
Abstract
The lack of effective disease-modifying therapies for the treatment of Amyotrophic Lateral Sclerosis (ALS) demands for major research investments aimed at investigating novel mechanistic hypotheses as well as at validating unprecedented cellular and molecular targets for therapeutic intervention. Within this framework, glial cells have recently acquire great importance in view of the growing body of evidence indicating that motor neuron degeneration involves non-cell autonomous mechanisms in ALS, including the interaction with various glial cell populations. These observations not only have drawn attention to the physiopathological changes glial cells undergo during ALS progression, but they have moved the focus of the investigations beyond the neuronal compartment towards glia-neuron interactions. With this in mind, in this chapter, we dissect the specific contribution of the different glial subtypes to the dreadful chain of events leading to motor neuron sufferance and death in various forms of ALS. Furthermore, we discuss the possibility of targeting specific molecular defects in glial cell physiology and glia-neuron communication for the treatment of this disorder.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others

Keywords
- Amyotrophic lateral sclerosis
- Glia
- Astrocytes
- Microglia
- Oligodendrocytes
- Schwann cells
- NG2+ cells
- Neurodegeneration
- Transgenic animals
11.1 Amyotrophic Lateral Sclerosis: A Brief Introduction
Amyotrophic Lateral Sclerosis (ALS) is a chronic and incurable disease characterized by the impairment of motor function due to weakness, atrophy and spasticity of voluntary muscles. Disease signs and symptoms stem from the combined degeneration of corticospinal and spinal motor neurons, leading to progressive muscle denervation.
ALS is the most common form of adult-onset motor neuron disease, with an incidence and prevalence of about 1–3 and 4–6 per 100,000 individuals each year, respectively. Generally, it manifests between 40 and 60 years of age, with symptoms reflecting the early involvement of spinal (fasciculation, tremors, muscle weakness) or bulbar (dysphagia and dysarthria) motor neurons.
The diagnosis of ALS is made upon meeting the El Escorial World Federation of Neurology Criteria, namely clinical signs of both upper and lower motor neuron degeneration, along with evidence of spreading of the motor syndrome within a region or to other regions. These criteria should be combined to the absence of electrophysiological and neuroimaging signs of other disease processes that might justify the observed symptoms (Brooks et al. 2000) .
Although cognitive functions are not affected in most instances, about 50 % of patients show behavioural and linguistic abnormalities. The cognitive decline is generally mild, but, in about 15 % of patients, the impairment becomes so severe to call for an additional diagnosis of Frontotemporal Lobe Dementia (FTLD) (Lomen-Hoerth et al. 2002; Ringholz et al. 2005) .
The progression of the disease is usually rapid, and it is monitored by the Revised ALS Functional Rating Scale (ALSFRS-R)(Hardiman et al. 2011) , a validated rating tool that allows to assess the progression of the patient disability. A decline in the ALSFRS-R score is basically considered as a predictor of reduced survival.
Symptomatic therapy can help to relief muscle spasticity, excessive drooling, body weight loss and depression, thus contributing, at least to some extent, to the preservation of quality of life. Yet, the sole drug approved by the US Food and Drug Administration (FDA) for the cure of ALS is currently riluzole, which can extend the patient survival only by 3–6 months (Hardiman et al. 2011). Thus, despite a certain variability, the patient death generally occurs within 3 to 5 years from the diagnosis, mostly by respiratory failure. As a consequence, the discovery of novel effective disease-modifying therapies for the treatment of this devastating condition has presently a high priority in the scientific community. In this context, the identification of novel cellular and molecular targets for therapeutic intervention is highly desirable, and may importantly contribute to design unprecedented strategies for the treatment of ALS patients.
In the vast majority of cases, ALS appears sporadically (sporadic ALS, sALS). Presumed risk factors include aging , environmental agents (e.g., exposure to heavy metals) and life habits (e.g., smoking). Yet, in about 5–10 % of instances, the disease is inherited, mostly as an autosomal dominant trait (familial ALS, fALS). Familial ALS has been linked to mutations in various genes, including Superoxide dismutase 1 (SOD1) (Rosen et al. 1993) , TAR DNA binding protein 43 (TARDBP) (Kabashi et al. 2008; Sreedharan et al. 2008) , Fused in sarcoma (FUS) (Kwiatkowski et al. 2009; Vance et al. 2009) , Alsin (Yang et al. 2001) , Optineurin (Maruyama et al. 2010) , C9orf72 (DeJesus-Hernandez et al. 2011; Renton et al. 2011) , Ubiquilin 2 (Deng et al. 2011) and Profilin 1 (Wu et al. 2012) .
During the last few years, several mechanistic hypotheses have been formulated to explain the origin of ALS. Among others, the involvement of oxidative stress, excitotoxicity , impaired axonal transport, mitochondrial dysfunction and/or alteration of RNA metabolism has been contemplated (reviewed in Ferraiuolo et al. 2011a) . While most of these mechanisms appear to suggest that neurodegeneration arises from intrinsic defects in motor neurons, there is accumulating evidence supporting the concept that non-neuronal cells of the central nervous system (CNS) contribute to the dreadful chain of events ultimately causing motor neuron demise.
This observation has brought attention to the abnormalities affecting glial cells in ALS. At the histopathological level, the loss of upper and lower motor neurons, in both sporadic and familial cases, is accompanied by massive activation of glial cells in the affected areas, a phenomenon commonly described as “reactive gliosis”. Furthermore, in ALS patients and transgenic animals , there is evidence of ubiquitinated protein inclusions in motor neurons as well as in glial cells (Bruijn et al. 1997; Pasinelli et al. 2000; Mendonca et al. 2006) . Noteworthy, the ubiquitinated inclusions identified in various forms of fALS and sALS have a distinct pattern of protein composition, suggesting that different ALS-linked gene products may trigger distinctive neurodegenerative mechanisms.
To predict the deleterious potential of glial cells, it should be, however, noted that neuroglia include distinct cell populations characterized by an independent embryological origin and exerting different functions (reviewed in Allen and Barres 2009) . These include astrocytes, which represent the main effectors of the brain homeostatic system; microglia, the immunocompetent and specialized brain macrophages; oligodendrocytes and Schwann cells , which form layers of myelin around neuronal axons in the central and peripheral nervous system, respectively; and NG2+ cells , a peculiar type of glial cells that express the NG2 proteoglycan and receive direct synaptic input from neurons.
In this chapter, we present recent mechanistic studies, performed in cellular and animal models of the disease, emphasizing the contribution of distinct glial cell populations to the development and progression of ALS. Furthermore, we recapitulate the evidence for glial cell dysfunction in the human pathology. Lastly, we highlight the importance of testing glia-targeted agents for their potential to slow down or halt the progression of this disorder (reviewed in Valori et al. 2013) .
11.2 ALS Genetics and Experimental Models
The landmark discovery, achieved in the early 1990s (Rosen et al. 1993) , that SOD1 presents various mutations in a certain percentage of fALS cases (ALS-SOD) raised the possibility to generate transgenic animal models of the disease, which express mutant forms of the human Copper, Zinc Superoxide Dismutase (hSOD1) enzyme. Several different hSOD1 mutations were introduced into these genetically modified animals, mostly causing the development of an ALS-like syndrome in vivo (reviewed in Turner and Talbot 2008) . Since the disease arose even in the presence of normal or increased dismutase activity, it was early proposed that mutant SOD1 toxicity is due to a gain-of-function rather than to the loss of the normal enzymatic activity of SOD1 mutants. The nature of such aberrant function of mutant SOD1s remains mostly elusive, though several mechanistic hypotheses have been proposed.
The first ALS mouse model to be produced was the one carrying a mutant form of hSOD1 harbouring a glycine with alanine substitution in position 93 of the amino acid sequence (Gly93→Ala; hSOD1G93A mice; Gurney et al. 1994) . Extensive characterization of these animals revealed that they exhibit a phenotype that recapitulates several aspects of the human condition, being characterized by tremor, progressing to muscular weakness, paralysis, and eventually premature death. On a neuropathological standpoint, the behavioural abnormalities of hSOD1G93A mice are strictly linked to motor neuron degeneration in the ventral horns of the spinal cord (Turner and Talbot 2008), an event that is accompanied by reactive astrocytosis and microgliosis (Hall et al. 1998) . Moreover, in this mouse model, there is evidence of the presence of Lewy body-like inclusions containing SOD1 and ubiquitin in motor neurons as well as in various glial subpopulations (Bruijn et al. 1997; Pasinelli et al. 2000; Stieber et al. 2000) . Such glial inclusions were originally proposed to be the earliest indicator of the disease in the hSOD1G85R mice (Gly85→Arg substitution), another transgenic line developing a late onset, ultrarapid disease progression (Bruijn et al. 1997). Similar inclusions, containing active caspase-3, were subsequently reported in the same hSOD1G85R mice, as well as in hSOD1G93A mice, which show earlier onset, but analogously fast disease progression (Pasinelli et al. 2000). Yet, the significance of these glial protein aggregates in the context of ALS pathology remained unclear until recently (Rossi et al. 2008) . The theory that glial cells can contribute to ALS pathogenesis was corroborated only in 2003 owing to the revolutionary discovery, achieved in chimeric mice, that motor neuron degeneration involves non-cell-autonomous events, including the interaction with mutant SOD1-expressing glial cells (Clement et al. 2003) . As we shall discuss in the next sections, this observation moved the focus of the investigations from intrinsic defects and weakening of motor neurons to glia-neuron interactions. This paved the way for a new series of experiments apt to clarify the specific contribution of the different glial cell types to both motor neuron cell death and ALS pathogenesis (reviewed in Ilieva et al. 2009) .
While most of the currently available mechanistic information on ALS is based on observations made on mutant SOD1-expressing experimental models, the landscape of ALS genetics has greatly expanded over the last few years, thus prompting new investigations apt to clarify the functions of the newly identified disease proteins. Thus, much effort is currently being invested to study TDP-43, the protein encoded by the TARDBP gene. This protein was originally discovered as a major component of ubiquitinated protein inclusions in FTLD and sALS cases in 2006 (Neumann et al. 2006) . Two years later, mutations in the TARDBP gene were identified in fALS patients (Kabashi et al. 2008; Sreedharan et al. 2008) , thus strengthening the hypothesis that TDP-43 may be involved in the development of the disease (ALS-TDP). At the cellular level, TDP-43 is normally concentrated in the nucleus, but can also shuttle back and forth between the nucleus and the cytoplasm (Ayala et al. 2008) . It is a global regulator of gene expression and is involved in the control of transcription as well as in multiple aspects of RNA processing and functioning. Besides, this protein can redistribute to cytosolic granules in response to neuronal stress, although nuclear localization is restored after recovery (Dewey et al. 2011; McDonald et al. 2011) . Concerning ALS, there is no clear consensus of how pathological TDP-43 operates within diseased cells (Halliday et al. 2012) .
In 2009, mutations in the FUS gene were also identified in about 4 % of fALS cases (ALS-FUS) (Kwiatkowski et al. 2009; Vance et al. 2009) . This gene codes for a ubiquitously expressed, predominantly nuclear, protein that is involved in DNA repair and regulation of transcription, RNA splicing, and export to the cytoplasm. Analyses of ALS-FUS cases, expressing different forms of the mutant protein, allowed to identify two distinct clinical and neuropathological patterns (Mackenzie et al. 2011) , which correlate with a different extent of cytoplasmic FUS mislocalization (Dormann et al. 2010) . Because many of the mutations identified in fALS patients cluster in the C-terminal domain of the FUS protein, and this includes a non-classic nuclear localization signal, it was shown that FUS mutations impair the physiological nuclear import of FUS (Dormann et al. 2010). However, similar to TDP-43, it is presently highly debated whether FUS mutations induce gain of toxic functions or loss of protective activities (Halliday et al. 2012). The discovery of these novel ALS-related genes led to a new generation of transgenic animal models suitable to explore the function of TDP-43 and FUS in vivo as well as to perform mechanistic studies (Lanson and Pandey 2012; Tsao et al. 2012) . As outlined below, some of these animals have been used to specifically investigate the impact of ALS glia on the disease outcome.
Because most of the currently available evidence on glial pathology relates to mutations/mislocalization of SOD1, TDP-43 or FUS, herein we specifically focus on the forms of ALS linked to these proteins, and we discuss the potential of glial cell dysfunction towards neurodegeneration and disease progression in these contexts.
11.3 Astrocytes
Considerable evidence indicates that astrocytes represent the main type of glia in the CNS and critically control the brain homeostasis, being responsible for all aspects of metabolic support, nutrition, control of ion and transmitter environment, regulation of brain-bloodbarrier (BBB), and defense of the CNS (reviewed in Rossi et al. 2011) . Considering the diversification and complexity of such astrocytic activities, it is reasonable to postulate that any astroglial dysfunction may impact the maintenance and performance of the CNS. This hypothesis has been corroborated by experimental evidence suggesting that astrocytes are critically involved in the development and progression of several neurological disorders, including ALS (reviewed in Parpura et al. 2012) . As we shall see in the next paragraphs, astroglial cells were shown to be implicated in various pathogenetic pathways in ALS, where they appear to exert a dual role: on the one hand, they were shown to actively contribute to motor neuron damage by secreting neurotoxic factors; on the other hand, astrocytes were reported to lose some of their physiological functions and, ultimately, undergo themselves degeneration. Thus, it seems that the pathological process of ALS transforms astrocytes from supportive friends for neurons into noxious foes, and ultimately eliminates them. The most straightforward consequence of these events is that neurons, deprived of their interactive partners, start suffering and eventually undergo accelerated cell death .
11.3.1 Direct Contribution of the Astrocytes to the Development of Neuronal Cell Death and Disease Manifestations
Several approaches in vivo and in vitro have been implemented in the last few years that aim to clarify the role of astrocytes in ALS. The initial generation of transgenic mice with restricted expression of the murine G86R SOD1 mutation (G85R in humans) in astrocytes resulted in astrocytosis, but failed to reproduce the neurodegenerative phenotype. This led to the early conclusion that the limited expression of mutant SOD1 in astrocytes is not sufficient to trigger the disease (Gong et al. 2000) . At variance with this, subsequent studies using chimeric mice revealed that wild-type non-neuronal cells, located in the microenvironment of mutant SOD1-expressing motor neurons, increase neuronal survival. In turn, the presence of mutant SOD1-expressing non-neuronal cells, in the motor neuronal neighbourhood, is sufficient to induce the formation of ubiquitinated protein inclusions and to elicit sufferance within wild-type motor neurons (Clement et al. 2003) . These seminal findings introduced the original idea that it is actually the interaction between motor neurons and their non-neuronal neighbours that critically impact the neurodegenerative process in ALS (Clement et al. 2003). In this context, alterations to astrocytes became very important, considering that these cells cover a wide range of functions that are critically involved in the maintenance and activity of neuronal cells. Several studies in vivo and in vitro have been then undertaken to clarify the impact of astroglial cells towards ALS-linked neurodegeneration and pathology. In particular, two distinct experimental strategies have been exploited to address these issues in vivo. Firstly, a variety of animal models have been generated by ablating mutant SOD1 expression selectively in astrocytes by means of the Cre/loxP recombination system (Yamanaka et al. 2008) . This genetic approach allowed to unveil that reducing the expression of the hSOD1G37R protein (Gly37→Arg substitution) within the astrocytes positively modulates the phenotype of mutant SOD1 transgenic mice by slowing down ALS progression (Yamanaka et al. 2008). Such results were soon expanded by the observation that depletion of the hSOD1G85R protein from astrocytes postponed the onset of the disease, in addition to extend the mouse lifespan (Wang et al. 2011) . Secondly, the neurotoxic potential of mutant SOD1 astroglial cells has been directly investigated by means of cell transplantation strategies. The introduction of hSOD1G93A-expressing astrocyte precursors into the spinal cord of wild-type rodents revealed that astrocytes alone can trigger motor neuron degeneration and ALS symptoms in vivo (Papadeas et al. 2011) . Complementary to this, the transplantation of wild-type astrocyte precursors into rodent models of ALS delayed disease progression and extended the animal life span, thus opening the perspective of astrocyte replacement-based therapeutic approaches (Lepore et al. 2008a) .
Although the vast majority of the investigations addressing the contribution of astrocytes to ALS pathogenesis was performed on experimental models of ALS-SOD, the recent generation of new transgenic animals , which mimic other forms of fALS, allowed to gain further insights into the impact of these cells on the disease outcome. Some important clues came from the recent characterization of a transgenic rat line showing tetracycline-inducible expression of the mutant TDP-43M337V (Met337→Val substitution) protein selectively in astrocytes (Tong et al. 2013) . Upon doxycycline withdrawal, these animals exhibited selective expression of the transgene in astrocytes, and this led to the first signs of motor weakness in as little as 20 days. The phenotype worsened rapidly resulting in complete paralysis within additional 20 days. Histopathological analysis revealed denervation atrophy of skeletal muscles, progressive loss of spinal cord motor neurons, microglial activation and ubiquitin-positive inclusions within astrocytes (Tong et al. 2013). In keeping with this, another recent report provided complementary information by investigating the impact of the expression of four different variants of human TDP-43 (D169G, G298S, A315T and N345K) in glia in vivo, using Drosophila as a model. Remarkably, glial expression of mutant TDP-43 resulted in significantly smaller neuromuscular junctions (NMJs), with a decreased number of synaptic boutons. This correlated with a considerable impairment in the locomotor performance (Estes et al. 2013) .
A distinct subpopulation of highly proliferating astrocytes, with an enhanced neurotoxic phenotype, has been then isolated from mutant hSOD1G93A-expressing transgenic rats (Diaz-Amarilla et al. 2011) . Increased proliferation of astroglial cells in the spinal cord of hSOD1G93A ALS mice was reported also in a different study, which suggested the implication of the Wnt signalling pathway in the development of the proliferating glial phenotype (Chen et al. 2012) . Wnt is a family of highly conserved signalling moleculesthat, in the CNS, play complex and diversified roles during development, but it is also involved in the maintenance of synaptic integrity (reviewed in Rosso and Inestrosa 2013) . Of note, alterations of this transduction pathway have been associated to several neurological disorders (reviewed in Al-Harthi 2012) . In the hSOD1G93A ALS mouse model, the expression of several members of the Wnt family was shown to be de-regulated (Yu et al. 2013) . In particular, some of these proteins resulted to be over-expressed in astrocytes, thus suggesting that the enhanced proliferative potential of astroglial cells might be a direct consequence of increased Wnt signalling (Li et al. 2013; Yu et al. 2013) . Yet, the relevance of proliferating astroglial cells towards the ALS phenotype was not corroborated in vivo, as the selective ablation of dividing astrocytes by genetic approaches was shown not to affect any measures of the disease outcome in mice (Lepore et al. 2008b) .
To gain new mechanistic insights into the contribution of astrocytes to motor neuron degeneration in vitro , several groups then established astrocyte-motor neuron co-culture systems, which allowed direct monitoring of the impact of ALS astroglia towards neuronal viability, with no interference by other neural cell types. By this means, they demonstrated that diseased astrocytes can impact motor neuron survival by releasing noxious factors (Di Giorgio et al. 2007; Nagai et al. 2007; Bilsland et al. 2008; Di Giorgio et al. 2008; Marchetto et al. 2008; Ferraiuolo et al. 2011b; Haidet-Phillips et al. 2011; Phatnani et al. 2013) . Although the identity of these toxic agent(s) remains mostly elusive, different molecules have been proposed to play a role in this dreadful intercellular cross-talk (Table 11.1). The first candidate, in terms of deleterious molecules, was initially indicated in the excitatory amino acid glutamate, which is well known to trigger neurodegeneration by excitotoxic mechanisms. This hypothesis was initially fuelled by multiple evidence pointing out a defect in the astrocyte-specific plasma membrane glutamate transporter EAAT2 (also known as GLT-1 in rodents) in both ALS patients and mutant SOD1 transgenic animals (Rothstein et al. 1992, 1995; Bruijn et al. 1997; Howland et al. 2002; Guo et al. 2003; Pardo et al. 2006) . EAAT2 is a high-affinity glutamate transporter that is critically involved in the clearance of glutamate from the synaptic cleft, and it is responsible for interrupting the activity of this excitatory neurotransmitter at the synapse (Danbolt 2001) . However, genetic studies revealed that EAAT2 plays a major role also in maintaining extracellular glutamate below excitotoxic levels (Rothstein et al. 1996; Tanaka et al. 1997) . Based on this, it was reasonably postulated that its impairment can cause abnormal increases in the extracellular concentration of glutamate, and this leads to neuronal cell death. Such hypothesis was boosted by the observation that abnormal levels of glutamate were detected in the cerebrospinal fluid (CSF) and spinal cord of sporadic ALS patients when compared to control individuals (Rothstein et al. 1990). Remarkably, defects in the glutamate uptake system were lately confirmed also in transgenic models of ALS, other than ALS-SOD. Thus, rats expressing the mutant M337V form of TDP-43 in astrocytes exhibited a progressive depletion of the astroglial glutamate transporters EAAT1 and EAAT2 in the spinal cord (Tong et al. 2013) . Furthermore, a recent study in Drosophila revealed that both depletion or overexpression of TBPH, the Drosophila homologue of human TDP-43, in glial cells caused detrimental effects in vivo. While the selective depletion of TBPH from glia led to age-related motor abnormalities, its over-expression caused premature lethality during the larval status. Importantly, both loss and gain of Drosophila TDP-43 were reported to alter the expression levels of the glutamate transporters EAAT1 and EAAT2 (Diaper et al. 2013) .
As yet, the mechanisms leading to EAAT2 depletion in ALS have not been fully elucidated, although several mechanistic hypotheses have been postulated. Initially, it was reported that the loss of this transporter was the consequence of aberrant splicing of its messenger RNA (mRNA) (Lin et al. 1998) . Yet, this event was later proved unspecific for ALS cases, as it was observed also in control and Alzheimer’s Disease patients (Honig et al. 2000) . EAAT2 was subsequently proposed to be a substrate of caspase-3 (Boston-Howes et al. 2006) , an enzyme executing the proteolytic phase of apoptotic programmed cell death. Cleavage generates a C-terminal fragment, and leads to inhibition of the transporter activity (Boston-Howes et al. 2006). The product of EAAT2 cleavage was also described to undergo additional post-translational modification, namely SUMOylation (Gibb et al. 2007) , and to accumulate in spinal astrocytes from hSOD1G93A mice at the symptomatic stage (Foran et al. 2011) . More recently, an additional theory has been proposed, based on the occurrence of aberrant editing events in intron 7 of the EAAT2 pre-mRNA in the affected areas of ALS patients (Flomen and Makoff 2011) . Such events appears to lead to alternative polyadenylation, ultimately causing premature transcriptional termination and reduced levels of EAAT2 (Flomen and Makoff 2011). Down-regulation of EAAT2 was also suggested to be the result of defective signalling from neurons. For example, presynaptic terminals were shown to transcriptionally control the expression of EAAT2 by modulating the astrocytic levels of the nuclear factor Kappa-B Motif-Binding Phosphoprotein (KBBP). The loss of presynaptic signalling, following axonal degeneration, resulted in diminished EAAT2 expression (Yang et al. 2009) . Finally, experiments in culture suggested that neuronal cells have the capacity to release exosomes containing the microRNA 124a (miR-124a) (Morel et al. 2013) . Such organelles can be internalized by astrocytes, where they increase the astrocytic miR-124a. In turn, the latter up-regulates EAAT2 expression through a yet unidentified mechanism (Morel et al. 2013). Since miR-124a expression is down-regulated in the spinal cord of hSOD1G93A mice, it was speculated that its reduced expression may be responsible for the diminished levels of EAAT2 in vivo (Morel et al. 2013). While this amount of evidence globally failed to provide definitive conclusions about the mechanism driving EAAT2 loss in ALS, it prompted the idea that the relevance of the transporter in ALS may be determined by directly modulating its expression in vivo. This hypothesis was pursued by genetic and pharmacological approaches. Thus, transgene-driven expression of EAAT2 in the hSOD1G93A mouse line was shown to delay neurodegeneration and disease onset (Guo et al. 2003) . Furthermore, double transgenic mice obtained by crossing hSOD1G93A ALS mice with animals showing ubiquitous over-expression of the Peroxisome Proliferator-Activated Receptor γ Coactivator 1α (PGC1α) exhibited enhanced EAAT2 expression, and this correlated with improved motor function (Liang et al. 2011) . From a pharmacological standpoint, the screening of a panel of US FDA-approved drugs revealed that beta-lactam antibiotics, and ceftriaxone in particular, are capable to enhance EAAT2 expression (Rothstein et al. 2005) . Chronic administration of ceftriaxone in hSOD1G93A transgenic mice extended the animal lifespan (Rothstein et al. 2005), thus raising great hope that this compound may successfully control disease progression in ALS patients. It is to mention that a clinical trial with ceftriaxone is currently ongoing, and results from Phase I and II indicate that this drug is generally well tolerated and can successfully reach the CNS at therapeutic doses (Berry et al. 2013) . In an attempt to identify other compounds that enhance EAAT2 expression, a further screening was undertaken by the same group, which led to the identification of harmine (Li et al. 2011) , a beta-carboline alkaloid, as a promising agent to be tested in vivo, in preclinical trials.
Further support to the excitotoxic hypothesis of ALS was provided by the landmark observation that mutant SOD1-expressing astrocytes can release high levels of D-serine, a co-activator of the N-methyl-D-aspartate (NMDA) receptors, and this exacerbates glutamate toxicity on motor neurons in vivo (Sasabe et al. 2007, 2012) . The relevance of this amino acid was corroborated also by the identification, in a fALS pedigree, of a unique mutation (Arg199→Trp substitution) in the gene coding for D-amino acid oxidase (DAOR199W), an enzyme that regulates the levels of D-serine (Mitchell et al. 2010) . In vitro characterization revealed that this mutation not only causes the loss of DAO enzymatic activity, but also promotes both the formation of ubiquitin aggregates and apoptosis in neuronal cells. Remarkably, the detrimental effect of the mutant protein was observed even when motor neurons were co-cultured on DAOR199W-expressing astrocytes, thus suggesting that the neurodegenerative process can be mediated by both cell autonomous and non-cell autonomous events (Mitchell et al. 2010).
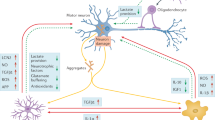
Increased production of reactive oxygen species (ROS), following mitochondrial dysfunction of mutant hSOD1G93A astrocytes, was also counted among the mechanisms contributing to motor neuron sufferance (Fig. 11.1). Oxidant species and Nitric Oxide Synthase (NOS) products were proposed to be involved in the neurotoxic process in vitro (Cassina et al. 2008) . In keeping, the over-production of ROS by human astrocytes expressing the hSOD1G37R mutant protein was corroborated by the observation that activation of NADPH oxidase (NOX2) contributes to the establishment of a condition of oxidative stress (Marchetto et al. 2008) . Interestingly, pharmacological inhibition of NOX2 activity by the drug apocynin reversed both ROS production and motor neuron cell death driven by mutant SOD1-expressing astrocytes (Marchetto et al. 2008). In addition, pharmacological (Cassina et al. 2008) or genetic (Vargas et al. 2008) manipulations apt to support the mitochondrial activity of astrocytes (Miquel et al. 2012) , or to potentiate the antioxidant defenses of the cells, (Vargas et al. 2008) showed a neuroprotective potential, thus establishing a direct link between the astrocytic production of free radicals and the process of neuronal cell death.

Impact of glial cells on motor neuron sufferance in ALS-SOD. In normal conditions, healthy astrocytes support the welfare of motor neurons by supplying trophic and metabolic factors as well as by scavenging potentially harmful agents. The transcription factor Nrf2 mediates the expression of several antioxidant proteins. Normally, its activity is repressed by the Keap1 protein in cytoplasm. However, when cells are exposed to chemical or oxidative stress, Nrf2 escapes Keap1-mediated repression, translocates into the nucleus, and promotes the expression of enzymes involved in the detoxification response. In the presence of mutant SOD1, the Nrf2-Keap1 pathway is down-regulated, and both oxidant species and Nitric Oxide Synthase (NOS) products are release from astrocytes following mitochondrial dysfunction (top, in green). During ALS, mutant SOD1 expression greatly impacts on the astroglial function, and some astrocytes (bottom, in green) display an enhanced susceptibility to physiological concentrations of the transmitter glutamate. This latter triggers a gliodegenerative process via the activation of its metabotropic receptor type 5 (mGluR5). mGluR5 activation induces aberrant Ca2+ release from the intracellular stores, mitochondrial disarrangement with cytochrome c release, and apoptotic cell death. In microglial cells (in blue), the activation of NADPH oxidase (NOX2) participates to the establishment of a condition of oxidative stress. SOD1 normally contributes to the regulation of NOX2 activity by binding to its regulatory protein Rac1. In the presence of mutant SOD1, a stronger interaction with Rac1 is established, and this leads to aberrant NOX2 activation with the consequent over-production of reactive oxygen species
The idea that targeting oxidative stress in astrocytes might be an efficient therapeutic option for ALS was further corroborated by a number of recent studies focused on the nuclear factor erythroid 2-related factor 2 (Nrf2). Nrf2 is a transcription factor that mediates important cellular defense mechanisms against oxidative stress, through its capacity to regulate the expression of an array of antioxidant proteins. Under normal condition, Nrf2-dependent transcription is repressed by the inhibitor Kelch-like ECH-associated protein 1 (Keap1), which sequesters Nrf2 in the cytoplasm and promotes its degradation by the ubiquitin proteasome pathway. However, when cells are exposed to chemical or oxidative stress, Nrf2 escapes Keap1-mediated repression and translocates into the nucleus. This allows its interaction with the antioxidant response element (ARE), a cis-acting regulatory sequence located in the promoter region of a number of genes. By this means, Nrf2 promotes the expression of enzymes involved in the detoxification and antioxidant response (Bryan et al. 2013) . The fact that the Nrf2-Keap1 signalling pathway may be involved in neuroprotective mechanisms in ALS was initially suggested by multiple investigations in vitro , in motor neuronal cell culture systems expressing either hSOD1G93A or various TDP-43 mutants (Kirby et al. 2005; Pehar et al. 2007; Duan et al. 2010) . Similar conclusions arose from additional studies investigating the expression of Nrf2 and Keap1 in post-mortem tissue samples from ALS patients (Sarlette et al. 2008) and hSOD1G93A transgenic animals (Mimoto et al. 2012) . Thus, the impact of different compounds acting on this signalling cascade was investigated in the hSOD1G93A animal model of the disease. Among the activators of the Nrf2/ARE system taken into consideration, there are DL-3-n-butylphthlide (Feng et al. 2012) , S[+]-Apomorphine (Mead et al. 2013) , and triterpenoids (Neymotin et al. 2011) . While all of these molecules resulted effective in improving the animal motor performance (Neymotin et al. 2011; Feng et al. 2012; Mead et al. 2013), the overall impact on survival was either modest (Neymotin et al. 2011; Feng et al. 2012) or even null (Mead et al. 2013). Consistent with this, genetic approaches apt to modulate neuronal Nrf2 expression in vivo, in the hSOD1G93A and hSOD1G85R mouse models, moderately delayed disease onset without affecting the animal lifespan (Vargas et al. 2013) . Similarly, a gene therapy protocol apt to induce Nrf2 over-expression in neurons failed to mitigate the detrimental phenotype in hSOD1G93A ALS mice (Nanou et al. 2013) . At variance with these results, Nrf2 activation in astrocytes gave more promising results. Firstly, increasing Nrf2 activity in astrocytes, by transfection or pharmacological methods, resulted neuroprotective in astrocyte-motor neuron co-culture experiments (Vargas et al. 2005, 2006). Secondly, transgene-driven expression of Nrf2 in astrocytes prevented motor neuron degeneration, delayed disease onset, and extended survival of hSOD1G93A ALS mice (Vargas et al. 2008) (Fig. 11.1).
Although not all studies agree (Guo et al. 2013) , these reports suggest that the Nrf2-Keap1 pathway may represent an attractive pharmacological target for therapeutic intervention in ALS. More importantly, they emphasize the concept that Nrf2 activation should be selectively achieved in the astrocytes.
Besides glutamate and oxidant species, additional neurotoxic candidate molecules of astrocytic origin were indicated in different inflammatory mediators . For example, abnormal signalling in astrocytes from different prostaglandins (PGs) was initially proposed as a likely contributor to motor neuron demise (Di Giorgio et al. 2008) . Analysis of the expression of the PGD2 and PGE2 receptors in fact revealed that both proteins are up-regulated in hSOD1G93A astrocytes (Di Giorgio et al. 2008; Liang et al. 2008) . Besides, the relevance of the PGE2 EP2 receptor was directly investigated in vivo. Importantly, its genetic ablation was reported to limit the pro-inflammatory response, to delay the onset of motor impairment, and to extend the lifespan of hSOD1G93A ALS mice (Liang et al. 2008). An abnormal signal transduction pathway, leading to motor neuron cell death, was identified also upon the release of interferon γ(IFNγ) from hSOD1G93A astrocytes in co-culture experiments (Aebischer et al. 2011) . More recently, an investigation of the gene expression profile of mutant SOD1-expressing astrocytes identified striking changes even in a panel of growth factors. Thus, analysis of hSOD1G93A astrocytes and motor neurons in co-culture revealed a complex and interconnected alteration in the transcriptome of these cells, suggesting a potential role for Transforming Growth Factor β (TGFβ) signalling in ALS (Phatnani et al. 2013) . Furthermore, an up-regulation of the TGFβ type II receptor (TGFβ–RII) was described in motor neurons from hSODG93A mice, with a peak of expression occurring before the onset of the disease. Interestingly, such increase in TGFβ-RII immunoreactivity temporally paralleled the progression of reactive astrocytosis, as indicated by the presence of thicker and hypertrophic astrocytes around TGFβ-RII-immunopositive motor neurons (Phatnani et al. 2013) . Transcription profile analysis of astrocytes isolated by laser-capture microdissection from the lumbar spinal cord of pre-symptomatic hSOD1G93A mice further revealed an up-regulation of Nerve Growth Factor β (β-NGF) (Ferraiuolo et al. 2011b) , a molecule that triggers apoptotic cell death in the neighbouring motor neurons through the activation of the p75 neurotrophin receptor (Pehar et al. 2007) . An increased release of β-NGF from astrocytes expressing the mutant hSOD1G93A protein was confirmed in vitro, and immunodepletion of this peptide was shown to rescue the neurotoxic effect of hSOD1G93A astrocytes in co-cultures (Ferraiuolo et al. 2011b). It should be, however, mentioned that the contribution of glial cells to motor neuron degeneration in vitro was not only shown in those forms of the disease linked to SOD1 mutations. In fact, TDP-43 was reported to interact with the Nuclear Factor-kB (NF-kB), a master regulator of several genes involved in the inflammatory response, in both neuronal and glial cells (Swarup et al. 2011) . The direct consequence of this event is that, under stress conditions, TDP-43-expressing microglia and astrocytes produce enhanced pro-inflammatory cytokines as well as neurotoxic mediators (Swarup et al. 2011). A role for the astroglial NF-kB signalling complex in mediating the inflammatory response was further supported by a recent gene expression study made on astrocytes from familial and sporadic ALS patients (Haidet-Phillips et al. 2011) .
Among the reported neurotoxic factors secreted by ALS astrocytes, there is also lipocalin 2 (lcn2), a protein characterized by the ability to bind and transport lipids and other hydrophobic molecules. While lcn2 is involved in diverse cellular processes, there is considerable evidence suggesting that this protein is able to induce cell death by stimulating an apoptotic program (Devireddy et al. 2001, 2005) . Remarkably, Bi and colleagues recently reported that lcn2 is released from reactive astrocytes in cultured organotypic brain slices from transgenic rats with neuron-specific expression of mutant human TDP43M337V. Furthermore, in the brain of transgenic rats expressing mutant forms of TDP-43, FUS, or SOD1, this protein was strongly induced in reactive astrocytes (Bi et al. 2013; Tong et al. 2013) . In vitro, lcn2 is cytotoxic to primary neurons, and toxicity is enhanced in cells that express disease genes, such as mutant FUS or TDP-43 (Bi et al. 2013).
Altogether, these observations suggest that the dynamic interactions between astrocytes and motor neurons become impaired at different levels during ALS progression.
11.3.2 Astrocyte Dysfunction and Sufferance
Several lines of evidence indicate that some of the neurosupportive functions of the astrocytes can be lost during the progression of the disease . As mentioned in the previous paragraph, astrocytes were consistently reported to exhibit a dysfunction of the glutamate uptake system in ALS patients and transgenic rodents, and this was proposed to contribute to excitotoxic motor neuron cell death (Rothstein et al. 1992, 1995; Bruijn et al. 1997; Howland et al. 2002; Guo et al. 2003; Pardo et al. 2006) . In addition, astroglia were reported to modulate the intrinsic susceptibility of motor neurons to glutamate by fine-tuning the expression of the GluA2 subunit of the glutamatergic α-amino-3-hydroxy-5-methyl-isoxazole propionate receptors (AMPARs) (Van Damme et al. 2007) . Remarkably, the presence of GluA2 in the AMPAR subunit composition critically impacts the biophysical properties of the receptor and determines its permeability to calcium ions (Ca2+). In particular, the expression of GluA2 normally renders AMPARs impermeable to Ca2+, because of an RNA editing event of the Q/R site (Q, unedited; R, edited) in the GluR2 mRNA (Sommer et al. 1991) . Yet, astrocyte expressing the mutant hSOD1G93A protein appear to lose their capacity to modulate the expression of the GluA2 subunit of the AMPARs in the neighbouring motor neurons, thereby exposing them to enhanced Ca2+ influx and cell death (Van Damme et al. 2007). Besides their involvement in excitotoxic mechanisms, ALS astroglia were described to hold additional functional deficits, including mitochondrial defects as well as a reduced capacity to release the metabolic substrate lactate (Cassina et al. 2008; Ferraiuolo et al. 2011b) . Furthermore, the early observations of ubiquitinated SOD1- and active caspase-3-immunopositive inclusions in these cells, in the two hSOD1G93A and hSOD1G85R mouse models of the disease, raised the original idea that mutant SOD1 can be toxic to astrocytes themselves (Bruijn et al. 1997; Pasinelli et al. 2000) . In subsequent experiments, our group investigated the significance of such astrocytic inclusions in hSOD1G93A- expressing transgenic mice. We found that these unusual cells are specifically located in the motor neuronal microenvironment in the ventral horns of the spinal cord (Fig. 11.2) . Furthermore, they display morphological and biochemical features reminiscent of degenerating cells, including an atypical spheroid morphology with an increased diameter and a reduced number or even the absence of cell processes immunopositive for the Glial Fibrillary Acid Protein (GFAP) (Rossi et al. 2008; Martorana et al. 2012) . Dying astrocytes were first observed at the pre-symptomatic stage (Rossi et al. 2008), i.e. at a time when motor neurons exhibit axonal damage, but their somas are still vital (Pun et al. 2006) . The number of spheroid astrocytes significantly increased concomitant with the onset of neuronal degeneration and the appearance of ALS symptoms (Rossi et al. 2008; Martorana et al. 2012). The relevance of this phenomenon was more recently reinforced in the context of the human disease by the identification of cells with an analogous aberrant morphology in the spinal cord of ALS patients (Martorana et al. 2012). Interestingly, we realized that degenerating astrocytes, in the spinal cord of hSOD1G93A mice, were surrounded by glutamatergic terminals (Fig. 11.2), as to indicate that these cells can sense the neurotransmitter glutamate spilled over during synaptic activity (Rossi et al. 2008; Martorana et al. 2012) . Mechanistic studies in vitro revealed that astrocytes expressing different SOD1 mutations, either G93A or G85R, displayed an enhanced susceptibility to physiological concentrations of glutamate. The glutamatergic mechanism responsible for the deleterious event was found to involve metabotropic glutamate receptor 5 (mGluR5) signalling, which triggers an apoptotic gliodegenerative pathway in culture (Rossi et al. 2008) . More recently, the signalling prompted by the activation of group I mGluRs, including mGluR5, was further characterized .

Degenerating astrocytes appear in the hSOD1G93A ALS mouse model at the pre-symptomatic stage of the disease. Arrows show ubiquitin-positive (in blue) astrocytes labelled with the astroglial marker Glial Fibrillary Acidic Protein (GFAP, in green). Degenerating astroglial cells are located in the motor neuronal microenvironment and became visible early during disease progression in the lumbar spinal cord of hSOD1G93A mice. They are enclosed by glutamate-containing pre-synaptic terminals immunolabelled for the Vesicular Glutamate Transporter 1 (VGLUT1, in red). Arrowheads indicate VGLUT1-positive glutamatergic vesicles and asterisks show motor neuronal somas. Scale bar, 20 μm
In normal astroglial cells, the activation of group I mGluRs is well known to cause the formation of inositol 1,4,5 trisphosphate (IP3), followed by IP3-mediatedrelease of Ca2+ from the endoplasmic reticulum (ER) stores, which results in intracellular Ca2+ oscillations (Zur Nieden and Deitmer 2006; Gunnarson et al. 2009) . Yet, mutant hSOD1G93A-expressing astrocytes responded to the receptor stimulation with an aberrant and persistent Ca2+ release from the intracellular stores. This correlated with mitochondrial disarrangement, cytochrome c release from mitochondria , and astrocyte degeneration (Martorana et al. 2012). These results are in line with the mitochondrial dysfunction previously described by Cassina and colleagues in mutant SOD1-expressing astrocytes (Cassina et al. 2008) .
The anti-apoptotic members of the Bcl-2 family, particularly Bcl-2 and Bcl-XL, were largely reported to confer cell death resistance by fine-tuning intracellular Ca2+ signalling through direct interaction with the IP3 receptor (IP3R) channels (Chen et al. 2004; White et al. 2005; Zhong et al. 2006; Li et al. 2007; Rong et al. 2008, 2009) . Interestingly, structure-function analysis of Bcl-2 homologues revealed that their N-terminal homology domain 4 (BH4) is essential for inhibition of apoptosis (Hunter et al. 1996; Lee et al. 1996; Huang et al. 1998) . Our group thus investigated the impact of the BH4 domain of Bcl-XL on astrocyte Ca2+ signalling by exploiting a biologically active BH4 peptide fused to the protein transduction domain of the HIV-1 TAT protein (TAT-BH4). We realized that TAT-BH4 modulates the IP3R-dependent Ca2+ release from the ER, and restores spontaneous Ca2+ oscillations in mutant SOD1-expressing astrocytes. This tight control of IP3Rs by the peptide prevents group I mGluR-driven aberrant release of Ca2+ from the intracellular stores, precludes the release of cytochrome c from mitochondria, and protects the cells from excitotoxic damage (Martorana et al. 2012) . Furthermore, chronic treatment of hSOD1G93A transgenic mice with TAT-BH4 reduces degeneration of spinal cord astrocytes and shows a positive impact on the disease manifestations (Martorana et al. 2012) .
Consistent with a specific vulnerability of ALS astrocytes, functional astroglia deriving from human induced pluripotent stem cells (iPSC) expressing the mutant TDP-43M337V protein were recently generated and shown to exhibit reduced cell survival (Serio et al. 2013) . Yet, in co-culture experiments, such cells were not harmful to wild-type motor neurons, possibly because in vitro differentiation may not have captured some of the detrimental properties that diseased astroglia harbor in vivo (Serio et al. 2013). In line with this conclusion, in transgenic rats expressing the same mutant form of TDP-43 in astrocytes, the loss of unhealthy astroglial cells paralleled progressive degeneration of motor neurons, denervation atrophy of skeletal muscles, and progressive paralysis (Tong et al. 2013) . Taken together, these findings support the view that glial cell sufferance can play a role in motor neuron degeneration also in ALS-TDP, though the evidence in this clinical subtype of the disease is still limited .
Another important function of the astrocytes that is worth mentioning concerns their association with the BBB, a complex structure tightly regulating the exchange of molecules between the brain parenchyma and the blood stream (reviewed in Daneman 2012) . On a structural standpoint, the BBB is made of endothelial cells, astroglia , pericytes and neurons, which globally establish a “neurovascular unit”. In this context, astrocytes project off their processes, and their endfeet ensheath the blood vessels. This strategic location suggests that they can importantly contribute to the maturation and maintenance of the BBB. Consistent with this vision, multiple studies in vitro have provided consistent evidence that astrocytes can influence the transendothelial resistance, a measure of BBB permeability; the organization of tight junctions between brain endothelial cells; and the luminal polarization of transporters, such the glucose transporter Glut-1 and the P-glycoprotein (Dehouck et al. 1990; Rubin et al. 1991; Hayashi et al. 1997; Sobue et al. 1999; Al Ahmad et al. 2011) . Although the integrity of the BBB is of outstanding importance for the maintenance of the optimal neuronal microenvironment, alterations of this structure have been described in a number of neurological disorders, including ALS (Daneman 2012). Early studies in the hSOD1G93A ALS mice identified ultrastructural damage to astrocytes and endothelial cells, leading to vascular leakage of intravenously injected Evans Blue (EB) dye already at the pre-symptomatic stage (Garbuzova-Davis et al. 2007) . EB extravasation abnormalities were found in both the cervical and lumbar spinal cord (Garbuzova-Davis et al. 2007), and correlated with altered expression of critical proteins for the regulation of the efflux of catabolites from the brain parenchyma, such as Glut-1 and P-glycoprotein (Garbuzova-Davis et al. 2007). Evidence of BBB damage and microhemorrhages was later extended to other transgenic mice, harboring the G37R and G85R SOD1 variants (Zhong et al. 2008) , and to sporadic ALS cases (Garbuzova-Davis et al. 2012; Winkler et al. 2013) . Given the importance of the BBB in tuning the access of drugs to the CNS, these findings should be taken in consideration when designing new therapies for ALS.
In conclusion, taken together, these studies confirm a multifaceted role for astrocytes in ALS, even though they fail to provide conclusive evidence as to whether the multiple and diverse contributions of astroglia are due to distinct astrocyte subpopulations or to the same population receiving different external inputs (Molofsky et al. 2012; Oberheim et al. 2012) . We suggest that a better understanding of the impact of the distinct astrocytic mechanisms to ALS pathogenesis will be certainly beneficial to develop targeted therapeutics .
11.4 Microglia
Microglia are the glial cell population deputed to the immune surveillance of the CNS. They typically respond to injury or disease with a massive activation in areas affected by neuronal degeneration. This condition is commonly known as “reactive microgliosis”. Reactive microglia have the capacity to release a wide variety of substances that can eitherlimit or exacerbate neuronal sufferance (reviewed in Aguzzi et al. 2013) . In both autoptic ALS cases and animal models, microgliosis was early recognized as a typical hallmark of the disease (Hall et al. 1998) . Yet, it was only with the recent advent of modern imaging technologies, coupled to the development of radiolabelled ligands, that the extent of this phenomenon could be assessed in vivo, in ALS patients, at the time of disease diagnosis. A number of positron emission tomography (PET) studies has been thus performed by neuroimaging the 18 kDa translocator protein (also known as the “peripheral benzodiazepine receptor”) (Turner et al. 2004; Corcia et al. 2012) , which is highly expressed in phagocytic inflammatory cells, including activated microglia (Papadopoulos et al. 2006) .
Several investigations, using mutant SOD1-expressing cellular and animal models of the disease, were then performed in order to elucidate the mechanism(s) that trigger the activation of such cells. These studies suggested that reactive microgliosis is prompted by the secretion of mutant SOD1 (Urushitani et al. 2006) , and the aggregated protein was described to be more efficient than the monomeric form in this activity (Roberts et al. 2013) . The relevance of microglia towards the disease manifestations was then tackled in vivo by genetic (Boillee et al. 2006; Gowing et al. 2008) and cell transplantation (Beers et al. 2006) approaches. Thus, reducing the expression of the mutant hSOD1G37R protein selectively in microglial cells was reported not to affect the disease onset, although it slowed down ALS progression in transgenic mice (Boillee et al. 2006). In agreement with these results, transplantation of donor-derived microglia from the spinal cord of hSOD1G93A mice into wild-type animals did not trigger ALS disease (Beers et al. 2006). Yet, transplantation of wild-type microglia induced neuroprotective effects on hSOD1G93A-expressing motor neurons and prolonged the survival of transgenic mice (Beers et al. 2006). Altogether, these studies strengthen the idea that microglial cells modulate ALS progression, rather than influencing the development of the disease .
At the molecular level, the mechanism(s) by which microglia contribute to motor neuron sufferance still needs to be elucidated in details. Yet, a transcriptional profile analysis recently performed on acutely isolated spinal cord microglia from hSOD1G93A transgenic mice revealed that these cells exhibit an ALS-specific phenotype characterized by the concomitant up-regulation of both potentially neurotoxic and neuroprotective factors (Chiu et al. 2013) . Among the molecules presumably mediating microglial toxicity, there are for example reactive oxidant species. In both human and mouse autoptic tissues, the ROS-generating NOX2 enzyme was in fact shown to be up-regulated specifically in microglial cells (Wu et al. 2006) . This event correlated with the appearance of oxidation products and protein carbonylation adducts, including oxidative modifications of insulin-like growth factor 1 (IGF1) receptors, which are critically involved in neuronal survival. The relevance of NOX2 in disease progression was then confirmed by the evidence that genetic ablation of its catalytic subunit ameliorated the phenotype and extended the lifespan of hSOD1G93A ALS mice (Wu et al. 2006). Further insights into the mechanism of NOX2- driven toxicity have been provided a few years later by Harraz and colleagues, who unveiled a new function for the SOD1 enzyme. Along with its dismutase activity, SOD1 was shown to participate to the regulation of NOX2 function by binding to its regulatory protein Rac1. Under reducing conditions, this interaction appears to stabilize Rac1 activation, thus leading to NOX2 activation and superoxide production. However, when the local concentration of oxidant species increases, SOD1 is displaced from Rac1, and NOX2 activity appears to be negatively regulated. Yet, certain mutant forms of SOD1 display a stronger interaction with Rac1, and this leads to both sustained NOX2 activation and excessive superoxide production. Because chronic treatment with the NOX2 inhibitor apocynin tremendously increased the lifespan of hSOD1G93A ALS mice, this mechanism was proposed to contribute to disease progression (Harraz et al. 2008) . The promising outcome of this study subsequently encouraged further investigations on NOX2 inhibitors in the context of ALS. Thus, the neuroprotective properties of diapocynin, a potent NOX2 inhibitor, were addressed in vitro and in vivo (Trumbull et al. 2012) . Despite this drug proved to prevent motor neuron death at lower doses in culture experiments when compared to apocynin, the treatment of hSOD1G93A transgenic mice failed to extend the animal lifespan. Moreover, the dramatic effect on mouse survival originally observed with apocynin could not be replicated in this study (Trumbull et al. 2012) , thus raising serious concerns about the therapeutic potential of these agents. Nonetheless, the pathway(s) leading to NOX2 activation, and their contribution to ALS pathogenesis, remain of great interest for the scientific community. In fact, the enzyme protein disulfide isomerase, which assists the oxidative protein folding in the ER, was recently shown to be up-regulated in microglia from hSOD1G93A mice. This suggests that the unfolded protein response (UPR) does not occur only in ALS motor neurons, but also in microglial cells (Jaronen et al. 2013) . Importantly, UPR was reported to trigger the activation of microglial NOX2 and, thus, to increase the production of superoxide in culture experiments (Jaronen et al. 2013). These findings suggest that UPR, initiated by protein misfolding, may lead to NOX2 activation and ROS generation. However, recent studies propose alternative pathways leading to the activation of NADPH oxidase. For example, experiments in vitro showed that stimulation of the purinergic receptor P2 × 7 in mutant hSOD1G93A-expressing microglia results in enhanced NOX2 activity and ROS production, thus suggesting a deleterious role for the P2 × 7 receptor in ALS (Apolloni et al. 2013b) . At variance with these results, genetic ablation of the P2 × 7 receptor in hSOD1G93A mice, however, exacerbated gliosis and motor neuron cell death; anticipated the clinical onset of the disease; and worsened ALS progression (Apolloni et al. 2013a). The protective effect of P2 × 7 suggested by these results in vivo was completely unexpected, given that stimulation of the P2 × 7 receptor had been linked also to the induction of a neurotoxic phenotype in hSOD1G93A astrocytes (Gandelman et al. 2010) and to motor neuron apoptosis (Gandelman et al. 2013). Altogether, these observations reveal that the P2 × 7 receptor exerts a complex dual role in ALS. Evidence in favor of a neuroprotective role for microglia in the context of ALS was provided by a recent study addressing the biological significance of the glycosaminoglycan keratan sulfate (KS) (Hirano et al. 2013) . In both autoptic ALS tissues and hSOD1G93Atransgenic mice, KS was found to be highly expressed by a specific subpopulation of microglial cells exerting anti-inflammatory functions during the early phase of the disease. Noteworthy, genetic ablation of KS in the CNS of hSOD1G93A mice resulted in marked reduction of anti-inflammatory microglia, and this correlated with acceleration of the clinical symptoms and shortening of the animal lifespan (Hirano et al. 2013) .
More recently, microglia was also describe to dialogue with the peripheral immunocompetent cells, an interaction that proved to be both beneficial and deleterious (reviewed in Appel et al. 2010) . Several lines of evidence indicate that T cells infiltrate the spinal cord of ALS patients (Troost et al. 1989; Kawamata et al. 1992; Engelhardt et al. 1993) and mutant SOD1 transgenic mice (Beers et al. 2008; Chiu et al. 2008) . Of note, infiltrating T lymphocytes were described to slow down disease progression in mice by modulating the microglial inflammatory response (Beers et al. 2008, 2011; Chiu et al. 2008; Henkel et al. 2013) . Yet, spinal cord microglia were also reported to recruit peripheral monocytes to the CNS, a process that critically impairs neuronal viability and the mouse survival (Butovsky et al. 2012) .
In conclusion, these findings globally suggest that the neuroinflammatory response driven by microglia includes both neuroprotective and neurotoxic aspects in ALS. Therefore, the possibility of establishing a successful therapeutic effect relies on the selective targeting of the toxic components of the immunoreaction, rather than on the non-specific suppression of the immunocompetent response .
11.5 Oligodendrocytes and Other Glial Cell Types
While astrocytes and microglia have been at the center of the investigations on ALS for almost a decade, other glial cell populations have attracted the attention in more recent times. Among others, these latter include oligodendrocytes and Schwann cells , the myelin-forming cells of the central and peripheral nervous system, respectively. Although early analyses of mutant SOD1 transgenic mice revealed SOD1-immunopositive inclusions in oligodendrocytes (Stieber et al. 2000) , this observation was not pursued immediately thereafter, and these cells have been involved in ALS pathogenesis quite recently. In both ALS patients and mutant hSOD1G93A transgenic mice, it has been consistently reported a loss of the monocarboxylate transporter 1 (MCT1), a protein that is highly expressed in oligodendrocytes, and which is deputed to provide motor neurons with the metabolic substrate lactate (Lee et al. 2012; Philips et al. 2013) . Noteworthy, ubiquitous genetic ablation of MCT1 in mice was described to cause axonopathy (Lee et al. 2012). Furthermore, selective reduction of its expression in oligodendrocytes resulted in axonal sufferance (Lee et al. 2012) . This suggests that the shuttling of lactate from oligodendrocytes, through MCT1, is crucial for the energy supply to axons, and any disturbance of this transport can lead to axon dysfunction and, eventually, to neuronal cell death.
More recently, oligodendrocytes themselves were described to be a direct target of the disease. An apoptotic oligodendrocytic phenotype was, in fact, detected in the ventral grey matter of the spinal cord from mutant hSOD1G93A transgenic mice before actual motor neuron loss became evident. Surprisingly, the overall number of oligodendrocytes was fully preserved (Philips et al. 2013). In demyelinating diseases, mature oligodendrocytes are typically replaced by the differentiation of NG2+ cells , which behave as precursors committed to the oligodendrocyte lineage (Chang et al. 2000) . In keeping with this, investigations of the NG2+ cell fate in symptomatic mutant SOD1 mice revealed that they exhibit an increased proliferation rate (Magnus et al. 2008; Kang et al. 2010, 2013; Philips et al. 2013) . The occurrence of this event is accompanied by a more frequent differentiation of NG2+ cells into oligodendrocytes (Magnus et al. 2008; Kang et al. 2010, 2013; Philips et al. 2013). Yet, at the time when mice show overt signs of the disease, the concomitant degeneration of early-born oligodendrocytes and, thus, the accelerated turnover of these cells, result in grey matter demyelination in ALS mice and human CNS (Kang et al. 2013). In addition, newly generated oligodendrocytes display reduced MCT1 expression, and thus fail to provide motor neuron with metabolic support (Kang et al. 2013; Philips et al. 2013). Since oligodendrocyte myelination is regulated by lactate (Rinholm et al. 2011) , it was proposed that reduced levels of MCT1 in oligodendrocytes may affect the process of myelination, in addition to depriving motor neurons of critical metabolic substrates (Magnus et al. 2008; Kang et al. 2010, 2013; Philips et al. 2013). A role for NG2+ cells and their oligodendrocyte progeny in ALS development was corroborated also by gene targeting experiments. Thus, diminishing mutant SOD1 expression in these cells was shown to delay the onset of the disease and to extend the life span of mutant hSOD1G37R transgenic mice (Kang et al. 2013).
In the peripheral nervous system (PNS), individual axons are myelinated by the Schwann cells , the major glial component of the PNS. An initial indication of the involvement of this glial cell population in the pathogenesis of ALS was provided by the early observation that femoral nerves from post-mortem cases displayed a certain degree of myelin disruption (Perrie et al. 1993) . Later studies in mutant hSOD1G93A mice provided further insights into the role of these cells in ALS by revealing that they exhibit signs of distress at the asymptomatic stage (Keller et al. 2009) . Yet, investigations addressing the impact of Schwann cells on ALS pathogenesis in vivo by cell-specific expression or ablation of mutant SOD1 provided slightly divergent outcomes. Thus, transgene-driven expression of mutant hSOD1G93A protein in Schwann cells resulted neither detrimental for motor neurons nor deleterious to the disease manifestations (Turner et al. 2010) . On the other hand, removal of mutant hSOD1G37R from Schwann cells by gene excision experiments accelerated disease progression in vivo (Lobsiger et al. 2009) . Considering the intimate relationship between motor neuronal axons and myelin, these results indicate that Schwann cells certainly deserve further attention in order to definitely elucidate their contribution to the disease.
11.6 Glial Abnormalities in Human Neuropathology
The growing awareness that non-cell autonomous mechanisms play a role in both the process of motor neuron degeneration and the manifestations of ALS disease prompted an in-depth assessment of glial abnormalities also in the human pathology. In addition to the specific alterations outlined in the previous paragraphs, there are a number of other neuropathological anomalies in glial cells related to the identification of misfolded protein inclusions in different molecular variants of the human disease. For example, misfolded SOD1 inclusions, the typical hallmark of ALS-SOD, have been recently described in various glial cell populations from both familial and sporadic ALS cases (Forsberg et al. 2011) . In addition, ubiquinated protein inclusions immunopositive for TDP-43 were found in neurons, but also in the cytoplasm of glial cells (Nishihira et al. 2008; Zhang et al. 2008) , particularly oligodendrocytes (Neumann et al. 2007; Seilhean et al. 2009) , of classic ALS, FTLD and ALS linked to mutant optineurin (ALS-OPTN) (Ito et al. 2011) . Similarly, abundant inclusions within oligodendrocytes have been identified in ALS-FUS, as revealed by double immunofluorescence labelling (Mackenzie et al. 2011) . FUS-related glial pathology appears to be a distinctive feature of patients harboring mutations that mildly impair the nuclear localization of the FUS protein (Hewitt et al. 2010; Yamamoto-Watanabe et al. 2010; Mackenzie et al. 2011; Robertson et al. 2011) . Remarkably, in the corticospinal tract of patients affected by ALS-OPTN, there are signs of demyelinization, further supporting the hypothesis of oligodendrocyte impairment in this form of fALS (Maruyama et al. 2010) . Taken together, this amount of evidence suggests that oligodendrocyte pathology might be a common feature of those forms of ALS characterized by misfolded/mislocalized protein inclusions. This lead to the speculation that factors responsible for protein aggregation might be expressed by both oligodendrocytes and motor neurons, and their identification may open new perspectives for therapeutic intervention.
11.7 Conclusions
Several lines of evidence indicate that ALS is a disease with a very complex and multifactorial etiopathogenesis. An effective therapeutic intervention on patients is still prevented by the lack of early biomarkers of the disease as well as by the absence of an effective pharmacological strategy. Among others, this situation is conditioned by the incomplete knowledge of the mutual regulation and interactions between the cellular structures affected by the disease, particularly motor neurons and glial cells. Yet, recent breakthroughs in neuroscience have led to the increasing awareness that neuron-glial interactions are much more critical for the correct brain functioning than previously thought, and disruption of such intercellular cross-talk impairs the performance of the CNS. The perception that glial cells are actively involved in CNS function and dysfunction clearly offers a wide range of new possibilities to unravel physiological and pathological mechanisms. In this chapter, we have summarized the currently available knowledge on the contribution of the different glial cell types to various forms of ALS. These observations convincingly demonstrate that the development of the disease involves the de-regulation of a finely tuned interplay between multiple neural cell populations. Thus, it seems that a substantial improvement of the outcome of ALS treatments may depend on a better understanding of the mechanisms governing the detrimental glial responses. The elucidation of such aspects may open new perspectives for innovative therapeutic strategies specifically targeting these cells.
References
Aebischer J, Cassina P, Otsmane B, Moumen A, Seilhean D, Meininger V, Barbeito L, Pettmann B, Raoul C (2011) IFNgamma triggers a LIGHT-dependent selective death of motoneurons contributing to the non-cell-autonomous effects of mutant SOD1. Cell Death Differ 18:754–768
Aguzzi A, Barres BA, Bennett ML (2013) Microglia: scapegoat, saboteur, or something else? Science 339:156–161
Al-Harthi L (2012) Wnt/beta-catenin and its diverse physiological cell signaling pathways in neurodegenerative and neuropsychiatric disorders. J Neuroimmune Pharmacol 7:725–730
Al Ahmad A, Taboada CB, Gassmann M, Ogunshola OO (2011) Astrocytes and pericytes differentially modulate blood-brain barrier characteristics during development and hypoxic insult. J Cereb Blood Flow Metab 31:693–705
Allen NJ, Barres BA (2009) Neuroscience: glia-more than just brain glue. Nature 457:675–677
Apolloni S, Amadio S, Montilli C, Volonte C, D’Ambrosi N (2013a) Ablation of P2 × 7 receptor exacerbates gliosis and motoneuron death in the SOD1-G93A mouse model of amyotrophic lateral sclerosis. Hum Mol Genet 22:4102–4116
Apolloni S, Parisi C, Pesaresi MG, Rossi S, Carri MT, Cozzolino M, Volonte C, D’Ambrosi N (2013b) The NADPH oxidase pathway is dysregulated by the P2 × 7 receptor in the SOD1-G93A microglia model of amyotrophic lateral sclerosis. J Immunol 190:5187–5195
Appel SH, Beers DR, Henkel JS (2010) T cell-microglial dialogue in Parkinson’s disease and amyotrophic lateral sclerosis: are we listening? Trends Immunol 31:7–17
Ayala YM, Zago P, D’Ambrogio A, Xu YF, Petrucelli L, Buratti E, Baralle FE (2008) Structural determinants of the cellular localization and shuttling of TDP-43. J Cell Sci 121:3778–3785
Beers DR, Henkel JS, Xiao Q, Zhao W, Wang J, Yen AA, Siklos L, McKercher SR, Appel SH (2006) Wild-type microglia extend survival in PU.1 knockout mice with familial amyotrophic lateral sclerosis. Proc Natl Acad Sci U S A 103:16021–16026
Beers DR, Henkel JS, Zhao W, Wang J, Appel SH (2008) CD4+ T cells support glial neuroprotection, slow disease progression, and modify glial morphology in an animal model of inherited ALS. Proc Natl Acad Sci U S A 105:15558–15563
Beers DR, Zhao W, Liao B, Kano O, Wang J, Huang A, Appel SH, Henkel JS (2011) Neuroinflammation modulates distinct regional and temporal clinical responses in ALS mice. Brain Behav Immun 25:1025–1035
Berry JD, Shefner JM, Conwit R, Schoenfeld D, Keroack M, Felsenstein D, Krivickas L, David WS, Vriesendorp F, Pestronk A, Caress JB, Katz J, Simpson E, Rosenfeld J, Pascuzzi R, Glass J, Rezania K, Rothstein JD, Greenblatt DJ, Cudkowicz ME (2013) Design and initial results of a multi-phase randomized trial of ceftriaxone in amyotrophic lateral sclerosis. PloS ONE 8:e61177
Bi F, Huang C, Tong J, Qiu G, Huang B, Wu Q, Li F, Xu Z, Bowser R, Xia XG, Zhou H (2013) Reactive astrocytes secrete lcn2 to promote neuron death. Proc Natl Acad Sci U S A 110:4069–4074
Bilsland LG, Nirmalananthan N, Yip J, Greensmith L, Duchen MR (2008) Expression of mutant SOD1 in astrocytes induces functional deficits in motoneuron mitochondria. J Neurochem 107:1271–1283
Boillee S, Yamanaka K, Lobsiger CS, Copeland NG, Jenkins NA, Kassiotis G, Kollias G, Cleveland DW (2006) Onset and progression in inherited ALS determined by motor neurons and microglia. Science 312:1389–1392
Boston-Howes W, Gibb SL, Williams EO, Pasinelli P, Brown RH Jr, Trotti D (2006) Caspase-3 cleaves and inactivates the glutamate transporter EAAT2. J Biol Chem 281:14076–14084
Brooks BR, Miller RG, Swash M, Munsat TL (2000) El Escorial revisited: revised criteria for the diagnosis of amyotrophic lateral sclerosis. Amyotroph Lateral Scler Other Motor Neuron Disord 1:293–299
Bruijn LI, Becher MW, Lee MK, Anderson KL, Jenkins NA, Copeland NG, Sisodia SS, Rothstein JD, Borchelt DR, Price DL, Cleveland DW (1997) ALS-linked SOD1 mutant G85R mediates damage to astrocytes and promotes rapidly progressive disease with SOD1-containing inclusions. Neuron 18:327–338
Bryan HK, Olayanju A, Goldring CE, Park BK (2013) The Nrf2 cell defence pathway: keap1-dependent and -independent mechanisms of regulation. Biochem Pharmacol 85:705–717
Butovsky O, Siddiqui S, Gabriely G, Lanser AJ, Dake B, Murugaiyan G, Doykan CE, Wu PM, Gali RR, Iyer LK, Lawson R, Berry J, Krichevsky AM, Cudkowicz ME, Weiner HL (2012) Modulating inflammatory monocytes with a unique microRNA gene signature ameliorates murine ALS. J Clin Investig 122:3063–3087
Cassina P, Cassina A, Pehar M, Castellanos R, Gandelman M, de Leon A, Robinson KM, Mason RP, Beckman JS, Barbeito L, Radi R (2008) Mitochondrial dysfunction in SOD1G93A-bearing astrocytes promotes motor neuron degeneration: prevention by mitochondrial-targeted antioxidants. J Neurosci 28:4115–4122
Chang A, Nishiyama A, Peterson J, Prineas J, Trapp BD (2000) NG2-positive oligodendrocyte progenitor cells in adult human brain and multiple sclerosis lesions. J Neurosci 20:6404–6412
Chen R, Valencia I, Zhong F, McColl KS, Roderick HL, Bootman MD, Berridge MJ, Conway SJ, Holmes AB, Mignery GA, Velez P, Distelhorst CW (2004) Bcl-2 functionally interacts with inositol 1,4,5-trisphosphate receptors to regulate calcium release from the ER in response to inositol 1,4,5-trisphosphate. J Cell Biol 166:193–203
Chen Y, Guan Y, Liu H, Wu X, Yu L, Wang S, Zhao C, Du H, Wang X (2012) Activation of the Wnt/beta-catenin signaling pathway is associated with glial proliferation in the adult spinal cord of ALS transgenic mice. Biochem Biophys Res Commun 420:397–403
Chiu IM, Chen A, Zheng Y, Kosaras B, Tsiftsoglou SA, Vartanian TK, Brown RH Jr, Carroll MC (2008) T lymphocytes potentiate endogenous neuroprotective inflammation in a mouse model of ALS. Proc Natl Acad Sci U S A 105:17913–17918
Chiu IM, Morimoto ET, Goodarzi H, Liao JT, O’Keeffe S, Phatnani HP, Muratet M, Carroll MC, Levy S, Tavazoie S, Myers RM, Maniatis T (2013) A neurodegeneration-specific gene-expression signature of acutely isolated microglia from an amyotrophic lateral sclerosis mouse model. Cell Rep 4:385–401
Clement AM, Nguyen MD, Roberts EA, Garcia ML, Boillee S, Rule M, McMahon AP, Doucette W, Siwek D, Ferrante RJ, Brown RH Jr, Julien JP, Goldstein LS, Cleveland DW (2003) Wild-type nonneuronal cells extend survival of SOD1 mutant motor neurons in ALS mice. Science 302:113–117
Corcia P, Tauber C, Vercoullie J, Arlicot N, Prunier C, Praline J, Nicolas G, Venel Y, Hommet C, Baulieu JL, Cottier JP, Roussel C, Kassiou M, Guilloteau D, Ribeiro MJ (2012) Molecular imaging of microglial activation in amyotrophic lateral sclerosis. PloS ONE 7:e52941
Danbolt NC (2001) Glutamate uptake. Prog Neurobiol 65:1–105
Daneman R (2012) The blood-brain barrier in health and disease. Ann Neurol 72:648–672
Dehouck MP, Meresse S, Delorme P, Fruchart JC, Cecchelli R (1990) An easier, reproducible, and mass-production method to study the blood-brain barrier in vitro. J Neurochem 54:1798–1801
DeJesus-Hernandez M et al (2011) Expanded GGGGCC hexanucleotide repeat in noncoding region of C9ORF72 causes chromosome 9p-linked FTD and ALS. Neuron 72:245–256
Deng HX et al (2011) Mutations in UBQLN2 cause dominant X-linked juvenile and adult-onset ALS and ALS/dementia. Nature 477:211–215
Devireddy LR, Teodoro JG, Richard FA, Green MR (2001) Induction of apoptosis by a secreted lipocalin that is transcriptionally regulated by IL-3 deprivation. Science 293:829–834
Devireddy LR, Gazin C, Zhu X, Green MR (2005) A cell-surface receptor for lipocalin 24p3 selectively mediates apoptosis and iron uptake. Cell 123:1293–1305
Dewey CM, Cenik B, Sephton CF, Dries DR, Mayer P 3rd, Good SK, Johnson BA, Herz J, Yu G (2011) TDP-43 is directed to stress granules by sorbitol, a novel physiological osmotic and oxidative stressor. Mol Cell Biol 31:1098–1108
Di Giorgio FP, Carrasco MA, Siao MC, Maniatis T, Eggan K (2007) Non-cell autonomous effect of glia on motor neurons in an embryonic stem cell-based ALS model. Nat Neurosci 10:608–614
Di Giorgio FP, Boulting GL, Bobrowicz S, Eggan KC (2008) Human embryonic stem cell-derived motor neurons are sensitive to the toxic effect of glial cells carrying an ALS-causing mutation. Cell Stem Cell 3:637–648
Diaper DC, Adachi Y, Lazarou L, Greenstein M, Simoes FA, Di Domenico A, Solomon DA, Lowe S, Alsubaie R, Cheng D, Buckley S, Humphrey DM, Shaw CE, Hirth F (2013) Drosophila TDP-43 dysfunction in glia and muscle cells cause cytological and behavioural phenotypes that characterize ALS and FTLD. Hum Mol Genet 22:3883–3893
Diaz-Amarilla P, Olivera-Bravo S, Trias E, Cragnolini A, Martinez-Palma L, Cassina P, Beckman J, Barbeito L (2011) Phenotypically aberrant astrocytes that promote motoneuron damage in a model of inherited amyotrophic lateral sclerosis. Proc Natl Acad Sci U S A 108:18126–18131
Dormann D, Rodde R, Edbauer D, Bentmann E, Fischer I, Hruscha A, Than ME, Mackenzie IR, Capell A, Schmid B, Neumann M, Haass C (2010) ALS-associated fused in sarcoma (FUS) mutations disrupt Transportin-mediated nuclear import. EMBO J 29:2841–2857
Duan W, Li X, Shi J, Guo Y, Li Z, Li C (2010) Mutant TAR DNA-binding protein-43 induces oxidative injury in motor neuron-like cell. Neuroscience 169:1621–1629
Engelhardt JI, Tajti J, Appel SH (1993) Lymphocytic infiltrates in the spinal cord in amyotrophic lateral sclerosis. Arch Neurol 50:30–36
Estes PS, Daniel SG, McCallum AP, Boehringer AV, Sukhina AS, Zwick RA, Zarnescu DC (2013) Motor neurons and glia exhibit specific, individualized responses to TDP-43 expression in a Drosophila model of ALS. Dis Models Mech 6:721–733
Feng X, Peng Y, Liu M, Cui L (2012) DL-3-n-butylphthalide extends survival by attenuating glial activation in a mouse model of amyotrophic lateral sclerosis. Neuropharmacology 62:1004–1010
Ferraiuolo L, Kirby J, Grierson AJ, Sendtner M, Shaw PJ (2011a) Molecular pathways of motor neuron injury in amyotrophic lateral sclerosis. Nat Rev Neurol 7:616–630
Ferraiuolo L, Higginbottom A, Heath PR, Barber S, Greenald D, Kirby J, Shaw PJ (2011b) Dysregulation of astrocyte-motoneuron cross-talk in mutant superoxide dismutase 1-related amyotrophic lateral sclerosis. Brain 134:2627–2641
Flomen R, Makoff A (2011) Increased RNA editing in EAAT2 pre-mRNA from amyotrophic lateral sclerosis patients: involvement of a cryptic polyadenylation site. Neurosci Lett 497:139–143
Foran E, Bogush A, Goffredo M, Roncaglia P, Gustincich S, Pasinelli P, Trotti D (2011) Motor neuron impairment mediated by a sumoylated fragment of the glial glutamate transporter EAAT2. Glia 59:1719–1731
Forsberg K, Andersen PM, Marklund SL, Brannstrom T (2011) Glial nuclear aggregates of superoxide dismutase-1 are regularly present in patients with amyotrophic lateral sclerosis. Acta Neuropathol 121:623–634
Gandelman M, Peluffo H, Beckman JS, Cassina P, Barbeito L (2010) Extracellular ATP and the P2 × 7 receptor in astrocyte-mediated motor neuron death: implications for amyotrophic lateral sclerosis. J Neuroinflamm 7:33
Gandelman M, Levy M, Cassina P, Barbeito L, Beckman JS (2013) P2 × 7 receptor-induced death of motor neurons by a peroxynitrite/FAS-dependent pathway. J Neurochem 126:382–388
Garbuzova-Davis S, Haller E, Saporta S, Kolomey I, Nicosia SV, Sanberg PR (2007) Ultrastructure of blood-brain barrier and blood-spinal cord barrier in SOD1 mice modeling ALS. Brain Res 1157:126–137
Garbuzova-Davis S, Hernandez-Ontiveros DG, Rodrigues MC, Haller E, Frisina-Deyo A, Mirtyl S, Sallot S, Saporta S, Borlongan CV, Sanberg PR (2012) Impaired blood-brain/spinal cord barrier in ALS patients. Brain Res 1469:114–128
Gibb SL, Boston-Howes W, Lavina ZS, Gustincich S, Brown RH Jr, Pasinelli P, Trotti D (2007) A caspase-3-cleaved fragment of the glial glutamate transporter EAAT2 is sumoylated and targeted to promyelocytic leukemia nuclear bodies in mutant SOD1-linked amyotrophic lateral sclerosis. J Biol Chem 282:32480–32490
Gong YH, Parsadanian AS, Andreeva A, Snider WD, Elliott JL (2000) Restricted expression of G86R Cu/Zn superoxide dismutase in astrocytes results in astrocytosis but does not cause motoneuron degeneration. J Neurosci 20:660–665
Gowing G, Philips T, Van Wijmeersch B, Audet JN, Dewil M, Van Den Bosch L, Billiau AD, Robberecht W, Julien JP (2008) Ablation of proliferating microglia does not affect motor neuron degeneration in amyotrophic lateral sclerosis caused by mutant superoxide dismutase. J Neurosci 28:10234–10244
Gunnarson E, Song Y, Kowalewski JM, Brismar H, Brines M, Cerami A, Andersson U, Zelenina M, Aperia A (2009) Erythropoietin modulation of astrocyte water permeability as a component of neuroprotection. Proc Natl Acad Sci U S A 106:1602–1607
Guo H, Lai L, Butchbach ME, Stockinger MP, Shan X, Bishop GA, Lin CL (2003) Increased expression of the glial glutamate transporter EAAT2 modulates excitotoxicity and delays the onset but not the outcome of ALS in mice. Hum Mol Genet 12:2519–2532
Guo Y, Zhang Y, Wen D, Duan W, An T, Shi P, Wang J, Li Z, Chen X, Li C (2013) The modest impact of transcription factor Nrf2 on the course of disease in an ALS animal model. Lab Invest 93:825–833
Gurney ME, Pu H, Chiu AY, Dal Canto MC, Polchow CY, Alexander DD, Caliendo J, Hentati A, Kwon YW, Deng HX et al (1994) Motor neuron degeneration in mice that express a human Cu, Zn superoxide dismutase mutation. Science 264:1772–1775
Haidet-Phillips AM, Hester ME, Miranda CJ, Meyer K, Braun L, Frakes A, Song S, Likhite S, Murtha MJ, Foust KD, Rao M, Eagle A, Kammesheidt A, Christensen A, Mendell JR, Burghes AH, Kaspar BK (2011) Astrocytes from familial and sporadic ALS patients are toxic to motor neurons. Nat Biotechnol 29:824–828
Hall ED, Oostveen JA, Gurney ME (1998) Relationship of microglial and astrocytic activation to disease onset and progression in a transgenic model of familial ALS. Glia 23:249–256
Halliday G, Bigio EH, Cairns NJ, Neumann M, Mackenzie IR, Mann DM (2012) Mechanisms of disease in frontotemporal lobar degeneration: gain of function versus loss of function effects. Acta Neuropathol 124:373–382
Hardiman O, van den Berg LH, Kiernan MC (2011) Clinical diagnosis and management of amyotrophic lateral sclerosis. Nature Rev Neurol 7:639–649
Harraz MM, Marden JJ, Zhou W, Zhang Y, Williams A, Sharov VS, Nelson K, Luo M, Paulson H, Schoneich C, Engelhardt JF (2008) SOD1 mutations disrupt redox-sensitive Rac regulation of NADPH oxidase in a familial ALS model. J Clin Investig 118:659–670
Hayashi Y, Nomura M, Yamagishi S, Harada S, Yamashita J, Yamamoto H (1997) Induction of various blood-brain barrier properties in non-neural endothelial cells by close apposition to co-cultured astrocytes. Glia 19:13–26
Henkel JS, Beers DR, Wen S, Rivera AL, Toennis KM, Appel JE, Zhao W, Moore DH, Powell SZ, Appel SH (2013) Regulatory T-lymphocytes mediate amyotrophic lateral sclerosis progression and survival. EMBO Mol Med 5:64–79
Hewitt C, Kirby J, Highley JR, Hartley JA, Hibberd R, Hollinger HC, Williams TL, Ince PG, McDermott CJ, Shaw PJ (2010) Novel FUS/TLS mutations and pathology in familial and sporadic amyotrophic lateral sclerosis. Arch Neurol 67:455–461
Hirano K, Ohgomori T, Kobayashi K, Tanaka F, Matsumoto T, Natori T, Matsuyama Y, Uchimura K, Sakamoto K, Takeuchi H, Hirakawa A, Suzumura A, Sobue G, Ishiguro N, Imagama S, Kadomatsu K (2013) Ablation of keratan sulfate accelerates early phase pathogenesis of ALS. PloS ONE 8:e66969
Honig LS, Chambliss DD, Bigio EH, Carroll SL, Elliott JL (2000) Glutamate transporter EAAT2 splice variants occur not only in ALS, but also in AD and controls. Neurology 55:1082–1088
Howland DS, Liu J, She Y, Goad B, Maragakis NJ, Kim B, Erickson J, Kulik J, DeVito L, Psaltis G, DeGennaro LJ, Cleveland DW, Rothstein JD (2002) Focal loss of the glutamate transporter EAAT2 in a transgenic rat model of SOD1 mutant-mediated amyotrophic lateral sclerosis (ALS). Proc Natl Acad Sci U S A 99:1604–1609
Huang DC, Adams JM, Cory S (1998) The conserved N-terminal BH4 domain of Bcl-2 homologues is essential for inhibition of apoptosis and interaction with CED-4. EMBO J 17:1029–1039
Hunter JJ, Bond BL, Parslow TG (1996) Functional dissection of the human Bc12 protein: sequence requirements for inhibition of apoptosis. Mol Cell Biology 16:877–883
Ilieva H, Polymenidou M, Cleveland DW (2009) Non-cell autonomous toxicity in neurodegenerative disorders: ALS and beyond. J Cell Biol 187:761–772
Ito H, Fujita K, Nakamura M, Wate R, Kaneko S, Sasaki S, Yamane K, Suzuki N, Aoki M, Shibata N, Togashi S, Kawata A, Mochizuki Y, Mizutani T, Maruyama H, Hirano A, Takahashi R, Kawakami H, Kusaka H (2011) Optineurin is co-localized with FUS in basophilic inclusions of ALS with FUS mutation and in basophilic inclusion body disease. Acta Neuropathol 121:555–557
Jaronen M, Vehvilainen P, Malm T, Keksa-Goldsteine V, Pollari E, Valonen P, Koistinaho J, Goldsteins G (2013) Protein disulfide isomerase in ALS mouse glia links protein misfolding with NADPH oxidase-catalyzed superoxide production. Hum Mol Genet 22:646–655
Kabashi E, Valdmanis PN, Dion P, Spiegelman D, McConkey BJ, Vande Velde C, Bouchard JP, Lacomblez L, Pochigaeva K, Salachas F, Pradat PF, Camu W, Meininger V, Dupre N, Rouleau GA (2008) TARDBP mutations in individuals with sporadic and familial amyotrophic lateral sclerosis. Nat Genet 40:572–574
Kang SH, Fukaya M, Yang JK, Rothstein JD, Bergles DE (2010) NG2+ CNS glial progenitors remain committed to the oligodendrocyte lineage in postnatal life and following neurodegeneration. Neuron 68:668–681
Kang SH, Li Y, Fukaya M, Lorenzini I, Cleveland DW, Ostrow LW, Rothstein JD, Bergles DE (2013) Degeneration and impaired regeneration of gray matter oligodendrocytes in amyotrophic lateral sclerosis. Nat Neurosci 16:571–579
Kawamata T, Akiyama H, Yamada T, McGeer PL (1992) Immunologic reactions in amyotrophic lateral sclerosis brain and spinal cord tissue. Am J Pathol 140:691–707
Keller AF, Gravel M, Kriz J (2009) Live imaging of amyotrophic lateral sclerosis pathogenesis: disease onset is characterized by marked induction of GFAP in Schwann cells. Glia 57:1130–1142
Kirby J, Halligan E, Baptista MJ, Allen S, Heath PR, Holden H, Barber SC, Loynes CA, Wood-Allum CA, Lunec J, Shaw PJ (2005) Mutant SOD1 alters the motor neuronal transcriptome: implications for familial ALS. Brain 128:1686–1706
Kwiatkowski TJ Jr et al (2009) Mutations in the FUS/TLS gene on chromosome 16 cause familial amyotrophic lateral sclerosis. Science 323:1205–1208
Lanson NA Jr, Pandey UB (2012) FUS-related proteinopathies: lessons from animal models. Brain Res 1462:44–60
Lee LC, Hunter JJ, Mujeeb A, Turck C, Parslow TG (1996) Evidence for alpha-helical conformation of an essential N-terminal region in the human Bcl2 protein. J Biol Chem 271:23284–23288
Lee Y, Morrison BM, Li Y, Lengacher S, Farah MH, Hoffman PN, Liu Y, Tsingalia A, Jin L, Zhang PW, Pellerin L, Magistretti PJ, Rothstein JD (2012) Oligodendroglia metabolically support axons and contribute to neurodegeneration. Nature 487:443–448
Lepore AC, Rauck B, Dejea C, Pardo AC, Rao MS, Rothstein JD, Maragakis NJ (2008a) Focal transplantation-based astrocyte replacement is neuroprotective in a model of motor neuron disease. Nat Neurosci 11:1294–1301
Lepore AC, Dejea C, Carmen J, Rauck B, Kerr DA, Sofroniew MV, Maragakis NJ (2008b) Selective ablation of proliferating astrocytes does not affect disease outcome in either acute or chronic models of motor neuron degeneration. Exp Neurol 211:423–432
Li C, Wang X, Vais H, Thompson CB, Foskett JK, White C (2007) Apoptosis regulation by Bcl-x(L) modulation of mammalian inositol 1,4,5-trisphosphate receptor channel isoform gating. Proc Natl Acad Sci U S A 104:12565–12570
Li Y, Sattler R, Yang EJ, Nunes A, Ayukawa Y, Akhtar S, Ji G, Zhang PW, Rothstein JD (2011) Harmine, a natural beta-carboline alkaloid, upregulates astroglial glutamate transporter expression. Neuropharmacology 60:1168–1175
Li X, Guan Y, Chen Y, Zhang C, Shi C, Zhou F, Yu L, Juan J, Wang X (2013) Expression of Wnt5a and its receptor Fzd2 is changed in the spinal cord of adult amyotrophic lateral sclerosis transgenic mice. Int J Clin Exp Pathol 6:1245–1260
Liang X, Wang Q, Shi J, Lokteva L, Breyer RM, Montine TJ, Andreasson K (2008) The prostaglandin E2 EP2 receptor accelerates disease progression and inflammation in a model of amyotrophic lateral sclerosis. Ann Neurol 64:304–314
Liang H, Ward WF, Jang YC, Bhattacharya A, Bokov AF, Li Y, Jernigan A, Richardson A, Van Remmen H (2011) PGC-1alpha protects neurons and alters disease progression in an amyotrophic lateral sclerosis mouse model. Muscle Nerve 44:947–956
Lin CL, Bristol LA, Jin L, Dykes-Hoberg M, Crawford T, Clawson L, Rothstein JD (1998) Aberrant RNA processing in a neurodegenerative disease: the cause for absent EAAT2, a glutamate transporter, in amyotrophic lateral sclerosis. Neuron 20:589–602
Lobsiger CS, Boillee S, McAlonis-Downes M, Khan AM, Feltri ML, Yamanaka K, Cleveland DW (2009) Schwann cells expressing dismutase active mutant SOD1 unexpectedly slow disease progression in ALS mice. Proc Natl Acad Sci U S A 106:4465–4470
Lomen-Hoerth C, Anderson T, Miller B (2002) The overlap of amyotrophic lateral sclerosis and frontotemporal dementia. Neurology 59:1077–1079
Mackenzie IR, Munoz DG, Kusaka H, Yokota O, Ishihara K, Roeber S, Kretzschmar HA, Cairns NJ, Neumann M (2011) Distinct pathological subtypes of FTLD-FUS. Acta Neuropathol 121:207–218
Magnus T, Carmen J, Deleon J, Xue H, Pardo AC, Lepore AC, Mattson MP, Rao MS, Maragakis NJ (2008) Adult glial precursor proliferation in mutant SOD1G93A mice. Glia 56:200–208
Marchetto MC, Muotri AR, Mu Y, Smith AM, Cezar GG, Gage FH (2008) Non-cell-autonomous effect of human SOD1 G37R astrocytes on motor neurons derived from human embryonic stem cells. Cell Stem Cell 3:649–657
Martorana F, Brambilla L, Valori CF, Bergamaschi C, Roncoroni C, Aronica E, Volterra A, Bezzi P, Rossi D (2012) The BH4 domain of Bcl-X(L) rescues astrocyte degeneration in amyotrophic lateral sclerosis by modulating intracellular calcium signals. Hum Mol Genet 21:826–840
Maruyama H et al (2010) Mutations of optineurin in amyotrophic lateral sclerosis. Nature 465:223–226
McDonald KK, Aulas A, Destroismaisons L, Pickles S, Beleac E, Camu W, Rouleau GA, Vande Velde C (2011) TAR DNA-binding protein 43 (TDP-43) regulates stress granule dynamics via differential regulation of G3BP and TIA-1. Hum Mol Genet 20:1400–1410
Mead RJ, Higginbottom A, Allen SP, Kirby J, Bennett E, Barber SC, Heath PR, Coluccia A, Patel N, Gardner I, Brancale A, Grierson AJ, Shaw PJ (2013) S[+] Apomorphine is a CNS penetrating activator of the Nrf2-ARE pathway with activity in mouse and patient fibroblast models of amyotrophic lateral sclerosis. Free Rad Biol Med 61C:438–452
Mendonca DM, Chimelli L, Martinez AM (2006) Expression of ubiquitin and proteasome in motorneurons and astrocytes of spinal cords from patients with amyotrophic lateral sclerosis. Neurosci Lett 404:315–319
Mimoto T, Miyazaki K, Morimoto N, Kurata T, Satoh K, Ikeda Y, Abe K (2012) Impaired antioxydative Keap1/Nrf2 system and the downstream stress protein responses in the motor neuron of ALS model mice. Brain Res 1446:109–118
Miquel E, Cassina A, Martinez-Palma L, Bolatto C, Trias E, Gandelman M, Radi R, Barbeito L, Cassina P (2012) Modulation of astrocytic mitochondrial function by dichloroacetate improves survival and motor performance in inherited amyotrophic lateral sclerosis. PloS ONE 7:e34776
Mitchell J, Paul P, Chen HJ, Morris A, Payling M, Falchi M, Habgood J, Panoutsou S, Winkler S, Tisato V, Hajitou A, Smith B, Vance C, Shaw C, Mazarakis ND, de Belleroche J (2010) Familial amyotrophic lateral sclerosis is associated with a mutation in D-amino acid oxidase. Proc Natl Acad Sci U S A 107:7556–7561
Molofsky AV, Krencik R, Ullian EM, Tsai HH, Deneen B, Richardson WD, Barres BA, Rowitch DH (2012) Astrocytes and disease: a neurodevelopmental perspective. Genes Dev 26:891–907
Morel L, Regan M, Higashimori H, Ng SK, Esau C, Vidensky S, Rothstein J, Yang Y (2013) Neuronal exosomal miRNA-dependent translational regulation of astroglial glutamate transporter GLT1. J Biol Chem 288:7105–7116
Nagai M, Re DB, Nagata T, Chalazonitis A, Jessell TM, Wichterle H, Przedborski S (2007) Astrocytes expressing ALS-linked mutated SOD1 release factors selectively toxic to motor neurons. Nat Neurosci 10:615–622
Nanou A, Higginbottom A, Valori CF, Wyles M, Ning K, Shaw P, Azzouz M (2013) Viral delivery of antioxidant genes as a therapeutic strategy in experimental models of amyotrophic lateral sclerosis. Mol Ther 21:1486–1496
Neumann M, Sampathu DM, Kwong LK, Truax AC, Micsenyi MC, Chou TT, Bruce J, Schuck T, Grossman M, Clark CM, McCluskey LF, Miller BL, Masliah E, Mackenzie IR, Feldman H, Feiden W, Kretzschmar HA, Trojanowski JQ, Lee VM (2006) Ubiquitinated TDP-43 in frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Science 314:130–133
Neumann M, Kwong LK, Truax AC, Vanmassenhove B, Kretzschmar HA, Van Deerlin VM, Clark CM, Grossman M, Miller BL, Trojanowski JQ, Lee VM (2007) TDP-43-positive white matter pathology in frontotemporal lobar degeneration with ubiquitin-positive inclusions. J Neuropathol Exp Neurol 66:177–183
Neymotin A, Calingasan NY, Wille E, Naseri N, Petri S, Damiano M, Liby KT, Risingsong R, Sporn M, Beal MF, Kiaei M (2011) Neuroprotective effect of Nrf2/ARE activators, CDDO ethylamide and CDDO trifluoroethylamide, in a mouse model of amyotrophic lateral sclerosis. Free Radic Biol Med 51:88–96
Nishihira Y, Tan CF, Onodera O, Toyoshima Y, Yamada M, Morita T, Nishizawa M, Kakita A, Takahashi H (2008) Sporadic amyotrophic lateral sclerosis: two pathological patterns shown by analysis of distribution of TDP-43-immunoreactive neuronal and glial cytoplasmic inclusions. Acta Neuropathol 116:169–182
Oberheim NA, Goldman SA, Nedergaard M (2012) Heterogeneity of astrocytic form and function. Methods Mol Biol 814:23–45
Papadeas ST, Kraig SE, O’Banion C, Lepore AC, Maragakis NJ (2011) Astrocytes carrying the superoxide dismutase 1 (SOD1G93A) mutation induce wild-type motor neuron degeneration in vivo. Proc Natl Acad Sci U S A 108:17803–17808
Papadopoulos V, Baraldi M, Guilarte TR, Knudsen TB, Lacapere JJ, Lindemann P, Norenberg MD, Nutt D, Weizman A, Zhang MR, Gavish M (2006) Translocator protein (18 kDa): new nomenclature for the peripheral-type benzodiazepine receptor based on its structure and molecular function. Trends Pharmacol Sci 27:402–409
Pardo AC, Wong V, Benson LM, Dykes M, Tanaka K, Rothstein JD, Maragakis NJ (2006) Loss of the astrocyte glutamate transporter GLT1 modifies disease in SOD1(G93A) mice. Exp Neurol 201:120–130
Parpura V, Heneka MT, Montana V, Oliet SH, Schousboe A, Haydon PG, Stout RF Jr, Spray DC, Reichenbach A, Pannicke T, Pekny M, Pekna M, Zorec R, Verkhratsky A (2012) Glial cells in (patho)physiology. J Neurochem 121:4–27
Pasinelli P, Houseweart MK, Brown RH Jr, Cleveland DW (2000) Caspase-1 and -3 are sequentially activated in motor neuron death in Cu, Zn superoxide dismutase-mediated familial amyotrophic lateral sclerosis. Proc Natl Acad Sci U S A 97:13901–13906
Pehar M, Vargas MR, Robinson KM, Cassina P, Diaz-Amarilla PJ, Hagen TM, Radi R, Barbeito L, Beckman JS (2007) Mitochondrial superoxide production and nuclear factor erythroid 2-related factor 2 activation in p75 neurotrophin receptor-induced motor neuron apoptosis. J Neurosci 27:7777–7785
Perrie WT, Lee GT, Curtis EM, Sparke J, Buller JR, Rossi ML (1993) Changes in the myelinated axons of femoral nerve in amyotrophic lateral sclerosis. J Neural Transm Suppl 39:223–233
Phatnani HP, Guarnieri P, Friedman BA, Carrasco MA, Muratet M, O’Keeffe S, Nwakeze C, Pauli-Behn F, Newberry KM, Meadows SK, Tapia JC, Myers RM, Maniatis T (2013) Intricate interplay between astrocytes and motor neurons in ALS. Proc Natl Acad Sci U S A 110:E756–E765
Philips T, Bento-Abreu A, Nonneman A, Haeck W, Staats K, Geelen V, Hersmus N, Kusters B, Van Den Bosch L, Van Damme P, Richardson WD, Robberecht W (2013) Oligodendrocyte dysfunction in the pathogenesis of amyotrophic lateral sclerosis. Brain 136:471–482
Pun S, Santos AF, Saxena S, Xu L, Caroni P (2006) Selective vulnerability and pruning of phasic motoneuron axons in motoneuron disease alleviated by CNTF. Nat Neurosci 9:408–419
Renton AE et al (2011) A hexanucleotide repeat expansion in C9ORF72 is the cause of chromosome 9p21-linked ALS-FTD. Neuron 72:257–268
Ringholz GM, Appel SH, Bradshaw M, Cooke NA, Mosnik DM, Schulz PE (2005) Prevalence and patterns of cognitive impairment in sporadic ALS. Neurology 65:586–590
Rinholm JE, Hamilton NB, Kessaris N, Richardson WD, Bergersen LH, Attwell D (2011) Regulation of oligodendrocyte development and myelination by glucose and lactate. J Neurosci 31:538–548
Roberts K, Zeineddine R, Corcoran L, Li W, Campbell IL, Yerbury JJ (2013) Extracellular aggregated Cu/Zn superoxide dismutase activates microglia to give a cytotoxic phenotype. Glia 61:409–419
Robertson J, Bilbao J, Zinman L, Hazrati LN, Tokuhiro S, Sato C, Moreno D, Strome R, Mackenzie IR, Rogaeva E (2011) A novel double mutation in FUS gene causing sporadic ALS. Neurobiol Aging 32 553:e527–e530
Rong YP, Aromolaran AS, Bultynck G, Zhong F, Li X, McColl K, Matsuyama S, Herlitze S, Roderick HL, Bootman MD, Mignery GA, Parys JB, De Smedt H, Distelhorst CW (2008) Targeting Bcl-2-IP3 receptor interaction to reverse Bcl-2’s inhibition of apoptotic calcium signals. Mol Cell 31:255–265
Rong YP, Bultynck G, Aromolaran AS, Zhong F, Parys JB, De Smedt H, Mignery GA, Roderick HL, Bootman MD, Distelhorst CW (2009) The BH4 domain of Bcl-2 inhibits ER calcium release and apoptosis by binding the regulatory and coupling domain of the IP3 receptor. Proc Natl Acad Sci U S A 106:14397–14402
Rosen DR, Siddique T, Patterson D, Figlewicz DA, Sapp P, Hentati A, Donaldson D, Goto J, O’Regan JP, Deng HX et al (1993) Mutations in Cu/Zn superoxide dismutase gene are associated with familial amyotrophic lateral sclerosis. Nature 362:59–62
Rossi D, Brambilla L, Valori CF, Roncoroni C, Crugnola A, Yokota T, Bredesen DE, Volterra A (2008) Focal degeneration of astrocytes in amyotrophic lateral sclerosis. Cell Death Differ 15:1691–1700
Rossi D, Martorana F, Brambilla L (2011) Implications of gliotransmission for the pharmacotherapy of CNS disorders. CNS Drugs 25:641–658
Rosso SB, Inestrosa NC (2013) WNT signaling in neuronal maturation and synaptogenesis. Front Cell Neurosci 7:103
Rothstein JD, Tsai G, Kuncl RW, Clawson L, Cornblath DR, Drachman DB, Pestronk A, Stauch BL, Coyle JT (1990) Abnormal excitatory amino acid metabolism in amyotrophic lateral sclerosis. Ann Neurol 28:18–25
Rothstein JD, Martin LJ, Kuncl RW (1992) Decreased glutamate transport by the brain and spinal cord in amyotrophic lateral sclerosis. N Engl J Med 326:1464–1468
Rothstein JD, Van Kammen M, Levey AI, Martin LJ, Kuncl RW (1995) Selective loss of glial glutamate transporter GLT-1 in amyotrophic lateral sclerosis. Ann Neurol 38:73–84
Rothstein JD, Dykes-Hoberg M, Pardo CA, Bristol LA, Jin L, Kuncl RW, Kanai Y, Hediger MA, Wang Y, Schielke JP, Welty DF (1996) Knockout of glutamate transporters reveals a major role for astroglial transport in excitotoxicity and clearance of glutamate. Neuron 16:675–686
Rothstein JD, Patel S, Regan MR, Haenggeli C, Huang YH, Bergles DE, Jin L, Dykes Hoberg M, Vidensky S, Chung DS, Toan SV, Bruijn LI, Su ZZ, Gupta P, Fisher PB (2005) Beta-lactam antibiotics offer neuroprotection by increasing glutamate transporter expression. Nature 433:73–77
Rubin LL, Hall DE, Porter S, Barbu K, Cannon C, Horner HC, Janatpour M, Liaw CW, Manning K, Morales J et al (1991) A cell culture model of the blood-brain barrier. J Cell Biol 115:1725–1735
Sarlette A, Krampfl K, Grothe C, Neuhoff N, Dengler R, Petri S (2008) Nuclear erythroid 2-related factor 2-antioxidative response element signaling pathway in motor cortex and spinal cord in amyotrophic lateral sclerosis. J Neuropathol Exp Neurol 67:1055–1062
Sasabe J, Chiba T, Yamada M, Okamoto K, Nishimoto I, Matsuoka M, Aiso S (2007) D-serine is a key determinant of glutamate toxicity in amyotrophic lateral sclerosis. EMBO J 26:4149–4159
Sasabe J, Miyoshi Y, Suzuki M, Mita M, Konno R, Matsuoka M, Hamase K, Aiso S (2012) D-amino acid oxidase controls motoneuron degeneration through D-serine. Proc Natl Acad Sci U S A 109:627–632
Seilhean D, Cazeneuve C, Thuries V, Russaouen O, Millecamps S, Salachas F, Meininger V, Leguern E, Duyckaerts C (2009) Accumulation of TDP-43 and alpha-actin in an amyotrophic lateral sclerosis patient with the K17I ANG mutation. Acta Neuropathol 118:561–573
Serio A, Bilican B, Barmada SJ, Ando DM, Zhao C, Siller R, Burr K, Haghi G, Story D, Nishimura AL, Carrasco MA, Phatnani HP, Shum C, Wilmut I, Maniatis T, Shaw CE, Finkbeiner S, Chandran S (2013) Astrocyte pathology and the absence of non-cell autonomy in an induced pluripotent stem cell model of TDP-43 proteinopathy. Proc Natl Acad Sci U S A 110:4697–4702
Sobue K, Yamamoto N, Yoneda K, Hodgson ME, Yamashiro K, Tsuruoka N, Tsuda T, Katsuya H, Miura Y, Asai K, Kato T (1999) Induction of blood-brain barrier properties in immortalized bovine brain endothelial cells by astrocytic factors. Neurosci Res 35:155–164
Sommer B, Kohler M, Sprengel R, Seeburg PH (1991) RNA editing in brain controls a determinant of ion flow in glutamate-gated channels. Cell 67:11–19
Sreedharan J, Blair IP, Tripathi VB, Hu X, Vance C, Rogelj B, Ackerley S, Durnall JC, Williams KL, Buratti E, Baralle F, de Belleroche J, Mitchell JD, Leigh PN, Al-Chalabi A, Miller CC, Nicholson G, Shaw CE (2008) TDP-43 mutations in familial and sporadic amyotrophic lateral sclerosis. Science 319:1668–1672
Stieber A, Gonatas JO, Gonatas NK (2000) Aggregates of mutant protein appear progressively in dendrites, in periaxonal processes of oligodendrocytes, and in neuronal and astrocytic perikarya of mice expressing the SOD1(G93A) mutation of familial amyotrophic lateral sclerosis. J Neurol Sci 177:114–123
Swarup V, Phaneuf D, Dupre N, Petri S, Strong M, Kriz J, Julien JP (2011) Deregulation of TDP-43 in amyotrophic lateral sclerosis triggers nuclear factor kappaB-mediated pathogenic pathways. J Exp Med 208:2429–2447
Tanaka K, Watase K, Manabe T, Yamada K, Watanabe M, Takahashi K, Iwama H, Nishikawa T, Ichihara N, Kikuchi T, Okuyama S, Kawashima N, Hori S, Takimoto M, Wada K (1997) Epilepsy and exacerbation of brain injury in mice lacking the glutamate transporter GLT-1. Science 276:1699–1702
Tong J, Huang C, Bi F, Wu Q, Huang B, Liu X, Li F, Zhou H, Xia XG (2013) Expression of ALS-linked TDP-43 mutant in astrocytes causes non-cell-autonomous motor neuron death in rats. EMBO J 32:1917–1926
Troost D, van den Oord JJ, de Jong JM, Swaab DF (1989) Lymphocytic infiltration in the spinal cord of patients with amyotrophic lateral sclerosis. Clin Neuropathol 8:289–294
Trumbull KA, McAllister D, Gandelman MM, Fung WY, Lew T, Brennan L, Lopez N, Morre J, Kalyanaraman B, Beckman JS (2012) Diapocynin and apocynin administration fails to significantly extend survival in G93A SOD1 ALS mice. Neurobiol Dis 45:137–144
Tsao W, Jeong YH, Lin S, Ling J, Price DL, Chiang PM, Wong PC (2012) Rodent models of TDP-43: recent advances. Brain Res 1462:26–39
Turner BJ, Talbot K (2008) Transgenics, toxicity and therapeutics in rodent models of mutant SOD1-mediated familial ALS. Prog Neurobiol 85:94–134
Turner MR, Cagnin A, Turkheimer FE, Miller CC, Shaw CE, Brooks DJ, Leigh PN, Banati RB (2004) Evidence of widespread cerebral microglial activation in amyotrophic lateral sclerosis: an [11C](R)-PK11195 positron emission tomography study. Neurobiol Dis 15:601–609
Turner BJ, Ackerley S, Davies KE, Talbot K (2010) Dismutase-competent SOD1 mutant accumulation in myelinating Schwann cells is not detrimental to normal or transgenic ALS model mice. Hum Mol Genet 19:815–824
Urushitani M, Sik A, Sakurai T, Nukina N, Takahashi R, Julien JP (2006) Chromogranin-mediated secretion of mutant superoxide dismutase proteins linked to amyotrophic lateral sclerosis. Nat Neurosci 9:108–118
Valori CF, Brambilla L, Martorana F, Rossi D (2013) The multifaceted role of glial cells in amyotrophic lateral sclerosis. Cell Mol Life Sci 71:287–297
Van Damme P, Bogaert E, Dewil M, Hersmus N, Kiraly D, Scheveneels W, Bockx I, Braeken D, Verpoorten N, Verhoeven K, Timmerman V, Herijgers P, Callewaert G, Carmeliet P, Van Den Bosch L, Robberecht W (2007) Astrocytes regulate GluR2 expression in motor neurons and their vulnerability to excitotoxicity. Proc Natl Acad Sci U S A 104:14825–14830
Vance C et al (2009) Mutations in FUS, an RNA processing protein, cause familial amyotrophic lateral sclerosis type 6. Science 323:1208–1211
Vargas MR, Pehar M, Cassina P, Martinez-Palma L, Thompson JA, Beckman JS, Barbeito L (2005) Fibroblast growth factor-1 induces heme oxygenase-1 via nuclear factor erythroid 2-related factor 2 (Nrf2) in spinal cord astrocytes: consequences for motor neuron survival. J Biol Chem 280:25571–25579
Vargas MR, Pehar M, Cassina P, Beckman JS, Barbeito L (2006) Increased glutathione biosynthesis by Nrf2 activation in astrocytes prevents p75NTR-dependent motor neuron apoptosis. J Neurochem 97:687–696
Vargas MR, Johnson DA, Sirkis DW, Messing A, Johnson JA (2008) Nrf2 activation in astrocytes protects against neurodegeneration in mouse models of familial amyotrophic lateral sclerosis. J Neurosci 28:13574–13581
Vargas MR, Burton NC, Gan L, Johnson DA, Schafer M, Werner S, Johnson JA (2013) Absence of Nrf2 or its selective overexpression in neurons and muscle does not affect survival in ALS-linked mutant hSOD1 mouse models. PloS ONE 8:e56625
Wang L, Gutmann DH, Roos RP (2011) Astrocyte loss of mutant SOD1 delays ALS disease onset and progression in G85R transgenic mice. Hum Mol Genet 20:286–293
White C, Li C, Yang J, Petrenko NB, Madesh M, Thompson CB, Foskett JK (2005) The endoplasmic reticulum gateway to apoptosis by Bcl-X(L) modulation of the InsP3R. Nat Cell Biol 7:1021–1028
Winkler EA, Sengillo JD, Sullivan JS, Henkel JS, Appel SH, Zlokovic BV (2013) Blood-spinal cord barrier breakdown and pericyte reductions in amyotrophic lateral sclerosis. Acta Neuropathol (Berl) 125:111–120
Wu CH et al (2012) Mutations in the profilin 1 gene cause familial amyotrophic lateral sclerosis. Nature 488:499–503
Wu DC, Re DB, Nagai M, Ischiropoulos H, Przedborski S (2006) The inflammatory NADPH oxidase enzyme modulates motor neuron degeneration in amyotrophic lateral sclerosis mice. Proc Natl Acad Sci U S A 103:12132–12137
Yamamoto-Watanabe Y, Watanabe M, Okamoto K, Fujita Y, Jackson M, Ikeda M, Nakazato Y, Ikeda Y, Matsubara E, Kawarabayashi T, Shoji M (2010) A Japanese ALS6 family with mutation R521C in the FUS/TLS gene: a clinical, pathological and genetic report. J Neurol Sci 296:59–63
Yamanaka K, Chun SJ, Boillee S, Fujimori-Tonou N, Yamashita H, Gutmann DH, Takahashi R, Misawa H, Cleveland DW (2008) Astrocytes as determinants of disease progression in inherited amyotrophic lateral sclerosis. Nat Neurosci 11:251–253
Yang Y, Hentati A, Deng HX, Dabbagh O, Sasaki T, Hirano M, Hung WY, Ouahchi K, Yan J, Azim AC, Cole N, Gascon G, Yagmour A, Ben-Hamida M, Pericak-Vance M, Hentati F, Siddique T (2001) The gene encoding alsin, a protein with three guanine-nucleotide exchange factor domains, is mutated in a form of recessive amyotrophic lateral sclerosis. Nat Genet 29:160–165
Yang Y, Gozen O, Watkins A, Lorenzini I, Lepore A, Gao Y, Vidensky S, Brennan J, Poulsen D, Won Park J, Li Jeon N, Robinson MB, Rothstein JD (2009) Presynaptic regulation of astroglial excitatory neurotransmitter transporter GLT1. Neuron 61:880–894
Yu L, Guan Y, Wu X, Chen Y, Liu Z, Du H, Wang X (2013) Wnt signaling is altered by spinal cord neuronal dysfunction in amyotrophic lateral sclerosis transgenic mice. Neurochem Res 38:1904–1913
Zhang H, Tan CF, Mori F, Tanji K, Kakita A, Takahashi H, Wakabayashi K (2008) TDP-43-immunoreactive neuronal and glial inclusions in the neostriatum in amyotrophic lateral sclerosis with and without dementia. Acta Neuropathol 115:115–122
Zhong F, Davis MC, McColl KS, Distelhorst CW (2006) Bcl-2 differentially regulates Ca2+ signals according to the strength of T cell receptor activation. J Cell Biol 172:127–137
Zhong Z, Deane R, Ali Z, Parisi M, Shapovalov Y, O’Banion MK, Stojanovic K, Sagare A, Boillee S, Cleveland DW, Zlokovic BV (2008) ALS-causing SOD1 mutants generate vascular changes prior to motor neuron degeneration. Nat Neurosci 11:420–422
Zur Nieden R, Deitmer JW (2006) The role of metabotropic glutamate receptors for the generation of calcium oscillations in rat hippocampal astrocytes in situ. Cereb Cortex 16:676–687
Acknowledgements
C.F.V receives a stipend by the Swiss National Science Foundation (grant 31003A-132864). D. R. received funds by the Telethon Foundation (grant GGP05244) to study the role of glial cells in ALS pathogenesis and progression. Figure 11.1 was produced using Servier Medical Art (http://www.servier.com).
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2014 Springer Science+Business Media New York
About this chapter
Cite this chapter
Valori, C., Brambilla, L., Rossi, D. (2014). Amyotrophic Lateral Sclerosis: A Glial Perspective. In: Parpura, V., Verkhratsky, A. (eds) Pathological Potential of Neuroglia. Springer, New York, NY. https://doi.org/10.1007/978-1-4939-0974-2_11
Download citation
DOI: https://doi.org/10.1007/978-1-4939-0974-2_11
Published:
Publisher Name: Springer, New York, NY
Print ISBN: 978-1-4939-0973-5
Online ISBN: 978-1-4939-0974-2
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)