
Abstract
Normal vision depends on signaling from photoreceptors to central visual areas via parallel pathways that are optimized for detecting increments (ON) or decrements (OFF) in light intensity. The divergence of these two pathways occurs at the first synapse. The OFF pathway is mediated via Off-bipolar cells that hyperpolarize in response to light increments because they utilize ionotropic glutamate receptors. On-bipolar cells that initiate the ON pathway utilize metabotropic glutamate receptors to signal via a G-protein cascade to the transient receptor potential melastatin 1 (TRPM1) channel, and depolarize in response to light increments. Several proteins (mGluR6, TRPM1, GPR179, RGS7, RGS11, nyctalopin, LRIT3, Gα0, Gβ3, Gβ5, and R9AP) have been shown to be required for normal functioning of the depolarizing bipolar cell cascade. Here, we use immunohistochemistry in mouse models that lack one or more of these proteins to understand their interdependency. The picture that evolves is that of a large complex, in which the removal of any one element results in either delocalization of or decreased expression of other elements.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others


Keywords
Light stimulation results in a graded change in sustained release of the neurotransmitter glutamate from photoreceptors. This chemical signal is converted into an electrical signal by two classes of postsynaptic bipolar cells (BCs) that either hyperpolarize (HBCs) or depolarize (DBCs) in response to a light increment. HBCs utilize ionotropic glutamate receptors and maintain the polarity of the photoreceptor signal. DBCs, in comparison, utilize the G-protein-coupled receptor (GPCR) metabotropic glutamate receptor type 6 (mGluR6) to modulate the transient receptor potential melastatin 1 (TRPM1) nonspecific cation channel, inverting the photoreceptor response. The members of this GPCR signal transduction cascade have been identified based on contributions from many laboratories. The focus of this review is to provide an overview of the interdependence of expression among these components of the DBC dendritic mGluR6 to TRPM1 signaling complex, or “signalplex,” based on analyses of multiple mutant mouse lines and cascade components by immunohistochemistry.
Defects in most components of the mGluR6–GPCR cascade have been identified in humans and animal models using the noninvasive assay of retinal function, the electroretinogram (ERG). All of these mutants share the same ERG phenotype, a normal a-wave reflecting the function of the photoreceptors themselves, but the absence of a b-wave reflecting the loss of DBC function. An example of an ERG series for the Nyx nob, no b-wave mouse mutant is shown in Fig. 5.1. The ERG defect indicates that nyctalopin (NYX) expression is required for normal DBC function not only in the mouse but also in human patients with complete congenital stationary night blindness (cCSNB) . Other members of the DBC signalplex subsequently have been identified based on a comparable loss of the ERG b-wave. These include in mouse models the genes: Grm6, Trpm1, Nyx, Gpr179 , Lrit3, Gα 0 , Gβ3, RGS7/RGS11 double knockouts, [10, 12, 14, 16, 19, 22, 23, 25, 26, 32, 34, 40, 41]; in cCSNB human patients: NYX, GRM6, TRPM1 , GPR179 , LRIT3, [1–3, 6, 11, 15, 18, 20, 21, 27, 35, 38, 39, 42, 44, 46–50] and in night-blind horse models, Trpm1 [5].

Comparison of WT (black traces) and Nyx nob (red traces) ERGs recorded to strobe flash stimuli presented under dark-adapted conditions (left) or superimposed upon a steady adapting field (right). WT mice generate a positive polarity b-wave that dominates the response to low luminance stimuli and follows the a-wave in response to high luminance flashes. The b-wave is absent in Nyx nob animals. Under dark-adapted condition, the removal of the b-wave reveals slow PIII, an ERG component of Müller cell origin. Values next to each pair of waveforms indicate strobe flash luminance (log cd s/m2)
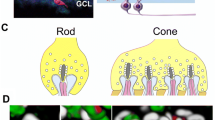
We know that the GPCR cascade begins when a change in photoreceptor glutamate release is detected by the mGluR6 receptor and ends with the gating of the TRPM1 channel and a depolarizing response in the DBCs. How the other parts of the DBC signalplex function remains incompletely understood, in part because the detailed interaction and stoichiometry of the proteins remain to be defined. In this chapter, we describe the known interdependencies that have been inferred from immunohistochemical analyses of key components in mouse mutants where expression of one or more members of the cascade is absent. We focus in particular on the expression of proteins that form puncta at the tips of DBC dendrites, presumed to be the locus of the DBC cascade. In the case of rod DBCs, discrete puncta are visible, and we focus our analysis on these structures, which can be visualized using immunohistochemistry with confidence (Fig. 5.2). We do not include a description of proteins such as Gαo or Gβ3 , which do not form distinct puncta in the OPL, rather they are expressed throughout the cell [14, 43]. This does not mean they are unimportant for DBC function, as the knockout models produced for each lack an ERG b-wave and DBC function [12–14]. Rather, this feature may indicate they interact with other signalplex components in a transient manner, or are simply not visible by immunostaining when localized to DBC puncta.

Punctate labeling of rod DBCs in OPL of the mouse retina. Transverse section of WT mouse retina stained for mGluR6 (green), which localizes to all DBC dendrites, and PNA (red) a marker for cone terminals. Note that GPR179 co-localizes with PNA. The green puncta in the merged image represent staining at the tips of rod DBCs
Table 5.1 summarizes the expression on the tips of the DBCs of the mGluR6– GPCR components localized to the DBC puncta that have been identified in mutant mouse models to date. Below, we review results obtained in mouse mutants for Grm6, Trpm1 , Gpr179 , Nyx, or Lrit3 in terms of the impact of a mutant allele on the expression of other signalplex components.
mGluR6 Expression is Required for the Localization of Multiple Signalplex Components
mGluR6 is encoded by Grm6, which was established as the DBC glutamate receptor using Grm6 knockout mice, created by gene targeting [23]. These mice lack the ERG b-wave and responses to light onset in the superior colliculus. Subsequently, two additional mutants for Grm6 were identified: Grm6 nob3 [22] and Grm6 nob4 [34]. Immunohistochemical results are very similar in all three lines, namely mGluR6 expression is absent from the DBC terminals. Using immunoprecipitation approaches, mGluR6 has been shown to interact with TRPM1 and GPR179 [29]. Figure 5.3 shows the consequences of the absence of mGluR6 on expression of TRPM1, GPR179, RGS11 , RGS7 , nyctalopin, and R9AP at the tips of DBCs. The impact on the protein level using western blots is similar (Fig. 5.4), although this does vary depending on the specific laboratory. These data and those summarized in Table 5.1 indicate that mGluR6 expression is required for the correct localization of TRPM1, RGS11, Gβ5, and R9AP to the dendritic tips of DBCs [8, 9]. The latter three components are proposed to form a trimeric GAP complex because the loss of any one results in the absence or significant reduction in expression of the other two. The western blot data indicate that the loss of mGluR6 has a relatively moderate impact on GPR179 and TRPM1 . In the case of TRPM1, this is because there is a large pool of TRPM1 in the other compartments of the cell, so loss of TRPM1 from the dendrites does not appear to have a major impact on total TRPM1 expression. RGS7 puncta remain in Grm6 nob4 mice [8], suggesting it is part of another complex, possibly GPR179/Gβ5/RGS7.

Interdependency of DBC signalplex components revealed by immunohistochemistry in WT, Grm6− −/−, GPR179 nob5 , and Trpm1− −/−mice. Staining in most panels was generated in the authors’ laboratory using published methods and antibodies [28, 32, 33]. The image of TRPM1 staining in the Grm6− −/−mice is adapted from Xu et al. [45]. Open box indicates that this particular experiment has not been done

Western blot of key DBC cascade proteins in mutant mouse lines. Western blots show the impact of eliminating three key elements of the DBC signal transduction cascade, GPR179, mGluR6, and TRPM1, on these proteins and the RGS7 and RG11 proteins critical to DBC function. In each mutant, the only protein lost is that produced by the targeted gene. Of particular note, the RGS proteins are present at near normal levels despite their abnormal localization to the DBC dendritic tips of GPR179 nob5 and Grm6− −/− mice (Fig. 5.2)
Localization of Other Signalplex Components is Independent of TRPM1 Expression
TRPM1 is the nonspecific cation channel modulated by the mGluR6 cascade . The first indication that TRPM1 was the channel required for DBC function came from studies in night-blind Appaloosa horses [4, 5]. In these horses, it was noted that the leopard spotting coat color and night blindness phenotypes were localized to a chromosomal region containing TRPM1. In addition, levels of trpm1–mRNA expression were significantly reduced (several hundred fold) in night-blind horses, as compared to animals with normal vision. The identity of the DBC cation channel as TRPM1 was confirmed by three groups independently using knockout mice [19, 25, 40]. Their data showed that: Trpm1− −/− mice lack the ERG b-wave and Trpm1− −/− DBCs lack mGlur6-mediated light evoked responses. TRPM1 colocalizes with mGluR6 in DBC puncta, together strongly suggesting that it is a critical component of the DBC signalplex. However, unlike some other components of the complex, TRPM1 is expressed both in the puncta and throughout the entire DBC, where it is located in intracellular compartments [33]. In Trpm1− −/− mice, mGluR6, GPR179 , RGS11 , RGS7, and nyctalopin are all expressed and normally localized (Fig. 5.3).
While the mechanism by which TRPM1 is gated remains to be firmly established (see Nawy chapter for review), the number of functional TRPM1 channels present in the DBC signalplex may be the limiting factor with respect to the maximal amplitude of the light-evoked DBC response. This conclusion was reached after using a TRPM1 mutant, Trpm1 tvrm27, resulting from an N-ethyl-N-nitrosourea (ENU) mutagenesis screen. The Trpm1 tvrm27 mutation is caused by a missense mutation, p.A1068T, in the predicted pore region of the channel, which is presumed to cause the lack of function. As predicted, mice homozygous for the Trpm1 tvrm27 share the same ERG phenotype with other Trpm1 knockout lines, namely the lack of a b-wave. In contrast to Trpm1− −/− mice, the mutant TRPM1tvrm27 protein is expressed and localized correctly into puncta on the DBC dendritic tips [31].
The most significant observations from this study arose from comparisons of mice heterozygous for either the Trpm1 tvrm27 or the Trpm1 knockout allele. In Trpm+ −/− mice heterozygotes, the b-wave was the same as WT controls, whereas the b-wave of Trpm1+ /tvrm27 heterozygous animals was about 32 % smaller. Patch-clamp recordings of Trpm1+ /tvrm27 heterozygous rod DBCs also showed mGluR6-mediated responses that were similarly reduced, a reduction that was not seen in heterozygous Trpm1+ −/− DBCs. These results suggest that the p.A1068T mutation acts as a dominant negative in the tetrameric TRPM1 channel and that the channel in the Trpm1+ /tvrm27 heterozygous DBCs is comprised of WT and mutant subunits. The quantitative reduction of DBC function is consistent with the hypothesis that channels with 0–2 mutant subunits retain function whereas those with 3–4 mutant subunits do not, although individual combinations of mutant and WT subunits also may have different kinetics. How the number of TRPM1 channels present in DBC puncta might be set is unclear but likely involves yet to be discovered scaffolding components of the signalplex.
Expression of GPR179 is Required for Localization of GAP Complexes to DBC Dendrites
GPR179 is a 7-transmembrane protein. Based on primary sequence data, it has been classified as a member of the GPCR superfamily. It was discovered as an important component of the mGluR6 transduction cascade independently and simultaneously from whole-exome sequencing of patients with cCSNB [2] and from mapping the gene involved in Gpr179 nob5, a naturally occurring mouse b-wave mutant [32] and it colocalizes with DBC signalplex components (Fig. 5.3). GPR179 expression localizes both RGS7 and RGS11 to the DBC terminals (Fig. 5.3 and Orlandi et al. [28]). RGS7 is likely to interact with Gβ5 , as DBCs in mice lacking Gβ5 expression also lack RGS7 expression [7, 8, 24, 36]. Because RGS7 does not interact with R9AP, it is possible that its interaction with GRP179 is critical to both its localization and perhaps function in the DBC cascade. While the specific functions of GPR179 remain to be determined, it is clear that GRP179 plays a critical role in assembling elements of the mGluR6–GPCR signalplex, although both mGluR6 and TRPM1 are localized to the tips of Gpr179 nob5 DBCs (Fig. 5.3). Our recently published data indicate that GPR179 sets the sensitivity of the TRPM1 channel, whereas RGS7/RGS11 sets the sensitivity of the mGluR6 cascade [37].
Nyctalopin is Required for TRPM1 Expression
In 1998, Pardue and colleagues identified a naturally occurring no b-wave mouse mutant. In this mouse, the phenotype was inherited as an X-linked trait [30] and subsequently we showed that it was caused by a deletion mutation in Nyx [17], the same gene that causes the X-linked form of human cCSNB [3, 35]. Nyctalopin is a member of the small leucine-rich repeat proteoglycan family of proteins. It is anchored to the cell membrane by either a single transmembrane domain or a GPI anchor in a species-dependent manner. Nyctalopin is comprised of a series of leucine-rich repeats, which are localized to the extracellular space. Efforts by several groups to make selective antibodies to nyctalopin have been unsuccessful, likely resulting from its extensive post-translational modifications. In view of this, we made a transgenic mouse line expressing an EYFP-nyctalopin fusion gene [17], which showed restricted and punctate expression of GFP to DBC terminals and colocalization with mGluR6 puncta. When these transgenic mice were crossed onto the Nyx nob background, the expression of the EYFP-nyctalopin fusion protein restored the ERG b-wave. In addition to the absence of the ERG b-wave, results show nyctalopin also interacts with TRPM1 [9, 33]. Together, these results suggest that Nyx nob DBC dysfunction is due to the loss of the TRPM1 channel from the signalplex (Fig. 5.5). How nyctalopin controls TRPM1 expression and localization to the DBC dendritic tips is unclear, but could reflect a role in trafficking TRPM1 or stabilization in the DBC membrane.

Interdependency of DBC signalplex components revealed by immunohistochemistry in WT, RGS7− −/− −/−RGS11 −/−, R9AP− −/−, and Nyx nob mice. Data for R9AP− −/− mice adapted from Cao et al. [8]. Open boxes indicate that these particular experiments have not been done
LRIT3 is Required for DBC Function
LRIT3 (leucine-rich-repeat, immunoglobulin-like and transmembrane-domain 3 (LRIT3) is the most recently identified member of the mGluR6 signalplex [47]. This discovery was made by whole-exome sequence analysis of cCSNB patients who did not harbor a mutation in any of the known members of the signalplex. Lrit3 knockout mice lack the ERG b-wave [26] supporting the results from cCSNB patients. While the function of LRIT3 is currently unknown, it is predicted to be an extracellular protein tethered to the membrane by a single transmembrane domain. The extracellular domain contains a LRR domain similar to nyctalopin. Whether LRIT3 interacts with nyctalopin via this domain will be of interest. This similarity also suggests it may be involved in trafficking and/or localization of some critical component of the DBC signalplex, similar to nyctalopin.
Interdependent Expression of RGS11, Gβ5, and R9AP
The DBC light evoked response requires the inactivation of the mGluR6 mediated G-protein cascade. Critical to this process are regulators of G-protein signaling (RGS), protein complexes that act as GTPase-activating proteins (GAPs). Two RGS proteins, RGS7 and RGS11 appear to have redundant function, as expression of both must be eliminated to produce a no b-wave ERG phenotype [10, 41]. In photoreceptors, RGS proteins form complexes with R9AP and Gβ5 . In DBCs, this is true at least for RGS11, since R9AP− −/− mice lack normal localization of RGS11 and reduced expression of Gβ5. Knockout of RGS7 and RGS11 or R9AP leave the primary members of the signalplex, mGluR6, TRPM1, and GPR179 localized correctly (Fig. 5.5).
General Conclusions
This review presents the analyses of a large number of knockout mouse lines using immunohistochemical approaches to examine the interdependent/independence of expression patterns of many components of the mGluR6–GPCR transduction cascade. We believe that the results suggest that the mGluR6–GPCR signalplex is comprised of several subdomains with clear hierarchies. One domain includes mGluR6, TRPM1, R9AP, RGS11, and Gβ5, which appear to form a complex with several interdependencies, but in which expression of all members is dependent on the presence of mGluR6. A second domain includes R9AP, RGS7, and Gβ5, as the elimination of any of these has no impact on mGluR6 expression. We think that RGS7 and Gβ5 form a third complex with GPR179 and that GPR179 appears to be a master regulator of the GAP complexes, as its absence causes mislocalization of RGS7/RGS11, R9AP, and Gβ5. Nyctalopin and TRPM1 form a separate subcomplex, and nyctalopin is essential for correct localization of the TRPM1 channel to the DBC membrane. In comparison, elimination of TRPM1 has the least impact on the expression of signalplex components, and its absence leaves the localized expression of every known component intact. This is consistent with the idea that TRPM1 gating is the final step in the DBC signal transduction process.
References
Audo I, Kohl S, Leroy BP, Munier FL, Guillonneau X et al (2009) TRPM1 is mutated in patients with autosomal-recessive complete congenital stationary night blindness. Am J Hum Genet 85:720–729
Audo I, Bujakowska K, Orhan E, Poloschek CM, Defoort-Dhellemmes S et al (2012) Whole-exome sequencing identifies mutations in gpr179 leading to autosomal-recessive complete congenital stationary night blindness. Am J Hum Genet 90:321–330
Bech-Hansen NT, Naylor MJ, Maybaum TA, Sparkes RL, Koop B et al (2000) Mutations in NYX, encoding the leucine-rich proteoglycan nyctalopin, cause X-linked complete congenital stationary night blindness. Nat Genet 26:319–323
Bellone RR, Brooks SA, Sandmeyer L, Murphy BA, Forsyth G et al (2008) Differential gene expression of TRPM1, the potential cause of congenital stationary night blindness and coat spotting patterns (LP) in the Appaloosa horse (Equus caballus). Genetics 179:1861–1870
Bellone RR, Forsyth G, Leeb T, Archer S, Sigurdsson S et al (2010) Fine-mapping and mutation analysis of TRPM1: a candidate gene for leopard complex (LP) spotting and congenital stationary night blindness in horses. Brief Funct Genomics 9:193–207
Bijveld MM, Florijn RJ, Bergen AA, van den Born LI, Kamermans M et al (2013) Genotype and phenotype of 101 Dutch patients with congenital stationary night blindness. Ophthalmology 120:2072–2081
Cao Y, Song H, Okawa H, Sampath AP, Sokolov M, Martemyanov KA (2008) Targeting of RGS7/Gbeta5 to the dendritic tips of ON-bipolar cells is independent of its association with membrane anchor R7BP. J Neurosci 28:10443–10449
Cao Y, Masuho I, Okawa H, Xie K, Asami J et al (2009) Retina-specific GTPase accelerator RGS11/G beta 5S/R9AP is a constitutive heterotrimer selectively targeted to mGluR6 in ON-bipolar neurons. J Neurosci 29:9301–9313
Cao Y, Posokhova E, Martemyanov KA (2011) TRPM1 forms complexes with nyctalopin in vivo and accumulates in postsynaptic compartment of ON-bipolar neurons in mGluR6-dependent manner. J Neurosci 31:11521–11526
Cao Y, Pahlberg J, Sarria I, Kamasawa N, Sampath AP, Martemyanov KA (2012) Regulators of G protein signaling RGS7 and RGS11 determine the onset of the light response in ON bipolar neurons. Proc Natl Acad Sci U S A 109:7905–7910
Devi S, Markandeya Y, Maddodi N, Dhingra A, Vardi N et al (2013) Metabotropic glutamate receptor 6 signaling enhances TRPM1 calcium channel function and increases melanin content in human melanocytes. Pigment Cell Melanoma Res 26:348–356
Dhingra A, Lyubarsky A, Jiang M, Pugh EN Jr, Birnbaumer L et al (2000) The light response of ON bipolar neurons requires G[alpha]o. J Neurosci 20:9053–9058
Dhingra A, Jiang M, Wang TL, Lyubarsky A, Savchenko A et al (2002) Light response of retinal ON bipolar cells requires a specific splice variant of Galpha(o). J Neurosci 22:4878–4884
Dhingra A, Ramakrishnan H, Neinstein A, Fina ME, Xu Y et al (2012) Gbeta3 is required for normal light ON responses and synaptic maintenance. J Neurosci 32:11343–11355
Dryja TP, McGee TL, Berson EL, Fishman GA, Sandberg MA et al (2005) Night blindness and abnormal cone electroretinogram ON responses in patients with mutations in the GRM6 gene encoding mGluR6. Proc Natl Acad Sci U S A 102:4884–4889
Gregg RG, Mukhopadhyay S, Candille SI, Ball SL, Pardue MT et al (2003) Identification of the gene and the mutation responsible for the mouse nob phenotype. Invest Ophthalmol Vis Sci 44:378–384
Gregg RG, Kamermans M, Klooster J, Lukasiewicz PD, Peachey NS et al (2007) Nyctalopin expression in retinal bipolar cells restores visual function in a mouse model of complete X-linked congenital stationary night blindness. J Neurophysiol 98:3023–3033
Jacobi FK, Andreasson S, Langrova H, Meindl A, Zrenner E et al (2002) Phenotypic expression of the complete type of X-linked congenital stationary night blindness in patients with different mutations in the NYX gene. Graefes Arch Clin Exp Ophthalmol 240:822–828
Koike C, Obara T, Uriu Y, Numata T, Sanuki R et al (2010) TRPM1 is a component of the retinal ON bipolar cell transduction channel in the mGluR6 cascade. Proc Natl Acad Sci U S A 107:332–337
Leroy BP, Budde BS, Wittmer M, De Baere E, Berger W, Zeitz C (2009) A common NYX mutation in Flemish patients with X linked CSNB. Br J Ophthalmol 93:692–696
Li Z, Sergouniotis PI, Michaelides M, Mackay DS, Wright GA et al (2009) Recessive mutations of the gene TRPM1 abrogate ON bipolar cell function and cause complete congenital stationary night blindness in humans. Am J Hum Genet 85:711–719
Maddox DM, Vessey KA, Yarbrough GL, Invergo BM, Cantrell DR et al (2008) Allelic variance between GRM6 mutants, Grm6nob3 and Grm6nob4 results in differences in retinal ganglion cell visual responses. J Physiol 586:4409–4424
Masu M, Iwakabe H, Tagawa Y, Miyoshi T, Yamashita M et al (1995) Specific deficit of the ON response in visual transmission by targeted disruption of the mGluR6 gene. Cell 80:757–765
Morgans CW, Weiwei L, Wensel TG, Brown RL, Perez-Leon JA et al (2007) Gbeta5-RGS complexes co-localize with mGluR6 in retinal ON-bipolar cells. Eur J Neurosci 26:2899–2905
Morgans CW, Zhang J, Jeffrey BG, Nelson SM, Burke NS et al (2009) TRPM1 is required for the depolarizing light response in retinal ON-bipolar cells. Proc Natl Acad Sci U S A 106:19174–19178
Neuille M, El Shamieh S, Orhan E, Michiels C, Antonio A et al (2014) Lrit3 Deficient Mouse (nob6): a novel model of complete congenital stationary night blindness (cCSNB). PLoS ONE 9:e90342
O’Connor E, Allen LE, Bradshaw K, Boylan J, Moore AT, Trump D (2006) Congenital stationary night blindness associated with mutations in GRM6 encoding glutamate receptor MGluR6. Br J Ophthalmol 90:653–654
Orlandi C, Posokhova E, Masuho I, Ray TA, Hasan N et al (2012) GPR158/179 regulate G protein signaling by controlling localization and activity of the RGS7 complexes. J Cell Biol 197:711–719
Orlandi C, Cao Y, Martemyanov KA (2013) Orphan receptor GPR179 forms macromolecular complexes with components of metabotropic signaling cascade in retina ON-bipolar neurons. Invest Ophthalmol Vis Sci 54:7153–7161
Pardue MT, McCall MA, LaVail MM, Gregg RG, Peachey NS (1998) A naturally occurring mouse model of X-linked congenital stationary night blindness. Invest Ophthalmol Vis Sci 39:2443–2449
Peachey NS, Pearring JN, Bojang P Jr, Hirschtritt ME, Sturgill-Short G et al (2012a) Depolarizing bipolar cell dysfunction due to a Trpm1 point mutation. J Neurophysiol 108:2442–2451
Peachey NS, Ray TA, Florijn R, Rowe LB, Sjoerdsma T et al (2012b) GPR179 is required for depolarizing bipolar cell function and is mutated in autosomal-recessive complete congenital stationary night blindness. Am J Hum Genet 90:331–339
Pearring JN, Bojang P, Shen Y, Koike C, Furukawa T et al (2011) A role for nyctalopin, a small leucine-rich repeat protein, in localizing the TRP melastatin 1 channel to retinal depolarizing bipolar cell dendrites. J Neurosci 31:10060–10066
Pinto LH, Vitaterna MH, Shimomura K, Siepka SM, Balannik V et al (2007) Generation, identification and functional characterization of the nob4 mutation of Grm6 in the mouse. Vis Neurosci 24:111–123
Pusch CM, Zeitz C, Brandau O, Pesch K, Achatz H et al (2000) The complete form of X-linked congenital stationary night blindness is caused by mutations in a gene encoding a leucine-rich repeat protein. Nat Genet 26:324–327
Rao A, Dallman R, Henderson S, Chen CK (2007) Gbeta5 is required for normal light responses and morphology of retinal ON-bipolar cells. J Neurosci 27:14199–14204
Ray TA, Heath KM, Hasan N, Noel JM, Samuels IS et al (2014) GPR179 is required for high sensitivity of the mGluR6 signaling cascade in depolarizing bipolar cells. J Neurosci 30:6334–6343
Scholl HP, Langrova H, Pusch CM, Wissinger B, Zrenner E, Apfelstedt-Sylla E (2001) Slow and fast rod ERG pathways in patients with X-linked complete stationary night blindness carrying mutations in the NYX gene. Invest Ophthalmol Vis Sci 42:2728–2736
Sergouniotis PI, Robson AG, Li Z, Devery S, Holder GE et al (2012) A phenotypic study of congenital stationary night blindness (CSNB) associated with mutations in the GRM6 gene. Acta Ophthalmol 90:e192–e197
Shen Y, Heimel JA, Kamermans M, Peachey NS, Gregg RG, Nawy S (2009) A transient receptor potential-like channel mediates synaptic transmission in rod bipolar cells. J Neurosci 29:6088–6093
Shim H, Wang CT, Chen YL, Chau VQ, Fu KG et al (2012) Defective retinal depolarizing bipolar cells in regulators of G protein signaling (RGS) 7 and 11 double null mice. J Biol Chem 287:14873–14879
van Genderen MM,B, Pearring JN et al (2009) Mutations in TRPM1 are a common cause of complete congenital stationary night blindness. Am J Hum Genet 85:730–736
Vardi N, Matesic DF, Manning DR, Liebman PA, Sterling P (1993) Identification of a G-protein in depolarizing rod bipolar cells. Vis Neurosci 10:473–478
Xu X, Li S, Xiao X, Wang P, Guo X, Zhang Q (2009) Sequence variations of GRM6 in patients with high myopia. Mol Vis 15:2094–2100
Xu Y, Dhingra A, Fina ME, Koike C, Furukawa T, Vardi N (2012) mGluR6 deletion renders the TRPM1 channel in retina inactive. J Neurophysiol 107:948–957
Zeitz C, Minotti R, Feil S, Matyas G, Cremers FP et al (2005a) Novel mutations in CACNA1F and NYX in Dutch families with X-linked congenital stationary night blindness. Mol Vis 11:179–183
Zeitz C, van Genderen M, Neidhardt J, Luhmann UF, Hoeben F et al (2005b) Mutations in GRM6 cause autosomal recessive congenital stationary night blindness with a distinctive scotopic 15-Hz flicker electroretinogram. Invest Ophthalmol Vis Sci 46:4328–4335
Zeitz C, Jacobson SG, Hamel CP, Bujakowska K, Neuille M et al (2013) Whole-exome sequencing identifies LRIT3 mutations as a cause of autosomal-recessive complete congenital stationary night blindness. Am J Hum Genet 92:67–75
Zhang Q, Xiao X, Li S, Jia X, Yang Z et al (2007) Mutations in NYX of individuals with high myopia, but without night blindness. Mol Vis 13:330–336
Zito I, Allen LE, Patel RJ, Meindl A, Bradshaw K et al (2003) Mutations in the CACNA1F and NYX genes in British CSNBX families. Hum Mutat 21:169
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2014 Springer Science+Business Media New York
About this chapter
Cite this chapter
Gregg, R., Ray, T., Hasan, N., McCall, M., Peachey, N. (2014). Interdependence Among Members of the mGluR6 G-protein Mediated Signalplex of Retinal Depolarizing Bipolar Cells. In: Martemyanov, K., Sampath, A. (eds) G Protein Signaling Mechanisms in the Retina. Springer Series in Vision Research, vol 3. Springer, New York, NY. https://doi.org/10.1007/978-1-4939-1218-6_5
Download citation
DOI: https://doi.org/10.1007/978-1-4939-1218-6_5
Published:
Publisher Name: Springer, New York, NY
Print ISBN: 978-1-4939-1217-9
Online ISBN: 978-1-4939-1218-6
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)