
Abstract
Strong association between alcohol abuse and an increase in both systemic and brain levels is evident from both humans and animal models. This chapter comprehensively reviews the interplay between progression of neuroinflammation and neurological disorders initiated by alcohol abuse. Recent observations of excitotoxicity associated with excessive neurotransmitter release, oxidative stress leading to free radical damage, and cell death through an enhanced inflammatory response provide important clues to the mechanisms that could mediate alcohol’s toxic effects on brain cells. Chronic alcoholics have the temporal hallmark of neurocognitive deficits, neuronal injury, and neurodegeneration. Studies suggest that the initiation and progression of alcohol-mediated neurodegeneration is driven in part by release of pro-inflammatory factors from activated microglia, oxidative stress, impairment of blood–brain barrier (BBB), and glutamate-associated neurotoxicity. Recent observation of strong associations between cannabinoid systems within the central nervous system in regulating neuroinflammation via the cannabinoid receptor 2 highlights the importance of this pathway in alcohol-driven neuroinflammation. Regulatory mechanisms that regulate alcohol-induced neuroinflammation, oxidative neuronal injury, and altered BBB are examined, as well as modalities to ameliorate these processes are discussed.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others


Keywords
Alcohol abuse continues to be a major morbidity factor, causing approximately 1.3 million deaths globally (3.2 % of all deaths) each year and accounts for approximately 4 % of the disease burden of all diseases [1]. While the effects of alcohol exposure on liver function and end-organ injury are well accepted, the significant association between neurodegeneration and alcohol exposure is less established. New intriguing data recently were acquired to suggest a relationship between chronic inflammatory responses as underlying causes for alcohol-associated neurodegeneration as well as elements of alcohol addiction [2]. Since alcohol dependence and abuse are important health problems, therapeutic strategies to overcome this addiction are urgently required to reduce the burden of such conditions on society.
Newer neuroimaging techniques have shown significant alteration of brain structure, including atrophy of subcortical and cortical areas, thalamus, corpus callosum, and cerebellum [3]. Substantial progress has been made in brain imaging in chronic alcoholics, indicating global reduction in gray matter and white matter and an increase in cerebral spinal fluid volume and diffusional abnormalities among alcoholics and heavy to moderate drinkers [4]. Chronic alcohol consumption has been shown to be related to shrinkage of different parts of the brain and impairment of the decision-making process. When compared to normal controls, two predictors (gray matter changes and decision-making measure) were significantly altered in alcoholics. Recent studies address the issue whether amelioration takes place during 2 weeks of abstinence from alcohol [5]. This study indicated gray and white matter recovery after few days of abstinence, but it varied between different brain regions. These findings offer a unique insight into potential therapeutic interventions, promoting structural changes in the CNS of alcoholics. The changes were attributed to a recovery of myelin in the corpus callosum [6].
According to Zahr et al. [3], brain tissue loss consists of two components, transient and permanent. It has been suggested that in tissue shrinkage secondary to neuronal loss, there is no complete brain tissue recovery. Magnetic resonance spectroscopy revealed that despite prolonged abstinence, individuals that chronically consume alcohol demonstrate persistent diminution of N-acetylaspartate (a neuronal marker) in the frontal lobe, thalamus, and cerebellum [7]. Other studies demonstrated improvement in the level of N-acetylaspartate and choline (a metabolite associated with re-myelination) during abstinence [8].
Corresponding neuropathology studies indicated a loss and destruction of white matter in the same brain regions. Structural changes in the brain and the associated functional consequences that occur with chronic alcohol exposure can be grouped into “uncomplicated alcohol-related brain damage.” [3] Neuropsychological manifestations of chronic uncomplicated alcohol exposure are characterized by the heterogeneity of severity and type of deficits. It has been shown that the pericerebral space with respect to intracranial cavity changed from 8.3 % of total intracranial volume in healthy controls to 11.3 % in patients with chronic alcohol exposure [9]. Previous stereologic studies indicated that this reduction occurred mainly due to decreased white matter volume. The morphologic substrate of white matter loss is currently unknown; however, this phenomenon is probably associated with a loss of myelin and axonal integrity. Common alcohol-associated CNS lesions encompass white matter loss (leukoencephalopathy), enlarged ventricles, cerebellar degeneration, and neuronal demise in the superior frontal association cortex, anterior cingulate area, hippocampus, entorhinal cortex, and hypothalamus, which contribute to cognitive and motor deficits [10, 11].
In patients with uncomplicated alcoholism, neuropathology studies reveal up to 25 % loss of pyramidal neurons in the superior frontal cortex [3]. Much is unknown about neuronal loss in the primary motor cortex in uncomplicated alcoholism; however, silver impregnation techniques showed that neurons in the superior frontal and motor cortex featured dendritic arbor shrinkage, indicating a compromise of interneuronal communications. No changes were detected in the number of neurons in the basal ganglia, hippocampus, or serotonergic raphe nuclei in uncomplicated alcoholism.
In recent years, genomic and proteomic analysis of samples of human frontal cortex identified several groups of alcohol-associated genes encoding myelination, synaptic structure, mitochondria, signal transduction and intracellular metabolism, protein trafficking, and transcriptional regulation [12–14]. The data acquired in these studies point to the involvement of multiple pathways in the effects of alcohol on the CNS. Changes in expression of proteolipid protein and myelin basic protein (participating in stabilization of the myelin sheath) could provide additional insights into white matter changes in chronic alcoholics. It has been suggested that chronic liver injury occurring in chronic alcoholism and its associated hepatitis results in production of toxic substances (such as ceramides, ammonia) and enhanced insulin resistance, promoting neurodegeneration [15].
A number of molecular mechanisms have been proposed for ethanol-associated brain injury. These encompass alcohol-specific effects, including toxic metabolites (production of acetaldehyde and fatty acid ethyl esters), defects in mitochondrial function, generation of reactive oxygen species, decrease in brain-derived neurotrophic factors, and effects on excessive glutamate on synaptic transmission (resulting in excitotoxicity) [3]. Increased gut permeability for bacterial byproducts, such as LPS (so-called bacterial translocation), and liver dysfunction can be additional factors leading to brain injury in chronic alcoholism [16]. The latter is of significant interest, providing links between alcohol exposure and development of systemic inflammatory responses.
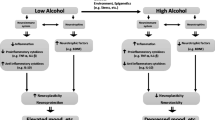
A significant body of evidence indicates that there is a close association between alcohol abuse and an increase in both systemic and brain levels of inflammation. The levels of cytokines in systemic circulation were increased in alcoholic subjects [17–19]. It has been shown that chronic alcohol consumption in humans is associated with increases in serum pro-inflammatory cytokines [20, 21]; monocytes isolated from the blood of alcoholics produce greater amounts of tumor necrosis factor α (TNFα) spontaneously and in response to endotoxin [22]. In experimental animals (rats), several months of alcohol administration increased the number of inflammatory factors [interleukin (IL-1β), inducible nitric oxide synthase (iNOS), cyclooxygenase 2 (COX-2)] [23]. Recent publications from the group of Dr. Crews suggested significant upregulation of pro-inflammatory genes in the brains of alcohol-exposed animals with secondary inflammatory stimulus (such as LPS administration) [19]. They found upregulation of the β-chemokine, CCL2, a key innate immune factor, in multiple regions of postmortem alcoholic brains as compared to age-matched controls. These changes are accompanied by profound microglial activation in different brain regions [16, 17]. An additional indication of inflammatory response was the upregulated expression of cannabinoid receptor 2 in the brain endothelium, changes seen in human brain tissues affected by encephalitis [16]. Chronic alcohol administration to mice caused a sustained increase in brain CCL2 and an increase in microglial activation paralleling human studies.
Studies indicating that genes involved in the immune stress response exhibited differential expression in the frontal cortex of human alcoholics and normal controls. It has been shown that expression of the same genes is also related to genetic predisposition of alcohol consumption in mouse strains, pointing to a role for pro-inflammatory mediators in the regulation of alcohol intake [24]. Gene expression data sets led to the selection of six genes related to inflammation, and behavioral testing of gene-deficient mouse strains showed that animals defective in such genes drink less alcohol. Conversely, injection of LPS prior to alcohol testing resulted in a sustained increase in alcohol consumption. It has been shown that one of the key inflammatory pathways, toll-like receptor (TLR) 4, is critical in enhanced neuroinflammation associated with alcohol exposure. Several labs found that alcohol can activate TLR4 receptors in the brain in different types of glial cells [25]. Furthermore, alcohol-exposed animals lacking these receptors showed evidence of protection from astrocyte and microglia activation, increased expression of pro-inflammatory factors, and neuronal injury. These animals did not show long-lasting memory impairment or anxiety-like behavior. These studies collectively point to the role of neuroinflammation, not only in neurodegeneration associated with alcohol exposure, but also to promotion of addictive behavior. Development of therapeutics diminishing inflammation, therefore, may protect the CNS from alcohol injury and mitigate addiction.
1 How Microglia Are Affected by Alcohol and Implications for Neurodegeneration
Maintaining the integrity of neurons and neuronal circuits in the CNS is imperative for proper signaling and communication. Neurodegeneration is characterized by the loss of structure or function of brain cells and comprises assembly of pathophysiological events that include cellular damage, disease development, and cellular death [26]. Glial cells, including astrocytes, microglia, and oligodendrocytes, are important to neuronal function as both supporting cells for proper neural circuit transmission and an alert mechanism during injury or infection. Over the years, glial cells have been implicated in modulating and influencing neuronal health. Microglia, the immunocompetent cells of the CNS, respond to any homeostatic modification and play a pivotal role in regulation of neuroinflammatory processes, including neurodegenerative disease [27]. Although they are historically considered to be quiescent in the healthy brain and active only during brain injury or disease, recent literature has demonstrated microglia as a dynamic entity undergoing morphologic changes to maintain healthy brain function. As the primary effector cells of the brain, microglia are indispensable for maintenance and clearance of foreign material and debris [28]. Microglia activation is a highly regulated process and is important in all aspects including proliferation, cell cycle, migration, and apoptosis during the response to inflammation, ischemia, trauma, injury, and neurodegenerative diseases [28]. Importantly, microglia can cause both neuroprotection and neurodegeneration in the CNS. Most of the known functions of microglia, including neurotoxic and neuroprotective properties, are attributed to morphologically activated microglia [29].
Microglial cell dysfunction plays an important role in pathogenesis of many neurodegenerative and neuroinflammatory diseases [30]. Constant alterations in the CNS lead to dynamic changes in microglial activation, contributing to inflammation and consequently leading to neurodegenerative outcomes. Prolonged microglial cell activation due to aberrant signaling alters the function of microglia in neurocognitive disorders, including Alzheimer’s disease and Parkinson’s disease [31]. Alcohol abuse and alcoholism represent substantial problems that affect a large portion of the general public [32]. Alcohol is a common dietary constituent that modulates the immune system [17]. It has been reported that the number of risk factors associated with chronic alcohol abuse has significantly increased, especially in disorders of the CNS [33]. Recent studies have enumerated the deleterious effects of alcohol on the brain, including the immune cells found within the CNS [32, 34–37]. Alcohol attenuates phagocytosis [34, 38, 39], proliferation [35], expression of brain-derived neurotrophic factor (BDNF) in the hippocampus [40], and apoptotic action in microglia [41]. Alcohol consumption has an impact on the structure and function of the brain [3].
In the mature brain, resting microglia exhibit a ramified morphology and are responsible for immune surveillance [42]. The classical phenotype of activated microglia is very important in the clearance of pathogens, as well as the release of soluble factors that act as signaling molecules to combat injury and infection [27]. When microglia are activated due to brain injury or immunological stimuli, they undergo dramatic morphologic alterations, from a ramified cell with a small soma and long processes to an activated amoeboid cell with a large soma and shorter processes. Microglia activation can be differentiated based on morphology, marker expression, and cytokine secretion [43]. Microglia phenotype varies with the type of insult, the extent of damage, and the time of recovery post-injury. Addressing the effects of alcohol on microglia-mediated neurodegeneration is important to understand their role in neuroinflammation. In the healthy brain, microglial cells are highly sensitive to changes in their microenvironment and readily become activated in response to infection or injury. Activated microglia upregulate a variety of surface receptors, including the major histocompatibility complex (MHC-II), CD11b, Iba-1, and 18 kDa mitochondrial translocator protein [43]. Expression of these surface molecules is one component of microglial cell activation associated with release of factors that are important for signaling in the brain during injury, including the release of pro-inflammatory cytokines.
Pro-inflammatory cytokine release is a hallmark of activated microglia and contributes to chronic neuroinflammation [17]. Studies by many groups have shown a significant increase in the release of these factors in alcohol models, both in vitro and in vivo, and excessive quantities of these individual factors produced by activated microglia can be deleterious to neurons [17, 44, 45]. Alcohol-related studies on microglia conclude that these surface receptors are upregulated in alcohol-treated brains as compared to normal non-treated brains [43]. CCL2, a member of the β-chemokine family that signals through G protein-coupled receptor CCR2, is important for neuroinflammation pathways in microglia. Crews et al. has demonstrated that the CCL2 signal is increased in the human alcoholic brain; this can potentially play an important role in driving microglia activation and therefore indirectly lead to excessive production of pro-inflammatory cytokines such as IL-1β and TNFα [17]. Lastly, CCL2 can act as a “priming” stimulus for microglia (lowering their “threshold sensitivity”), enhancing their synthesis of pro-inflammatory cytokines in response to subsequent stimulation [43, 46]. From these data, we can speculate that alcohol can dramatically affect the release of CCL2 in microglia, potentially orchestrate the release of cytokines, and generate a prolonged pro-inflammatory immune response.
While several groups have demonstrated neuroinflammation in microglial cell reaction to blood–brain barrier (BBB) injury [47], Marshall and colleagues [43] suggested that activated microglia produced anti-inflammatory cytokines (such as IL-10 and TGFβ) and were not fully activated in the binge drinking model (7 days of alcohol exposure). They were unable to demonstrate increased permeability of the BBB in their in vivo model. These investigators proposed that partial activation of microglia following binge ethanol exposure suggests that microglia have homeostatic roles, rather than directly contributing to neurodegeneration, and are a consequence of alcohol-induced damage, rather than the source of the damage.
Specialized functions such as migration and phagocytosis are the main characterizing features of microglia, and the integrity of neurons in the CNS depends on their proper function. Alcohol alters microglial cell migration and phagocytosis [34, 48]; however, the exact mechanisms remain elusive. Likewise, classically activated microglia show increased phagocytic activity as seen in postmortem alcoholic brains stained for ED-1, classically used to detect phagocytic microglia [49]. The level of activation achieved and cytokines released influences whether microglia exacerbate injury or promote recovery. The effects of alcohol on microglia are poorly understood; however, there are two potential mechanisms by which alcohol may influence microglial neurodegeneration: toll-like receptors (TLRs) and purinergic receptors.
TLRs are a family of pattern-recognition receptors that enable the recognition of conserved structural motifs in a wide array of pathogens. TLRs recognize components released from stressed or damaged host cells including ATP, aggregated β-amyloid, and heat shock proteins [42]. Activation of TLRs triggers the downstream stimulation of nuclear factor-kB (NF-kB) encoding molecules associated with inflammation [50]. Multiple studies have shown that alcohol induces microglial cell activation in vitro by stimulating TLR4 enhancing phagocytosis and leading to neuronal death, indicating that activation of the TLR4 response by alcohol can be an important mechanism of alcohol-induced neuroinflammation and neurodegeneration [51].
Purinergic receptors (P2R), also known as purinoceptors, play a unique role in integrating neuronal and glial cellular circuits, as virtually every type of glial cell possesses receptors for purines and pyrimidines [52]. These receptors are ubiquitously expressed and mediate a remarkable variety of physiological and pathophysiological reactions [52–56]. Several signaling pathways are coupled to P2R in the CNS, including the MAPK/ERK pathway, NGF expression, and calcium mobilization [40, 53, 57–61]. P2R have been implicated in alcohol abuse disorders and shown to affect signaling in the CNS [62, 63]. Recent literature has shown the involvement of P2R in alcohol’s action in microglia [63]. It is now generally accepted that microglia contribute to the neurodegenerative process through the release of a variety of neurotoxic factors that exacerbate the degeneration of neurons. It remains to be determined, however, how alcohol triggers microglial activation and if P2R have a role in regulating microglial activity.
Microglia, the CNS representatives of macrophages, partake in neuroinflammation in response to various intrinsic or extrinsic stimuli. It has been recently suggested that microglial signal transduction is one of the main targets of alcohol action in the brain: alcohol exposure selectively modulates intracellular signal transduction in microglia rather than globally inhibiting signaling pathways in a nonspecific manner. Deregulation of the inflammatory activation signaling of microglia by alcohol may contribute to the derangement of CNS immune and inflammatory responses [36]. Inflammation is a common denominator among the diverse list of neurodegenerative diseases. Previously, inflammation was considered to be a passive response to neuronal damage; however, an increasing number of reports demonstrate that prolonged inflammation in the CNS contributes to neuronal death. The importance of microglia, as inflammatory mediators of neurodegeneration, and their mechanisms require further study.
An ongoing controversy exists regarding whether microglia are neuroprotective or neurotoxic when activated. In their resting state, microglia provide “checks and balances” and safeguard mechanisms in the CNS, ensuring that the brain functions properly. Likewise, if a pathogen has breached the CNS or an injury has occurred, microglia assume a more central role in releasing pro-inflammatory cytokines and chemokines to combat the damage. Alcohol has been shown to play a role in modulating the activation of microglia and affecting their normal function, which may be potentially harmful in neuronal death. The exact mechanisms by which alcohol influences microglial cell activation are currently unknown; however, recent studies have shown evidence to support alcohol’s effect on microglia-mediated neurodegeneration. Understanding the balance between neuroprotection and neurodegeneration is important in understanding the diseases of the CNS. Microglia possess a myriad of functions within the CNS, and emergence of their role in health and disease has become of interest in studying neurodegenerative diseases. Rather than classifying microglia as exclusively beneficial or deleterious, it is more likely that microglia function in both roles.
2 Alcohol Effects on the BBB
While a substantial amount of data has been acquired regarding the role of glia in alcohol-driven neurodegeneration, only recently it has become obvious that BBB compromise could be part of this process. Alcohol exposure (25–50 mM) of human brain endothelial cells results in a rapid (20–30 min) decrease of BBB tightness (measured by transendothelial electrical resistance and permeability to tracers of different molecular weights), formation of small gaps in monolayers, and redistribution of tight junction (TJ) proteins [64]. These effects are secondary to oxidative stress due to alcohol metabolism via induction of catalytic activity and expression of alcohol-metabolizing enzymes (CYP2E1 and alcohol dehydrogenase), which parallel enhanced generation of reactive oxygen species (ROS) in BMVEC. These changes lead to Ca2+ release (via stimulation of inositol 1,4,5-triphosphate receptor), activation of myosin light chain (MLC) kinase, and phosphorylation of MLC and TJ proteins [64–66]. These effects are reversible after alcohol withdrawal or inhibition of specific intracellular pathways. In addition, BBB compromise enhances migration of mononuclear cells across endothelial monolayers in vitro.
Longer periods of exposure (24–48 h) to alcohol stimulated the activity/expression of matrix metalloproteinases (MMP-1, MMP-2, and MMP-9) and decreased the levels of tissue inhibitors of MMPs (TIMP-1, TIMP-2) via activation of protein tyrosine kinase, modifications of TJ proteins, and disruption of basement membrane integrity [67, 68]. All these effects could be reproduced by exposure to acetaldehyde or donors of oxidative stress, indicating the importance of such effects by products of alcohol metabolism in the effects of alcohol on the BBB. Our more recent work indicated a compromise of antioxidative protective mechanisms in BMVEC exposed to alcohol and suggested protective approaches for the BBB. We studied whether stabilization of antioxidant enzyme activity would prevent ROS generation that results in barrier disruption. We determined the effects of alcohol on the kinetic profile of superoxide dismutase (SOD), catalase activity, and ROS/nitric oxide (NO) generation in primary human brain endothelial cells. Alcohol simultaneously augmented ROS generation and the activity of the antioxidative enzymes, SOD and catalase. SOD activity was increased for a much longer period of time than was catalase activity [69]. A decline in SOD activity and protein levels preceded elevation of oxidant levels. SOD stabilization by the antioxidant and mitochondria-protecting agent, N-acetyl-l-carnitine (ALC), and the anti-inflammatory agent, rosiglitazone, suppressed ROS levels. Mitochondrial membrane protein damage and decrease in membrane potential after alcohol exposure indicated mitochondrial injury. These changes were prevented by ALC. Importantly, a rapid increase in permeability can be demonstrated in animal models (mice, rabbits) exposed to pathophysiologically relevant doses of alcohol (1–2 h). In addition, alcohol promotes the pro-inflammatory phenotype in the brain endothelium: upregulation of COX-2, increased generation of prostaglandin E2, and enhanced expression of cannabinoid 2 receptor (unpublished observations).
In addition to structural tightness, alcohol exposure (50 mM) decreased glucose uptake and correlated with the reduction of glucose transporter protein 1 (GLUT1) in BMVEC [70]. In vivo, chronic alcohol intake inhibited the transport of glucose into the frontal and occipital regions of the brain. These changes paralleled a marked decrease in GLUT1 protein expression in the BBB. In parallel, alcohol intake impaired BBB TJ proteins in the brain microvessels and enhanced permeability (measured by sodium fluorescein and Evans blue accumulation in brain tissue), thus confirming the leakiness of the BBB. The antioxidant, ALC, attenuated these effects of alcohol on glucose uptake and BBB. Such changes occurring on a repeated basis after exposure to alcohol could be one of the underlying mechanisms of neurodegeneration that warrants further investigation as a potential target for therapeutic interventions.
3 Neuronal Injury and Astrocyte Dysfunction Caused by Alcohol Exposure
Alcohol abuse-related neuronal injury and dysfunction are associated with increases in oxidative stress in the brain that coincide with the induction of pro-inflammatory cytokines and oxidative enzymes. We found that the metabolism of alcohol in primary human neurons by alcohol dehydrogenase (ADH) and cytochrome P450 2E1 generated ROS. In addition, alcohol metabolites further augment ROS/NO levels via induction of NADPH/xanthine oxidase (NOX/XOX) and nitric oxide synthase (NOS) in human neurons [71]. A marked increase in lipid peroxidation and a decrease in a neuronal-specific marker paralleled ROS generation. Increase in iNOS protein correlated with an upregulation of 3-nitrotyrosine protein levels in the frontal cortex of alcohol-fed mice [72]. Colocalization of neurofilaments and iNOS protein confirmed that iNOS was mostly expressed in neurons. Of note, neither astrocytes nor microglia exhibited colocalization of iNOS/3-NT in this brain region, further confirming not only that iNOS induction is a major source of peroxynitrite but also that the enzyme is not responsive in astrocytes and microglia. These findings indicate that chronic alcohol ingestion preferentially modulates iNOS protein levels in neurons, but not in astrocytes or microglia, validating our recent findings that alcohol/acetaldehyde exposure increased the level of iNOS protein in cultured primary human neurons [71].
It is accepted that alcohol administered acutely in a pathophysiologically relevant dose can selectively and potently suppress the function of N-methyl-d-aspartate (NMDA) receptors [73]. Until now, the exact mechanism or site of action is unknown. Prolonged administration of alcohol leads to an adaptive increase in the sensitivity of NMDA receptors in vivo and in vitro. Such changes potentially can result in an enhanced vulnerability for glutamate-induced cytotoxic response (excitotoxicity) [74]. Animal studies suggest that chronic alcohol exposure and withdrawal are accompanied by a hyper-glutamatergic state, leading to neurotoxicity [75]. Preclinical models have shown that “anti-glutamatergic” compounds can reduce neuronal cell death. Increased sensitivity of neurons to excitotoxic insults is one of the mechanisms underlying alcohol-induced brain damage. NMDA stimulation results in increased calcium influx that is associated with uptake into mitochondria and causes the production of ROS that interfere with the function of mitochondria and plasma membranes. Direct suppression of the mitochondrial respiratory chain also indirectly induces further NMDA receptor stimulation. If the suppressive effect of alcohol on NMDA receptors is removed during withdrawal, the possibility of neuronal damage is significantly augmented through this receptor system, more so when increased and/or prolonged withdrawal signs after repeated withdrawal [76]. Alcohol-induced brain damage is mediated by glutamate-mediated transmission. Recently, the sulfur-containing amino acid, homocysteine, has been suggested to be neurotoxic in alcoholism [77]. The catabolism of homocysteine to methionine, a key step in detoxifying homocysteine, requires folate as a cofactor. Chronic alcoholics often have a low intake of folate resulting in a sustained hyperhomocysteinemia. Homocysteine is a partial or complete agonist at the glutamate and glycine binding sites within the NMDA receptor complex, respectively. Enhanced levels of homocysteine may lead to a pathological increase in receptor activity and subsequent excitotoxicity. From a clinical perspective, increased levels of plasma homocysteine can be used as a marker to predict alcohol withdrawal symptoms, so that therapeutic intervention can be initiated [78]. To date, little is known regarding gender differences in alcohol-mediated neuroinflammation and neurodegeneration. Recently acquired data suggest that there is more pronounced glial reaction (reflective of inflammation) and neuronal injury in female versus male mice in a binge model of alcohol administration. Alfonso-Loeches et al. [79] showed that chronic alcohol treatment induces inflammatory mediators (iNOS and COX-2), cytokines (IL-1β, TNFα), gliosis (GFAP), caspase-3 activation, and greater neuronal loss in the cerebral cortex of female mice when compared to male animals.
Astrocytes are altered by alcohol exposure in vitro and in vivo reflecting putative direct and indirect effects. Astrocytes play a significant role in supporting the function of neurons and brain endothelium. It has been reported that glial fibrillary acidic protein, (GFAP), a marker for reactive astrocytes, and vimentin (detecting hyperactive astrocytes) are substantially increased in animals subjected to chronic alcohol administration [80]. These changes appeared to be related to neuronal cell death in the same areas. Mechanisms underlying the effect of alcohol on astrocytes remain the subject of debate; however, several groups reported complimentary data pointing to TLR4 activation as one possible pathway [81]. Blanco and colleagues demonstrated that astrocyte activation with IL-1β or alcohol (10 and 50 mM) resulted in the translocation of IL-1 receptor, IL-1R, and/or TLR4 into lipid raft–caveolae-enriched fractions and the recruitment of signaling molecules (phospho-IL-1R-associated kinase and phospho-extracellular-regulated kinase) into these microdomains. Using cellular imaging techniques, they demonstrated that IL-1R was internalized by caveolar endocytosis via enlarged caveosomes after IL-1β or alcohol treatment, which sorted their IL-1R cargo into the endoplasmic reticulum–Golgi compartment and into the nucleus.
Using primary human astrocytes, we showed that activation of cytosolic phospholipase A2 (cPLA2) and cyclooxygenase (COX-2) by alcohol in astrocytes enhanced the secretion of inflammatory agents via the interactive tyrosine phosphorylation of TLR4 and Src kinase [82]. Alcohol exposure (20 mM for 48 h) increased the activity of cytochrome P450 2E1, ROS levels, and secretion of prostaglandin E2 (PGE2). PGE2 generation was dependent on induction of cPLA2 activity/protein as well as COX-2 protein level. Src phosphorylation was necessary for these effects of alcohol. The interactive tyrosine phosphorylation of TLR4–Src complex at the cell membrane triggered the activation of cPLA2 and COX-2 in the cytoplasm through a Src signaling intermediate. Inhibition of alcohol metabolism and blockage of Src activity or TLR4 prevented the activation of cPLA2 and COX-2 as well as diminished PGE2 production, suggesting that interactive phosphorylation of TLR4–Src regulated the pro-inflammatory response in astrocytes. Alcohol-driven changes were reduced in TLR4 knockout mice underscoring its involvement in CNS alcohol effects [79].
Another possibility of alcohol-induced astrocyte dysfunction is its effects on adenosine signaling. Lee et al. [83] demonstrated that mice lacking the ethanol-sensitive adenosine transporter, type 1 equilibrative nucleoside transporter (ENT1), consumed more alcohol compared with wild-type mice and had elevated striatal glutamate levels. ENT1 inhibition or knockdown reduces glutamate transporter expression in cultured astrocytes. Inhibition or deletion of ENT1 reduced the expression of type 2 excitatory amino acid transporter (EAAT2) and the astrocyte-specific water channel, aquaporin 4 (AQP4). EAAT2 and AQP4 colocalization was reduced in the striatum of ENT1 null mice. Ceftriaxone, an antibiotic increasing EAAT2 function, elevated not only EAAT2 but also AQP4 expression in the striatum. Furthermore, ceftriaxone reduced alcohol drinking, suggesting that ENT1-mediated downregulation of EAAT2 and AQP4 expression contributes to excessive alcohol consumption in a mouse model. These observations have significant implications as AQP4 regulates water content in the brain and could be another factor contributing to neurodegeneration.
Concentrations of extracellular glutamate were increased in animals exposed to alcohol for 4–8 days, suggesting deficits in glutamate transport [84]. Increased gene expression for EAAT1 was shown in the brains (frontal cortex) of alcoholics, while no results were presented for EAAT2 [85]. In contrast to discrepant experimental results, a number of clinical studies showed efficacy of anti-glutamatergic approaches for treating alcohol withdrawal symptoms [86] and dependence [87]. Furthermore, increased glutamate levels in animals with defective glutamate transporters enhanced their alcohol consumption [88]. Taken together, these data indicate multifaceted effects of alcohol on astrocyte function and suggest potential interventions.
4 Summary
It is clear that prolonged and excessive alcohol exposure affects all cell types in the brain via both direct and indirect effects. Importantly, new data suggest that inflammatory responses play a significant role in alcohol-associated neurodegeneration and alcohol addiction.
References
WHO: Alcohol and injury in emergency departments: Summary of the report from the WHO Collaborative Study on Alcohol and Injuries. World Health Organization, Geneva 2007.
Wu Y, Lousberg EL, Moldenhauer LM, Hayball JD, Robertson SA, Coller JK, Watkins LR, Somogyi AA, Hutchinson MR. Attenuation of microglial and IL-1 signaling protects mice from acute alcohol-induced sedation and/or motor impairment. Brain Behav Immun. 2011;25 Suppl 1:S155–64.
Zahr NM, Kaufman KL, Harper CG. Clinical and pathological features of alcohol-related brain damage. Nat Rev Neurol. 2011;7:284–94.
Pfefferbaum A, Rosenbloom M, Rohlfing T, Sullivan EV. Degradation of association and projection white matter systems in alcoholism detected with quantitative fiber tracking. Biol Psychiatry. 2009;65:680–90.
van Eijk J, Demirakca T, Frischknecht U, Hermann D, Mann K, Ende G. Rapid partial regeneration of brain volume during the first 14 days of abstinence from alcohol. Alcohol Clin Exp Res. 2013;37:67–74.
Alhassoon OM, Sorg SF, Taylor MJ, Stephan RA, Schweinsburg BC, Stricker NH, Gongvatana A, Grant I. Callosal white matter microstructural recovery in abstinent alcoholics: a longitudinal diffusion tensor imaging study. Alcohol Clin Exp Res. 2012;36:1922–31.
Carlen PL, Wilkinson DA, Wortzman G, Holgate R. Partially reversible cerebral atrophy and functional improvement in recently abstinent alcoholics. Can J Neurol Sci. 1984;11:441–6.
Gazdzinski S, Durazzo TC, Meyerhoff DJ. Temporal dynamics and determinants of whole brain tissue volume changes during recovery from alcohol dependence. Drug Alcohol Depend. 2005;78:263–73.
Harper CG, Kril JJ, Holloway RL. Brain shrinkage in chronic alcoholics: a pathological study. Br Med J (Clin Res Ed). 1985;290:501–4.
Harper C. The neuropathology of alcohol-specific brain damage, or does alcohol damage the brain? J Neuropathol Exp Neurol. 1998;57:101–10.
Harper C, Dixon G, Sheedy D, Garrick T. Neuropathological alterations in alcoholic brains. Studies arising from the New South Wales Tissue Resource Centre. Prog Neuropsychopharmacol Biol Psychiatry. 2003;27:951–61.
Mayfield RD, Lewohl JM, Dodd PR, Herlihy A, Liu J, Harris RA. Patterns of gene expression are altered in the frontal and motor cortices of human alcoholics. J Neurochem. 2002;81: 802–13.
Liu J, Lewohl JM, Harris RA, Iyer VR, Dodd PR, Randall PK, Mayfield RD. Patterns of gene expression in the frontal cortex discriminate alcoholic from nonalcoholic individuals. Neuropsychopharmacology. 2006;31:1574–82.
Etheridge N, Lewohl JM, Mayfield RD, Harris RA, Dodd PR. Synaptic proteome changes in the superior frontal gyrus and occipital cortex of the alcoholic brain. Proteomics Clin Appl. 2009;3:730–42.
de la Monte SM, Longato L, Tong M, DeNucci S, Wands JR. The liver-brain axis of alcohol-mediated neurodegeneration: role of toxic lipids. Int J Environ Res Public Health. 2009;6: 2055–75.
Persidsky Y, Ho W, Ramirez SH, Potula R, Abood ME, Unterwald E, Tuma R. HIV-1 infection and alcohol abuse: neurocognitive impairment, mechanisms of neurodegeneration and therapeutic interventions. Brain Behav Immun. 2011;25 Suppl 1:S61–70.
He J, Crews FT. Increased MCP-1 and microglia in various regions of the human alcoholic brain. Exp Neurol. 2008;210:349–58.
Achur RN, Freeman WM, Vrana KE. Circulating cytokines as biomarkers of alcohol abuse and alcoholism. J Neuroimmune Pharmacol. 2010;5:83–91.
Qin L, He J, Hanes RN, Pluzarev O, Hong JS, Crews FT. Increased systemic and brain cytokine production and neuroinflammation by endotoxin following ethanol treatment. J Neuroinflammation. 2008;5:10.
McClain CJ, Barve S, Deaciuc I, Kugelmas M, Hill D. Cytokines in alcoholic liver disease. Semin Liver Dis. 1999;19:205–19.
Kiefer F, Jahn H, Schick M, Wiedemann K. Alcohol intake, tumour necrosis factor-alpha, leptin and craving: factors of a possibly vicious circle? Alcohol Alcohol. 2002;37:401–4.
McClain CJ, Song Z, Barve SS, Hill DB, Deaciuc I. Recent advances in alcoholic liver disease. IV. Dysregulated cytokine metabolism in alcoholic liver disease. Am J Physiol Gastrointest Liver Physiol. 2004;287:G497–502.
Valles SL, Blanco AM, Pascual M, Guerri C. Chronic ethanol treatment enhances inflammatory mediators and cell death in the brain and in astrocytes. Brain Pathol. 2004;14:365–71.
Blednov YA, Benavidez JM, Geil C, Perra S, Morikawa H, Harris RA. Activation of inflammatory signaling by lipopolysaccharide produces a prolonged increase of voluntary alcohol intake in mice. Brain Behav Immun. 2011;25 Suppl 1:S92–105.
Corrigan F, Hutchinson M. Are the effects of alcohol on the CNS influenced by Toll-like receptor signaling? Expert Rev Clin Immunol. 2012;8:201–3.
Jaskova K, Pavlovicova M, Jurkovicova D. Calcium transporters and their role in the development of neuronal disease and neuronal damage. Gen Physiol Biophys. 2012;31:375–82.
Zhao YN, Wang F, Fan YX, Ping GF, Yang JY, Wu CF. Activated microglia are implicated in cognitive deficits, neuronal death, and successful recovery following intermittent ethanol exposure. Behav Brain Res. 2013;236:270–82.
Kettenmann H, Hanisch UK, Noda M, Verkhratsky A. Physiology of microglia. Physiol Rev. 2011;91:461–553.
Vinet J, Weering HR, Heinrich A, Kalin RE, Wegner A, Brouwer N, Heppner FL, Rooijen N, Boddeke HW, Biber K. Neuroprotective function for ramified microglia in hippocampal excitotoxicity. J Neuroinflammation. 2012;9:27.
Rock RB, Gekker G, Hu S, Sheng WS, Cheeran M, Lokensgard JR, Peterson PK. Role of microglia in central nervous system infections. Clin Microbiol Rev. 2004;17:942–64. table of contents.
Lindl KA, Marks DR, Kolson DL, Jordan-Sciutto KL. HIV-associated neurocognitive disorder: pathogenesis and therapeutic opportunities. J Neuroimmune Pharmacol. 2010;5: 294–309.
Deehan Jr GA, Brodie MS, Rodd ZA. What is in that drink: the biological actions of ethanol, acetaldehyde, and salsolinol. Curr Top Behav Neurosci. 2013;13:163–84.
Nelson S, Kolls JK. Alcohol, host defence and society. Nat Rev Immunol. 2002;2:205–9.
Karavitis J, Murdoch EL, Deburghgraeve C, Ramirez L, Kovacs EJ. Ethanol suppresses phagosomal adhesion maturation, Rac activation, and subsequent actin polymerization during FcgammaR-mediated phagocytosis. Cell Immunol. 2012;274:61–71.
Nixon K, Kim DH, Potts EN, He J, Crews FT. Distinct cell proliferation events during abstinence after alcohol dependence: microglia proliferation precedes neurogenesis. Neurobiol Dis. 2008;31:218–29.
Suk K. Microglial signal transduction as a target of alcohol action in the brain. Curr Neurovasc Res. 2007;4:131–42.
Szabo G. Alcohol’s contribution to compromised immunity. Alcohol Health Res World. 1997;21:30–41.
Sabino KR, Petroianu A, Alberti LR. Influence of the acute alcoholism on the phagocytic function of the mononuclear phagocytic system. J Med Life. 2011;4:421–3.
Fang KM, Yang CS, Sun SH, Tzeng SF. Microglial phagocytosis attenuated by short-term exposure to exogenous ATP through P2X receptor action. J Neurochem. 2009;111:1225–37.
Majumder P, Trujillo CA, Lopes CG, Resende RR, Gomes KN, Yuahasi KK, Britto LRG, Ulrich H. New insights into purinergic receptor signaling in neuronal differentiation, neuroprotection, and brain disorders. Purinergic Signal. 2007;3:317–31.
James G, Butt AM. P2Y and P2X purinoceptor mediated Ca2+ signalling in glial cell pathology in the central nervous system. Eur J Pharmacol. 2002;447:247–60.
Brown GC, Neher JJ. Inflammatory neurodegeneration and mechanisms of microglial killing of neurons. Mol Neurobiol. 2010;41:242–7.
Marshall SA, McClain JA, Kelso ML, Hopkins DM, Pauly JR, Nixon K. Microglial activation is not equivalent to neuroinflammation in alcohol-induced neurodegeneration: The importance of microglia phenotype. Neurobiol Dis. 2013;54:239–51.
Chao TS, Byron KL, Lee KM, Villereal M, Rosner MR. Activation of MAP kinases by calcium-dependent and calcium-independent pathways. Stimulation by thapsigargin and epidermal growth factor. J Biol Chem. 1992;267:19876–83.
Fernandez-Lizarbe S, Pascual M, Guerri C. Critical role of TLR4 response in the activation of microglia induced by ethanol. J Immunol. 2009;183:4733–44.
Raivich G, Jones LL, Werner A, Bluthmann H, Doetschmann T, Kreutzberg GW. Molecular signals for glial activation: pro- and anti-inflammatory cytokines in the injured brain. Acta Neurochir Suppl. 1999;73:21–30.
Pan W, Barron M, Hsuchou H, Tu H, Kastin AJ. Increased leptin permeation across the blood-brain barrier after chronic alcohol ingestion. Neuropsychopharmacology. 2008;33:859–66.
Karavitis J, Kovacs EJ. Macrophage phagocytosis: effects of environmental pollutants, alcohol, cigarette smoke, and other external factors. J Leukoc Biol. 2011;90:1065–78.
Graeber MB, Streit WJ. Microglia: biology and pathology. Acta Neuropathol. 2010;119: 89–105.
Alfonso-Loeches S, Pascual-Lucas M, Blanco AM, Sanchez-Vera I, Guerri C. Pivotal role of TLR4 receptors in alcohol-induced neuroinflammation and brain damage. J Neurosci. 2010;30:8285–95.
Fernandez-Lizarbe S, Montesinos J, Guerri C. Ethanol induces TLR4/TLR2 association, triggering an inflammatory response in microglial cells. J Neurochem. 2013;126(2):261–73.
Verkhratsky A, Krishtal OA, Burnstock G. Purinoceptors on neuroglia. Mol Neurobiol. 2009;39:190–208.
Potucek YD, Crain JM, Watters JJ. Purinergic receptors modulate MAP kinases and transcription factors that control microglial inflammatory gene expression. Neurochem Int. 2006;49:204–14.
Burnstock G. Purinergic signalling and disorders of the central nervous system. Nat Rev Drug Discov. 2008;7:575–90.
Dooley R, Mashukova A, Toetter B, Hatt H, Neuhaus EM. Purinergic receptor antagonists inhibit odorant-mediated CREB phosphorylation in sustentacular cells of mouse olfactory epithelium. BMC Neurosci. 2011;12:86.
Inoue K, Tsuda M. P2X4 receptors of microglia in neuropathic pain. CNS Neurol Disord Drug Targets. 2012;11:699–704.
Mei L, Du W, Gao W, Mei QB. Purinergic signaling: a novel mechanism in immune surveillance. Acta Pharmacol Sin. 2010;31:1149–53.
Koshimizu TA, Van Goor F, Tomic M, Wong AO, Tanoue A, Tsujimoto G, Stojilkovic SS. Characterization of calcium signaling by purinergic receptor-channels expressed in excitable cells. Mol Pharmacol. 2000;58:936–45.
Ko WH, Au CL, Yip CY. Multiple purinergic receptors lead to intracellular calcium increases in cultured rat Sertoli cells. Life Sci. 2003;72:1519–35.
James G, Butt AM. Adenosine 5′ triphosphate evoked mobilization of intracellular calcium in central nervous system white matter of adult mouse optic nerve. Neurosci Lett. 1999;268: 53–6.
Guthrie PB, Knappenberger J, Segal M, Bennett MV, Charles AC, Kater SB. ATP released from astrocytes mediates glial calcium waves. J Neurosci. 1999;19:520–8.
Asatryan L, Nam HW, Lee MR, Thakkar MM, Saeed Dar M, Davies DL, Choi DS. Implication of the purinergic system in alcohol use disorders. Alcohol Clin Exp Res. 2011;35:584–94.
Ostrovskaya O, Asatryan L, Wyatt L, Popova M, Li K, Peoples RW, Alkana RL, Davies DL. Ethanol is a fast channel inhibitor of P2X4 receptors. J Pharmacol Exp Ther. 2011;337: 171–9.
Haorah J, Heilman D, Knipe B, Chrastil J, Leibhart J, Ghorpade A, Miller DW, Persidsky Y. Ethanol-induced activation of myosin light chain kinase leads to dysfunction of tight junctions and blood-brain barrier compromise. Alcohol Clin Exp Res. 2005;29:999–1009.
Haorah J, Knipe B, Leibhart J, Ghorpade A, Persidsky Y. Alcohol-induced oxidative stress in brain endothelial cells causes blood-brain barrier dysfunction. J Leukoc Biol. 2005;78: 1223–32.
Haorah J, Knipe B, Gorantla S, Zheng J, Persidsky Y. Alcohol-induced blood-brain barrier dysfunction is mediated via inositol 1,4,5-triphosphate receptor (IP3R)-gated intracellular calcium release. J Neurochem. 2007;100:324–36.
Haorah J, Ramirez SH, Schall K, Smith D, Pandya R, Persidsky Y. Oxidative stress activates protein tyrosine kinase and matrix metalloproteinases leading to blood-brain barrier dysfunction. J Neurochem. 2007;101:566–76.
Haorah J, Schall K, Ramirez SH, Persidsky Y. Activation of protein tyrosine kinases and matrix metalloproteinases causes blood-brain barrier injury: Novel mechanism for neurodegeneration associated with alcohol abuse. Glia. 2008;56:78–88.
Haorah J, Floreani NA, Knipe B, Persidsky Y. Stabilization of superoxide dismutase by acetyl-l-carnitine in human brain endothelium during alcohol exposure: novel protective approach. Free Radic Biol Med. 2011;51:1601–9.
Abdul Muneer PM, Alikunju S, Szlachetka AM, Haorah J. Inhibitory effects of alcohol on glucose transport across the blood-brain barrier leads to neurodegeneration: preventive role of acetyl-L: -carnitine. Psychopharmacology (Berl). 2011;214:707–18.
Haorah J, Ramirez SH, Floreani N, Gorantla S, Morsey B, Persidsky Y. Mechanism of alcohol-induced oxidative stress and neuronal injury. Free Radic Biol Med. 2008;45:1542–50.
Rump TJ, Abdul Muneer PM, Szlachetka AM, Lamb A, Haorei C, Alikunju S, Xiong H, Keblesh J, Liu J, Zimmerman MC, Jones J, Donohue Jr TM, Persidsky Y, Haorah J. Acetyl-L-carnitine protects neuronal function from alcohol-induced oxidative damage in the brain. Free Radic Biol Med. 2011;49:1494–504.
Harper C, Matsumoto I. Ethanol and brain damage. Curr Opin Pharmacol. 2005;5:73–8.
Dodd PR, Beckmann AM, Davidson MS, Wilce PA. Glutamate-mediated transmission, alcohol, and alcoholism. Neurochem Int. 2000;37:509–33.
Thomson AD, Guerrini I, Bell D, Drummond C, Duka T, Field M, Kopelman M, Lingford-Hughes A, Smith I, Wilson K, Marshall EJ. Alcohol-related brain damage: report from a Medical Council on Alcohol Symposium, June 2010. Alcohol Alcohol. 2012;47:84–91.
Matsumoto I, Burke L, Inoue Y, Wilce PA. Two models of ethanol withdrawal kindling. Nihon Arukoru Yakubutsu Igakkai Zasshi. 2001;36:53–64.
Bleich S, Bandelow B, Javaheripour K, Muller A, Degner D, Wilhelm J, Havemann-Reinecke U, Sperling W, Ruther E, Kornhuber J. Hyperhomocysteinemia as a new risk factor for brain shrinkage in patients with alcoholism. Neurosci Lett. 2003;335:179–82.
Bleich S, Degner D, Bandelow B, von Ahsen N, Ruther E, Kornhuber J. Plasma homocysteine is a predictor of alcohol withdrawal seizures. Neuroreport. 2000;11:2749–52.
Alfonso-Loeches S, Pascual M, Guerri C. Gender differences in alcohol-induced neurotoxicity and brain damage. Toxicology. 2013;311(1–2):27–34.
Kelso ML, Liput DJ, Eaves DW, Nixon K. Upregulated vimentin suggests new areas of neurodegeneration in a model of an alcohol use disorder. Neuroscience. 2011;197:381–93.
Blanco AM, Perez-Arago A, Fernandez-Lizarbe S, Guerri C. Ethanol mimics ligand-mediated activation and endocytosis of IL-1RI/TLR4 receptors via lipid rafts caveolae in astroglial cells. J Neurochem. 2008;106:625–39.
Floreani NA, Rump TJ, Muneer PM, Alikunju S, Morsey BM, Brodie MR, Persidsky Y, Haorah J. Alcohol-induced interactive phosphorylation of Src and toll-like receptor regulates the secretion of inflammatory mediators by human astrocytes. J Neuroimmune Pharmacol. 2010;5:533–45.
Lee MR, Ruby CL, Hinton DJ, Choi S, Adams CA, Young Kang N, Choi DS. Striatal adenosine signaling regulates EAAT2 and astrocytic AQP4 expression and alcohol drinking in mice. Neuropsychopharmacology. 2013;38:437–45.
Melendez RI, Hicks MP, Cagle SS, Kalivas PW. Ethanol exposure decreases glutamate uptake in the nucleus accumbens. Alcohol Clin Exp Res. 2005;29:326–33.
Flatscher-Bader T, van der Brug M, Hwang JW, Gochee PA, Matsumoto I, Niwa S, Wilce PA. Alcohol-responsive genes in the frontal cortex and nucleus accumbens of human alcoholics. J Neurochem. 2005;93:359–70.
Krupitsky EM, Rudenko AA, Burakov AM, Slavina TY, Grinenko AA, Pittman B, Gueorguieva R, Petrakis IL, Zvartau EE, Krystal JH. Antiglutamatergic strategies for ethanol detoxification: comparison with placebo and diazepam. Alcohol Clin Exp Res. 2007;31:604–11.
Ma JZ, Ait-Daoud N, Johnson BA. Topiramate reduces the harm of excessive drinking: implications for public health and primary care. Addiction. 2006;101:1561–8.
Spanagel R, Pendyala G, Abarca C, Zghoul T, Sanchis-Segura C, Magnone MC, Lascorz J, Depner M, Holzberg D, Soyka M, Schreiber S, Matsuda F, Lathrop M, Schumann G, Albrecht U. The clock gene Per2 influences the glutamatergic system and modulates alcohol consumption. Nat Med. 2005;11:35–42.
Acknowledgements
This work was supported in part by NIH grants AA017398, AA015913, MH65151, and DA025566.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2014 Springer Science+Business Media New York
About this chapter
Cite this chapter
Persidsky, Y., Gofman, L., Potula, R. (2014). Alcohol and Neurodegeneration. In: Peterson, P., Toborek, M. (eds) Neuroinflammation and Neurodegeneration. Springer, New York, NY. https://doi.org/10.1007/978-1-4939-1071-7_24
Download citation
DOI: https://doi.org/10.1007/978-1-4939-1071-7_24
Published:
Publisher Name: Springer, New York, NY
Print ISBN: 978-1-4939-1070-0
Online ISBN: 978-1-4939-1071-7
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)