
Abstract
This chapter gives an overview over the biology and the clinical consequences of infection with the human polyomavirus JCV. Current data as well as those aspects that are not yet fully understood are explained.
JCV virus is a human polyomavirus that leads to an asymptomatic infection in a large percentage of healthy individuals and can lead to a latent infection of the kidneys or the bone marrow. JCV reactivation and lytic infection of oligodendrocytes in the brain lead to a devastating demyelinating disease called progressive multifocal leukoencephalopathy (PML). PML typically occurs in patients with an impaired cellular immune response due to an underlying disease such as human immunodeficiency virus (HIV) infection or a systemic malignancy or due to treatment with immunomodulatory therapies. While supporting the recovery of the patient’s immune system (either by giving antiretroviral therapy in HIV-infected patients or by stopping the immunomodulatory therapies leading to PML) is currently the only known effective therapeutic intervention, the response of the immune system can lead to clinical and radiographic worsening known as immune reconstitution inflammatory syndrome (IRIS). The clinical and biological consequences of neuroinflammation in this viral infection are presented in this chapter.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others


Keywords
- Progressive multifocal leukoencephalopathy
- JC virus
- Polyomavirus
- Human immunodeficiency virus (HIV)
- Multiple sclerosis
- Natalizumab
- Immune reconstitution inflammatory syndrome (IRIS)
1 Introduction
In 1972, two different human polyomaviruses were isolated from a lymphoma patient with a CNS demyelinating disease: JC virus (JCV) from the brain of a patient with progressive multifocal leukoencephalopathy (PML) and BK virus (BKV) from a kidney transplant recipient with fulminant polyomavirus-associated nephropathy (PVAN). Both viruses were named from the initials of the patients from whom they were isolated. Although JCV and BKV were shown to be nearly 75 % homologous at both the nucleotide and amino acid level, these new polyomaviruses demonstrated very different characteristics for kinetics of growth, host range, and pathologies [1]. Both JCV and BKV infections are almost exclusively found in individuals with impaired cell-mediated immune responses. Since then, there have been at least four more human polyomaviruses identified including Merkel cell from rare skin carcinomas, Ki and Wu from the respiratory tract, and Trp from trichoplasia skin lesions [2]. All human polyomaviruses, however, are globally ubiquitous, infecting the majority of the population in the early years and maintaining latency in nearly half the population as evidence of viral DNA identification in urinary excretions. This is a common feature of all primate, rodent, and avian polyomaviruses. JCV however is the only human polyomavirus that is predominantly neurotropic and neurovirulent, targeting the myelin-producing cell in the human brain causing a slowly progressing lytic infection of the oligodendrocyte resulting in widespread loss of myelin in the brain. Although many attempts have been made to model PML in rodents and nonhuman primates, there are currently no animal models to study JCV lytic infection. Also, there are no antiviral agents to inhibit JCV infection nor effective treatments for PML. Studies of the biology and pathogenesis of JCV in its human host remain difficult challenges for laboratory and clinical investigations.
2 The Biology of the JC Virus
2.1 Cellular Host Range and Molecular Factors for Productive Infection
Epidemiological studies indicate that seroconversion to positive antibody status occurs early in childhood starting at 4–5 years with increasing prevalence in the population. By the fifth decade, 60–75 % of the population is seropositive although antibody levels can rise or decline over time [3]. It is obvious, however, that initial infection that accounts for seroconversion or maintaining high titers of antibody does not occur in the brain. JCV can establish latency in uroepithelial cells and be excreted into the urine at high concentrations without pathogenic consequences unlike BKV. There is no clear understanding of this observation. What is interesting, however, is the genotype of the urine-excreted variant. In Fig. 1, the JCV genome is shown as a closed circular, double-stranded DNA of 5.13 kilobases, packaged into a naked, icosahedral 40-nm virion particle. There are two protein-coding regions for the early proteins, T and t, that have multiple functions for DNA replication and control over cellular metabolism that are synthesized from transcripts on one strand. Capsid proteins VP1, VP2, and VP3 are synthesized from transcripts on the opposite strand. The intergenic region between the 5′ start sequences of the early and late genes is the noncoding regulatory region or NCRR whose nucleotides have the origin for DNA replication, the transcription binding protein sites, and enhancer sequences for transcription efficiency also seen in Fig. 2. In the urine-excreted or archetype variant, the nucleotide sequences show a linear 200-base-pair arrangement. In pathological tissues such as the brain and cerebrospinal fluid, the nucleotide sequences show direct tandem repeats of 98 or fewer nucleotides with frequent deletions and duplication known as the prototype variant. A number of DNA-binding proteins have been identified on the NCRR as shown in Fig. 1 extending from the origin of DNA replication to nucleotide 50 that are essential for virus growth including those for the TATA-binding proteins and NF-1 class X [4].

JC virus characteristics. On the left, an electron microscopic picture of a virion particle is shown. The structure of the JC viral genome is depicted on the right upper part of this figure with the regions encoding for the early T protein, the noncoding regulatory region (promoter enhancer), and the late capsid proteins VP1, VP2, and VP3. The lower right corner of the picture shows lesions caused by JCV as seen on an MRI

Viral factors that may favor JCV infection. Part (a) shows the amino acid residues for the viral capsid protein VP1, which is responsible for binding to host cells; sequences for VP1 can be altered following latency. Part (b) shows two variations of the noncoding regulatory sequences. The nonpathogenic archetype, which is typically found in kidney tissue and contains no tandem repeats, is depicted above, whereas the pathogenic prototype, which is found in affected tissues in PML and which contains direct tandem repeat structures, is shown below
There has been considerable interest in determining the contributions to cellular host range between the NCRR and the capsid VP1 protein that binds cell receptors. The comparison between these two regions of the viral genome is shown in Fig. 2. The three-dimensional crystallographic structure shows the amino acid domains in color that interact with sialic residues on cell surface membranes for virion attachment [5]. In addition to sialic acids, the key receptor for JCV, the 5HT2A serotonin receptor plays a secondary role in attachments and particle entry [6]. Virions enter the cell cytoplasm and traffic to endosomes using clathrin-coated pits and are then transported to the nuclear membrane as disrupted particles. Viral DNA enters the nucleus that is then transcribed using host DNA-binding factors starting with early region for T proteins. The NCRR plays a key role in viral susceptibility since it contains the transcription binding sites for factors that initiate T protein mRNA and synthesis. The T protein then binds the origin of DNA replication on the 5′ region of the NCRR that interacts with cellular DNA alpha polymerase to form protein complexes for viral DNA synthesis. It is thought that an increase in the amount of viral DNA then allows transcription of mRNA for viral capsid proteins and then virion assembly—all in the nucleus. The current working models suggest that those factors that are necessary for JCV growth in uroepithelial cells bind to the archetype arrangement of the NCRR and those factors for JCV growth in glial cells and some immune system cells bind to prototype arrangement of the NCRR that leads to lytic infection and PML [7].
In cell culture studies and in clinical tissue samples, JCV multiplication is evident in oligodendrocytes and astrocytes in the brain and in CD34+ and CD19+ cells in the bone marrow and peripheral blood, as well as stromal cells in tonsils and B cells in the spleen [8, 9].
2.2 Presence of JCV in Infected Individuals
Without an animal model of JCV lytic infection resulting in PML, tracking virus in tissues requires samples mostly from infected patients, notably those with PML. Nothing is known about the initial site or timing of infection. However, since the majority of the population worldwide demonstrates antibodies to JCV, it is assumed that respiratory inhalation and/or ingestion is the primary route of infection. There are several major techniques used to identify JCV in tissues: quantitative polymerase chain reaction, qPCR, and in situ DNA hybridization that detects the viral genome and immunocytochemistry for viral proteins [10]. For the laboratory confirmatory diagnosis of PML, real-time qPCR assays are done detecting viral DNA in the cerebrospinal fluid of suspected PML patients. One assay currently used at the National Institute of Neurological Disorders and Stroke (NINDS), a validated/certified assay in the Laboratory of Molecular Medicine and Neuroscience (LMMN), is the multiplex qPCR that not only quantitates the level of viral DNA but also distinguishes whether the JCV variant is the more pathogenic prototype from the nonpathogenic archetype genotype [11]. Typically in PML patients, the brain and CSF will show the prototype, while the urine will show the archetype. Plasma or serum of patients may have both but, during the course of active infection leading to PML, will shift from archetype to prototype. In HIV patients, approximately 20 % can be viremic at some time although only 3 % will develop PML. Approximately 2 % of healthy individuals may be viremic at some time point and seem to clear infection. This observation is not surprising since JCV is ubiquitous in all parts of the world. It is surprising, however, that nearly 30 % of the population excretes JCV in urine without pathological consequences.
3 The Pathogenesis of JCV Demyelinating Infection
3.1 Viral Variants and Latency
The principal cells in which JCV DNA has been detected during infection are the uroepithelial cells in the kidney, CD34+ cells in the bone marrow, CD20+ cells in peripheral blood, and oligodendrocytes in the brain. Figure 3 shows nuclear location of replicating DNA using in situ DNA hybridization with a highly specific viral DNA probe and diaminobenzidine as the chromophore (brown) staining [10]. The bone marrow biopsy shown in Fig. 3 was taken 4 years prior to the development of PML in a Wiskott-Aldrich syndrome patient. The NCRR of the variant in the bone marrow was the prototype and nearly identical to what was seen in the B cells and brain taken at the time of PML diagnosis. Observations such as these and in other cases have led to the hypothesis that bone marrow may harbor latent JCV in hematopoietic cells for long periods [12]. If such latently infected cells migrate into the peripheral circulation and differentiate toward a B-cell lineage, then it is possible that JCV can reactivate to slowly multiply as such cells begin the expression of factors that favor viral growth [4]. This pathway is thought to account for the development of PML in multiple sclerosis patients treated with natalizumab, a monoclonal antibody that blocks the alpha 4 cell surface integrin VLA 4 to VCAM, vascular cell adhesion molecule, preventing homing of CD+ 34-positive cells in the bone marrow and extravasation of inflammatory cells into the brain [13]. Certainly, JCV maintains decades-long periods of latency in the kidney with the NCRR archetype variant. If that is the predecessor to the pathogenic prototype, then virions released from the kidney may find their way to lymphoid tissues in which genotypic alterations or rearrangements occur to produce the prototype NCRR. This is also a plausible pathway that may occur in some patients [13].

Possible pathway of JCV in the body. Depicted are tissues shown to contain JC virus. (a) Shows the kidney tissue (which usually contains archetype JCV), b shows the bone marrow (which usually contains prototype JCV), c shows the peripheral blood (which can contain archetype and/or prototype), and d shows oligodendrocytes in the brain (which contain prototype)
3.2 Trafficking of Virus from Sites of Latency to the Brain
There is little evidence of how JCV enters the brain from peripheral sites of infection. Two possibilities are possible and not mutually exclusive. Since cell-free virus can be found in the blood, it is possible that JCV can cross the blood-brain barrier and enter the brain parenchyma, directly infecting astrocytes and its primary target, the oligodendrocyte. JCV may also be carried into the brain within a cell such as B cells seen in blood. JCV does not seem to infect monocytes or T lymphocytes that bring other viruses into the brain like HIV-1 and cytomegalovirus (CMV). There have been reports of viral latency in the brain that reactivates due to lack of immune surveillance [14]. However, that observation does not address the low incidence of PML in the many thousands of patients with substantial immune suppression like AIDS, organ transplants, cancer, and rheumatic diseases. Mechanisms of viral entry into the brain remain mostly unknown, requiring better cell culture and clinical studies.
3.3 Predictive Markers for Active JCV Infection
Although PML is considered a rare disease using the NIH criterion for prevalence, of 200,000 cases, the reports of PML have increased substantially over the last 10 years. In fact, PML is now considered a complication of not only HIV-1 infection and an AIDS defining illness but also a risk associated with a number of therapies for other diseases [15]. Consequently assessing that risk becomes a critical issue for patient treatment. Investigations have focused on defining biomarkers that identify active JCV infection in high-risk patients. Three JCV-linked factors for active infection that have well-developed assays have been described including the presence and rise of antiviral antibodies, viremia during the course of a risk treatment, and identification of T-cell responses to JCV multiple antigens [16]. In addition to these markers, molecular factors needed for infection in specific target cells like DNA-binding proteins for viral mRNA transcription may have a useful role, but their detection has not been reduced to a practical assay methodology. Detection of specific antibodies to JCV is a marker for prior exposure. Any increase in the antibody levels would indicate either an active or recent infection. ELISA, enzyme-linked immunosorbent assay, is the most routine technique used to measure antibodies and their levels. There are several such assays in clinical use for JCV that both detect antibody and measure its levels using the viral VP1 capsid protein as the antigen [17, 18]. The VP1 protein makes up the majority of virion structure and receptor for cell attachment. Plasma/serum levels of antibody can be monitored over time in patients to determine not only serostatus but also whether there is a rise in antibody levels. Most frequently, patients about to enter treatment with a drug or biological with a PML risk would have antibody levels determined. Samples that test seronegative however can be confounding since antibody levels can change over time from negative to positive. And in some cases, patients may not develop detectable antibodies but still have been exposed or have an active infection [19, 20]. Consequently, other markers should be used to assess those patients’ JCV status.
Recently, CD4 and CD8 T-cell responses have been measured using viral peptides across the entire viral genome. Healthy individuals and MS patients treated with natalizumab showed both T-cell responses to all viral proteins including the nonstructural T proteins and agno and all three capsid proteins. Some of these individuals who had T-cell responses did not have antibody [21]. Assays for T-cell responses require more blood than antibody assays and use flow cytometric analysis and cytokine measurement following viral peptide contact. However, these assays have become routine in many laboratories and so do not present a difficult obstacle for assessment. In combination with serology assays, measurement of cell immune response to JCV provides a more complete analysis of not only exposure but also the ability of the patient to mount an appropriate immune response.
The advantage of measuring immune responses is their relative stability over time. However, they do not reflect direct measurement of JCV infection at times of greatest risk for PML. The advances in qPCR methods that are specific to ultrasensitive detection of viral DNA, ten copies of the genome per ml of sample, have led to the laboratory confirmation of PML in many cases that may not have been accurately diagnosed. The current multiplex qPCR assay developed at NINDS requires small volumes of blood, CSF, urine, or cells in tissue samples. As a risk-monitoring assay, detection of viral DNA in the plasma or serum in a patient with an underlying disease that is a risk for PML can lead to substantial concern. If the individual remains viremic over weeks or months, particularly with the pathogenic variant, and shows any increase in the amount of viral DNA, then the risk for PML is elevated. At present there are no data that statistically measures that risk, i.e., percent of patients with viremia who develop PML. However, most patients show viremia at the time of PML diagnosis. It is possible to monitor patients then over time using these markers, antibody levels, T-cell responses, and viremia measured from one blood sample. As more data are collected on these patient populations, it may be possible to use a checklist of factors that reflect the risk in which combinations of “checks” can become a quantifiable number.
4 The Clinical Pattern of JCV-Induced PML
4.1 Patients at Risk for PML: Underlying Diseases and Medications
Exposure to JCV occurs in a large proportion of the population [3, 22], but the virus is thought to lead to PML almost exclusively in individuals with an impaired immune system. Some case reports of patients with PML without any apparent immunodeficiency exist [23], but they are the exception, and in most cases, an occult immunodeficiency-like idiopathic CD4+ lymphocytopenia or liver cirrhosis is eventually discovered. However, not all immunocompromised patients develop PML. Some diseases or medications affecting the immune system are more commonly associated with PML than others, and understanding these connections may help us in our understanding of the pathogenesis of PML. The initial description of PML was in a patient with lymphoma [24], and the majority of published cases in the mid-1980s described lymphoproliferative diseases as the underlying disease [25]. The incidence of PML increased significantly with the beginning of the HIV epidemic in the 1990s, and despite antiretroviral therapy, PML is still seen in up to 5 % of HIV-infected individuals [26].
Additionally, since 2004, a number of drugs have been found to be associated with the development of PML. Natalizumab, a monoclonal anti-integrin antibody used for the treatment of MS, is perhaps the most prominent of these immunomodulatory agents found to lead to an increased risk of developing PML [27]. However, other immunomodulatory medications used for the treatment of rheumatological, neurological, or oncological diseases, especially monoclonal antibodies like rituximab, efalizumab, and brentuximab, have been reported to be associated with PML [12, 28]. Patients undergoing bone marrow or solid organ transplant have also been described to develop PML. These patients are often immunocompromised due to the disease leading to transplant (e.g., kidney or liver failure, leukemia) and are furthermore often on multiple immunosuppressive agents. The exact rate of PML in these patients is difficult to assess. One multicenter study reported a rate of 1 in 1,000 patients undergoing lung or heart transplant [29]. It is challenging to get an accurate assessment of the actual risk associated with each of these underlying diseases or predisposing medications as PML is not a reportable disease, and our knowledge about the incidence and prevalence of PML is limited.
4.2 Diagnosis
An accurate diagnosis of PML can be challenging, especially in patients with other underlying neurological diseases like MS, which like PML is a multifocal demyelinating disease. In the early years of understanding the disease, the diagnosis of PML was usually based on brain biopsy showing typical histological findings (demyelination, multinuclear bizarre astrocytes, oligodendroglial nuclear inclusions; see below). Since JCV was identified as the virus causing PML, laboratory methods have been established that together with clinical and radiological findings can help establish the diagnosis of PML without the need for an invasive brain biopsy. A recent consensus statement from the American Academy of Neurology (AAN) Neuroinfectious Disease Section recommends to base the diagnosis of PML on a triad of compatible clinical features, compatible imaging findings, and a presence of JCV in the CSF by PCR. In cases where JCV can be detected in the CSF but the clinical or imaging findings are not typical, the AAN assigns a diagnosis of probable PML. If only clinical and imaging factors are present but the JCV in the CSF is non-detectable or if the CSF is positive but both clinical and MRI findings are atypical, a diagnosis of possible PML is suggested.
The typical clinical findings seen in PML are discussed in the next section of this chapter. Imaging used for the diagnosis of PML is mainly magnetic resonance imaging (MRI) of the brain. A recent study evaluating the MRIs of 22 patients with natalizumab-associated PML identified typical PML lesions as large (>3 cm), subcortical, T2 or fluid-attenuated inversion recovery (FLAIR) hyperintense, T1 hypointense, and diffusion hyperintense lesions, with a sharp border toward the gray matter and an ill-defined border toward the white matter on T2-weighted images [30]. An example of these typical MRI findings can be seen in Fig. 4. While PML lesions in HIV-infected individuals are usually not contrast enhancing, 41 % of the natalizumab-associated PML cases in this study were found to have contrast enhancement on the first scan at clinical presentation. However, these patients all had clinically diagnosed immune reconstitution inflammatory syndrome (IRIS). This complication is described in more detail in Sect. 5.1 of this chapter. Magnetic resonance spectroscopy of the brain has been suggested by some to add information, but most studies to date have not found this technique helpful [31, 32]. Positron emission tomography can in some cases help to differentiate between lymphoma and PML but is also not commonly used in the diagnosis of PML [32].

MRI findings in PML. Panel (a) shows an axial T2-weighted image with large multifocal hyperintense lesions with an ill-defined border toward the white matter. Panel (b) shows a coronal T1-weighted post-contrast image which demonstrates faint, patchy contrast enhancement in multifocal PML lesions in a patient with IRIS
4.3 Clinical Manifestations
Symptoms in this multifocal disease can be varied, depending on the location of the lesions and the extent of demyelination. As in other brain diseases, patients can present with hemiparesis, ataxia, or visual changes. In contrast to other multifocal demyelinating CNS diseases like MS, optic neuritis and spinal cord involvement are not typically seen [33]. However, there are some rare reports of PML lesions in the spinal cord either by imaging or by histology at autopsy. Unlike MS but similar to acute disseminated encephalomyelitis (ADEM), another demyelinating syndrome more commonly seen in children, changes in alertness, behavior, and cognition are common in PML. In fact, about 50 % of patients with PML exhibit mental status changes at diagnosis [34]. Seizures are also frequently seen in PML and can be the initial presenting feature. About 20 % of patients in multiple different case series describing PML associated with HIV, lymphoproliferative disease, or immunomodulatory medications develop seizures, typically within the first few months, if not at presentation [34, 35]. This is somewhat surprising, since seizures are generally thought to arise from the cortical gray matter but PML patients with seizures are often found to have lesions immediately adjacent to the cortex.
4.4 Histopathology
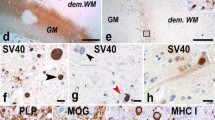
While the development of JCV PCR detection in the CSF has replaced brain biopsy as the most important diagnostic tool in PML care, evaluation of brain tissue is still done in cases of diagnostic uncertainty, especially when recurrence of lymphoma is a differential consideration or in cases of IRIS. Analysis of brain tissue can also increase our pathophysiological understanding of the disease. Biopsy or autopsy material of patients with PML is characterized by the combination of multifocal demyelination, bizarre astrocytes with hyperchromatic nuclei, and nuclear inclusions in oligodendrocytes in the white matter. When demyelination is extensive, necrosis can also be seen [36]. Lymphocyte infiltrates are not typically seen in PML lesions, unless there is presence of immune reconstitution (see the following section). Immunostaining can demonstrate presence of JCV capsid antigen, primarily in the white matter and typically in oligodendrocytes and to a lesser degree in astrocytes. Detection of JCV DNA in infected tissue with in situ hybridization or in situ PCR may be more sensitive and specific than immunohistochemistry [36]. In addition to causing the infection of oligodendrocytes characteristic for PML, JCV has also been reported by some to infect neurons, especially the granule cell neurons in the cerebellum. Histopathologically, granule cell neuropathy is characterized by hypochromatic granule cells with enlarged nuclei [37]. Significant gliosis is seen throughout the cerebellar cortex with relative sparing of the Purkinje cell and molecular layers [38]. Examples of typical demyelination and immunohistochemistry of autopsy brain tissue of a patient with PML are depicted in Fig. 5.

Histopathology in PML. Panel (a) shows multifocal demyelination of the brain (hematoxylin-eosin staining). Panel (b) shows evidence of JCV detected by immunohistochemistry
5 Treatment and Prognosis of PML
5.1 Inflammation and IRIS
In some patients with PML, rapidly worsening neurological symptoms, fever, and seizures develop. This clinical worsening is thought to be due to an improvement of immune function called immune reconstitution inflammatory syndrome (IRIS). IRIS occurs in 10–20 % of HIV patients that are started on antiretrovirals and in most patients with natalizumab (up to 90 %). HIV patients are at an increased risk of developing IRIS if they are antiretroviral naïve and have a CD4 count of less than 50 cells/mm3 [39]. IRIS typically develops about 3–12 weeks after antiretrovirals or plasma exchange to remove natalizumab are initiated, but in some cases, IRIS can develop up to 6 months after the initiation of antiretroviral therapy [42]. On MRI, there may be enhancement detected due to the local inflammation and breakdown of the blood-brain barrier. However, only in about 50 % of clinically or biopsy-diagnosed IRIS have contrast enhancement on MRI [39]. Edema or mass effect may also be seen on imaging. In the most severe cases, the inflammation and subsequent edema can lead to cerebral herniation and death. Analysis of brain biopsy samples of PML patients with and without IRIS shows an increase in cytotoxic CD8 T cells in patients with IRIS. This is associated with a better control of JCV dissemination but at the cost of oligodendrocyte cell death and demyelination [40, 41].
While there are no randomized trials assessing the best management of IRIS, most physicians use corticosteroids to dampen the immune response and avoid deleterious cerebral edema. A retrospective analysis of 54 patients with PML-IRIS in the setting of HIV infection showed that corticosteroids, especially if administered early and for a prolonged period of time, may improve survival [42].
5.2 Immune Responsiveness: T and B Cells
The immune system encompasses humoral, mainly B cell driven, as well as cellular, mainly T cell driven, immunity. The role of these components of the immune system in the development of and the recovery from PML is not fully understood, but there are many clues that can help answer this question. T-cell-mediated immune response appears to be a significant risk factor for developing PML as a deficit in CD4+ T cells is a prominent component of HIV infection. Rituximab, a drug that depletes B cells but not T cells, however, has also been found to be associated with PML risk, albeit at a lower rate than HIV infection [12]. A recent study showed that natalizumab-treated MS patients with PML have absent or aberrant JCV-specific T-cell responses compared with non-PML MS patients, indicating that changes in T-cell-mediated control of JCV replication may contribute to the risk of developing PML [21].
While brain biopsy or autopsy samples of PML without IRIS generally do not reveal many lymphocytes, in patients with IRIS, there are usually infiltrates with predominately cytotoxic CD8 T cells present. Plasma cells or macrophages can also be seen, but B cells or immunoglobulin deposits are uncommon, suggesting a mainly cellular immune response to PML at least in the case of IRIS [40, 41].
5.3 Treatment Targets: Failures and Future Directions
Several agents have been proposed as treatments for PML, but no specific anti-JCV therapy has been proven to have clinical efficacy to date. On the basis of in vitro experiments demonstrating inhibition of JCV replication as well as anecdotal case reports, intravenous and intrathecal cytarabine were tested in a clinical trial but neither form of application was found to be beneficial [43, 44]. Although cidofovir is not effective against JCV in cell culture, case reports and retrospective case series implicated efficacy in both HIV-positive and HIV-negative patients with PML. However, subsequent studies demonstrated no survival benefit and no improvement in residual disability at 12 months [45, 46]. Since JCV infection of glial cells is at least partially mediated through the serotonergic receptor 5-hydroxytryptamine receptor 2A (5HT2A) and several 5HT2A receptor antagonists blocked JCV infection of glial cells in vivo [6], the serotonin receptor blocker mirtazapine has been used off-label in a number of cases. No rigorous placebo-controlled trials have been undertaken, but analysis of the existing case series does not show any statistically significant clinical benefit [47]. A screen of chemical compounds indicated that the antimalarial drug mefloquine can inhibit JCV replication in vitro [48]. However, despite anecdotal reports of the beneficial effect of mefloquine, a multicenter clinical trial could not demonstrate an effect of mefloquine on CSF JCV titers, clinical, or MRI findings [49]. None of these agents is therefore recommended for the therapy of PML.
Since no specific therapy for PML has been identified, the main approach to treatment currently consists of a reversal of the immune suppression interfering with the normal host response to JCV. Treatment strategies depend on the patient’s underlying predisposing condition. In HIV-infected individuals, antiretroviral therapy (ART) is the most important aspect of PML management. In patients who are not on ART, this should be started immediately. For patients on ART but with detectable HIV viral load, antiretroviral resistance should be investigated, and their ART regimen should be optimized to accomplish viral suppression. More problematic are patients who develop PML despite successful viral suppression on ART. In these patients, intensification of their antiretroviral regimen with special attention to the CNS penetrance of their ART should be considered, though the effectiveness of this approach requires further study. While no study directly comparing the outcome of patients receiving ART and those not receiving ART in the setting of PML has been done, the comparison of the clinical outcomes of patients receiving ART and historic controls prior to the availability of ART shows a dramatic improvement in survival from 10 to 50 % when antiretrovirals were given [50]. In patients who develop PML due to treatment with immunomodulatory medications like natalizumab, removal of the immunomodulatory drug with plasmapheresis or immunoabsorption is generally recommended [34]. In patients with other underlying immune deficits (like idiopathic CD4+ lymphocytopenia or hematologic malignancy), however, restoring the immune system in a timely manner can be challenging or impossible. Immunomodulation with interferon alpha has been implicated after a retrospective analysis suggested improved mortality in HIV-infected patients with PML, though a subsequent study did not support a benefit of this treatment. Anecdotal reports suggested a benefit of interleukin-2 therapy, though no controlled trials have substantiated this claim. In summary, only initiation of antiretroviral therapy in HIV-infected individuals and discontinuation or removal of immunomodulatory or immunosuppressive medications can currently be recommended given the existing evidence.
As discussed above, immune reconstitution achieved by initiation of ART or removal of immunomodulatory medications is often associated with the development of IRIS. Corticosteroids are currently the mainstay of management of clinically significant IRIS, though further data are needed to establish the optimal dose and duration of this therapy.
5.4 PML Prognosis and JCV Persistence
The prognosis of PML is generally thought to be poor, though survival depends on the underlying condition. Before antiretrovirals were available, the mortality in HIV-infected individuals was about 90 %, whereas the outcome is significantly improved when viral load suppression is achieved with antiretrovirals [50]. A retrospective study assessing 87 patients with PML, mainly associated with HIV, showed no survival benefit for the 27 individuals who developed IRIS [39]. In patients who develop PML due to immunomodulatory therapy, mortality can be close to 90 % as in the reported cases of rituximab [12] or as low as 22 % as reported for natalizumab. This may be in part due to the fact that most PML cases associated with rituximab occur in patients with lymphoma and therefore worse general heath at baseline compared to MS patients who receive natalizumab. However, outside of survival, it is important to note that many natalizumab-treated patients who survive PML have significant residual disability [34].
Since we do not currently have specific treatments for PML and our main approach is to promote restoration of the immune system (while managing IRIS if it occurs), what happens with JCV in the brain is an interesting question. A small retrospective study analyzed serial CSF samples of a cohort with natalizumab-associated PML and found persistence of JCV in the CSF for 3 years or longer in more than 50 % of patients [51].
5.5 Epilogue
As summarized in this chapter, PML is a devastating demyelinating disease of the brain that typically occurs in individuals with an impaired immune system. The natural history of this disease and the role different risk factors play in the development and in the course of this disease are not sufficiently understood. As several new monoclonal antibodies and other immunomodulatory drugs are currently being developed and new medications have recently been implicated as risk factors for developing PML, the epidemiology of PML may be changing. Since PML is not a reportable disease, there are no publically available data to better study this disease. An additional challenge to the development of a unified approach to the diagnosis and management of PML is that patients with PML can be seen by physicians from different specialties like neurology, hematology-oncology, dermatology, gastroenterology, transplant medicine, or rheumatology. To improve our understanding of the epidemiology and underlying pathophysiology of PML, a group of experts from different institutions (Mayo Clinic, Rochester, MN; Cleveland Clinic, Cleveland, OH; Massachusetts General Hospital, Cambridge, MS; Center for Disease Control and Surveillance, Atlanta, GA; Washington University, St. Louis, MO; and NINDS, NIH, Bethesda, MD) has formed a steering committee and initiated and implemented a web-based disease registry for PML (https://pmlregistry.ninds.nih.gov). Health-care providers from around the world can enter anonymized clinical, radiographic, laboratory, and demographic data as well as treatment strategies and outcomes on their patients with PML, regardless of the underlying etiology. The data in this registry is stored on a secure server and administered by a team at the NIH. Researchers can request access to the data by contacting the steering committee, and a summary of the epidemiological data will be posted annually on the registry website. Additionally, the website serves as an informational source for providers as well as for patients and their families.
References
Imperiale M, Major EO. Polyomaviruses. In: Knipe DM, Howley PM, editors. Fields virology. 5th ed. Philadelphia, PA: Wolters Kluwer/Lippincott Williams and Wilkins; 2007. p. 2263–98.
Feltkamp MC, Kazem S, van der Meijden E, Lauber C, Gorbalenya AE. From Stockholm to Malawi: recent developments in studying human polyomaviruses. J Gen Virol. 2013;94(Pt 3):482–96. PubMed PMID: 23255626.
Egli A, Infanti L, Dumoulin A, Buser A, Samaridis J, Stebler C, et al. Prevalence of polyomavirus BK and JC infection and replication in 400 healthy blood donors. J Infect Dis. 2009;199(6):837–46.
Ferenczy MW, Marshall LJ, Nelson CD, Atwood WJ, Nath A, Khalili K, et al. Molecular biology, epidemiology, and pathogenesis of progressive multifocal leukoencephalopathy, the JC virus-induced demyelinating disease of the human brain. Clin Microbiol Rev. 2012;25(3):471–506. PubMed PMID: 22763635. Pubmed Central PMCID: 3416490.
Maginnis MS, Stroh LJ, Gee GV, O’Hara BA, Derdowski A, Stehle T, et al. Progressive multifocal leukoencephalopathy-associated mutations in the JC polyomavirus capsid disrupt lactoseries tetrasaccharide c binding. MBio. 2013;4(3):e00247–13. PubMed PMID: 23760462. Pubmed Central PMCID: 3685208.
Elphick GF, Querbes W, Jordan JA, Gee GV, Eash S, Manley K, et al. The human polyomavirus, JCV, uses serotonin receptors to infect cells. Science. 2004;306(5700):1380–3. PubMed PMID: 15550673.
Major EO, Elder G, Houff SA. Glial cells of the human developing brain and B cells of the immune system share a common DNA binding factor for recognition of the regulatory sequences of the human polyomavirus JCV. J Neurosci Res. 1990;27(4):461–71.
Monaco MC, Gravell M, Tornatore CS, Major EO. JC virus infection of hematopoietic progenitor cells, primary B lymphocytes, and tonsillar stromal cells: implications for viral latency. J Virol. 1996;70(10):7004–12.
Monaco MC, Sabath BF, Durham LC, Major EO. JC virus multiplication in human hematopoietic progenitor cells requires the NF-1 class D transcription factor. J Virol. 2001;75(20):9687–95. PubMed PMID: 11559801. Pubmed Central PMCID: 114540.
Tan CS, Dezube BJ, Bhargava P, Autissier P, Wuthrich C, Miller J, et al. Detection of JC virus DNA and proteins in the bone marrow of HIV-positive and HIV-negative patients: implications for viral latency and neurotropic transformation. J Infect Dis. 2009;199(6):881–8. PubMed PMID: 19434914. Pubmed Central PMCID: 2893283.
Ryschkewitsch CF, Jensen PN, Major EO. Multiplex qPCR assay for ultra sensitive detection of JCV DNA with simultaneous identification of genotypes that discriminates non-virulent from virulent variants. J Clin Virol. 2013;57(3):243–8. PubMed PMID: 23619054. Pubmed Central PMCID: 3698945.
Carson KR, Focosi D, Major EO, Petrini M, Richey EA, West DP, et al. Monoclonal antibody-associated progressive multifocal leucoencephalopathy in patients treated with rituximab, natalizumab, and efalizumab: a Review from the Research on Adverse Drug Events and Reports (RADAR) Project. Lancet Oncol. 2009;10:816–24.
Major EO. Progressive multifocal leukoencephalopathy in patients on immunomodulatory therapies. Annu Rev Med. 2010;61:35–47. PubMed PMID: 19719397.
Tan CS, Ellis LC, Wuthrich C, Ngo L, Broge Jr TA, Saint-Aubyn J, et al. JC virus latency in the brain and extraneural organs of patients with and without progressive multifocal leukoencephalopathy. J Virol. 2010;84(18):9200–9. PubMed PMID: 20610709. Pubmed Central PMCID: 2937633.
Major EO, Douek DC. Risk factors for rare diseases can be risky to define: PML and natalizumab. Neurology. 2013;81(10):858–9. PubMed PMID: 23925759.
Ryschkewitsch CF, Jensen PN, Monaco MC, Major EO. JC virus persistence following progressive multifocal leukoencephalopathy in multiple sclerosis patients treated with natalizumab. Ann Neurol. 2010;68(3):384–91. PubMed PMID: 20818792. Pubmed Central PMCID: 3739486.
Hamilton RS, Gravell M, Major EO. Comparison of antibody titers determined by hemagglutination inhibition and enzyme immunoassay for JC virus and BK virus. J Clin Microbiol. 2000;38:105–9.
Gorelik L, Lerner M, Bixler S, Crossman M, Schlain B, Simon K, et al. Anti-JC virus antibodies: implications for PML risk stratification. Ann Neurol. 2010;68(3):295–303. PubMed PMID: 20737510.
Berger JR, Houff SA, Gurwell J, Vega N, Miller CS, Danaher RJ. JC virus antibody status underestimates infection rates. Ann Neurol. 2013;74(1):84–90. PubMed PMID: 23526716. Pubmed Central PMCID: 3737275.
Major EO, Frohman E, Douek D. JC viremia in natalizumab treated patients with multiple sclerosis. New Engl J Med. 2013;368:2240–1.
Perkins MR, Ryschkewitsch C, Liebner JC, Monaco MC, Himelfarb D, Ireland S, et al. Changes in JC virus-specific T cell responses during natalizumab treatment and in natalizumab-associated progressive multifocal leukoencephalopathy. PLoS Pathog. 2012;8(11):e1003014. PubMed PMID: 23144619. Pubmed Central PMCID: 3493478.
Kean JM, Rao S, Wang M, Garcea RL. Seroepidemiology of human polyomaviruses. PLoS Pathog. 2009;5(3):e1000363. PubMed PMID: 19325891. Pubmed Central PMCID: 2655709.
Gheuens S, Pierone G, Peeters P, Koralnik IJ. Progressive multifocal leukoencephalopathy in individuals with minimal or occult immunosuppression. J Neurol Neurosurg Psychiatry. 2010;81(3):247–54. PubMed PMID: 19828476. Pubmed Central PMCID: 2889486.
Astrom K-E, Mancall Jr SL, EPR. Progressive multifocal leuko-encephalopathy a hitherto unrecognized complication of chronic lymphatic leukemia and Hodgkin’s disease. Brain. 1958;81(1):93–111.
Berger JR, Pall L, Lanska D, Whiteman M. Progressive multifocal leukoencephalopathy in patients with HIV infection. J Neurovirol. 1998;4:59–68.
Sacktor N. The epidemiology of human immunodeficiency virus-associated neurological disease in the era of highly active antiretroviral therapy. J Neurovirol. 2002;8 Suppl 2:115–21. PubMed PMID: 12491162.
Langer-Gould A, Green AJ, Bollen AW, Pelletier D. Progressive multifocal leukoencephalopathy in a patient treated with natalizumab. N Engl J Med. 2005;353:375–81.
von Geldern G, Calabresi PA, Newsome SD. PML-IRIS in a patient treated with Brentuximab. Neurology. 2012;79:2075–7.
Mateen FJ, Muralidharan R, Carone M, van de Beek D, Harrison DM, Aksamit AJ, et al. Progressive multifocal leukoencephalopathy in transplant recipients. Ann Neurol. 2011;70(2):305–22. PubMed PMID: 21823157.
Yousry TA, Pelletier D, Cadavid D, Gass A, Richert ND, Radue EW, et al. Magnetic resonance imaging pattern in natalizumab-associated progressive multifocal leukoencephalopathy. Ann Neurol. 2012;72(5):779–87. PubMed PMID: 23280794.
Berghoff M, Dassinger B, Iwinska-Zelder J, Giraldo M, Bilgin S, Kaps M, et al. A case of natalizumab-associated progressive multifocal leukoencephalopathy-role for advanced MRI? Clin Neuroradiol. Accessed on 27, 2013. PubMed PMID: 23532437.
Westwood TD, Hogan C, Julyan PJ, Coutts G, Bonington S, Carrington B, et al. Utility of FDG-PETCT and magnetic resonance spectroscopy in differentiating between cerebral lymphoma and non-malignant CNS lesions in HIV-infected patients. Eur J Radiol. 2013;82(8):e374–9. PubMed PMID: 23578921.
Boster A, Hreha S, Berger JR, Bao F, Penmesta F, Tselis A, Endress C, Zak I, Perumal J, Caon C, Vazquez J, Tyler KL, Racke MK, Millis S, Khan O. Progressive multifocal leukoencephalopathy and relapsing-remitting multiple sclerosis. Arch Neurol. 2009;66(5):593–9.
DA Clifford DB, Simpson DM, Arendt G, Giovannoni G, Nath A. Natalizumab-associated progressive multifocal leukoencephalopathy in patients with multiple sclerosis: lessons from 28 cases. Lancet Neurol. 2011;9(4):438–46.
Lima MA, Drislane FW, Koralnik IJ. Seizures and their outcome in progressive multifocal leukoencephalopathy. Neurology. 2006;66(2):262–4. PubMed PMID: 16434670.
Aksamit AJ, Gendelman HE, Orenstein JM, Pezeshkpour GH. AIDS-associated progressive multifocal leukoencephalopathy (PML): comparison to non-AIDS PML with in situ hybridization and immunohistochemistry. Neurology. 1990;40(7):1073–8.
Tan CS, Koralnik IJ. Progressive multifocal leukoencephalopathy and other disorders caused by JC virus: clinical features and pathogenesis. Lancet Neurol. 2010;9(4):425–37.
Keith J, Bilbao J, Baskind R. JC virus granular neuronopathy and rhombencephalic progressive multifocal leukoencephalopathy: case report and review of the literature. Neuropathology. 2012;32(3):280–4. PubMed PMID: 21981108.
Harrison DM, Newsome SD, Skolasky RL, McArthur JC, Nath A. Immune reconstitution is not a prognostic factor in progressive multifocal leukoencephalopathy. J Neuroimmunol. 2011;238(1–2):81–6. PubMed PMID: 21840066.
Martin-Blondel G, Bauer J, Cuvinciuc V, Uro-Coste E, Debard A, Massip P, et al. In situ evidence of JC virus control by CD8+ T cells in PML-IRIS during HIV infection. Neurology. 2013;81(11):964–70.
Metz I, Radue EW, Oterino A, Kumpfel T, Wiendl H, Schippling S, et al. Pathology of immune reconstitution inflammatory syndrome in multiple sclerosis with natalizumab-associated progressive multifocal leukoencephalopathy. Acta Neuropathol. 2012;123(2):235–45. PubMed PMID: 22057786. Pubmed Central PMCID: 3259335.
Tan K, Roda R, Ostrow L, McArthur J, Nath A. PML-IRIS in patients with HIV infection—clinical manifestations and treatment with steroids. Neurology. 2009;72:1458–64.
Hall CD, Dafni U, Simpson D, Clifford D, Wetherill PE, Cohen B, McArthur J, Hollander H, Yainnoutsos C, Major E, Millar L, Timpone J, and the AIDS Clinical Trials Group 243 Team. Failure of cytarabine in progressive multifocal leukoencephalopathy associated with human immunodeficiency virus infection. N Engl J Med. 1998;338:1345–51.
De Luca A, Giancola ML, Cingolani A, Ammassari A, Gillini L, Murri R, Antinori A. Clinical and virological monitoring during treatment with intrathecal cytarabine in patients with AIDS-associated progressive multifocal leukoencephalopathy. Clin Infect Dis. 1999;28:624–8.
De Luca A, Ammassari A, Pezzotti P, Cinque P, Gasnault J, Berenguer J, et al. Cidofovir in addition to antiretroviral treatment is not effective for AIDS-associated progressive multifocal leukoencephalopathy: a multicohort analysis. AIDS. 2008;22(14):1759–67. PubMed PMID: 18753934.
Marra CM, Rajicic N, Barker DE, Cohen BA, Clifford D, Donovan Post MJ, Ruiz A, Bowen BC, Huang M, Queen-Baker J, Andersen J, Kelly S, Shriver S, and the Adult AIDS Clinical Trials Group 363 Team. A pilot study of cidofovir for progressive multifocal leukoencephalopathy in AIDS. AIDS. 2002;16:1791–7.
Marzocchetti A, Tompkins T, Clifford DB, Gandhi RT, Kesari S, Berger JR, et al. Determinants of survival in progressive multifocal leukoencephalopathy. Neurology. 2009;73(19):1551–8. PubMed PMID: 19901246. Pubmed Central PMCID: 2777072.
Brickelmaier M, Lugovskoy A, Kartikeyan R, Reviriego-Mendoza MM, Allaire N, Simon K, et al. Identification and characterization of mefloquine efficacy against JC virus in vitro. Antimicrob Agents Chemother. 2009;53(5):1840–9. PubMed PMID: 19258267. Pubmed Central PMCID: 2681498.
Clifford DB, Nath A, Cinque P, Brew BJ, Zivadinov R, Gorelik L, et al. A study of mefloquine treatment for progressive multifocal leukoencephalopathy: results and exploration of predictors of PML outcomes. J Neurovirol. 2013;19(4):351–8. PubMed PMID: 23733308.
Clifford DB, Yiannoutsos C, Glicksman M, Simpson DM, Singer EJ, Piliero PJ, et al. HAART improves prognosis in HIV-associated progressive multifocal leukoencephalopathy. Neurology. 1999;52:623–5.
Ryschkewitsch CFJP, Monaco MC, Major EO. JC virus persistence following progressive multifocal leukoencephalopathy in multiple sclerosis patients treated with natalizumab. Ann Neurol. 2010;68:384–91.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2014 Springer Science+Business Media New York
About this chapter
Cite this chapter
von Geldern, G., Barhams, M.J., Major, E.O. (2014). The Biology and Clinical Consequence of Infection with the Human Polyomavirus JCV. In: Peterson, P., Toborek, M. (eds) Neuroinflammation and Neurodegeneration. Springer, New York, NY. https://doi.org/10.1007/978-1-4939-1071-7_16
Download citation
DOI: https://doi.org/10.1007/978-1-4939-1071-7_16
Published:
Publisher Name: Springer, New York, NY
Print ISBN: 978-1-4939-1070-0
Online ISBN: 978-1-4939-1071-7
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)