2006, 2009; Vance et al. 2009). These findings suggest that common molecular pathways may be disrupted by a number of different ALS/FTD-linked genetic variants. In addition to potential effects on RNA processing, RNA editing may be disrupted in ALS due to loss of ADAR2 responsible for A-to-I RNA editing (Aizawa et al. 2010; Kawahara et al. 2004).
The C9ORF72 expansion mutation is surprisingly common among the genetic variants linked to ALS/FTD, and can cause both sporadic and familial forms of ALS and FTD, strongly supporting the disease spectrum hypothesis. Although the distributions of normal and disease-associated G4C2 repeat lengths are still being actively investigated, thus far it appears that unaffected individuals have <30 repeats, generally with 2–5 repeats, whereas ALS/FTD patients have considerably longer expansions of 700–1,600 repeats (DeJesus-Hernandez et al. 2011; Dobson-Stone et al. 2012; Gijselinck et al. 2012; Majounie et al. 2012; Robberecht and Philips 2013) (Fig. 10.1b). The expansion is located in the first intron of the C9ORF72 gene, but this region is also the promoter of an alternative isoform (Fig. 10.4). The gene encodes three mRNA, and two protein, isoforms. One possibility is that the expansion alters activity of the promoter resulting in decreased gene expression levels. In support of this hypothesis, some studies have found that ALS patients with the G4C2 exp mutation have reduced levels of C9ORF72 mRNA (DeJesus-Hernandez et al. 2011; Gijselinck et al. 2012). This observation suggests that the repeat could interfere with expression of the C9ORF72 protein and cause haploinsufficiency, however no clear reduction in protein levels has been reported in these ALS/FTD patients.

Toxic RNAs and peptides in C9ORF72 ALS/FTD. The GGGCCexp mutation (red box) in C9ORF72 intron 1a blocks transcription initiation (arrow) from exon 1b but promotes initiation from exon 1a. RNA processing generates at least two mRNAs and a released intron 1a lariat (or intron 1a might be retained during alternative splicing) that may fold into a G-quadruplex and accumulate in nuclear foci (pink ellipsoid) together with multiple nuclear RNA-binding proteins (RBM45, Purα, SFRS1, hnRNP A2, hnRNP A3). Further repeat expansion may lead to failure of this retention mechanism due to titration of rGGGCCexp-binding factors leading to nuclear export of intron 1a-containing mRNA, RAN translation, and RAN protein aggregation
While the function of the C9ORF72 protein remains obscure, homology searches reveal that it is structurally related to DENN (differentially expressed in normal and neoplasia) domain proteins (Levine et al. 2013). DENN domain proteins are GDP/GTP exchange factors (GEFs) for Rab-GTPases. Therefore, based upon homology, C9ORF72 might be involved in membrane trafficking related to Rab-GTPase switches. C9ORF72 mRNA is expressed in a wide variety of cell types and the C9ORF72 protein is primarily cytoplasmic in neurons (DeJesus-Hernandez et al. 2011). Several immunohistochemical studies have revealed that the G4C2 exp causes no apparent changes in cellular distribution of the C9ORF72 protein and no abnormal C9ORF72 aggregates, suggesting that altered function of this protein may not be the primary molecular mechanism in ALS/FTD (Rademakers et al. 2012). Although patients have no C9ORF72 protein aggregates, many studies have revealed that patients with C9ORF72 mutations have the neuropathological hallmark of TDP-43 positive inclusions in the brain and spinal cord, as seen in other ALS/FTD patients. In addition to this characteristic TDP-43 pathology, these patients have a unique feature of neuronal inclusions that are negative for TDP-43, yet positive for UPS-associated proteins such as ubiquitin, ubiquilins, and sequestosome-1 (SQSTM1), also known as ubiquitin-binding protein p62 (p62) (Al-Sarraj et al. 2011). The presence of UPS suggests aberrant accumulation of unidentified molecules marked for degradation, possibly unique to patients with C9ORF72 mutations.
Several recent reports have shown that RAN translation may be an important pathogenic feature of C9ORF72 ALS/FTD. The G4C2 exp was initially predicted to form hairpins but may also fold into G-quadruplex structures (Ash et al. 2013; Fratta et al. 2012; Reddy et al. 2013) (Fig. 10.4). While G-quadruplexes in 5′UTRs often suppress canonical cap-dependent translation, these structures have also been reported to aid in noncanonical IRES-mediated translation initiation (Morris et al. 2010). To address the question of whether RAN translation initiates from rG4C2 exp RNA, antibodies against the predicted RAN translation products were used to assess the presence of these products in C9ORF72 patient tissues (Ash et al. 2013; Mori et al. 2013b). As hypothesized, RAN translation products are detectable from all three ORFs resulting in the dipeptide-repeat (DPR) proteins poly(Gly-Ala), poly(Gly-Pro), and poly(Gly-Arg). These DPR proteins form insoluble nuclear and cytoplasmic inclusions in C9ORF72 patient neurons and are not detectable in healthy controls, ALS/FTD patients negative for the G4C2 exp mutations or in patients with other neurodegenerative diseases. These inclusions are also distinct from TDP-43 inclusions but colocalize with p62-positive inclusions, suggesting UPS involvement in the turnover of RAN translation products (Ash et al. 2013; Mori et al. 2013b). Whether these protein products are neurotoxic and contribute significantly to ALS/FTD pathology remains a critical question.
Another molecular mechanism potentially involved in C9ORF72 disease pathogenesis is rG4C2 exp toxicity, since these mutant RNAs accumulate in nuclear foci in ALS/FTD tissues and patient-derived iPS cells (Almeida et al. 2013; DeJesus-Hernandez et al. 2011) (Fig. 10.4). While several reports have indicated that the total levels of C9ORF72 mRNA are reduced approximately twofold in patient lymphoblasts (DeJesus-Hernandez et al. 2011), frontal lobe (Gijselinck et al. 2012), and cerebellum (Mori et al. 2013b), levels of both sense and antisense intron 1a-containing RNAs increase seven- to eightfold (Mori et al. 2013b). The latter result suggests that C9ORF72 splicing is impaired and/or unspliced, or partially spliced, pre-mRNAs accumulate in RNA foci in C9ORF72 ALS/FTD, similar to the accumulation of CNBP intron 1 CCUGexp RNAs in DM2 cells (Margolis et al. 2006). Several candidates for factors sequestered by rG4C2 exp RNAs have been proposed including hnRNP A2/B1 (DeJesus-Hernandez et al. 2011), RBM45 (Collins et al. 2012), hnRNP A3 (Mori et al. 2013a), SRSF1 (ASF/SF2) (Reddy et al. 2013), and Purα (Xu et al. 2013). The next critical step for validation of these putative sequestered factors is to demonstrate that they bind to rG4C2 exp RNAs in patient’s cells and affected tissues. Moreover, appropriate loss-of-function mammalian models must be developed that recapitulate distinct ALS/FTD phenotypes.
6.2 Conclusion and Perspective
While the functions of microsatellites within the normal repeat range remain obscure, several common themes have emerged from studies on microsatellite expansions and disease. First, expansions occur frequently, but not exclusively, in GC-rich microsatellites due to their inherent tendency to form imperfect hairpins, quadraplex-like and slipped-stranded structures setting the stage for error-prone DNA replication, recombination and repair. Expansion mechanisms for repeat expansions that are not GC-rich, such as the SCA10 ATTCTexp and the GAAexp in Friedreich’s ataxia (FRDA), a recessively inherited neurological disorder caused by frataxin loss-of-function, are less studied. However, abnormal DNA structures may also be involved in these diseases with potential triplexes in FRDA and replication-associated template switching in SCA10 (Cherng et al. 2011; Lopez Castel et al. 2010; Mirkin 2007). Second, microsatellite expansions preferentially cause neurological and neuromuscular diseases, even when the affected gene is expressed ubiquitously, suggesting that different cell types possess varying sensitivities to these mutations. For example, why does a small increase in a polyalanine stretch in the N-terminal region of the PABPN1 protein cause a late-onset disease characterized by eyelid drooping, swallowing difficulty, and proximal limb weakness? Third, studies on expansion disorders have revealed novel disease mechanisms, including RNA toxicity and RAN translation. RAN translation, or the production of unusual reiterated (homopolymeric and heteropolymeric) proteins from repetitive RNA templates, is reminiscent of the cell-free experiments that employed synthetic RNA polynucleotides to decipher the genetic code (Nirenberg 2004). However, RAN translation produces homopolymeric proteins in all three frames from trinucleotide repeat expansions without frameshifting (Zu et al. 2011). Fourth, bidirectional transcription of repeat expansions generates multiple potentially toxic RNAs as well as conventional mutant, and RAN, proteins. Delineating the relative toxicities of each of these pathogenic entities will be a difficult, although an essential, step towards the development of effective new therapies for these diseases.
Many questions and experimental challenges remain in the unstable microsatellite field. How many additional familial and “sporadic” diseases are caused by tandem repeat expansion mutations? Why are so many of these disorders characterized by a late-onset clinical profile and why are muscle and nervous systems particularly vulnerable? Why are alterations in RNA binding protein functions and aggregation states so prominent in this group of diseases? Are RNA foci and protein inclusions pathogenic, protective, or innocent bystanders? Technological and computational advances in RNA and protein analyses, and the development of more informative cell and animal disease models, should provide additional experimental surprises and mechanistic insights into how microsatellite expansions perturb normal cellular pathways.
References
Abu-Baker A, Messaed C, Laganiere J, Gaspar C, Brais B, Rouleau GA (2003) Involvement of the ubiquitin-proteasome pathway and molecular chaperones in oculopharyngeal muscular dystrophy. Hum Mol Genet 12:2609–2623
Adereth Y, Dammai V, Kose N, Li R, Hsu T (2005) RNA-dependent integrin alpha3 protein localization regulated by the Muscleblind-like protein MLP1. Nat Cell Biol 7:1240–1247
Aizawa H, Sawada J, Hideyama T, Yamashita T, Katayama T, Hasebe N, Kimura T, Yahara O, Kwak S (2010) TDP-43 pathology in sporadic ALS occurs in motor neurons lacking the RNA editing enzyme ADAR2. Acta Neuropathol 120:75–84
Akins MR, Berk-Rauch HE, Fallon JR (2009) Presynaptic translation: stepping out of the postsynaptic shadow. Front Neural Circuits 3:17
Al-Chalabi A, Jones A, Troakes C, King A, Al-Sarraj S, van den Berg LH (2012) The genetics and neuropathology of amyotrophic lateral sclerosis. Acta Neuropathol 124:339–352
Al-Sarraj S, King A, Troakes C, Smith B, Maekawa S, Bodi I, Rogelj B, Al-Chalabi A, Hortobagyi T, Shaw CE (2011) p62 positive, TDP-43 negative, neuronal cytoplasmic and intranuclear inclusions in the cerebellum and hippocampus define the pathology of C9orf72-linked FTLD and MND/ALS. Acta Neuropathol 122:691–702
Albrecht M, Golatta M, Wullner U, Lengauer T (2004) Structural and functional analysis of ataxin-2 and ataxin-3. Eur J Biochem 271:3155–3170
Almeida S, Gascon E, Tran H, Chou HJ, Gendron TF, Degroot S, Tapper AR, Sellier C, Charlet-Berguerand N, Karydas A, Seeley WW, Boxer AL, Petrucelli L, Miller BL, Gao FB (2013) Modeling key pathological features of frontotemporal dementia with C9ORF72 repeat expansion in iPSC-derived human neurons. Acta Neuropathol 126(3):385–99
Anvar SY, Hoen PA, Venema A, van der Sluijs B, van Engelen B, Snoeck M, Vissing J, Trollet C, Dickson G, Chartier A, Simonelig M, van Ommen GJ, van der Maarel SM, Raz V (2011) Deregulation of the ubiquitin-proteasome system is the predominant molecular pathology in OPMD animal models and patients. Skelet Muscle 1:15
Apponi LH, Leung SW, Williams KR, Valentini SR, Corbett AH, Pavlath GK (2010) Loss of nuclear poly(A)-binding protein 1 causes defects in myogenesis and mRNA biogenesis. Hum Mol Genet 19:1058–1065
Ash PE, Bieniek KF, Gendron TF, Caulfield T, Lin WL, Dejesus-Hernandez M, van Blitterswijk MM, Jansen-West K, Paul JW 3rd, Rademakers R, Boylan KB, Dickson DW, Petrucelli L (2013) Unconventional translation of C9ORF72 GGGGCC expansion generates insoluble polypeptides specific to c9FTD/ALS. Neuron 77:639–646
Banerjee A, Apponi LH, Pavlath GK, Corbett AH (2013) PABPN1: molecular function and muscle disease. FEBS J 280:4230–4250
Bassell GJ, Warren ST (2008) Fragile X syndrome: loss of local mRNA regulation alters synaptic development and function. Neuron 60:201–214
Batra R, Charizanis K, Swanson MS (2010) Partners in crime: bidirectional transcription in unstable microsatellite disease. Hum Mol Genet 19:R77–R82
Beaulieu YB, Kleinman CL, Landry-Voyer AM, Majewski J, Bachand F (2012) Polyadenylation-dependent control of long noncoding RNA expression by the poly(A)-binding protein nuclear 1. PLoS Genet 8:e1003078
Berman RF, Willemsen R (2009) Mouse models of fragile X-associated tremor ataxia. J Investig Med 57:837–841
Bhakar AL, Dolen G, Bear MF (2012) The pathophysiology of fragile X (and what it teaches us about synapses). Annu Rev Neurosci 35:417–443
Bhogal B, Jongens TA (2010) Fragile X syndrome and model organisms: identifying potential routes of therapeutic intervention. Dis Model Mech 3:693–700
Bomsztyk K, Denisenko O, Ostrowski J (2004) hnRNP K: one protein multiple processes. Bioessays 26:629–638
Botta A, Caldarola S, Vallo L, Bonifazi E, Fruci D, Gullotta F, Massa R, Novelli G, Loreni F (2006) Effect of the [CCTG]n repeat expansion on ZNF9 expression in myotonic dystrophy type II (DM2). Biochim Biophys Acta 1762:329–334
Brais B (2009) Oculopharyngeal muscular dystrophy: a polyalanine myopathy. Curr Neurol Neurosci Rep 9:76–82
Brais B, Bouchard JP, Xie YG, Rochefort DL, Chretien N, Tome FM, Lafreniere RG, Rommens JM, Uyama E, Nohira O, Blumen S, Korczyn AD, Heutink P, Mathieu J, Duranceau A, Codere F, Fardeau M, Rouleau GA (1998) Short GCG expansions in the PABP2 gene cause oculopharyngeal muscular dystrophy. Nat Genet 18:164–167
Brook JD, McCurrach ME, Harley HG, Buckler AJ, Church D, Aburatani H, Hunter K, Stanton VP, Thirion JP, Hudson T et al (1992) Molecular basis of myotonic dystrophy: expansion of a trinucleotide (CTG) repeat at the 3′ end of a transcript encoding a protein kinase family member. Cell 68:799–808
Budworth H, McMurray CT (2013) Bidirectional transcription of trinucleotide repeats: roles for excision repair. DNA Repair (Amst) 12:672–684
Buschiazzo E, Gemmell NJ (2010) Conservation of human microsatellites across 450 million years of evolution. Genome Biol Evol 2:153–165
Cardani R, Mancinelli E, Giagnacovo M, Sansone V, Meola G (2009) Ribonuclear inclusions as biomarker of myotonic dystrophy type 2, even in improperly frozen or defrozen skeletal muscle biopsies. Eur J Histochem 53:107–111
Cardani R, Mancinelli E, Rotondo G, Sansone V, Meola G (2006) Muscleblind-like protein 1 nuclear sequestration is a molecular pathology marker of DM1 and DM2. Eur J Histochem 50:177–182
Charizanis K, Lee KY, Batra R, Goodwin M, Zhang C, Yuan Y, Shiue L, Cline M, Scotti MM, Xia G, Kumar A, Ashizawa T, Clark HB, Kimura T, Takahashi MP, Fujimura H, Jinnai K, Yoshikawa H, Gomes-Pereira M, Gourdon G, Sakai N, Nishino S, Foster TC, Ares M Jr, Darnell RB, Swanson MS (2012) Muscleblind-like 2-mediated alternative splicing in the developing brain and dysregulation in myotonic dystrophy. Neuron 75:437–450
Chartier A, Benoit B, Simonelig M (2006) A Drosophila model of oculopharyngeal muscular dystrophy reveals intrinsic toxicity of PABPN1. EMBO J 25:2253–2262
Chen IC, Lin HY, Lee GC, Kao SH, Chen CM, Wu YR, Hsieh-Li HM, Su MT, Lee-Chen GJ (2009) Spinocerebellar ataxia type 8 larger triplet expansion alters histone modification and induces RNA foci. BMC Mol Biol 10:9
Chen W, Wang Y, Abe Y, Cheney L, Udd B, Li YP (2007) Haploinsuffciency for Znf9 in Znf9+/− mice is associated with multiorgan abnormalities resembling myotonic dystrophy. J Mol Biol 368:8–17
Cherng N, Shishkin AA, Schlager LI, Tuck RH, Sloan L, Matera R, Sarkar PS, Ashizawa T, Freudenreich CH, Mirkin SM (2011) Expansions, contractions, and fragility of the spinocerebellar ataxia type 10 pentanucleotide repeat in yeast. Proc Natl Acad Sci U S A 108:2843–2848
Ciechanover A, Brundin P (2003) The ubiquitin proteasome system in neurodegenerative diseases: sometimes the chicken, sometimes the egg. Neuron 40:427–446
Cleary JD, Ranum LP (2013) Repeat-associated non-ATG (RAN) translation in neurological disease. Hum Mol Genet 22(R1):R45–R51
Coffee B, Zhang F, Warren ST, Reines D (1999) Acetylated histones are associated with FMR1 in normal but not fragile X-syndrome cells. Nat Genet 22:98–101
Collins M, Riascos D, Kovalik T, An J, Krupa K, Krupa K, Hood BL, Conrads TP, Renton AE, Traynor BJ, Bowser R (2012) The RNA-binding motif 45 (RBM45) protein accumulates in inclusion bodies in amyotrophic lateral sclerosis (ALS) and frontotemporal lobar degeneration with TDP-43 inclusions (FTLD-TDP) patients. Acta Neuropathol 124:717–732
Corbeil-Girard LP, Klein AF, Sasseville AM, Lavoie H, Dicaire MJ, Saint-Denis A, Page M, Duranceau A, Codere F, Bouchard JP, Karpati G, Rouleau GA, Massie B, Langelier Y, Brais B (2005) PABPN1 overexpression leads to upregulation of genes encoding nuclear proteins that are sequestered in oculopharyngeal muscular dystrophy nuclear inclusions. Neurobiol Dis 18:551–567
Costa Mdo C, Paulson HL (2012) Toward understanding Machado-Joseph disease. Prog Neurobiol 97:239–257
Cziko AM, McCann CT, Howlett IC, Barbee SA, Duncan RP, Luedemann R, Zarnescu D, Zinsmaier KE, Parker RR, Ramaswami M (2009) Genetic modifiers of dFMR1 encode RNA granule components in Drosophila. Genetics 182:1051–1060
Darnell JC, Jensen KB, Jin P, Brown V, Warren ST, Darnell RB (2001) Fragile X mental retardation protein targets G quartet mRNAs important for neuronal function. Cell 107:489–499
Darnell JC, Van Driesche SJ, Zhang C, Hung KY, Mele A, Fraser CE, Stone EF, Chen C, Fak JJ, Chi SW, Licatalosi DD, Richter JD, Darnell RB (2011) FMRP stalls ribosomal translocation on mRNAs linked to synaptic function and autism. Cell 146:247–261
Daughters RS, Tuttle DL, Gao W, Ikeda Y, Moseley ML, Ebner TJ, Swanson MS, Ranum LP (2009) RNA gain-of-function in spinocerebellar ataxia type 8. PLoS Genet 5:e1000600
Davies JE, Sarkar S, Rubinsztein DC (2006) Trehalose reduces aggregate formation and delays pathology in a transgenic mouse model of oculopharyngeal muscular dystrophy. Hum Mol Genet 15:23–31
Davies JE, Wang L, Garcia-Oroz L, Cook LJ, Vacher C, O’Donovan DG, Rubinsztein DC (2005) Doxycycline attenuates and delays toxicity of the oculopharyngeal muscular dystrophy mutation in transgenic mice. Nat Med 11:672–677
Davis BM, McCurrach ME, Taneja KL, Singer RH, Housman DE (1997) Expansion of a CUG trinucleotide repeat in the 3′ untranslated region of myotonic dystrophy protein kinase transcripts results in nuclear retention of transcripts. Proc Natl Acad Sci U S A 94:7388–7393
de Klerk E, Venema A, Anvar SY, Goeman JJ, Hu O, Trollet C, Dickson G, den Dunnen JT, van der Maarel SM, Raz V, t Hoen PA (2012) Poly(A) binding protein nuclear 1 levels affect alternative polyadenylation. Nucleic Acids Res 40:9089–9101
DeJesus-Hernandez M, Mackenzie IR, Boeve BF, Boxer AL, Baker M, Rutherford NJ, Nicholson AM, Finch NA, Flynn H, Adamson J, Kouri N, Wojtas A, Sengdy P, Hsiung GY, Karydas A, Seeley WW, Josephs KA, Coppola G, Geschwind DH, Wszolek ZK, Feldman H, Knopman DS, Petersen RC, Miller BL, Dickson DW, Boylan KB, Graff-Radford NR, Rademakers R (2011) Expanded GGGGCC hexanucleotide repeat in noncoding region of C9ORF72 causes chromosome 9p-linked FTD and ALS. Neuron 72:245–256
Dennissen FJ, Kholod N, van Leeuwen FW (2012) The ubiquitin proteasome system in neurodegenerative diseases: culprit, accomplice or victim? Prog Neurobiol 96:190–207
Dobson-Stone C, Hallupp M, Bartley L, Shepherd CE, Halliday GM, Schofield PR, Hodges JR, Kwok JB (2012) C9ORF72 repeat expansion in clinical and neuropathologic frontotemporal dementia cohorts. Neurology 79:995–1001
Doolittle WF, Sapienza C (1980) Selfish genes, the phenotype paradigm and genome evolution. Nature 284:601–603
Du H, Cline MS, Osborne RJ, Tuttle DL, Clark TA, Donohue JP, Hall MP, Shiue L, Swanson MS, Thornton CA, Ares M Jr (2010) Aberrant alternative splicing and extracellular matrix gene expression in mouse models of myotonic dystrophy. Nat Struct Mol Biol 17:187–193
Echeverria GV, Cooper TA (2012) RNA-binding proteins in microsatellite expansion disorders: mediators of RNA toxicity. Brain Res 1462:100–111
Elden AC, Kim HJ, Hart MP, Chen-Plotkin AS, Johnson BS, Fang X, Armakola M, Geser F, Greene R, Lu MM, Padmanabhan A, Clay-Falcone D, McCluskey L, Elman L, Juhr D, Gruber PJ, Rub U, Auburger G, Trojanowski JQ, Lee VM, Van Deerlin VM, Bonini NM, Gitler AD (2010) Ataxin-2 intermediate-length polyglutamine expansions are associated with increased risk for ALS. Nature 466:1069–1075
Finkbeiner S (2011) Huntington’s disease. Cold Spring Harb Perspect Biol 3
Fogel BL, Wexler E, Wahnich A, Friedrich T, Vijayendran C, Gao F, Parikshak N, Konopka G, Geschwind DH (2012) RBFOX1 regulates both splicing and transcriptional networks in human neuronal development. Hum Mol Genet 21:4171–4186
Fratta P, Mizielinska S, Nicoll AJ, Zloh M, Fisher EM, Parkinson G, Isaacs AM (2012) C9orf72 hexanucleotide repeat associated with amyotrophic lateral sclerosis and frontotemporal dementia forms RNA G-quadruplexes. Sci Rep 2:1016
Friedman JE (2011) Anticipation in hereditary disease: the history of a biomedical concept. Hum Genet 130:705–714
Fu Y, Ramisetty SR, Hussain N, Baranger AM (2012) MBNL1-RNA recognition: contributions of MBNL1 sequence and RNA conformation. ChemBioChem 13:112–119
Fu YH, Pizzuti A, Fenwick RG Jr, King J, Rajnarayan S, Dunne PW, Dubel J, Nasser GA, Ashizawa T, de Jong P et al (1992) An unstable triplet repeat in a gene related to myotonic muscular dystrophy. Science 255:1256–1258
Gambardella S, Rinaldi F, Lepore SM, Viola A, Loro E, Angelini C, Vergani L, Novelli G, Botta A (2010) Overexpression of microRNA-206 in the skeletal muscle from myotonic dystrophy type 1 patients. J Transl Med 8:48
Gehman LT, Stoilov P, Maguire J, Damianov A, Lin CH, Shiue L, Ares M Jr, Mody I, Black DL (2011) The splicing regulator Rbfox1 (A2BP1) controls neuronal excitation in the mammalian brain. Nat Genet 43:706–711
Gemayel R, Vinces MD, Legendre M, Verstrepen KJ (2010) Variable tandem repeats accelerate evolution of coding and regulatory sequences. Annu Rev Genet 44:445–477
Gijselinck I, Van Langenhove T, van der Zee J, Sleegers K, Philtjens S, Kleinberger G, Janssens J, Bettens K, Van Cauwenberghe C, Pereson S, Engelborghs S, Sieben A, De Jonghe P, Vandenberghe R, Santens P, De Bleecker J, Maes G, Baumer V, Dillen L, Joris G, Cuijt I, Corsmit E, Elinck E, Van Dongen J, Vermeulen S, Van den Broeck M, Vaerenberg C, Mattheijssens M, Peeters K, Robberecht W, Cras P, Martin JJ, De Deyn PP, Cruts M, Van Broeckhoven C (2012) A C9orf72 promoter repeat expansion in a Flanders-Belgian cohort with disorders of the frontotemporal lobar degeneration-amyotrophic lateral sclerosis spectrum: a gene identification study. Lancet Neurol 11:54–65
Giordana MT, Ferrero P, Grifoni S, Pellerino A, Naldi A, Montuschi A (2011) Dementia and cognitive impairment in amyotrophic lateral sclerosis: a review. Neurol Sci 32:9–16
Goers ES, Purcell J, Voelker RB, Gates DP, Berglund JA (2010) MBNL1 binds GC motifs embedded in pyrimidines to regulate alternative splicing. Nucleic Acids Res 38:2467–2484
Goti D, Katzen SM, Mez J, Kurtis N, Kiluk J, Ben-Haiem L, Jenkins NA, Copeland NG, Kakizuka A, Sharp AH, Ross CA, Mouton PR, Colomer V (2004) A mutant ataxin-3 putative-cleavage fragment in brains of Machado-Joseph disease patients and transgenic mice is cytotoxic above a critical concentration. J Neurosci 24:10266–10279
Graff-Radford NR, Woodruff BK (2007) Frontotemporal dementia. Semin Neurol 27:48–57
Greco CM, Hagerman RJ, Tassone F, Chudley AE, Del Bigio MR, Jacquemont S, Leehey M, Hagerman PJ (2002) Neuronal intranuclear inclusions in a new cerebellar tremor/ataxia syndrome among fragile X carriers. Brain 125:1760–1771
Gros-Louis F, Gaspar C, Rouleau GA (2006) Genetics of familial and sporadic amyotrophic lateral sclerosis. Biochim Biophys Acta 1762:956–972
Ha AD, Fung VS (2012) Huntington’s disease. Curr Opin Neurol 25:491–498
Haacke A, Broadley SA, Boteva R, Tzvetkov N, Hartl FU, Breuer P (2006) Proteolytic cleavage of polyglutamine-expanded ataxin-3 is critical for aggregation and sequestration of non-expanded ataxin-3. Hum Mol Genet 15:555–568
Hagerman PJ, Hagerman RJ (2004) Fragile X-associated tremor/ataxia syndrome (FXTAS). Ment Retard Dev Disabil Res Rev 10:25–30
Hashem V, Galloway JN, Mori M, Willemsen R, Oostra BA, Paylor R, Nelson DL (2009) Ectopic expression of CGG containing mRNA is neurotoxic in mammals. Hum Mol Genet 18:2443–2451
Hernandez RN, Feinberg RL, Vaurio R, Passanante NM, Thompson RE, Kaufmann WE (2009) Autism spectrum disorder in fragile X syndrome: a longitudinal evaluation. Am J Med Genet A 149A:1125–1137
Hersheson J, Haworth A, Houlden H (2012) The inherited ataxias: genetic heterogeneity, mutation databases, and future directions in research and clinical diagnostics. Hum Mutat 33:1324–1332
Ho TH, Charlet BN, Poulos MG, Singh G, Swanson MS, Cooper TA (2004) Muscleblind proteins regulate alternative splicing. EMBO J 23:3103–3112
Ho TH, Savkur RS, Poulos MG, Mancini MA, Swanson MS, Cooper TA (2005) Colocalization of muscleblind with RNA foci is separable from mis-regulation of alternative splicing in myotonic dystrophy. J Cell Sci 118:2923–2933
Hokkanen S, Feldmann HM, Ding H, Jung CK, Bojarski L, Renner-Muller I, Schuller U, Kretzschmar H, Wolf E, Herms J (2012) Lack of Pur-alpha alters postnatal brain development and causes megalencephaly. Hum Mol Genet 21:473–484
Holmes SE, O’Hearn E, Rosenblatt A, Callahan C, Hwang HS, Ingersoll-Ashworth RG, Fleisher A, Stevanin G, Brice A, Potter NT, Ross CA, Margolis RL (2001) A repeat expansion in the gene encoding junctophilin-3 is associated with Huntington disease-like 2. Nat Genet 29:377–378
Hsu RJ, Hsiao KM, Lin MJ, Li CY, Wang LC, Chen LK, Pan H (2011) Long tract of untranslated CAG repeats is deleterious in transgenic mice. PLoS One 6:e16417
Huichalaf C, Schoser B, Schneider-Gold C, Jin B, Sarkar P, Timchenko L (2009) Reduction of the rate of protein translation in patients with myotonic dystrophy 2. J Neurosci 29:9042–9049
Huynh DP, Figueroa K, Hoang N, Pulst SM (2000) Nuclear localization or inclusion body formation of ataxin-2 are not necessary for SCA2 pathogenesis in mouse or human. Nat Genet 26:44–50
Ikeda Y, Daughters RS, Ranum LP (2008) Bidirectional expression of the SCA8 expansion mutation: one mutation, two genes. Cerebellum 7:150–158
Iwahashi CK, Yasui DH, An HJ, Greco CM, Tassone F, Nannen K, Babineau B, Lebrilla CB, Hagerman RJ, Hagerman PJ (2006) Protein composition of the intranuclear inclusions of FXTAS. Brain 129:256–271
Jansen A, Gemayel R, Verstrepen KJ (2012) Unstable microsatellite repeats facilitate rapid evolution of coding and regulatory sequences. Genome Dyn 7:108–125
Jansen G, Mahadevan M, Amemiya C, Wormskamp N, Segers B, Hendriks W, O’Hoy K, Baird S, Sabourin L, Lennon G et al (1992) Characterization of the myotonic dystrophy region predicts multiple protein isoform-encoding mRNAs. Nat Genet 1:261–266
Jenal M, Elkon R, Loayza-Puch F, van Haaften G, Kuhn U, Menzies FM, Oude Vrielink JA, Bos AJ, Drost J, Rooijers K, Rubinsztein DC, Agami R (2012) The poly(A)-binding protein nuclear 1 suppresses alternative cleavage and polyadenylation sites. Cell 149:538–553
Jin P, Duan R, Qurashi A, Qin Y, Tian D, Rosser TC, Liu H, Feng Y, Warren ST (2007) Pur alpha binds to rCGG repeats and modulates repeat-mediated neurodegeneration in a Drosophila model of fragile X tremor/ataxia syndrome. Neuron 55:556–564
Johnson EM, Kinoshita Y, Weinreb DB, Wortman MJ, Simon R, Khalili K, Winckler B, Gordon J (2006) Role of Pur alpha in targeting mRNA to sites of translation in hippocampal neuronal dendrites. J Neurosci Res 83:929–943
Junghans RP (2009) Dystrophia myotonia: why focus on foci? Eur J Hum Genet 17:543–553
Kanadia RN, Johnstone KA, Mankodi A, Lungu C, Thornton CA, Esson D, Timmers AM, Hauswirth WW, Swanson MS (2003a) A muscleblind knockout model for myotonic dystrophy. Science 302:1978–1980
Kanadia RN, Urbinati CR, Crusselle VJ, Luo D, Lee YJ, Harrison JK, Oh SP, Swanson MS (2003b) Developmental expression of mouse muscleblind genes Mbnl1, Mbnl2 and Mbnl3. Gene Expr Patterns 3:459–462
Kawahara Y, Ito K, Sun H, Aizawa H, Kanazawa I, Kwak S (2004) Glutamate receptors: RNA editing and death of motor neurons. Nature 427:801
Kenneson A, Zhang F, Hagedorn CH, Warren ST (2001) Reduced FMRP and increased FMR1 transcription is proportionally associated with CGG repeat number in intermediate-length and premutation carriers. Hum Mol Genet 10:1449–1454
Kim DH, Langlois MA, Lee KB, Riggs AD, Puymirat J, Rossi JJ (2005) HnRNP H inhibits nuclear export of mRNA containing expanded CUG repeats and a distal branch point sequence. Nucleic Acids Res 33:3866–3874
Klein AF, Ebihara M, Alexander C, Dicaire MJ, Sasseville AM, Langelier Y, Rouleau GA, Brais B (2008) PABPN1 polyalanine tract deletion and long expansions modify its aggregation pattern and expression. Exp Cell Res 314:1652–1666
Koob MD, Moseley ML, Schut LJ, Benzow KA, Bird TD, Day JW, Ranum LP (1999) An untranslated CTG expansion causes a novel form of spinocerebellar ataxia (SCA8). Nat Genet 21:379–384
Koyano S, Uchihara T, Fujigasaki H, Nakamura A, Yagishita S, Iwabuchi K (1999) Neuronal intranuclear inclusions in spinocerebellar ataxia type 2: triple-labeling immunofluorescent study. Neurosci Lett 273:117–120
Krzyzosiak WJ, Sobczak K, Wojciechowska M, Fiszer A, Mykowska A, Kozlowski P (2012) Triplet repeat RNA structure and its role as pathogenic agent and therapeutic target. Nucleic Acids Res 40:11–26
Kuyumcu-Martinez NM, Wang GS, Cooper TA (2007) Increased steady-state levels of CUGBP1 in myotonic dystrophy 1 are due to PKC-mediated hyperphosphorylation. Mol Cell 28:68–78
Kwiatkowski TJ Jr, Bosco DA, Leclerc AL, Tamrazian E, Vanderburg CR, Russ C, Davis A, Gilchrist J, Kasarskis EJ, Munsat T, Valdmanis P, Rouleau GA, Hosler BA, Cortelli P, de Jong PJ, Yoshinaga Y, Haines JL, Pericak-Vance MA, Yan J, Ticozzi N, Siddique T, McKenna-Yasek D, Sapp PC, Horvitz HR, Landers JE, Brown RH Jr (2009) Mutations in the FUS/TLS gene on chromosome 16 cause familial amyotrophic lateral sclerosis. Science 323:1205–1208
La Spada AR, Wilson EM, Lubahn DB, Harding AE, Fischbeck KH (1991) Androgen receptor gene mutations in X-linked spinal and bulbar muscular atrophy. Nature 352:77–79
Laurent FX, Sureau A, Klein AF, Trouslard F, Gasnier E, Furling D, Marie J (2012) New function for the RNA helicase p68/DDX5 as a modifier of MBNL1 activity on expanded CUG repeats. Nucleic Acids Res 40:3159–3171
Leehey MA, Hagerman PJ (2012) Fragile X-associated tremor/ataxia syndrome. Handb Clin Neurol 103:373–386
Lemay JF, Lemieux C, St-Andre O, Bachand F (2010) Crossing the borders: poly(A)-binding proteins working on both sides of the fence. RNA Biol 7:291–295
Levine TP, Daniels RD, Gatta AT, Wong LH, Hayes MJ (2013) The product of C9orf72, a gene strongly implicated in neurodegeneration, is structurally related to DENN Rab-GEFs. Bioinformatics 29:499–503
Li LB, Yu Z, Teng X, Bonini NM (2008) RNA toxicity is a component of ataxin-3 degeneration in Drosophila. Nature 453:1107–1111
Li Y, Jin P (2012) RNA-mediated neurodegeneration in fragile X-associated tremor/ataxia syndrome. Brain Res 1462:112–117
Li YR, King OD, Shorter J, Gitler AD (2013) Stress granules as crucibles of ALS pathogenesis. J Cell Biol 201:361–372
Lin X, Miller JW, Mankodi A, Kanadia RN, Yuan Y, Moxley RT, Swanson MS, Thornton CA (2006) Failure of MBNL1-dependent post-natal splicing transitions in myotonic dystrophy. Hum Mol Genet 15:2087–2097
Ling SC, Polymenidou M, Cleveland DW (2013) Converging Mechanisms in ALS and FTD: Disrupted RNA and Protein Homeostasis. Neuron 79:416–438
Liquori CL, Ricker K, Moseley ML, Jacobsen JF, Kress W, Naylor SL, Day JW, Ranum LP (2001) Myotonic dystrophy type 2 caused by a CCTG expansion in intron 1 of ZNF9. Science 293:864–867
Liu J, Tang TS, Tu H, Nelson O, Herndon E, Huynh DP, Pulst SM, Bezprozvanny I (2009) Deranged calcium signaling and neurodegeneration in spinocerebellar ataxia type 2. J Neurosci 29:9148–9162
Lomen-Hoerth C, Anderson T, Miller B (2002) The overlap of amyotrophic lateral sclerosis and frontotemporal dementia. Neurology 59:1077–1079
Lopez Castel A, Cleary JD, Pearson CE (2010) Repeat instability as the basis for human diseases and as a potential target for therapy. Nat Rev Mol Cell Biol 11:165–170
Lukong KE, Richard S (2008) Motor coordination defects in mice deficient for the Sam68 RNA-binding protein. Behav Brain Res 189:357–363
Magana JJ, Velazquez-Perez L, Cisneros B (2013) Spinocerebellar ataxia type 2: clinical presentation, molecular mechanisms, and therapeutic perspectives. Mol Neurobiol 47:90–104
Mahadevan M, Tsilfidis C, Sabourin L, Shutler G, Amemiya C, Jansen G, Neville C, Narang M, Barcelo J, O’Hoy K et al (1992) Myotonic dystrophy mutation: an unstable CTG repeat in the 3′ untranslated region of the gene. Science 255:1253–1255
Mahadevan MS (2012) Myotonic dystrophy: is a narrow focus obscuring the rest of the field? Curr Opin Neurol 25:609–613
Majounie E, Renton AE, Mok K, Dopper EG, Waite A, Rollinson S, Chio A, Restagno G, Nicolaou N, Simon-Sanchez J, van Swieten JC, Abramzon Y, Johnson JO, Sendtner M, Pamphlett R, Orrell RW, Mead S, Sidle KC, Houlden H, Rohrer JD, Morrison KE, Pall H, Talbot K, Ansorge O, Chromosome ALSFTDC, French research network on FFA, Consortium I, Hernandez DG, Arepalli S, Sabatelli M, Mora G, Corbo M, Giannini F, Calvo A, Englund E, Borghero G, Floris GL, Remes AM, Laaksovirta H, McCluskey L, Trojanowski JQ, Van Deerlin VM, Schellenberg GD, Nalls MA, Drory VE, Lu CS, Yeh TH, Ishiura H, Takahashi Y, Tsuji S, Le Ber I, Brice A, Drepper C, Williams N, Kirby J, Shaw P, Hardy J, Tienari PJ, Heutink P, Morris HR, Pickering-Brown S, Traynor BJ (2012) Frequency of the C9orf72 hexanucleotide repeat expansion in patients with amyotrophic lateral sclerosis and frontotemporal dementia: a cross-sectional study. Lancet Neurol 11:323–330
Mankodi A, Logigian E, Callahan L, McClain C, White R, Henderson D, Krym M, Thornton CA (2000) Myotonic dystrophy in transgenic mice expressing an expanded CUG repeat. Science 289:1769–1773
Margolis JM, Schoser BG, Moseley ML, Day JW, Ranum LP (2006) DM2 intronic expansions: evidence for CCUG accumulation without flanking sequence or effects on ZNF9 mRNA processing or protein expression. Hum Mol Genet 15:1808–1815
Margolis RL, Holmes SE, Rosenblatt A, Gourley L, O’Hearn E, Ross CA, Seltzer WK, Walker RH, Ashizawa T, Rasmussen A, Hayden M, Almqvist EW, Harris J, Fahn S, MacDonald ME, Mysore J, Shimohata T, Tsuji S, Potter N, Nakaso K, Adachi Y, Nakashima K, Bird T, Krause A, Greenstein P (2004) Huntington’s disease-like 2 (HDL2) in North America and Japan. Ann Neurol 56:670–674
Martin GM (2005) Genetic modulation of senescent phenotypes in Homo sapiens. Cell 120:523–532
Massa R, Panico MB, Caldarola S, Fusco FR, Sabatelli P, Terracciano C, Botta A, Novelli G, Bernardi G, Loreni F (2010) The myotonic dystrophy type 2 (DM2) gene product zinc finger protein 9 (ZNF9) is associated with sarcomeres and normally localized in DM2 patients’ muscles. Neuropathol Appl Neurobiol 36:275–284
Matilla-Duenas A, Corral-Juan M, Volpini V, Sanchez I (2012) The spinocerebellar ataxias: clinical aspects and molecular genetics. Adv Exp Med Biol 724:351–374
McLennan Y, Polussa J, Tassone F, Hagerman R (2011) Fragile x syndrome. Curr Genomics 12:216–224
Messaed C, Dion PA, Abu-Baker A, Rochefort D, Laganiere J, Brais B, Rouleau GA (2007) Soluble expanded PABPN1 promotes cell death in oculopharyngeal muscular dystrophy. Neurobiol Dis 26:546–557
Messaed C, Rouleau GA (2009) Molecular mechanisms underlying polyalanine diseases. Neurobiol Dis 34:397–405
Miller JW, Urbinati CR, Teng-Umnuay P, Stenberg MG, Byrne BJ, Thornton CA, Swanson MS (2000) Recruitment of human muscleblind proteins to (CUG)(n) expansions associated with myotonic dystrophy. EMBO J 19:4439–4448
Mirkin SM (2007) Expandable DNA repeats and human disease. Nature 447:932–940
Mori K, Lammich S, Mackenzie IR, Forne I, Zilow S, Kretzschmar H, Edbauer D, Janssens J, Kleinberger G, Cruts M, Herms J, Neumann M, Van Broeckhoven C, Arzberger T, Haass C (2013a) hnRNP A3 binds to GGGGCC repeats and is a constituent of p62-positive/TDP43-negative inclusions in the hippocampus of patients with C9orf72 mutations. Acta Neuropathol 125:413–423
Mori K, Weng SM, Arzberger T, May S, Rentzsch K, Kremmer E, Schmid B, Kretzschmar HA, Cruts M, Van Broeckhoven C, Haass C, Edbauer D (2013b) The C9orf72 GGGGCC repeat is translated into aggregating dipeptide-repeat proteins in FTLD/ALS. Science 339:1335–1338
Morris MJ, Negishi Y, Pazsint C, Schonhoft JD, Basu S (2010) An RNA G-quadruplex is essential for cap-independent translation initiation in human VEGF IRES. J Am Chem Soc 132:17831–17839
Moseley ML, Zu T, Ikeda Y, Gao W, Mosemiller AK, Daughters RS, Chen G, Weatherspoon MR, Clark HB, Ebner TJ, Day JW, Ranum LP (2006) Bidirectional expression of CUG and CAG expansion transcripts and intranuclear polyglutamine inclusions in spinocerebellar ataxia type 8. Nat Genet 38:758–769
Mrazek J, Guo X, Shah A (2007) Simple sequence repeats in prokaryotic genomes. Proc Natl Acad Sci U S A 104:8472–8477
Muslimov IA, Patel MV, Rose A, Tiedge H (2011) Spatial code recognition in neuronal RNA targeting: role of RNA-hnRNP A2 interactions. J Cell Biol 194:441–457
Mykowska A, Sobczak K, Wojciechowska M, Kozlowski P, Krzyzosiak WJ (2011) CAG repeats mimic CUG repeats in the misregulation of alternative splicing. Nucleic Acids Res 39:8938–8951
Nakamori M, Sobczak K, Puwanant A, Welle S, Eichinger K, Pandya S, Dekdebrun J, Heatwole CR, McDermott MP, Chen T, Cline M, Tawil R, Osborne RJ, Wheeler TM, Swanson M, Moxley RT 3rd, Thornton CA (2013) Splicing biomarkers of disease severity in myotonic dystrophy. Ann Neurol 59(3):474–477
Nakamoto M, Nakano S, Kawashima S, Ihara M, Nishimura Y, Shinde A, Kakizuka A (2002) Unequal crossing-over in unique PABP2 mutations in Japanese patients: a possible cause of oculopharyngeal muscular dystrophy. Arch Neurol 59:474–477
Nelson DL, Orr HT, Warren ST (2013) The unstable repeats—three evolving faces of neurological disease. Neuron 77:825–843
Neumann M, Rademakers R, Roeber S, Baker M, Kretzschmar HA, Mackenzie IR (2009) A new subtype of frontotemporal lobar degeneration with FUS pathology. Brain 132:2922–2931
Neumann M, Sampathu DM, Kwong LK, Truax AC, Micsenyi MC, Chou TT, Bruce J, Schuck T, Grossman M, Clark CM, McCluskey LF, Miller BL, Masliah E, Mackenzie IR, Feldman H, Feiden W, Kretzschmar HA, Trojanowski JQ, Lee VM (2006) Ubiquitinated TDP-43 in frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Science 314:130–133
Nirenberg M (2004) Historical review: deciphering the genetic code—a personal account. Trends Biochem Sci 29:46–54
Ohashi S, Kobayashi S, Omori A, Ohara S, Omae A, Muramatsu T, Li Y, Anzai K (2000) The single-stranded DNA- and RNA-binding proteins pur alpha and pur beta link BC1 RNA to microtubules through binding to the dendrite-targeting RNA motifs. J Neurochem 75:1781–1790
Ohno S (1972) So much “junk” DNA in our genome. Brookhaven Symp Biol 23:366–370
Orgel LE, Crick FH (1980) Selfish DNA: the ultimate parasite. Nature 284:604–607
Orr HT (2012a) Cell biology of spinocerebellar ataxia. J Cell Biol 197:167–177
Orr HT (2012b) Polyglutamine neurodegeneration: expanded glutamines enhance native functions. Curr Opin Genet Dev 22:251–255
Orr HT, Zoghbi HY (2007) Trinucleotide repeat disorders. Annu Rev Neurosci 30:575–621
Paul S, Dansithong W, Jog SP, Holt I, Mittal S, Brook JD, Morris GE, Comai L, Reddy S (2011) Expanded CUG repeats dysregulate RNA splicing by altering the stoichiometry of the muscleblind 1 complex. J Biol Chem 286:38427–38438
Paul S, Dansithong W, Kim D, Rossi J, Webster NJ, Comai L, Reddy S (2006) Interaction of muscleblind, CUG-BP1 and hnRNP H proteins in DM1-associated aberrant IR splicing. EMBO J 25:4271–4283
Paulson HL, Perez MK, Trottier Y, Trojanowski JQ, Subramony SH, Das SS, Vig P, Mandel JL, Fischbeck KH, Pittman RN (1997) Intranuclear inclusions of expanded polyglutamine protein in spinocerebellar ataxia type 3. Neuron 19:333–344
Pearson CE (2011) Repeat associated non-ATG translation initiation: one DNA, two transcripts, seven reading frames, potentially nine toxic entities! PLoS Genet 7:e1002018
Peprah E, He W, Allen E, Oliver T, Boyne A, Sherman SL (2010) Examination of FMR1 transcript and protein levels among 74 premutation carriers. J Hum Genet 55:66–68
Perbellini R, Greco S, Sarra-Ferraris G, Cardani R, Capogrossi MC, Meola G, Martelli F (2011) Dysregulation and cellular mislocalization of specific miRNAs in myotonic dystrophy type 1. Neuromuscul Disord 21:81–88
Philips AV, Timchenko LT, Cooper TA (1998) Disruption of splicing regulated by a CUG-binding protein in myotonic dystrophy. Science 280:737–741
Poulos MG, Batra R, Charizanis K, Swanson MS (2011) Developments in RNA splicing and disease. Cold Spring Harb Perspect Biol 3:a000778
Querido E, Gallardo F, Beaudoin M, Menard C, Chartrand P (2011) Stochastic and reversible aggregation of mRNA with expanded CUG-triplet repeats. J Cell Sci 124:1703–1714
Rademakers R, Neumann M, Mackenzie IR (2012) Advances in understanding the molecular basis of frontotemporal dementia. Nat Rev Neurol 8:423–434
Raheem O, Olufemi SE, Bachinski LL, Vihola A, Sirito M, Holmlund-Hampf J, Haapasalo H, Li YP, Udd B, Krahe R (2010) Mutant (CCTG)n expansion causes abnormal expression of zinc finger protein 9 (ZNF9) in myotonic dystrophy type 2. Am J Pathol 177:3025–3036
Ralser M, Albrecht M, Nonhoff U, Lengauer T, Lehrach H, Krobitsch S (2005) An integrative approach to gain insights into the cellular function of human ataxin-2. J Mol Biol 346:203–214
Ramakrishnan P, Baltimore D (2011) Sam68 is required for both NF-kappaB activation and apoptosis signaling by the TNF receptor. Mol Cell 43:167–179
Ranum LP, Cooper TA (2006) RNA-mediated neuromuscular disorders. Annu Rev Neurosci 29:259–277
Rau F, Freyermuth F, Fugier C, Villemin JP, Fischer MC, Jost B, Dembele D, Gourdon G, Nicole A, Duboc D, Wahbi K, Day JW, Fujimura H, Takahashi MP, Auboeuf D, Dreumont N, Furling D, Charlet-Berguerand N (2011) Misregulation of miR-1 processing is associated with heart defects in myotonic dystrophy. Nat Struct Mol Biol 18:840–845
Ravel-Chapuis A, Belanger G, Yadava RS, Mahadevan MS, DesGroseillers L, Cote J, Jasmin BJ (2012) The RNA-binding protein Staufen1 is increased in DM1 skeletal muscle and promotes alternative pre-mRNA splicing. J Cell Biol 196:699–712
Raz V, Abraham T, van Zwet EW, Dirks RW, Tanke HJ, van der Maarel SM (2011) Reversible aggregation of PABPN1 pre-inclusion structures. Nucleus 2:208–218
Raz V, Butler-Browne G, van Engelen B, Brais B (2013) 191st ENMC international workshop: recent advances in oculopharyngeal muscular dystrophy research: from bench to bedside 8–10 June 2012, Naarden, The Netherlands. Neuromuscul Disord 23:516–523
Reddy K, Zamiri B, Stanley SY, Macgregor RB Jr, Pearson CE (2013) The disease-associated r(GGGGCC)n repeat from the C9orf72 gene forms tract length-dependent uni- and multimolecular RNA G-quadruplex structures. J Biol Chem 288:9860–9866
Renton AE, Majounie E, Waite A, Simon-Sanchez J, Rollinson S, Gibbs JR, Schymick JC, Laaksovirta H, van Swieten JC, Myllykangas L, Kalimo H, Paetau A, Abramzon Y, Remes AM, Kaganovich A, Scholz SW, Duckworth J, Ding J, Harmer DW, Hernandez DG, Johnson JO, Mok K, Ryten M, Trabzuni D, Guerreiro RJ, Orrell RW, Neal J, Murray A, Pearson J, Jansen IE, Sondervan D, Seelaar H, Blake D, Young K, Halliwell N, Callister JB, Toulson G, Richardson A, Gerhard A, Snowden J, Mann D, Neary D, Nalls MA, Peuralinna T, Jansson L, Isoviita VM, Kaivorinne AL, Holtta-Vuori M, Ikonen E, Sulkava R, Benatar M, Wuu J, Chio A, Restagno G, Borghero G, Sabatelli M, Consortium I, Heckerman D, Rogaeva E, Zinman L, Rothstein JD, Sendtner M, Drepper C, Eichler EE, Alkan C, Abdullaev Z, Pack SD, Dutra A, Pak E, Hardy J, Singleton A, Williams NM, Heutink P, Pickering-Brown S, Morris HR, Tienari PJ, Traynor BJ (2011) A hexanucleotide repeat expansion in C9ORF72 is the cause of chromosome 9p21-linked ALS-FTD. Neuron 72:257–268
Richter JD (2007) CPEB: a life in translation. Trends Biochem Sci 32:279–285
Robberecht W, Philips T (2013) The changing scene of amyotrophic lateral sclerosis. Nat Rev Neurosci 14:248–264
Robinson DO, Wills AJ, Hammans SR, Read SP, Sillibourne J (2006) Oculopharyngeal muscular dystrophy: a point mutation which mimics the effect of the PABPN1 gene triplet repeat expansion mutation. J Med Genet 43:e23
Rohrer JD, Guerreiro R, Vandrovcova J, Uphill J, Reiman D, Beck J, Isaacs AM, Authier A, Ferrari R, Fox NC, Mackenzie IR, Warren JD, de Silva R, Holton J, Revesz T, Hardy J, Mead S, Rossor MN (2009) The heritability and genetics of frontotemporal lobar degeneration. Neurology 73:1451–1456
Rub U, Schols L, Paulson H, Auburger G, Kermer P, Jen JC, Seidel K, Korf HW, Deller T (2013) Clinical features, neurogenetics and neuropathology of the polyglutamine spinocerebellar ataxias type 1, 2, 3, 6 and 7. Prog Neurobiol 104:38–66
Rudnicki DD, Holmes SE, Lin MW, Thornton CA, Ross CA, Margolis RL (2007) Huntington’s disease–like 2 is associated with CUG repeat-containing RNA foci. Ann Neurol 61:272–282
Rudnicki DD, Pletnikova O, Vonsattel JP, Ross CA, Margolis RL (2008) A comparison of huntington disease and huntington disease-like 2 neuropathology. J Neuropathol Exp Neurol 67:366–374
Satterfield TF, Pallanck LJ (2006) Ataxin-2 and its Drosophila homolog, ATX2, physically assemble with polyribosomes. Hum Mol Genet 15:2523–2532
Schneider-Gold C, Timchenko LT (2010) CCUG repeats reduce the rate of global protein synthesis in myotonic dystrophy type 2. Rev Neurosci 21:19–28
Seixas AI, Holmes SE, Takeshima H, Pavlovich A, Sachs N, Pruitt JL, Silveira I, Ross CA, Margolis RL, Rudnicki DD (2012) Loss of junctophilin-3 contributes to Huntington disease-like 2 pathogenesis. Ann Neurol 71:245–257
Sellier C, Freyermuth F, Tabet R, Tran T, He F, Ruffenach F, Alunni V, Moine H, Thibault C, Page A, Tassone F, Willemsen R, Disney MD, Hagerman PJ, Todd PK, Charlet-Berguerand N (2013) Sequestration of DROSHA and DGCR8 by expanded CGG RNA repeats alters microRNA processing in fragile X-associated tremor/ataxia syndrome. Cell Rep 3:869–880
Sellier C, Rau F, Liu Y, Tassone F, Hukema RK, Gattoni R, Schneider A, Richard S, Willemsen R, Elliott DJ, Hagerman PJ, Charlet-Berguerand N (2010) Sam68 sequestration and partial loss of function are associated with splicing alterations in FXTAS patients. EMBO J 29:1248–1261
Serrano-Munuera C, Corral-Juan M, Stevanin G, San Nicolas H, Roig C, Corral J, Campos B, de Jorge L, Morcillo-Suarez C, Navarro A, Forlani S, Durr A, Kulisevsky J, Brice A, Sanchez I, Volpini V, Matilla-Duenas A (2013) New subtype of spinocerebellar ataxia with altered vertical eye movements mapping to chromosome 1p32. JAMA Neurol 70:764–771
Shibata H, Huynh DP, Pulst SM (2000) A novel protein with RNA-binding motifs interacts with ataxin-2. Hum Mol Genet 9:1303–1313
Shieh SY, Bonini NM (2011) Genes and pathways affected by CAG-repeat RNA-based toxicity in Drosophila. Hum Mol Genet 20:4810–4821
Sicot G, Gomes-Pereira M (2013) RNA toxicity in human disease and animal models: from the uncovering of a new mechanism to the development of promising therapies. Biochim Biophys Acta 1832:1390–1409
Simon-Sanchez J, Hanson M, Singleton A, Hernandez D, McInerney A, Nussbaum R, Werner J, Gallardo M, Weiser R, Gwinn-Hardy K, Singleton AB, Clarimon J (2005) Analysis of SCA-2 and SCA-3 repeats in Parkinsonism: evidence of SCA-2 expansion in a family with autosomal dominant Parkinson’s disease. Neurosci Lett 382:191–194
Siomi H, Siomi MC, Nussbaum RL, Dreyfuss G (1993) The protein product of the fragile X gene, FMR1, has characteristics of an RNA-binding protein. Cell 74:291–298
Sofola OA, Jin P, Qin Y, Duan R, Liu H, de Haro M, Nelson DL, Botas J (2007) RNA-binding proteins hnRNP A2/B1 and CUGBP1 suppress fragile X CGG premutation repeat-induced neurodegeneration in a Drosophila model of FXTAS. Neuron 55:565–571
Sun S, Zhang Z, Fregoso O, Krainer AR (2012) Mechanisms of activation and repression by the alternative splicing factors RBFOX1/2. RNA 18:274–283
Sutcliffe JS, Nelson DL, Zhang F, Pieretti M, Caskey CT, Saxe D, Warren ST (1992) DNA methylation represses FMR-1 transcription in fragile X syndrome. Hum Mol Genet 1:397–400
Taneja KL, McCurrach M, Schalling M, Housman D, Singer RH (1995) Foci of trinucleotide repeat transcripts in nuclei of myotonic dystrophy cells and tissues. J Cell Biol 128:995–1002
Tassone F, Hagerman R (2012) The fragile X-associated tremor ataxia syndrome. Results Probl Cell Differ 54:337–357
Tassone F, Hagerman RJ, Garcia-Arocena D, Khandjian EW, Greco CM, Hagerman PJ (2004a) Intranuclear inclusions in neural cells with premutation alleles in fragile X associated tremor/ataxia syndrome. J Med Genet 41:e43
Tassone F, Hagerman RJ, Loesch DZ, Lachiewicz A, Taylor AK, Hagerman PJ (2000a) Fragile X males with unmethylated, full mutation trinucleotide repeat expansions have elevated levels of FMR1 messenger RNA. Am J Med Genet 94:232–236
Tassone F, Hagerman RJ, Taylor AK, Gane LW, Godfrey TE, Hagerman PJ (2000b) Elevated levels of FMR1 mRNA in carrier males: a new mechanism of involvement in the fragile-X syndrome. Am J Hum Genet 66:6–15
Tassone F, Hagerman RJ, Taylor AK, Mills JB, Harris SW, Gane LW, Hagerman PJ (2000c) Clinical involvement and protein expression in individuals with the FMR1 premutation. Am J Med Genet 91:144–152
Tassone F, Iwahashi C, Hagerman PJ (2004b) FMR1 RNA within the intranuclear inclusions of fragile X-associated tremor/ataxia syndrome (FXTAS). RNA Biol 1:103–105
Teive HA, Munhoz RP, Arruda WO, Raskin S, Werneck LC, Ashizawa T (2011) Spinocerebellar ataxia type 10—a review. Parkinsonism Relat Disord 17:655–661
Teplova M, Patel DJ (2008) Structural insights into RNA recognition by the alternative-splicing regulator muscleblind-like MBNL1. Nat Struct Mol Biol 15:1343–1351
Timchenko LT, Miller JW, Timchenko NA, DeVore DR, Datar KV, Lin L, Roberts R, Caskey CT, Swanson MS (1996) Identification of a (CUG)n triplet repeat RNA-binding protein and its expression in myotonic dystrophy. Nucleic Acids Res 24:4407–4414
Timchenko NA, Patel R, Iakova P, Cai ZJ, Quan L, Timchenko LT (2004) Overexpression of CUG triplet repeat-binding protein, CUGBP1, in mice inhibits myogenesis. J Biol Chem 279:13129–13139
Todd PK, Oh SY, Krans A, He F, Sellier C, Frazer M, Renoux AJ, Chen KC, Scaglione KM, Basrur V, Elenitoba-Johnson K, Vonsattel JP, Louis ED, Sutton MA, Taylor JP, Mills RE, Charlet-Berguerand N, Paulson HL (2013) CGG repeat-associated translation mediates neurodegeneration in fragile X tremor ataxia syndrome. Neuron 78:440–455
Tome FM, Chateau D, Helbling-Leclerc A, Fardeau M (1997) Morphological changes in muscle fibers in oculopharyngeal muscular dystrophy. Neuromuscul Disord 7(Suppl 1):S63–S69
Tome FM, Fardeau M (1980) Nuclear inclusions in oculopharyngeal dystrophy. Acta Neuropathol 49:85–87
Udd B, Krahe R (2012) The myotonic dystrophies: molecular, clinical, and therapeutic challenges. Lancet Neurol 11:891–905
Underwood JG, Boutz PL, Dougherty JD, Stoilov P, Black DL (2005) Homologues of the Caenorhabditis elegans Fox-1 protein are neuronal splicing regulators in mammals. Mol Cell Biol 25:10005–10016
Van Damme P, Robberecht W (2013) Clinical implications of recent breakthroughs in amyotrophic lateral sclerosis. Curr Opin Neurol 26(5):466–472
Vance C, Rogelj B, Hortobagyi T, De Vos KJ, Nishimura AL, Sreedharan J, Hu X, Smith B, Ruddy D, Wright P, Ganesalingam J, Williams KL, Tripathi V, Al-Saraj S, Al-Chalabi A, Leigh PN, Blair IP, Nicholson G, de Belleroche J, Gallo JM, Miller CC, Shaw CE (2009) Mutations in FUS, an RNA processing protein, cause familial amyotrophic lateral sclerosis type 6. Science 323:1208–1211
Verkerk AJ, Pieretti M, Sutcliffe JS, Fu YH, Kuhl DP, Pizzuti A, Reiner O, Richards S, Victoria MF, Zhang FP et al (1991) Identification of a gene (FMR-1) containing a CGG repeat coincident with a breakpoint cluster region exhibiting length variation in fragile X syndrome. Cell 65:905–914
Wakamiya M, Matsuura T, Liu Y, Schuster GC, Gao R, Xu W, Sarkar PS, Lin X, Ashizawa T (2006) The role of ataxin 10 in the pathogenesis of spinocerebellar ataxia type 10. Neurology 67:607–613
Walker RH, Morgello S, Davidoff-Feldman B, Melnick A, Walsh MJ, Shashidharan P, Brin MF (2002) Autosomal dominant chorea-acanthocytosis with polyglutamine-containing neuronal inclusions. Neurology 58:1031–1037
Wang ET, Cody NA, Jog S, Biancolella M, Wang TT, Treacy DJ, Luo S, Schroth GP, Housman DE, Reddy S, Lecuyer E, Burge CB (2012a) Transcriptome-wide regulation of pre-mRNA splicing and mRNA localization by muscleblind proteins. Cell 150:710–724
Wang LC, Chen KY, Pan H, Wu CC, Chen PH, Liao YT, Li C, Huang ML, Hsiao KM (2011) Muscleblind participates in RNA toxicity of expanded CAG and CUG repeats in Caenorhabditis elegans. Cell Mol Life Sci 68:1255–1267
Wang T, Bray SM, Warren ST (2012b) New perspectives on the biology of fragile X syndrome. Curr Opin Genet Dev 22:256–263
Warrick JM, Paulson HL, Gray-Board GL, Bui QT, Fischbeck KH, Pittman RN, Bonini NM (1998) Expanded polyglutamine protein forms nuclear inclusions and causes neural degeneration in Drosophila. Cell 93:939–949
Wellington CL, Ellerby LM, Hackam AS, Margolis RL, Trifiro MA, Singaraja R, McCutcheon K, Salvesen GS, Propp SS, Bromm M, Rowland KJ, Zhang T, Rasper D, Roy S, Thornberry N, Pinsky L, Kakizuka A, Ross CA, Nicholson DW, Bredesen DE, Hayden MR (1998) Caspase cleavage of gene products associated with triplet expansion disorders generates truncated fragments containing the polyglutamine tract. J Biol Chem 273:9158–9167
White M, Xia G, Gao R, Wakamiya M, Sarkar PS, McFarland K, Ashizawa T (2012) Transgenic mice with SCA10 pentanucleotide repeats show motor phenotype and susceptibility to seizure: a toxic RNA gain-of-function model. J Neurosci Res 90:706–714
White MC, Gao R, Xu W, Mandal SM, Lim JG, Hazra TK, Wakamiya M, Edwards SF, Raskin S, Teive HA, Zoghbi HY, Sarkar PS, Ashizawa T (2010) Inactivation of hnRNP K by expanded intronic AUUCU repeat induces apoptosis via translocation of PKCdelta to mitochondria in spinocerebellar ataxia 10. PLoS Genet 6:e1000984
Wilburn B, Rudnicki DD, Zhao J, Weitz TM, Cheng Y, Gu X, Greiner E, Park CS, Wang N, Sopher BL, La Spada AR, Osmand A, Margolis RL, Sun YE, Yang XW (2011) An antisense CAG repeat transcript at JPH3 locus mediates expanded polyglutamine protein toxicity in Huntington’s disease-like 2 mice. Neuron 70:427–440
Winter R, Kuhn U, Hause G, Schwarz E (2012) Polyalanine-independent conformational conversion of nuclear poly(A)-binding protein 1 (PABPN1). J Biol Chem 287:22662–22671
Wojciechowska M, Krzyzosiak WJ (2011) Cellular toxicity of expanded RNA repeats: focus on RNA foci. Hum Mol Genet 20:3811–3821
Xu Z, Poidevin M, Li X, Li Y, Shu L, Nelson DL, Li H, Hales CM, Gearing M, Wingo TS, Jin P (2013) Expanded GGGGCC repeat RNA associated with amyotrophic lateral sclerosis and frontotemporal dementia causes neurodegeneration. Proc Natl Acad Sci U S A 110:7778–7783
Zoghbi HY, Orr HT (2009) Pathogenic mechanisms of a polyglutamine-mediated neurodegenerative disease, spinocerebellar ataxia type 1. J Biol Chem 284:7425–7429
Zu T, Gibbens B, Doty NS, Gomes-Pereira M, Huguet A, Stone MD, Margolis J, Peterson M, Markowski TW, Ingram MA, Nan Z, Forster C, Low WC, Schoser B, Somia NV, Clark HB, Schmechel S, Bitterman PB, Gourdon G, Swanson MS, Moseley M, Ranum LP (2011) Non-ATG-initiated translation directed by microsatellite expansions. Proc Natl Acad Sci U S A 108:260–265
Acknowledgements
Our studies on microsatellite expansion disease mechanisms are supported by the NIH (AR046799, NS058901), the Keck Foundation, and the Muscular Dystrophy Association. The authors thank T. Ashizawa, J. Cleary, and L. Ranum for comments on the manuscript.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2014 Springer Science+Business Media New York
About this chapter
Cite this chapter
Goodwin, M., Swanson, M.S. (2014). RNA-Binding Protein Misregulation in Microsatellite Expansion Disorders. In: Yeo, G. (eds) Systems Biology of RNA Binding Proteins. Advances in Experimental Medicine and Biology, vol 825. Springer, New York, NY. https://doi.org/10.1007/978-1-4939-1221-6_10
Download citation
DOI: https://doi.org/10.1007/978-1-4939-1221-6_10
Published:
Publisher Name: Springer, New York, NY
Print ISBN: 978-1-4939-1220-9
Online ISBN: 978-1-4939-1221-6
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)