
Abstract
Drugs of abuse stimulate the pleasure centers of the brain to initiate addiction. During the beginning stages of addiction, the rewarding or reinforcing properties of abused drugs drive intake. However, as addiction develops drug intake is more likely to be dominated by negative reinforcement. The main reward center of the brain is the mesolimbic pathway which consists of dopaminergic neurons originating in the ventral tegmental area that project to the nucleus accumbens. Most, if not all, abused drugs stimulate this circuit resulting in increased release of the neurotransmitter, dopamine, in the nucleus accumbens, a phenomenon intimately associated with reward and reinforcement. Neuronal nAChRs are robustly expressed within the microcircuitry of this reward pathway. Drugs of abuse such as nicotine and alcohol directly interact with nAChRs expressed within the mesolimbic circuit to affect drug reward sensitivity, whereas with other drugs of abuse such as the psychostimulants and opioids, nAChRs play a more indirect, modulatory role on drug reward. In this chapter, the expression and function of nAChRs in the reinforcing/rewarding properties of drugs of abuse are explored.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others


Keywords
1 Introduction
Species that learned to respond to natural rewards (such as when and where they could obtain food, have the opportunity to mate) ensured their survival. Achieving these goals function as rewards [1]. Consequently, many neural substrates that modulate reward systems are conserved across species from Drosophila, mice, and rats to humans and include conserved circuitry, neurotransmitters, receptors, signaling molecules, and transcription factors [2]. Not surprisingly, this endogenous system can be exogenously altered via drugs that have potential to become abused. We now know that responses to natural rewards and addictive drugs have many similarities and shared pathways within the central nervous system (CNS). For example, studies in rats have shown a cross-sensitization between the natural reward sugar and the drug amphetamine [3]. In addition, a recent study found similar neuroadaptations in reward circuitry between chronic exposure of abused drugs and high-energy palatable food [4].
A common effect of natural rewards and most drugs of abuse is an enhancement of activity in the mesolimbic dopamine (DA) system (discussed in more detail below), leading to an increase of DA release in the nucleus accumbens (NAc) [5–7]. While it is widely accepted that the epicenter of reward stimuli processing within the brain, whether natural or drug, is the mesolimbic DA circuitry, much controversy exists regarding the precise role of DA in modulating goal-directed behavior. Mesolimbic DA is critical for a variety of physiological and affective behaviors such as movement, motivation, reward, learning, arousal, attention, and emotion [8]. Indeed, each of these individual behavioral components is necessary for the outward, measurable behavior of reward (i.e., an organism must locate a reward, pay attention, learn where to find it, like it, and have a desire to return to it).
Most of what is known regarding the underlying circuitry and molecular underpinnings of reward in addiction stems from pharmacological and genetic manipulations in rodent models. How does one measure the rewarding properties of drugs in animal models of dependence? The rewarding properties of drugs of abuse are typically measured via operant self-administration and/or conditioned place preference assays (CPP). In the former assay, an animal learns to self-administer a drug by pressing an active lever or nose poke that delivers a fixed dose to the animal by way of intravenous catheter, cannula to the brain, or, in the case of ethanol, a consumable liquid [9]. If a drug is reinforcing, then the animal will press on the active lever to self-administer the drug while ignoring a second inactive lever which yields no drug. In the CPP assay, an animal prefers a chamber where it received drug over the chamber where it received vehicle (i.e., the drug conditions a place preference as a measure of reward [10]).
Current theories on drug addiction suggest that the acute, rewarding properties of abused drugs drive intake during the initial stages of dependence; whereas drug intake in later stages is motivated by negative reinforcement (i.e., drugs are taken to predominantly alleviate negative affective states precipitated by withdrawal) [11]. This chapter focuses on nAChRs in the acute rewarding properties of drugs of abuse, while chapter 18 will focus on nAChRs in negative reinforcement, aversion, and withdrawal. It is important to point out that the circuitry underlying positive reinforcement (i.e., reward) and negative reinforcement (i.e., aversion) likely interact. However, the most well-studied circuit in the context of reward, addiction, and nAChRs is the mesolimbic pathway.
2 The Mesolimbic DA Pathway
It is widely accepted that the mesolimbic DA system plays a central role in modulating the rewarding effects of drugs of abuse [12, 13]. Olds and Milner first identified this pathway in 1954. Using brain stimulation reward (BSR) they discovered that rats returned to the same region of a testing apparatus where they had received electrical stimulation to the septal area of the brain [14]. Upon further examination using mapping and lesion studies, it was determined that the most sensitive sites in the brain (i.e., lowest stimulation threshold) were along the medial forebrain bundle (MFB) which connects the ventral tegmental area (VTA) to the basal forebrain [14–16]. Next, using pharmacology, studies showed that DAergic receptor blockade attenuated brain stimulation reward [17, 18], suggesting that specific neurotransmitter systems were involved in reward mechanisms [19].
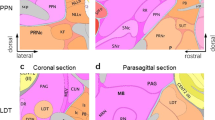
Flash-forward almost 60 years and what was once commonly referred to as the “reward circuit” is now known as the mesolimbic DA pathway. This pathway consists of DAergic neurons whose cell bodies originate in the ventral tegmental area (VTA), a region of the midbrain, and project to regions of the limbic system including the NAc, amygdala, and hippocampus among other regions. An additional DAergic pathway, the mesocortical pathway, also originates in the VTA and project to regions of the prefrontal cortex. These pathways are shown in a simplified diagram in Fig. 15.1.

Neuronal nAChR expression in the mesolimbic and mesocortical pathways. A sagittal rodent brain section depicting a simplified circuit diagram of the mesolimbic and mesocortical pathways is shown. The VTA (yellow box) consists of DAergic neurons projecting to the NAc (purple box) and prefrontal cortex (orange box). VTA GABAergic neurons provide local inhibition within the VTA and also project to the NAc. Glutamatergic neurons provide excitatory input into the VTA. Cholinergic, GABAergic, and glutamatergic VTA inputs also stem from laterodorsal tegmental (LTD) and pedunculopontine (PPTg) afferents. Drugs of abuse ultimately increase release of DA into the NAc to affect medium spiny projection neuron (MSN) activity. DA release at DAergic neuron presynaptic terminals is modulated by endogenous ACh provided by large aspiny cholinergic interneurons. Location of nAChR expression within the mesolimbic and mesocortical circuitry is indicated by the receptor icons
3 The Ventral Tegmental Area
The VTA is known to at least partially mediate the rewarding effects of nicotine, opiates, psychostimulants, ethanol, and cannabinoids [20]. For example, rats and mice will self-administer opiates [21], cannabinoids [22], cocaine [23], nicotine [24], or ethanol [25, 26] directly into the VTA. Additionally, intravenous nicotine self-administration is attenuated by either selective lesions of VTA DAergic neurons in rats [27] or a local VTA infusion of a nicotinic receptor antagonist [28]. The VTA is located in the midbrain, medial to the substantia nigra and ventral to the red nucleus [29]. It is referred to as an “area” and not considered to be a “nucleus” because the cryoarchitecture of the region is not well defined such that the boundaries of the VTA are determined by its neighboring structures [20, 30]. Within the VTA are two main cell populations, the A10 DAergic projection neurons, which comprise ~60 % of cells in this region [31], as well as local GABAergic interneurons [32, 33]. Although data are emerging indicating that different subpopulations of neurons within the VTA exist including DAergic neurons that also co-release glutamate, GABAergic projection neurons, and a small number of purely glutamatergic neurons [34, 35], the expression and function of nAChRs in these neuronal subpopulations as they relate to reward are unknown. The VTA receives inputs from regions throughout the CNS [36] including glutamatergic projections from the prefrontal cortex [37], as well as glutamatergic, cholinergic, and GABAergic projections from two groups of mesopontine tegmental area neurons, the pedunculopontine tegmental nucleus (PPTg) and the laterodorsal tegmental nucleus (LDT) [38–40]. Other regions that project to the VTA include the NAc, amygdala, ventral pallidum, superior colliculus, and lateral hypothalamus [30]. Additionally, the lateral habenula, a small nucleus that is a part of the epithalamus, has been shown to project to and stimulate midbrain areas that inhibit the release of DA from the VTA and substantia nigra pars compacta [41–43].
Projections from the VTA are primarily to the ventromedial striatum including the NAc shell and core as well as smaller projections to the prefrontal cortex (PFC), hippocampus, entorhinal cortex, and lateral septal areas [30]. Furthermore, studies using retrograde markers have shown that distinct groups of neurons originating in the VTA project to specific forebrain regions [44, 45]. Projections to the NAc contain the largest proportion of DA neurons, with 65–85 % being DAergic, while the PFC projections are only 30–40 % DAergic [31, 45]. The remaining component of VTA afferents to the NAc and PFC contain GABAergic neurons [32]. Although the VTA consists of two predominant neuronal subtypes, there is mounting evidence that this brain structure is not homogenous but can be divided into discrete subregions including anterior (aVTA), posterior (pVTA), and tail (tVTA) [20, 46–48]. Recent data indicate that the aVTA and pVTA project to distinct regions of the ventral striatum and are differentially responsive to various drugs of abuse suggesting functional heterogeneity [22, 49–52]. For example, rats will self-administer nicotine and ethanol directly in the pVTA but not the aVTA although the mechanistic basis of this regional selectivity is unknown [49].
4 The Nucleus Accumbens
For decades, the NAc has been a main focus of mesolimbic DA in studies of natural and drug reward [8]. It is located in the ventromedial striatum and is primarily composed of GABAergic medium spiny neurons (~95 %) and to a lesser extent a variety of interneurons (1–2 %) including cholinergic, fast-spiking GABAergic and low-threshold spiking. Two distinct regions of the NAc have been described, the core and shell, based on differences in functions and anatomical connectivity [53, 54]. Additionally, studies have shown that the response to extracellular DA release of these two regions differs. For example, it has been shown that the DA release induced by a food reward is rapidly habituated in the shell, but not the core [55]. Another study showed differential NAc shell and core Fos immunolabeling (a marker of neuronal activation) of cholinergic interneurons after cocaine self-administration [56]. These and other data suggest the possibility that the shell may act to modulate the initiation of drug-seeking behavior by mediating the hedonic states associated with reward [57, 58] while the core may modulate acquisition and maintenance of drug seeking [59].
The extracellular DA concentration in the NAc is regulated by two main factors: (1) the rate of release of DA from DAergic neurons that originate in the VTA and (2) dopamine uptake through dopamine transporters located in perisynaptic areas [60]. DAergic neurons of the VTA are known to be the main input source of extracellular DA in the NAc. Under normal conditions, the action potential (AP) firing rate of DAergic neurons is tonic with spike activity at 1–5 Hz [61]. However, when an unexpected presentation of a primary reward or a reward-predicting stimulus occurs, the firing rate increases to 2–10 APs at 10–30 Hz [62, 63].
4.1 Neuronal nAChR Expression in Reward Circuitry
Neuronal nicotinic acetylcholine receptors (nAChRs) are ligand-gated cation channels that, under normal conditions, are activated by the endogenous neurotransmitter, acetylcholine (ACh) [64, 65]. Eleven mammalian genes encoding nAChR subunits have been identified (α2–α7, α9–α10, β2–β4) and five subunits coassemble to form a functional receptor [64, 66]. The majority of nAChRs with high affinity for agonist are heteromeric consisting of two or three alpha subunits coassembled with two or three beta subunits while a subset of low-affinity receptors are homomeric, consisting of predominantly α7 subunits [64]. The subunit composition of the receptor determines the biophysical and pharmacological properties of each receptor subtype. Given the large number of nAChR subunits, the potential for a vast array of nAChR subtypes exists.
Multiple studies have examined nAChR expression and function within the VTA [67–72]. Klink et al. compared nAChR expression and function in DAergic and GABAergic neurons between the VTA and substantia nigra pars compacta (SNc). Utilizing β2, α4, and α7 KO mice in combination with nAChR antagonists, they concluded that most DAergic neurons express nAChRs containing α4, α5, α6, β2, and β3 subunits while most GABAergic neurons express nAChRs containing α4 and β2 subunits [67]. Using a similar strategy, Wooltorton et al. determined that α7 expression was more prevalent in VTA neurons than SNc neurons while nAChRs containing the β2 subunit (denoted β2*) are prevalent in DAergic and non-DAergic neurons throughout both brain regions [72]. The α6 nAChR subunit is predominantly expressed in DAergic neurons (although it may also be expressed in GABAergic terminal boutons) and can coassemble with β2, β3, and α4 subunits [70, 71, 73–76]. Using immunoprecipitation approaches in ventral midbrain, Gotti et al. deduced that at least five distinct nAChR subtypes were expressed in DAergic neurons at the level of soma/dendrites including α4β2, α2α4β2, α4α5β2, α4β2β3, and α4α6β2β3 nAChRs [77]. Within the NAc, the majority of nAChRs are expressed in DAergic presynaptic terminals where they modulate the probability of DA release by endogenous ACh and DAergic neuron firing frequency [78, 79]. DAergic neuron terminal nAChRs consist of α4β2, α4α5β2, α4β2β3, α4α6β2β3, and α6β2β3 subtypes [77]. Of these subtypes, α4α6β2β3 appears to dominate control of DA release at least in the NAc core [80].
4.2 Nicotinic Receptor Subtypes Involved in Nicotine Reward/Reinforcement
Smoking is the primary cause of preventable mortality in the world [81]. When volatized, nicotine, the addictive component of tobacco smoke, is absorbed into the bloodstream via the lungs and rapidly, on the order of seconds, crosses the blood-brain barrier [65]. Although nAChRs are expressed throughout the CNS, nicotine-induced activation of the mesocorticolimbic reward circuitry likely initiates addiction [66]. Indeed, pharmacological blockade of DA receptors or destruction of DA neurons or lesioning of the NAC reduces nicotine self-administration [27, 82]. Within this pathway, nicotine ultimately drives activity of DAergic neurons originating in the VTA resulting in increased DA release in the NAc and prefrontal cortex (PFC) [83]. More recently, nicotine has been found to also increase DA release in the hippocampus where it facilitates memory formation of nicotine reward [84].
With the great diversity of potential nAChR subunit combinations possible in nAChR subtypes within the VTA, a major goal of nicotine dependence research is to identify nAChR subunit combinations that are critical for the rewarding properties of nicotine. The majority of insights into reward circuitry nAChRs in reward and reinforcement stems from pharmacological and genetic studies in rodent models. Infusion of the nonspecific nAChR antagonist, mecamylamine, into the VTA reduces self-administration of nicotine in rodents while also blocking nicotine-mediated increases in NAc DA [49, 85]. In addition, the β2*-selective antagonist dihydro-β-erythroidine (dhβe) also reduces nicotine self-administration in rats when infused into the VTA [28]. Finally, infusion of the α6β2-selective antagonist, α-conotoxin MII, into the VTA or NAc reduces nicotine self-administration [77, 86].
Because of the limited nAChR subtype selectivity of most pharmacological agents, a more direct approach to address nAChR subunit composition in nicotine reward is through the use of genetically engineered mouse models. To date, several studies have utilized traditional knockout mice, which do not express a given nAChR subunit, or mice that express “gain-of-function” receptors that harbor a mutated subunit hypersensitive to nicotine, to examine the role of individual nAChR subunits in nicotine reward and reinforcement [87, 88]. Mice that do not express the β2 subunit fail to maintain nicotine self-administration indicating that nAChRs containing β2 are necessary for nicotine reinforcement [89]. These knockout mice also do not condition a place preference to nicotine consistent with a critical role for β2* nAChRs in nicotine reward [90]. In addition, mice that express a single-point mutation in the gene encoding the α4 subunit (a leucine residue mutated to an alanine residue in the pore forming transmembrane domain of the α4 subunit) that renders α4* nAChRs supersensitive to agonist condition a place preference to nicotine at sub-reward-threshold doses indicating that selective activation of α4* nAChRs is sufficient for nicotine reward [91]. In addition, mice harboring a distinct mutation within the α4 subunit also resulting in nicotine-hypersensitive α4* nAChRs self-administer nicotine at lower doses [92] than mice with non-mutated receptors. Knockout mice that do not express β2, α4, or α6* nAChRs fail to self-administer nicotine but nicotine intake can be rescued via viral mediated expression of these subunits in the VTA, indicating that expression of nAChRs specifically in the VTA is sufficient to support nicotine reinforcement [93, 94]. Thus, the emerging consensus across laboratories, based on a combination of pharmacology and mouse genetics, is that expression of α4β2* and α6* nAChRs in the VTA is necessary and sufficient for nicotine reward and reinforcement.
The identification of α4β2* nAChRs as critical for nicotine reward has led to rational design of small-molecule compounds to target these receptors in an effort to facilitate smoking cessation. The most successful smoking cessation aid to date is varenicline. Varenicline was designed as a high-affinity partial agonist at α4β2* nAChRs [95]. Studies in rodent midbrain slices indicate that varenicline activates α4β2* nAChRs in the mesolimbic circuitry modestly increasing DA release in the NAc while blocking further stimulation by the full agonist, nicotine [96]. In doing so, it is hypothesized that, in smokers, varenicline will alleviate affective withdrawal symptoms through increasing mesolimbic DA stimulation but also block the pleasurable effects of nicotine achieved through smoking.
4.3 Mechanisms of VTA DAergic Neuron Activation by Nicotine
VTA DAergic neurons fire tonically and also fire bursts [97, 98]. Recent studies using optogenetics to precisely depolarize DAergic neurons through light activation of the cationic ion channel, channelrhodopsin, indicate that bursting, but not tonic, DAergic neuron firing is sufficient to condition a place preference [99]. Conversely optogenetic activation of VTA GABAergic neurons alone inhibits DAergic neurons and signal aversion [100]. Acutely, nicotine elicits both an increase in baseline DAergic neuron firing frequency and an increase in burst firing that can persist up to an hour after a single bolus of nicotine [101, 102]. Previous studies indicate that nicotine can directly activate DAergic neurons in rodent midbrain slices [103, 104] and neuronal α4β2* nAChR subunits are critical for this effect. Indeed, nicotine fails to condition a place preference in mice that do not express α4* nAChRs selectively in DAergic neurons [105]. However, how VTA GABAergic neurons, which make up as many as half the neurons in the VTA [106] and also robustly express α4β2* nAChRs [67, 68, 70, 89, 91], contribute to shaping nicotine responses in DAergic neurons is emerging. In rat midbrain slices, nicotine may desensitize α4β2* nAChRs on GABAergic neurons, thereby disinhibiting DAergic neurons, increasing their activation [107]. In addition, blood nicotine concentrations achieved by smoking rapidly and persistently desensitize a portion of nAChRs on both DAergic and GABAergic neurons [102, 107]. Low-affinity α7 nAChRs, which are expressed on glutamatergic terminals that innervate the VTA, may rapidly recover from desensitization and drive glutamate release, thereby allowing for persistent activation of DAergic neurons by nicotine [107]. This is consistent with previous data indicating that glutamate release into the VTA is critical for nicotine reinforcement [108]. More recently, Tolu et al. found that nicotine, at least acutely, activates both DAergic and GABAergic VTA neurons in vivo [109]. Using viral mediated gene delivery to selectively re-express β2 nAChR subunits in VTA DAergic neurons of β2 KO mice was insufficient to restore nicotine self-administration and nicotine-mediated DA release in NAc. Surprisingly, β2 expression in both VTA DAergic and GABAergic neurons was required for rescue of nicotine self-administration. Remarkably, β2 expression in GABAergic neurons was critical for nicotine-mediated burst firing of DAergic neurons. These data indicate that nicotine activation of GABAergic interneurons in concert with activation of DAergic neurons may shape the firing pattern of DAergic neurons and modulate nicotine reward and reinforcement. Finally, recent studies have identified a unique nAChR subtype in VTA DAergic neurons consisting of both α4 and α6 subunits. These α4α6* nAChRs remain active with prolonged exposure to nicotine, and cause persistent depolarization of DAergic neurons [110, 111]. This persistent activation leads to changes in NMDA/AMPA receptor expression which may underlie sensitization to repeated nicotine exposure and enhance nicotine reward over time [111].
5 Neuronal nAChRs in Alcohol Reward
Alcohol abuse is the third largest cause of preventable mortality in the world [112]. As with nicotine, the rewarding or reinforcing properties of alcohol are associated with an increase in DA release in the NAc [113–117]. Ethanol-induced release of DA is critical for the onset and maintenance of dependence [118–121].
Multiple mechanisms underlying alcohol-mediated activation of VTA DAergic neurons have been proposed including modulation of intrinsic ion channels within these neurons, as well as alcohol-mediated alterations in synaptic input, both excitatory and inhibitory [122–128]. However, cholinergic signaling through nAChRs also contributes to NAc DA release and ethanol reinforcement [129–132]. For example, in rats, ethanol-mediated DA elevation in the NAc is inhibited by systemic or VTA but not NAc infusion of the noncompetitive, nonselective, nAChR antagonist, mecamylamine [130, 131, 133–136]. Blocking midbrain nAChRs via mecamylamine also decreases ethanol consumption and sensitization in rats. In addition, patients administered mecamylamine report reduced pleasurable effects of alcoholic beverages [137].
As discussed above, neuronal nAChR subtypes are expressed throughout the VTA in both DAergic neurons projecting to the NAc and in local GABAergic interneurons [67, 72]. How does ethanol interact with these receptors? Systemic ethanol has been shown to increase ACh concentrations in the VTA, presumably activating nAChRs in this area [135]. In addition, ethanol can directly modulate nAChR activity depending on the subtype of nicotinic receptor expressed [138–140]. In ventral midbrain slices containing the VTA, acetylcholine-induced activation of DAergic neurons is potentiated by ethanol and blocked by mecamylamine. In addition, the effects of ethanol on VTA DAergic neuron activity is reduced in α4 KO mice and enhanced in gain-of-function α4 knock-in mice [141]. Finally, potentiation is also blocked by an α6* nAChR-selective antagonist and reduced in α6 KO mice [142]. Thus, α4, α6, and/or α4α6* nAChRs may contribute to activation of VTA DAergic neurons by ethanol.
5.1 What Are the nAChR Subtypes Involved in Ethanol Reward and Reinforcement?
Identifying the nAChR subtype(s) that may underlie ethanol reward and consumption is necessary as they may represent therapeutic targets to reduce alcohol consumption. This endeavor is complicated by the fact that ethanol physiological and behavioral effects involve additional non-cholinergic mechanisms. In an effort to tease out individual nAChR subunits in ethanol-related behaviors, several studies have utilized pharmacology. As mentioned above, the nonspecific nAChR antagonist, mecamylamine, when injected systemically or locally within the VTA blocks ethanol consumption [132, 143, 144]. Alcohol consumption and alcohol-mediated DA release in the NAc are resistant to dhβe [133–135, 145–149]. In addition, the α7-selective antagonist, methyllycaconitine (MLA), does not affect alcohol-mediated behaviors precluding a role for homomeric α7 nAChRs [133, 144, 150]. On the other hand, the α3β2*, β3*, and α6* subtype-selective antagonist, α-conotoxin MII, does inhibit ethanol consumption and DA release in the NAc [151, 152]. Importantly, recent data indicate that approximately half of α-conotoxin MII-sensitive nAChRs in the striatum contain the α4 subunit [74, 153] and deletion of β2* nAChRs nearly abolishes α-conotoxin MII binding in the VTA [68]. Varenicline, an α4β2 partial agonist clinically approved as a smoking cessation therapeutic [95, 154–156], can reduce both ethanol intake and seeking in rats [155] and acute alcohol consumption in mice [157]. However, at high concentrations, varenicline is also a partial agonist at α6β2* nAChRs, a full agonist at α3β4 and α7 nAChRs, as well as at 5-hydroxytryptophan-3 receptors, which may also explain some of its effects on alcohol consumption [158–161]. Sazetidine-A, an α4β2* nAChR-selective “desensitizer,” can also reduce alcohol consumption in rats [162]. Cytisine, a partial agonist that preferentially activates high-affinity β2* nAChRs at low doses but also is a full β4* nAChR agonist at high doses, also reduces alcohol consumption [163–165]. Novel partial agonists targeting α3β4* nAChRs reduce alcohol consumption and seeking in rats [166]. However, infusion of the α3β4* nAChR antagonist 18-methoxycoronaridine into the VTA fails to reduce alcohol consumption [167] consistent with data indicating low expression of β4* nAChRs in VTA DAergic neurons [76, 77].
Behavioral studies in genetically engineered mice have also been used to glean information on nAChR subtypes that are involved in alcohol consumption. To date, mice that do not express α6, α4, α7, β2, or β3 subunits have been evaluated in a two-bottle alcohol consumption assay. α6, β2, and β3 nAChR subunit KO mice consume and prefer alcohol similarly to WT controls [157, 168, 169], whereas α7 KO mice consume less alcohol at high concentrations [157]. In addition, α4 KO mice consume acutely less alcohol in a binge-drinking assay compared to WT littermates and are less sensitive to ethanol reward as measured in the CPP assay. In contrast, ethanol conditions a place preference at low doses in gain-of-function α4 knock-in mice (i.e., mice that are hypersensitive to acetylcholine) compared to WT mice [141]. Similarly, mice expressing gain-of-function α6* nAChRs consume more ethanol than WT mice and are sensitive to ethanol reward at sub-reward-threshold doses [170]. Thus, consistent with a potential role in activation of VTA DAergic neurons by ethanol, α4 and/or α6 or α4α6* nAChRs within the VTA may be inherently critical for the rewarding properties of ethanol, although additional experiments are needed to identify the precise brain region and circuitry where these nAChRs are expressed.
6 Neuronal nAChRs in Psychostimulant Reward
Whereas nicotine and ethanol interact with nAChRs directly to modulate function of the mesolimbic reward circuitry, the interaction between nAChRs and psychostimulant is likely indirect occurring at the circuit level. Indeed, psychostimulants such as cocaine and amphetamine bind to the dopamine transporter (DAT), which, under basal conditions, takes up DA at the synaptic cleft from the presynaptic side where it can be recycled to help terminate DA receptor signaling [171]. Cocaine blocks DAT while amphetamine reverses transport resulting in increased NAc DA and reward. Neuronal nAChRs modulate the rewarding and reinforcing properties of psychostimulants. Nicotine preexposure potentiates self-administration of low doses of cocaine in rats and augments conditioned place preference in mice [172, 173], whereas mecamylamine reduces cocaine self-administration in rats and reduces low-dose cocaine place preference in mice [173–175]. Neuronal nAChRs that influence psychostimulant reward are likely expressed at DAergic presynaptic terminals where they modulate DA release through cholinergic input from large aspiny cholinergic interneurons within the NAc. Cholinergic neuron activity, and hence cholinergic signaling, is critical for cocaine reward as the drug fails to condition a place preference if these interneurons are silenced [176]. Supporting a role for NAc DAergic presynaptic terminal nAChRs on cocaine reinforcement, infusion of mecamylamine or dhβe and MLA into the NAc reduces DA release elicited by an i.p. injection of cocaine in rats [177]. While the precise nAChR subtype involved in cocaine reward has not been fully elucidated, they most likely contain the β2 subunit, as β2 KO show reduced CPP in response to low doses of cocaine [173].
7 Neuronal nAChRs in Opioid Reward
Morphine and commonly abused prescription opioids are opioid receptor agonists. Like the psychostimulants, opioids do not interact with nAChRs directly. However, they do indirectly stimulate VTA DAergic neurons in the mesolimbic pathway by binding to and activating mu opioid receptors on VTA GABAergic interneurons and reducing interneuron activity [178]. Infusion of nicotine in the VTA potentiates morphine-conditioned place preference, whereas infusion of mecamylamine into the VTA inhibits morphine CPP suggesting a role for VTA nAChRs in opioid reward [179]. In addition, dhβe or MLA blocks drug priming-induced reinstatement of morphine CPP [180]. However, few studies have directly examined the role of nAChRs in the mesolimbic pathway in opioid reward. Thus, further studies to identify the mechanism of action of nAChRs in opioid reward are needed.
8 Conclusions
Although neuronal nAChRs are expressed throughout the CNS, most studies examining the role of nAChRs in drug reward have focused on the DAergic mesolimbic reward circuitry. Indeed, nAChRs are robustly expressed within the mesolimbic circuitry in multiple neuronal subpopulation including DAergic projection neurons and GABAergic interneuron among others. Direct stimulation of α4β2, α6, and/or α4α6* nAChRs within the VTA by nicotine underlies the acute rewarding properties of the drug. Neuronal nAChRs containing the α4 and/or α6 subunit also contribute to alcohol reward. Ethanol potentiates the response to ACh at these receptors. In addition, ethanol may enhance release of ACh in the VTA to activate DAergic neurons in this pathway through indirect nAChR activation. Emerging evidence indicates that nAChRs within the mesolimbic pathway may also modestly affect psychostimulant and opioid reward through modulation of DA release in the NAc. Identification of nAChR subtypes involved in drug reward may provide novel molecular targets for therapeutics designed to help treat drug addiction.
References
Hyman SE, Malenka RC, Nestler EJ. Neural mechanisms of addiction: the role of reward-related learning and memory. Annu Rev Neurosci. 2006;29:565–98.
Kelley AE, Berridge KC. The neuroscience of natural rewards: relevance to addictive drugs. J Neurosci. 2002;22(9):3306–11.
Avena NM, Hoebel BG. A diet promoting sugar dependency causes behavioral cross-sensitization to a low dose of amphetamine. Neuroscience. 2003;122(1):17–20.
Johnson PM, Kenny PJ. Dopamine D2 receptors in addiction-like reward dysfunction and compulsive eating in obese rats. Nat Neurosci. 2010;13(5):635–41.
Di Chiara G, Imperato A. Preferential stimulation of dopamine release in the nucleus accumbens by opiates, alcohol, and barbiturates: studies with transcerebral dialysis in freely moving rats. Ann N Y Acad Sci. 1986;473:367–81.
Koob GF, Bloom FE. Cellular and molecular mechanisms of drug dependence. Science. 1988;242(4879):715–23.
Wise RA. Drug-activation of brain reward pathways. Drug Alcohol Depend. 1998;51(1–2):13–22.
Gonzales RA, Job MO, Doyon WM. The role of mesolimbic dopamine in the development and maintenance of ethanol reinforcement. Pharmacol Ther. 2004;103(2):121–46.
Fowler CD, Kenny PJ. Intravenous nicotine self-administration and cue-induced reinstatement in mice: effects of nicotine dose, rate of drug infusion and prior instrumental training. Neuropharmacology. 2011;61(4):687–98.
Cunningham CL, Gremel CM, Groblewski PA. Drug-induced conditioned place preference and aversion in mice. Nat Protoc. 2006;1(4):1662–70.
Koob GF, Le Moal M. Review. Neurobiological mechanisms for opponent motivational processes in addiction. Philos Trans R Soc Lond B Biol Sci. 2008;363(1507):3113–23.
Koob GF. Drugs of abuse: anatomy, pharmacology and function of reward pathways. Trends Pharmacol Sci. 1992;13(5):177–84.
Wise RA, Bozarth MA. A psychomotor stimulant theory of addiction. Psychol Rev. 1987;94(4):469–92.
Olds J, Milner P. Positive reinforcement produced by electrical stimulation of septal area and other regions of rat brain. J Comp Physiol Psychol. 1954;47(6):419–27.
Corbett D, Wise RA. Intracranial self-stimulation in relation to the ascending dopaminergic systems of the midbrain: a moveable electrode mapping study. Brain Res. 1980;185(1):1–15.
Wise RA. Intracranial self-stimulation: mapping against the lateral boundaries of the dopaminergic cells of the substantia nigra. Brain Res. 1981;213(1):190–4.
Wise RA. Catecholamine theories of reward: a critical review. Brain Res. 1978;152(2):215–47.
Liebman JM. Discriminating between reward and performance: a critical review of intracranial self-stimulation methodology. Neurosci Biobehav Rev. 1983;7(1):45–72.
Wise RA. The role of reward pathways in the development of drug dependence. Pharmacol Ther. 1987;35(1–2):227–63.
Ikemoto S. Dopamine reward circuitry: two projection systems from the ventral midbrain to the nucleus accumbens-olfactory tubercle complex. Brain Res Rev. 2007;56(1):27–78.
Bozarth MA, Wise RA. Intracranial self-administration of morphine into the ventral tegmental area in rats. Life Sci. 1981;28(5):551–5.
Zangen A, et al. Two brain sites for cannabinoid reward. J Neurosci. 2006;26(18):4901–7.
Rodd ZA, et al. Intracranial self-administration of cocaine within the posterior ventral tegmental area of Wistar rats: evidence for involvement of serotonin-3 receptors and dopamine neurons. J Pharmacol Exp Ther. 2005;313(1):134–45.
David V, et al. Reinforcing effects of nicotine microinjections into the ventral tegmental area of mice: dependence on cholinergic nicotinic and dopaminergic D1 receptors. Neuropharmacology. 2006;50(8):1030–40.
Gatto GJ, et al. Ethanol self-infusion into the ventral tegmental area by alcohol-preferring rats. Alcohol. 1994;11(6):557–64.
Rodd-Henricks ZA, et al. Regional heterogeneity for the intracranial self-administration of ethanol within the ventral tegmental area of female Wistar rats. Psychopharmacology (Berl). 2000;149(3):217–24.
Corrigall WA, et al. The mesolimbic dopaminergic system is implicated in the reinforcing effects of nicotine. Psychopharmacology (Berl). 1992;107(2–3):285–9.
Corrigall WA, Coen KM, Adamson KL. Self-administered nicotine activates the mesolimbic dopamine system through the ventral tegmental area. Brain Res. 1994;653(1–2):278–84.
Paxinos G, Franklin KBJ. The mouse brain in stereotaxic coordinates. 2nd ed. San Diego: Academic; 2000. p. 296.
Fields HL, et al. Ventral tegmental area neurons in learned appetitive behavior and positive reinforcement. Annu Rev Neurosci. 2007;30:289–316.
Swanson LW. The projections of the ventral tegmental area and adjacent regions: a combined fluorescent retrograde tracer and immunofluorescence study in the rat. Brain Res Bull. 1982;9(1–6):321–53.
Carr DB, Sesack SR. GABA-containing neurons in the rat ventral tegmental area project to the prefrontal cortex. Synapse. 2000;38(2):114–23.
Margolis EB, et al. The ventral tegmental area revisited: is there an electrophysiological marker for dopaminergic neurons? J Physiol. 2006;577(Pt 3):907–24.
Stuber GD, et al. Dopaminergic terminals in the nucleus accumbens but not the dorsal striatum corelease glutamate. J Neurosci. 2010;30(24):8229–33.
Hnasko TS, et al. Vesicular glutamate transport promotes dopamine storage and glutamate corelease in vivo. Neuron. 2010;65(5):643–56.
Geisler S, Zahm DS. Afferents of the ventral tegmental area in the rat-anatomical substratum for integrative functions. J Comp Neurol. 2005;490(3):270–94.
Sesack SR, Pickel VM. Prefrontal cortical efferents in the rat synapse on unlabeled neuronal targets of catecholamine terminals in the nucleus accumbens septi and on dopamine neurons in the ventral tegmental area. J Comp Neurol. 1992;320(2):145–60.
Cornwall J, Cooper JD, Phillipson OT. Afferent and efferent connections of the laterodorsal tegmental nucleus in the rat. Brain Res Bull. 1990;25(2):271–84.
Oakman SA, et al. Distribution of pontomesencephalic cholinergic neurons projecting to substantia nigra differs significantly from those projecting to ventral tegmental area. J Neurosci. 1995;15(9):5859–69.
Semba K, Fibiger HC. Afferent connections of the laterodorsal and the pedunculopontine tegmental nuclei in the rat: a retro- and antero-grade transport and immunohistochemical study. J Comp Neurol. 1992;323(3):387–410.
Herkenham M, Nauta WJ. Efferent connections of the habenular nuclei in the rat. J Comp Neurol. 1979;187(1):19–47.
Ji H, Shepard PD. Lateral habenula stimulation inhibits rat midbrain dopamine neurons through a GABA(A) receptor-mediated mechanism. J Neurosci. 2007;27(26):6923–30.
Matsumoto M, Hikosaka O. Lateral habenula as a source of negative reward signals in dopamine neurons. Nature. 2007;447(7148):1111–5.
Margolis EB, et al. Kappa opioids selectively control dopaminergic neurons projecting to the prefrontal cortex. Proc Natl Acad Sci U S A. 2006;103(8):2938–42.
Fallon JH, et al. Neurons in the ventral tegmentum have separate populations projecting to telencephalon and inferior olive, are histochemically different, and may receive direct visual input. Brain Res. 1984;321(2):332–6.
Ikemoto S, Murphy JM, McBride WJ. Regional differences within the rat ventral tegmental area for muscimol self-infusions. Pharmacol Biochem Behav. 1998;61(1):87–92.
Perrotti LI, et al. DeltaFosB accumulates in a GABAergic cell population in the posterior tail of the ventral tegmental area after psychostimulant treatment. Eur J Neurosci. 2005;21(10):2817–24.
Kaufling J, et al. Afferents to the GABAergic tail of the ventral tegmental area in the rat. J Comp Neurol. 2009;513(6):597–621.
Ikemoto S, Qin M, Liu ZH. Primary reinforcing effects of nicotine are triggered from multiple regions both inside and outside the ventral tegmental area. J Neurosci. 2006;26(3):723–30.
Boehm 2nd SL, et al. Ventral tegmental area region governs GABA(B) receptor modulation of ethanol-stimulated activity in mice. Neuroscience. 2002;115(1):185–200.
Ericson M, et al. Nicotinic acetylcholine receptors in the anterior, but not posterior, ventral tegmental area mediate ethanol-induced elevation of accumbal dopamine levels. J Pharmacol Exp Ther. 2008;326(1):76–82.
Shabat-Simon M, et al. Dissociation between rewarding and psychomotor effects of opiates: differential roles for glutamate receptors within anterior and posterior portions of the ventral tegmental area. J Neurosci. 2008;28(34):8406–16.
Heimer L, et al. Specificity in the projection patterns of accumbal core and shell in the rat. Neuroscience. 1991;41(1):89–125.
Zahm DS. Functional-anatomical implications of the nucleus accumbens core and shell subterritories. Ann N Y Acad Sci. 1999;877:113–28.
Bassareo V, De Luca MA, Di Chiara G. Differential expression of motivational stimulus properties by dopamine in nucleus Accumbens shell versus core and prefrontal cortex. J Neurosci. 2002;22(11):4709–19.
Berlanga ML, et al. Cholinergic interneurons of the nucleus accumbens and dorsal striatum are activated by the self-administration of cocaine. Neuroscience. 2003;120(4):1149–56.
Pecina S, Berridge KC. Opioid site in nucleus accumbens shell mediates eating and hedonic ‘liking’ for food: map based on microinjection Fos plumes. Brain Res. 2000;863(1–2):71–86.
Rodd-Henricks ZA, et al. Cocaine is self-administered into the shell but not the core of the nucleus accumbens of Wistar rats. J Pharmacol Exp Ther. 2002;303(3):1216–26.
Ito R, Robbins TW, Everitt BJ. Differential control over cocaine-seeking behavior by nucleus accumbens core and shell. Nat Neurosci. 2004;7(4):389–97.
Nirenberg MJ, et al. The dopamine transporter: comparative ultrastructure of dopaminergic axons in limbic and motor compartments of the nucleus accumbens. J Neurosci. 1997;17(18):6899–907.
Grace AA, Bunney BS. The control of firing pattern in nigral dopamine neurons: single spike firing. J Neurosci. 1984;4(11):2866–76.
Pan WX, et al. Dopamine cells respond to predicted events during classical conditioning: evidence for eligibility traces in the reward-learning network. J Neurosci. 2005;25(26):6235–42.
Schultz W. Predictive reward signal of dopamine neurons. J Neurophysiol. 1998;80(1):1–27.
Albuquerque EX, et al. Mammalian nicotinic acetylcholine receptors: from structure to function. Physiol Rev. 2009;89(1):73–120.
Tapper AR, Nashmi R, Lester HA. Neuronal nicotinic acetylcholine receptors and nicotine dependence. In: Cell biology of addiction. Cold Spring Harbor: Cold Spring Harbor Laboratory Press; 2006. p. 179–91.
Laviolette SR, van der Kooy D. The neurobiology of nicotine addiction: bridging the gap from molecules to behaviour. Nat Rev Neurosci. 2004;5(1):55–65.
Klink R, et al. Molecular and physiological diversity of nicotinic acetylcholine receptors in the midbrain dopaminergic nuclei. J Neurosci. 2001;21(5):1452–63.
Marubio LM, et al. Effects of nicotine in the dopaminergic system of mice lacking the alpha4 subunit of neuronal nicotinic acetylcholine receptors. Eur J Neurosci. 2003;17(7):1329–37.
Champtiaux N, Changeux JP. Knockout and knock-in mice to investigate the role of nicotinic receptors in the central nervous system. Prog Brain Res. 2004;145:235–51.
Champtiaux N, et al. Subunit composition of functional nicotinic receptors in dopaminergic neurons investigated with knock-out mice. J Neurosci. 2003;23(21):7820–9.
Champtiaux N, et al. Distribution and pharmacology of alpha 6-containing nicotinic acetylcholine receptors analyzed with mutant mice. J Neurosci. 2002;22(4):1208–17.
Wooltorton JR, et al. Differential desensitization and distribution of nicotinic acetylcholine receptor subtypes in midbrain dopamine areas. J Neurosci. 2003;23(8):3176–85.
Drenan RM, et al. In vivo activation of midbrain dopamine neurons via sensitized, high-affinity alpha6* nicotinic acetylcholine receptors. Neuron. 2008;60(1):123–36.
Salminen O, et al. Pharmacology of alpha-conotoxin MII-sensitive subtypes of nicotinic acetylcholine receptors isolated by breeding of null mutant mice. Mol Pharmacol. 2007;71(6):1563–71.
Salminen O, et al. Subunit composition and pharmacology of two classes of striatal presynaptic nicotinic acetylcholine receptors mediating dopamine release in mice. Mol Pharmacol. 2004;65(6):1526–35.
Zhao-Shea R, et al. Nicotine-mediated activation of dopaminergic neurons in distinct regions of the ventral tegmental area. Neuropsychopharmacology. 2011;36(5):1021–32.
Gotti C, et al. Nicotinic acetylcholine receptors in the mesolimbic pathway: primary role of ventral tegmental area alpha6beta2* receptors in mediating systemic nicotine effects on dopamine release, locomotion, and reinforcement. J Neurosci. 2010;30(15):5311–25.
Zhou FM, Liang Y, Dani JA. Endogenous nicotinic cholinergic activity regulates dopamine release in the striatum. Nat Neurosci. 2001;4(12):1224–9.
Zhang H, Sulzer D. Frequency-dependent modulation of dopamine release by nicotine. Nat Neurosci. 2004;7(6):581–2.
Exley R, et al. Distinct contributions of nicotinic acetylcholine receptor subunit {alpha}4 and subunit {alpha}6 to the reinforcing effects of nicotine. Proc Natl Acad Sci U S A. 2011;108(18):7577–82.
Harris DS, Anthenelli RM. Expanding treatment of tobacco dependence. Curr Psychiatry Rep. 2005;7(5):344–51.
Corrigall WA, Coen KM. Selective dopamine antagonists reduce nicotine self-administration. Psychopharmacology (Berl). 1991;104(2):171–6.
Dani JA. Roles of dopamine signaling in nicotine addiction. Mol Psychiatry. 2003;8(3):255–6.
Tang J, Dani JA. Dopamine enables in vivo synaptic plasticity associated with the addictive drug nicotine. Neuron. 2009;63(5):673–82.
Nisell M, Nomikos GG, Svensson TH. Systemic nicotine-induced dopamine release in the rat nucleus accumbens is regulated by nicotinic receptors in the ventral tegmental area. Synapse. 1994;16(1):36–44.
Brunzell DH, et al. Alpha-conotoxin MII-sensitive nicotinic acetylcholine receptors in the nucleus accumbens shell regulate progressive ratio responding maintained by nicotine. Neuropsychopharmacology. 2010;35(3):665–73.
Drenan RM, Lester HA. Insights into the neurobiology of the nicotinic cholinergic system and nicotine addiction from mice expressing nicotinic receptors harboring gain-of-function mutations. Pharmacol Rev. 2012;64(4):869–79.
Champtiaux N, Changeux JP. Knock-out and knock-in mice to investigate the role of nicotinic receptors in the central nervous system. Curr Drug Target CNS Neurol Disord. 2002;1(4):319–30.
Picciotto MR, et al. Acetylcholine receptors containing the beta2 subunit are involved in the reinforcing properties of nicotine. Nature. 1998;391(6663):173–7.
Walters CL, et al. The beta2 but not alpha7 subunit of the nicotinic acetylcholine receptor is required for nicotine-conditioned place preference in mice. Psychopharmacology (Berl). 2006;184(3–4):339–44.
Tapper AR, et al. Nicotine activation of alpha4* receptors: sufficient for reward, tolerance, and sensitization. Science. 2004;306(5698):1029–32.
Cahir E, et al. The necessity of alpha4* nicotinic receptors in nicotine-driven behaviors: dissociation between reinforcing and motor effects of nicotine. Neuropsychopharmacology. 2011;36(7):1505–17.
Maskos U, et al. Nicotine reinforcement and cognition restored by targeted expression of nicotinic receptors. Nature. 2005;436(7047):103–7.
Pons S, et al. Crucial role of alpha4 and alpha6 nicotinic acetylcholine receptor subunits from ventral tegmental area in systemic nicotine self-administration. J Neurosci. 2008;28(47):12318–27.
Gonzales D, et al. Varenicline, an alpha4beta2 nicotinic acetylcholine receptor partial agonist, vs sustained-release bupropion and placebo for smoking cessation: a randomized controlled trial. JAMA. 2006;296(1):47–55.
Rollema H, et al. Pharmacological profile of the alpha4beta2 nicotinic acetylcholine receptor partial agonist varenicline, an effective smoking cessation aid. Neuropharmacology. 2007;52(3):985–94.
Li W, Doyon WM, Dani JA. Acute in vivo nicotine administration enhances synchrony among dopamine neurons. Biochem Pharmacol. 2011;82(8):977–83.
De Biasi M, Dani JA. Reward, addiction, withdrawal to nicotine. Annu Rev Neurosci. 2011;34:105–30.
Tsai HC, et al. Phasic firing in dopaminergic neurons is sufficient for behavioral conditioning. Science. 2009;324(5930):1080–4.
Tan KR, et al. GABA neurons of the VTA drive conditioned place aversion. Neuron. 2012;73(6):1173–83.
Mansvelder HD, et al. Cholinergic modulation of dopaminergic reward areas: upstream and downstream targets of nicotine addiction. Eur J Pharmacol. 2003;480(1–3):117–23.
Mansvelder HD, McGehee DS. Cellular and synaptic mechanisms of nicotine addiction. J Neurobiol. 2002;53(4):606–17.
Fisher JL, Pidoplichko VI, Dani JA. Nicotine modifies the activity of ventral tegmental area dopaminergic neurons and hippocampal GABAergic neurons. J Physiol Paris. 1998;92(3–4):209–13.
Pidoplichko VI, et al. Nicotine activates and desensitizes midbrain dopamine neurons. Nature. 1997;390(6658):401–4.
McGranahan TM, et al. alpha4beta2 nicotinic acetylcholine receptors on dopaminergic neurons mediate nicotine reward and anxiety relief. J Neurosci. 2011;31(30):10891–902.
Nashmi R, et al. Chronic nicotine cell specifically upregulates functional alpha 4* nicotinic receptors: basis for both tolerance in midbrain and enhanced long-term potentiation in perforant path. J Neurosci. 2007;27(31):8202–18.
Mansvelder HD, Keath JR, McGehee DS. Synaptic mechanisms underlie nicotine-induced excitability of brain reward areas. Neuron. 2002;33(6):905–19.
Kenny PJ, et al. NMDA receptors regulate nicotine-enhanced brain reward function and intravenous nicotine self-administration: role of the ventral tegmental area and central nucleus of the amygdala. Neuropsychopharmacology. 2009;34(2):266–81.
Tolu S, et al. Co-activation of VTA DA and GABA neurons mediates nicotine reinforcement. Mol Psychiatry. 2013;18(3):382–93.
Liu L, et al. Nicotine persistently activates ventral tegmental area dopaminergic neurons via nicotinic acetylcholine receptors containing alpha4 and alpha6 subunits. Mol Pharmacol. 2012;81(4):541–8.
Engle SE, et al. alpha4alpha6beta2* nAChR activation on VTA DA neurons is sufficient to stimulate a depolarizing conductance and enhance surface AMPA receptor function. Mol Pharmacol. 2013;84(3):393–406.
W.H.O. Global status report on alcohol. Geneva: World Health Organization; 2011.
Samson HH, et al. Alcohol self-administration: role of mesolimbic dopamine. Ann N Y Acad Sci. 1992;654:242–53.
Diana M, Rossetti ZL, Gessa G. Rewarding and aversive effects of ethanol: interplay of GABA, glutamate and dopamine. Alcohol Alcohol Suppl. 1993;2:315–9.
Lanca AJ. Reduction of voluntary alcohol intake in the rat by modulation of the dopaminergic mesolimbic system: transplantation of ventral mesencephalic cell suspensions. Neuroscience. 1994;58(2):359–69.
Lewis MJ, June HL. Neurobehavioral studies of ethanol reward and activation. Alcohol. 1990;7(3):213–9.
Weiss F, et al. Oral alcohol self-administration stimulates dopamine release in the rat nucleus accumbens: genetic and motivational determinants. J Pharmacol Exp Ther. 1993;267(1):250–8.
Rassnick S, Stinus L, Koob GF. The effects of 6-hydroxydopamine lesions of the nucleus accumbens and the mesolimbic dopamine system on oral self-administration of ethanol in the rat. Brain Res. 1993;623(1):16–24.
Ikemoto S, et al. 6-OHDA-lesions of the nucleus accumbens disrupt the acquisition but not the maintenance of ethanol consumption in the alcohol-preferring P line of rats. Alcohol Clin Exp Res. 1997;21(6):1042–6.
Kiianmaa K. Decreased intoxicating effect of ethanol in rats after 6-hydroxydopamine-induced degeneration of ascending dopamine pathways. Pharmacol Biochem Behav. 1978;9(3):391–3.
Koob GF, Weiss F. Pharmacology of drug self-administration. Alcohol. 1990;7(3):193–7.
Okamoto T, Harnett MT, Morikawa H. Hyperpolarization-activated cation current (Ih) is an ethanol target in midbrain dopamine neurons of mice. J Neurophysiol. 2006;95(2):619–26.
Rodd ZA, et al. Serotonin-3 receptors in the posterior ventral tegmental area regulate ethanol self-administration of alcohol-preferring (P) rats. Alcohol. 2010;44(3):245–55.
Xiao C, Ye JH. Ethanol dually modulates GABAergic synaptic transmission onto dopaminergic neurons in ventral tegmental area: role of mu-opioid receptors. Neuroscience. 2008;153(1):240–8.
Xiao C, et al. Ethanol facilitates glutamatergic transmission to dopamine neurons in the ventral tegmental area. Neuropsychopharmacology. 2009;34(2):307–18.
Guan Y, et al. GABAergic actions mediate opposite ethanol effects on dopaminergic neurons in the anterior and posterior ventral tegmental area. J Pharmacol Exp Ther. 2012;341(1):33–42.
Theile JW, et al. GABAergic transmission modulates ethanol excitation of ventral tegmental area dopamine neurons. Neuroscience. 2011;172:94–103.
Job MO, et al. Mu (mu) opioid receptor regulation of ethanol-induced dopamine response in the ventral striatum: evidence of genotype specific sexual dimorphic epistasis. Biol Psychiatry. 2007;62(6):627–34.
Soderpalm B, et al. Nicotinic mechanisms involved in the dopamine activating and reinforcing properties of ethanol. Behav Brain Res. 2000;113(1–2):85–96.
Blomqvist O, et al. The mesolimbic dopamine-activating properties of ethanol are antagonized by mecamylamine. Eur J Pharmacol. 1993;249(2):207–13.
Blomqvist O, et al. Voluntary ethanol intake in the rat: effects of nicotinic acetylcholine receptor blockade or subchronic nicotine treatment. Eur J Pharmacol. 1996;314(3):257–67.
Blomqvist O, Soderpalm B, Engel JA. Ethanol-induced locomotor activity: involvement of central nicotinic acetylcholine receptors? Brain Res Bull. 1992;29(2):173–8.
Larsson A, et al. Role of different nicotinic acetylcholine receptors in mediating behavioral and neurochemical effects of ethanol in mice. Alcohol. 2002;28(3):157–67.
Le AD, et al. Involvement of nicotinic receptors in alcohol self-administration. Alcohol Clin Exp Res. 2000;24(2):155–63.
Ericson M, et al. Ethanol elevates accumbal dopamine levels via indirect activation of ventral tegmental nicotinic acetylcholine receptors. Eur J Pharmacol. 2003;467(1–3):85–93.
Blomqvist O, et al. Accumbal dopamine overflow after ethanol: localization of the antagonizing effect of mecamylamine. Eur J Pharmacol. 1997;334(2–3):149–56.
Chi H, de Wit H. Mecamylamine attenuates the subjective stimulant-like effects of alcohol in social drinkers. Alcohol Clin Exp Res. 2003;27(5):780–6.
Forman SA, Zhou Q. Nicotinic receptor pore mutations create a sensitive inhibitory site for ethanol. Alcohol Clin Exp Res. 2000;24(9):1363–8.
Zhou QL, Zhou Q, Forman SA. The n-alcohol site in the nicotinic receptor pore is a hydrophobic patch. Biochemistry. 2000;39(48):14920–6.
Zuo Y, et al. Alcohol modulation of neuronal nicotinic acetylcholine receptors is alpha subunit dependent. Alcohol Clin Exp Res. 2002;26(6):779–84.
Liu L, et al. Nicotinic acetylcholine receptors containing the alpha4 subunit modulate alcohol reward. Biol Psychiatry. 2013;73(8):738–46.
Liu L, et al. Nicotinic acetylcholine receptors containing the alpha6 subunit contribute to ethanol activation of ventral tegmental area dopaminergic neurons. Biochem Pharmacol. 2013;86(8):1194–200.
Ericson M, et al. Voluntary ethanol intake in the rat and the associated accumbal dopamine overflow are blocked by ventral tegmental mecamylamine. Eur J Pharmacol. 1998;358(3):189–96.
Hendrickson LM, Zhao-Shea R, Tapper AR. Modulation of ethanol drinking-in-the-dark by mecamylamine and nicotinic acetylcholine receptor agonists in C57BL/6J mice. Psychopharmacology (Berl). 2009;204(4):563–72.
Harvey SC, Luetje CW. Determinants of competitive antagonist sensitivity on neuronal nicotinic receptor beta subunits. J Neurosci. 1996;16(12):3798–806.
Harvey SC, Maddox FN, Luetje CW. Multiple determinants of dihydro-beta-erythroidine sensitivity on rat neuronal nicotinic receptor alpha subunits. J Neurochem. 1996;67(5):1953–9.
Kamens HM, Phillips TJ. A role for neuronal nicotinic acetylcholine receptors in ethanol-induced stimulation, but not cocaine- or methamphetamine-induced stimulation. Psychopharmacology (Berl). 2008;196(3):377–87.
Lof E, et al. Nicotinic acetylcholine receptors in the ventral tegmental area mediate the dopamine activating and reinforcing properties of ethanol cues. Psychopharmacology (Berl). 2007;195(3):333–43.
Moroni M, et al. alpha4beta2 nicotinic receptors with high and low acetylcholine sensitivity: pharmacology, stoichiometry, and sensitivity to long-term exposure to nicotine. Mol Pharmacol. 2006;70(2):755–68.
Kuzmin A, et al. Effects of subunit selective nACh receptors on operant ethanol self-administration and relapse-like ethanol-drinking behavior. Psychopharmacology (Berl). 2009;203(1):99–108.
Jerlhag E, et al. Role of the subunit composition of central nicotinic acetylcholine receptors for the stimulatory and dopamine-enhancing effects of ethanol. Alcohol Alcohol. 2006;41(5):486–93.
Larsson A, et al. Is an alpha-conotoxin MII-sensitive mechanism involved in the neurochemical, stimulatory, and rewarding effects of ethanol? Alcohol. 2004;34(2–3):239–50.
Grady SR, et al. The subtypes of nicotinic acetylcholine receptors on dopaminergic terminals of mouse striatum. Biochem Pharmacol. 2007;74(8):1235–46.
Coe JW, et al. Varenicline: an alpha4beta2 nicotinic receptor partial agonist for smoking cessation. J Med Chem. 2005;48(10):3474–7.
Steensland P, et al. Varenicline, an alpha4beta2 nicotinic acetylcholine receptor partial agonist, selectively decreases ethanol consumption and seeking. Proc Natl Acad Sci U S A. 2007;104(30):12518–23.
Tonstad S, et al. Effect of maintenance therapy with varenicline on smoking cessation: a randomized controlled trial. JAMA. 2006;296(1):64–71.
Kamens HM, Andersen J, Picciotto MR. Modulation of ethanol consumption by genetic and pharmacological manipulation of nicotinic acetylcholine receptors in mice. Psychopharmacology (Berl). 2010;208(4):613–26.
Mihalak KB, Carroll FI, Luetje CW. Varenicline is a partial agonist at alpha4beta2 and a full agonist at alpha7 neuronal nicotinic receptors. Mol Pharmacol. 2006;70(3):801–5.
Lummis SC, et al. Varenicline is a potent agonist of the human 5-hydroxytryptamine3 receptor. J Pharmacol Exp Ther. 2011;339(1):125–31.
Bordia T, et al. Varenicline is a potent partial agonist at alpha6beta2* nicotinic acetylcholine receptors in rat and monkey striatum. J Pharmacol Exp Ther. 2012;342(2):327–34.
Papke RL, Wecker L, Stitzel JA. Activation and inhibition of mouse muscle and neuronal nicotinic acetylcholine receptors expressed in Xenopus oocytes. J Pharmacol Exp Ther. 2010;333(2):501–18.
Rezvani AH, et al. Effects of sazetidine-A, a selective alpha4beta2 nicotinic acetylcholine receptor desensitizing agent on alcohol and nicotine self-administration in selectively bred alcohol-preferring (P) rats. Psychopharmacology (Berl). 2010;211(2):161–74.
Sajja RK, Rahman S. Neuronal nicotinic receptor ligands modulate chronic nicotine-induced ethanol consumption in C57BL/6J mice. Pharmacol Biochem Behav. 2012;102(1):36–43.
Sajja RK, Rahman S. Lobeline and cytisine reduce voluntary ethanol drinking behavior in male C57BL/6J mice. Prog Neuropsychopharmacol Biol Psychiatry. 2011;35(1):257–64.
Bell RL, et al. Nicotinic receptor ligands reduce ethanol intake by high alcohol-drinking HAD-2 rats. Alcohol. 2009;43(8):581–92.
Chatterjee S, et al. Partial agonists of the alpha3beta4* neuronal nicotinic acetylcholine receptor reduce ethanol consumption and seeking in rats. Neuropsychopharmacology. 2011;36(3):603–15.
Carnicella S, et al. Noribogaine, but not 18-MC, exhibits similar actions as ibogaine on GDNF expression and ethanol self-administration. Addict Biol. 2010;15(4):424–33.
Kamens HM, et al. The alpha6 nicotinic acetylcholine receptor subunit influences ethanol-induced sedation. Alcohol. 2012;46(5):463–71.
Dawson A, Miles MF, Damaj MI. The beta2 nicotinic acetylcholine receptor subunit differentially influences ethanol behavioral effects in the mouse. Alcohol. 2013;47(2):85–94.
Powers MS, et al. Nicotinic acetylcholine receptors containing alpha6 subunits contribute to alcohol reward-related behaviors. Genes Brain Behav. 2013;12(5):543–53.
Williams JM, Galli A. The dopamine transporter: a vigilant border control for psychostimulant action. Handb Exp Pharmacol. 2006;175:215–32.
Horger BA, Giles MK, Schenk S. Preexposure to amphetamine and nicotine predisposes rats to self-administer a low dose of cocaine. Psychopharmacology (Berl). 1992;107(2–3):271–6.
Zachariou V, et al. Nicotine receptor inactivation decreases sensitivity to cocaine. Neuropsychopharmacology. 2001;24(5):576–89.
Hansen ST, Mark GP. The nicotinic acetylcholine receptor antagonist mecamylamine prevents escalation of cocaine self-administration in rats with extended daily access. Psychopharmacology (Berl). 2007;194(1):53–61.
Levin ED, et al. The nicotinic antagonist mecamylamine preferentially inhibits cocaine vs. food self-administration in rats. Physiol Behav. 2000;71(5):565–70.
Witten IB, et al. Cholinergic interneurons control local circuit activity and cocaine conditioning. Science. 2010;330(6011):1677–81.
Zanetti L, Picciotto MR, Zoli M. Differential effects of nicotinic antagonists perfused into the nucleus accumbens or the ventral tegmental area on cocaine-induced dopamine release in the nucleus accumbens of mice. Psychopharmacology (Berl). 2007;190(2):189–99.
Kreek MJ, et al. Opiate and cocaine addiction: from bench to clinic and back to the bench. Curr Opin Pharmacol. 2009;9(1):74–80.
Rezayof A, et al. Morphine-induced place preference: involvement of cholinergic receptors of the ventral tegmental area. Eur J Pharmacol. 2007;562(1–2):92–102.
Feng B, et al. Blocking alpha4beta2 and alpha7 nicotinic acetylcholine receptors inhibits the reinstatement of morphine-induced CPP by drug priming in mice. Behav Brain Res. 2011;220(1):100–5.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2014 Springer Science+Business Media New York
About this chapter
Cite this chapter
Hendrickson, L.M., Tapper, A.R. (2014). Neuronal Nicotinic Acetylcholine Receptors in Reward and Addiction. In: Lester, R. (eds) Nicotinic Receptors. The Receptors, vol 26. Humana Press, New York, NY. https://doi.org/10.1007/978-1-4939-1167-7_15
Download citation
DOI: https://doi.org/10.1007/978-1-4939-1167-7_15
Published:
Publisher Name: Humana Press, New York, NY
Print ISBN: 978-1-4939-1166-0
Online ISBN: 978-1-4939-1167-7
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)