
Abstract
To replicate their genomes in cells and generate new progeny, viruses typically require factors provided by the cells that they have infected. Subversion of the cellular machinery that controls replication of the infected host cell is a common activity of many viruses. Viruses employ different strategies to deregulate cell cycle checkpoint controls and modulate cell proliferation pathways. A number of DNA and RNA viruses encode proteins that target critical cell cycle regulators to achieve cellular conditions that are beneficial for viral replication. Many DNA viruses induce quiescent cells to enter the cell cycle; this is thought to increase pools of deoxynucleotides and thus, facilitate viral replication. In contrast, some viruses can arrest cells in a particular phase of the cell cycle that is favorable for replication of the specific virus. Cell cycle arrest may inhibit early cell death of infected cells, allow the cells to evade immune defenses, or help promote virus assembly. Although beneficial for the viral life cycle, virus-mediated alterations in normal cell cycle control mechanisms could have detrimental effects on cellular physiology and may ultimately contribute to pathologies associated with the viral infection, including cell transformation and cancer progression and maintenance. In this chapter, we summarize various strategies employed by DNA and RNA viruses to modulate the replication cycle of the virus-infected cell. When known, we describe how these virus-associated effects influence replication of the virus and contribute to diseases associated with infection by that specific virus.
You have full access to this open access chapter, Download protocol PDF
Similar content being viewed by others


Key words
1 Introduction
Viruses are obligate intracellular parasites that depend on the infected host cell for the resources that are required to replicate the viral genome; viruses have evolved multiple mechanisms to manipulate the environment of infected cells in order to replicate more efficiently [1]. Viral genomes can be composed of single- or double-stranded DNA or single- or double-stranded RNA, hereafter referred to as DNA or RNA viruses, respectively. While many viruses replicate their genomes by directly generating an exact DNA or RNA copy of the genome, other viruses, such as retroviruses or hepadnaviruses, use reverse transcription to generate intermediates that are required for their replication [2]. Subversion of the host cell replication cycle, hereafter referred to as the “cell cycle,” is a common strategy employed by many viruses to create a cellular environment that is favorable for viral replication [1]. Examples of virus-induced alterations in cellular replication processes have been identified as consequences of infection by both DNA and RNA viruses.
DNA viruses have been studied more extensively in regard to effects on cell cycle control. Many DNA viruses primarily infect quiescent or differentiated cells, which contain rate-limiting levels of deoxynucleotides and may not represent an ideal environment for viral replication. It is thought that these viruses can induce quiescent cells to enter the cell cycle in order to create an environment that generates factors, such as nucleotides, that are required for viral replication [3]. Some small DNA tumor viruses can promote entry into the S phase in order to activate the host cell DNA replication machinery and provide the resources necessary for viral replication. In contrast, some larger DNA viruses such as Herpesviruses can elicit a cell cycle arrest to limit the competition between the virus and the host for cellular DNA replication resources. Retroviruses and other RNA viruses can also interfere with the host cell cycle [1, 4–7]. There are various speculations regarding the advantages associated with regulation of the cell cycle by RNA viruses; these include increasing the efficiency of replication, translation, and virus assembly [8, 9]. Cell cycle arrest may also help delay the apoptosis of infected cells [10]. Additionally, a G2/M arrest induced by the human immunodeficiency virus (HIV) type-1 is thought to help HIV-1 avoid human immune defenses by preventing new cell production [8]. Overall, both DNA and RNA viruses manipulate the cell cycle to generate resources and cellular conditions that favor viral replication.
An unfortunate consequence of virus-mediated deregulation of normal cell cycle control mechanisms is that these effects may ultimately generate an environment that promotes disease, including the development, progression, or maintenance of certain types of cancer [11]. Some viruses encode proteins that deregulate normal cell cycle controls and manipulate cell proliferation pathways, and some of these proteins can directly influence the oncogenic potential of that virus. Viruses that cause human cancers include Hepatitis B virus (HBV), Hepatitis C virus (HCV), Human T-cell lymphotropic virus type I, Kaposi’s sarcoma-associated herpesvirus (KSHV), Epstein–Barr virus (EBV), and Human papillomavirus (HPV), and viral infections may account for approximately 20 % of all human cancers worldwide [12–14]. Deregulation of the cell cycle and alteration in the expression levels and activities of the cell cycle regulatory proteins are frequently observed in transformed cells; consequently, disruption of normal mechanisms that regulate the cell cycle is thought to contribute to the development of many cancers [15]. The study of viral regulation of the cell cycle has contributed to our understanding of viral replication processes and mechanisms that regulate the cell cycle and are altered in cancers. Moreover, analyses of the dynamic regulation of cell cycle by viruses have helped highlight key regulators of cell cycle progression. The cell cycle factors that are targeted by specific viral gene products to deregulate the cell cycle can be potential therapeutic targets for antiviral interventions and prevention of associated cancers [1, 16, 17].
In this chapter, we focus on different strategies employed by viruses to manipulate the host cell cycle in order to create an environment conducive for viral replication. A description of all viral factors that influence the cell cycle is beyond the scope of this chapter. Instead, examples of how some DNA and RNA viruses regulate different stages of the cell cycle are discussed to illustrate various viral strategies. Viral regulation of the G0/G1 transition, the G1 and S phases, and the G2/M checkpoint will be the focus of this review. In each section, we provide examples of viruses that can regulate the specific phase of the cell cycle, describe viral proteins that are involved in the virus-mediated deregulation of the cell cycle and mechanisms associated with the effects of these viral proteins, and discuss known or proposed consequences of the virus-mediated cell cycle stimulation and/or arrest for the virus life cycle and virus-associated diseases. Regulation of the cell cycle by certain viruses, such as the small DNA tumor viruses, has been studied for decades and has been reviewed extensively [18, 19]. While we briefly describe how these viruses modulate the host cell cycle, we emphasize more recently discovered effects of the Influenza A virus, HCV, HBV, and KSHV on the cell cycle. Overall, we aim to summarize key mechanisms that are used by viruses to manipulate the cell cycle and to provide insights into the consequences of these viral protein-mediated effects on the cell cycle for both the virus and the host cell.
2 The Cell Cycle
2.1 An Overview of the Cell Cycle
The eukaryotic cell cycle is composed of an ordered and tightly regulated series of events that can be controlled by intracellular and extracellular factors. The cell cycle also includes checkpoints that ensure normal cell cycle progression. The eukaryotic cell cycle consists of 4 phases: Gap 1 (G1), Synthesis (S), Gap 2 (G2), and Mitosis (M) (Fig. 1) [20, 21]. Differentiated cells are usually maintained in a nondividing state, known as the quiescent or G0 phase [22]. Quiescent cells must receive a growth signal in order to exit the G0 phase and enter the cell cycle [21, 23]. Binding of external factors such as mitogens to their cell surface receptors can activate signaling pathways, such as the Ras/mitogen-activated protein kinase (MAPK) pathway, which play a major role in cell entry into the G1 phase. When quiescent cells receive a growth signal, they enter into the G1 phase. During G1, the cell prepares to replicate its DNA; synthesis of the mRNAs and proteins necessary for DNA synthesis also occurs. The first major checkpoint of the cell cycle, which is present at the G1/S border, is known as the restriction point; if this checkpoint is not activated and the growth signal is still present, the cell proceeds into S phase, the stage during which DNA synthesis and duplication of the cell genome occurs. Once the cell enters S phase, DNA replication is completed regardless of the removal of the growth signal or the presence of DNA damage. After DNA replication is completed, the cell enters the G2 phase and prepares for mitosis, cell division. The G2 phase provides an opportunity for the cellular machinery to check for any DNA damage that may have accumulated during DNA replication. Therefore, cell cycle progression into the S phase and mitosis is controlled by the checkpoints at G1 and G2, respectively. Once the appropriate signals that are required for cell cycle progression are present, the cell enters into the M phase [20, 21]. A third checkpoint, referred to as the spindle checkpoint, exists after metaphase and prior to anaphase, which are steps during mitosis that are required for cell division. At this checkpoint, the cell employs strategies to detect improper alignment of chromosomes on the mitotic spindle. If improper alignment of chromosomes is detected, the cell cycle is stopped in metaphase; however, if the chromosomes are properly attached to the spindle apparatus, the cell continues into anaphase, completes the cell cycle, and eventually generates two daughter cells [20, 24].

Overview of the eukaryotic cell cycle. The eukaryotic cell cycle consists of 4 phases; G1, S, G2, and M. Progression through the cell cycle is tightly controlled; both positive and negative regulators of the cell cycle are shown. See text and references for details
2.2 Mechanisms That Control the Cell Cycle
2.2.1 Positive Regulators of Cell Cycle Progression
Various cellular proteins regulate the transition from one phase of the cell cycle to the next phase. Key regulatory proteins that control cell cycle progression are cyclins and cyclin-dependent kinases (CDKs). CDKs are a family of serine/threonine protein kinases that are activated at specific points in the cell cycle. There are five CDKs that have been associated with cell cycle progression in mammalian cells: CDKs 4 and 6, which are active during the early G1 phase; CDK2, which is active in the late G1 and S phase; CDK1, which is active during the G2 and M phases; and CDK7, which acts in combination with cyclin H as a CDK-activating kinase (CAK) (Fig. 1). The activity of CDKs is highly regulated and requires the expression of activating cyclins and phosphorylation of the cyclin-CDK complex. CDK expression levels remain stable throughout the cell cycle. In contrast to CDK expression, cyclin levels rise and fall depending on the phase of the cell cycle, enabling cyclins to periodically activate the CDKs [20, 21]. The D type cyclins, cyclin D1, cyclin D2, and cyclin D3, bind to CDK4 and CDK6 to activate these CDKs. Activation of CDK4 and CDK6 is required for entry into the G1 phase [25, 26]. Cyclin D is synthesized as long as the growth factor stimulation is present [27]. Cyclin E associates with CDK2 to regulate progression from G1 into S phase [28]. During the S phase, cyclin A binds to CDK2 to regulate S-phase progression, and during the G2 and M phases, cyclin A binds to CDK1 to promote entry into the M phase [29, 30]. An additional cyclin, cyclin B, is expressed during mitosis; cyclin B binds to CDK1 to regulate the remainder of mitosis. Cyclins are rapidly degraded by proteasomes when the cell cycle has progressed beyond the phase during which their expression is required [20].
Complete CDK activity is dependent upon cyclin expression and binding to the CDK as well as the phosphorylation of the CDK by the cyclin H-CDK7 complex, also referred to as the CAK. CAK phosphorylation of the CDKs occurs on conserved threonine residues and induces conformational changes, which can enhance the binding of cyclins to further regulate CDK activity. CDK4 activation requires phosphorylation of threonine 172 of CDK4, activation of CDK2 requires phosphorylation of threonine 160 of CDK2, and CDK1 activation requires phosphorylation of threonine 161 of CDK1 [20]. Phosphorylation of the cyclin-CDK complexes can also inhibit CDK activity. The cyclin A-CDK1 complex can be inhibited by phosphorylation of CDK1 at tyrosine 15 and/or threonine 14 by the kinases Wee1 and Myt1. The enzyme Cdc25 phosphatase can remove this inhibitory phosphate, and this dephosphorylation of CDK1 is required for the full activation of CDK1 and subsequent progression through the cell cycle [31].
Active CDKs induce downstream signaling events by phosphorylating target proteins that regulate cell cycle progression [32, 33]. One of the most frequently studied CDK substrates is the retinoblastoma tumor suppressor protein (pRB), which the CDK4/6-cyclin D complex phosphorylates to inactivate (Fig. 1). In its active state, pRb is in a complex with the histone deacetylase (HDAC) protein and the transcription factors E2F-1 and DP-1. During the G1 phase, pRb is phosphorylated, which results in its inactivation and the subsequent release of E2F-1 and DP-1. E2F-1 activates transcription of genes required for S-phase progression, including the cyclin E, cyclin A, and Cdc25 genes [34–36]. E2F also regulates the expression of genes encoding enzymes that are involved in nucleotide synthesis, such as dihydrofolate reductase, thymidine kinase, and thymidylate synthetase [37]. pRb remains hyperphosphorylated for the rest of the cell cycle, and the cyclin E-CDK2 complex stabilizes this hyperphosphorylated state. During the G1/S phase, the cyclin E-CDK2 complex also phosphorylates p27, a negative regulator of CDK2-containing complexes; this phosphorylation results in degradation of p27 [38, 39]. The cyclin E-CDK2 complexes also phosphorylate histone H1, which may be important for chromosome condensation that is required during DNA replication. Histone H1 is also a substrate for cyclin B-CDK1 complexes. Finally, the cyclin A-dependent kinases regulate initiation of DNA replication by phosphorylating the DNA polymerase alpha primase [20].
2.2.2 Negative Regulators of Cell Cycle Progression
The cell cycle is also controlled by negative regulators; these typically inhibit CDK activity. CDK activity can be negatively regulated by interacting with cellular proteins referred to as CDK inhibitors, or CKIs (Fig. 1). CKIs can either bind to isolated CDKs or to the cyclin-CDK complex to prevent activation of CDKs. There are two families of CKIs, the INK4 (inhibitor of CDK4) family and the Cip (CDK-interacting protein)/Kip (kinase inhibitor protein) family. The INK4 family includes p15 (INK4b), p16 (INK4a), p18 (INK4c), and p19 (INK4d). Members of the INK4 family of CKIs inactivate the CDKs by forming stable complexes with the isolated CDKs prior to cyclin binding. Binding of INK4 family members to CDK4 and CDK6 blocks their association with cyclin D and prevents entry into the G1 phase [20, 21]. Members of the Cip/Kip family include p21 (Waf1, Cip1), p27 (Kip1), and p57 (Kip2). These inhibitors contain a conserved region that is involved in cyclin binding and kinase inhibition [21, 40]. Members of the Cip/Kip family display a broader specificity than the INK4 family and can bind and inhibit the activities of the cyclin E-CDK2, cyclin A-CDK2, and cyclin B-CDK1 complexes [20, 21]. Interestingly, members of the Cip/Kip family of CKIs can participate in activation of the G1 phase by assisting in the assembly of the cyclin D-CDK4/6 complexes in the early G1 phase and by stabilizing this complex throughout G1 [41]. p21 expression is controlled by the transcription factor and tumor suppressor p53. In response to cellular stresses, p53 receives signals from various cellular factors such as Ataxia Telangiectasia Mutated (ATM), Ataxia Telangiectasia and Rad3-related protein (ATR), Chk1, and Chk2, members of a signaling cascade network that responds to the detection of damaged DNA, and stimulates p21 expression and associated inhibition of cell cycle progression beyond the G1 phase [42]. Finally, PP2A phosphatases can dephosphorylate pRB, thus activating pRB so that it can bind with E2F, inhibiting E2F activity and progression of the cell cycle [43].
2.2.3 Control of the G2/M Checkpoint
Additional factors not described above are involved in the control of the G2/M checkpoint. Control of the G2/M checkpoint and progression through the G2 and M phases are critical for the replication of some viruses and are therefore summarized here.
Before the cell enters mitosis, the G2 phase allows a delay in cell cycle progression to ensure that no DNA damage has occurred and that the entire cellular genome has been replicated to generate two copies. The G2/M checkpoint, which is activated in response to DNA damage and incomplete genome replication, induces a G2 arrest and prevents entry into mitosis [44–46]. The ultimate goal of the G2/M checkpoint is to inhibit the cyclin B-CDK1 complex, which is referred to as the mitosis-promoting kinase complex (Fig. 1). Progression of the cell cycle from the G2 phase to mitosis, M phase, requires the activation of CDK1. During the G2 phase, cyclin B accumulates and forms a complex with CDK1. The cyclin B-CDK1 complex, also referred to as the M-CDK complex, is kept inactivated by phosphorylation of a pair of inhibitory sites on CDK1; phosphorylation is catalyzed by the Wee1 kinase. During the late G2 phase, the dephosphorylation of CDK1 by the Cdc25C phosphatase activates the cyclin B-CDK1 complex, which triggers entry into the M phase (Fig. 1). Thus, a balance between the activities of the Wee1 kinase and the Cdc25C phosphatase can regulate the entry of cells into mitosis. Interestingly, Cdc25C can be partly activated by CDK1, and the inhibitory Wee1 kinase may be inhibited by the active M-CDK complex. Since M-CDK can activate its own activator and inhibit its own inhibitor, this suggests that the activation of M-CDK in mitosis involves positive feedback loops [42, 44]. The cyclin B-CDK1 complex must be in the nucleus to phosphorylate the substrates that are required during mitosis [47]. The cyclin B-CDK1 complex can enter the nucleus in the G2 phase; however, since its rate of nuclear export exceeds its rate of nuclear import, the cyclin B-CDK1 complex is predominantly localized in the cytoplasm. The inhibition of nuclear export of the cyclin B-CDK1 complex leads to nuclear accumulation of the active complex, which promotes entry into mitosis [8, 44]. The cyclin B-CDK1 complex can be inactivated by the E3 ubiquitin ligase anaphase-promoting complex (APC), which targets cyclin B for degradation. The ubiquitination of cyclin B by APC, which leads to its degradation, is essential for the cells to exit mitosis [44, 48]. The activity of APC is regulated by interaction with either of two coactivator proteins, cell division cycle protein 20 (Cdc20) or Cdc20 homologue 1 (Cdh1), both of which act on different phases of the cell cycle [49–52]. PP2A can inhibit APC through its interaction with Cdc20 [53–55]. Finally, progression through mitosis requires that spindle fibers attach to chromatids via a complex of proteins called the kinetochores that help pull the sister chromatids apart, which is essential for chromosome segregation [8, 50].
Cdc25C is usually cytoplasmic but translocates to the nucleus before the M phase. However, when Cdc25C is bound to 14-3-3 proteins, Cdc25C is sequestered in the cytoplasm, which prevents it from activating the cyclin B-CDK1 complex [56]. In the presence of DNA damage or stalled DNA replication forks, it is critical for the cells to prevent mitotic entry. Depending upon the type of DNA damage, ATM or ATR is activated, which in turn phosphorylates Chk2 or Chk1, leading to their activation. Both Chk1 and Chk2 phosphorylate Cdc25C on serine residue 216 of Cdc25C, which facilitates binding of Cdc25C to 14-3-3 proteins. Thus, Chk1- and Chk2-mediated phosphorylation of Cdc25C causes cytoplasmic sequestration of Cdc25C and prevents the activation of CDK1. The checkpoint control regulators, ATM-Chk2 or ATR-Chk1, respond to conditions such as DNA damage or inhibition of DNA replication and arrest cells in the G2 phase. These checkpoint pathways can also prevent the nuclear accumulation of cyclin B-CDK1 complexes. Finally, the cyclin B-CDK1 complex can be inhibited following activation of the p53 tumor suppressor pathway. p53 upregulates the expression of p21, which can bind to cyclin B1-CDK1 complexes and inhibit their kinase activity. Further, p53 can also inhibit CDK1 through the activation of 14-3-3σ and DNA damage-inducible 45 (GADD45) [8, 42, 44, 57, 58].
3 Viral Regulation of the G0/G1 Transition
3.1 Influenza A Virus Induces a G0/G1 Phase Cell Cycle Arrest
Influenza A virus (IAV) is an important pathogenic virus that causes influenza in humans. IAV is the most virulent human pathogen among the three types of influenza viruses and causes contagious respiratory illnesses [59–61]. There have been three human IAV pandemics during the last century, with the 1918 flu pandemic, referred to as the Spanish flu pandemic, resulting in about 50–100 million deaths worldwide [62, 63]. IAV belongs to the Orthomyxovirus family; viruses in this family are enveloped and have a single-stranded, negative-sense, segmented RNA genome. Orthomyxoviruses are unique among RNA viruses because Orthomyxoviruses replicate their genomes inside the nucleus of an infected host cell [2, 64–66].
IAVs induce a G0/G1 arrest to create favorable conditions for viral replication (Fig. 2) [10, 67–69]. Influenza A H1N1 virus (a subtype of IAV) can cause a G0/G1 phase accumulation of infected A549 cells, a human lung adenocarcinoma epithelial cell line. This G0/G1-phase arrest was caused by prevention of entry of virus-infected cells into the S phase [10]. Infection with the H1N1 virus decreased the levels of hyperphosphorylated pRb, which is critical for progression of cells from late G1 to S phase. Additionally, HIN1 IAV-infected cells showed a significant increase in levels of the CDK inhibitor, p21 and a decrease in levels of the G1/S cyclins, cyclin D and cyclin E. Interestingly, cells synchronized in the G0/G1 phase and subsequently infected with H1N1 IAV had increased viral protein accumulation and progeny virus production as compared to unsynchronized cells or those synchronized in the G2/M phase. The G0/G1 arrest was also observed in cells infected with different strains of IAV, indicating that the G0/G1 arrest may be a common strategy employed by IAVs to facilitate their own replication [10]. These results were also consistent with other studies demonstrating that influenza viruses cause an increase in the expression of the tumor suppressor, p53 [67, 69]. Since p21 expression is upregulated by p53, it was speculated that influenza virus replication might induce a G0/G1-phase arrest by regulating the p53-p21 signaling axis [10]. However, conflicting results were obtained in a different study where the p53 pathway was found to be downregulated in IAV-infected A549 cells. This study demonstrated that infection with IAV decreased the expression levels of p21 and that inhibition of p53 was important for IAV replication (Fig. 2) [70]. The reasons for these contradictory observations are unknown, and the role of p53 in IAV replication remains incompletely understood.

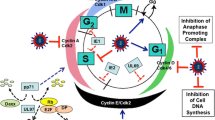
Viral regulation of G0/G1 transition. Examples of viruses that can regulate the G0/G1 transition are shown. Influenza A Virus (IAV), Coronaviruses (SARS-CoA and MHV), and the Herpesvirus (EBV) encode proteins that induce a G0/G1 arrest. The mechanisms by which these viruses induce a G0/G1 arrest are shown. In contrast, some viral proteins (MT-5, encoded by the Myxoma virus and HBx, encoded by the Hepatitis B Virus (HBV)) induce progression from G0 to G1. See text and references for details
IAV-mediated G0/G1 arrest has also been linked to expression of the IAV nonstructural protein 1 (NS1) (Fig. 2) [68]. NS1 is a nonessential IAV protein that has a plethora of accessory functions during viral infection [71]. Of particular importance to cell cycle regulation, the NS1 protein was shown to downregulate the expression and activity of the Ras homologue gene family member A (RhoA) kinase [68]. RhoA is a small GTPase that is critical for the G1/S phase transition. RhoA inhibition can affect G1/S progression by increasing the expression levels of p21 and p27, the accumulation of p16, and decreasing cyclin D1 levels [72–75]. Overexpression of NS1 increased the levels of the CDK inhibitors, p16 and p21, whereas cyclin D1 levels decreased [68]. NS1 also decreased the phosphorylation levels of pRb, a downstream mediator of RhoA. Consistent with the observation that NS1 mediates hypophosphorylation of pRb, CDK4 and CDK6 activities were also reduced. In summary, NS1 protein was found to arrest the host cell cycle at G0/G1 via inhibition of the RhoA-pRb signaling cascade, and this was linked to the enhanced viral protein accumulation and replication [68].
While it is clear that IAV proteins can regulate the expression levels and activities of key host factors that are involved in G1/S phase transition of IAV-infected cells, precisely how the G0/G1 arrest affects IAV replication remains unclear. Although still speculative, there have been some proposed reasons for why IAV induces cells to arrest in G0/G1. For example, IAV transcription requires DNA-dependent RNA polymerase II (Pol II) [76], and the transcriptional activity of Pol II is significantly higher in the G0/G1 phase as compared to the S and G2/M phases [77]. It is therefore possible that IAV arrests cells in the G0/G1 phase to increase the transcriptional activity of Pol II, which would consequently lead to enhanced viral transcription and replication [10]. Secondly, there is evidence that the translation of influenza viral proteins is linked to host cap-dependent translation activity [78, 79]; cap-dependent translation is optimal in the G0/G1 phase and is suppressed in mitosis [80]. Therefore, a G0-G1 arrest of IAV-infected cells would prevent progression into mitosis and could enhance cap-dependent translation of viral proteins and ultimately favor viral replication [10]. Thirdly, since cell cycle arrest can delay induction of apoptosis under certain conditions [81, 82], it is possible that IAV-mediated modulation of the G0/G1 phase prevents early death of infected cells, which would likely benefit IAV genome replication [10].
In summary, the results of many studies suggest that infection with different strains of IAV can alter initiation of the host cell cycle to maintain cells in the G0/G1 phase; retention of IAV-infected cells in the G0/G1 phase is thought to provide optimal conditions for IAV replication. In future studies, it would be interesting to determine the impact of an IAV infection on cell cycle initiation in primary human lung epithelial cells. This would provide valuable information for defining mechanisms that link IAV-dependent modulation of the cell cycle to enhanced IAV replication in the normal site of an IAV infection.
3.2 Severe Acute Respiratory Syndrome Coronavirus and the Murine Coronavirus Arrest Cells in the G0/G1 Phase of the Cell Cycle
Severe acute respiratory syndrome coronavirus (SARS-CoV) and the murine coronavirus mouse hepatitis virus (MHV) can induce a G0/G1-phase arrest of infected host cells (Fig. 2) [83–86]. SARS-CoV and MHV belong to the Coronavirus family of viruses [87]. Members of the Coronavirus family are enveloped viruses with a positive-sense, single-stranded RNA genome [2]. SARS-CoV is the causative agent of the severe acute respiratory syndrome (SARS) [87]. The genome of SARS-CoV encodes a replicase, four major structural proteins, and a number of nonstructural proteins [87–89]. The SARS-CoV 3b nonstructural protein can induce cell cycle arrest at the G0/G1 phase (Fig. 2) [83]. Additionally, the SARS-CoV 7a nonstructural protein can inhibit cell growth and induce a G0/G1-phase arrest (Fig. 2); expression of 7a was shown to decrease the levels of cyclin D3 and inhibit phosphorylation of pRb [84]. Unfortunately the effects of 3b and 7a have not been assessed in the context of SARS-CoV infection, and it remains unclear whether these effects are apparent during an authentic SARS-CoV infection. Further, the molecular mechanisms that underlie the effects of 3b and 7a on the host cell cycle remain undefined.
MHV can also modulate the cell cycle of infected cells [85, 86]. MHV causes various diseases in mice, including hepatitis and enteritis [90, 91]. The results of one study demonstrated that MHV infection inhibited cellular DNA synthesis and caused an accumulation of the infected cells in the G0/G1 phase (Fig. 2) [85]. When quiescent cells were infected with MHV and then serum stimulated, these cells failed to enter S phase. MHV infection led to a reduction in the levels of Cdk4, Cdk6, and G1 cyclins in infected cell, which led to insufficient phosphorylation of pRb and caused the cells to arrest at the G0/G1 phase [85]. Results from another study demonstrated that the MHV nonstructural protein p28 can also induce a G0/G1-phase arrest (Fig. 2) [86]. The expression of p28 induced the stabilization and accumulation of p53, which elevated transcription of p21. The increased levels of p21 suppressed cyclin E-CDK2 activity and resulted in an accumulation of hypo- and/or unphosphorylated Rb. Whether similar effects are apparent in the context of a natural MHV infection was not determined, and future studies could focus on defining the effect of p28, in the presence of other MHV proteins and in the context of MHV replication, on the infected host cell cycle.
The effect of MHV-induced cell cycle arrest on MHV replication remains incompletely understood; however, various possibilities have been proposed [85]. The first proposed possibility is that cell cycle arrest in the G0/G1 phase may provide greater amounts of ribonucleotides for the synthesis of MHV RNA. Since ribonucleotides are precursors for synthesis of deoxyribonucleotides, an inhibition of host cellular DNA synthesis could increase the availability of ribonucleotides in cells and promote efficient MHV RNA synthesis [85]. A second proposed reason for why MHV causes cell cycle arrest is that this may prevent the induction and execution of early cell death in the MHV-infected cells [85]. MHV replication in cultured cells has been shown to lead to cell death; however, the onset of apoptosis occurs when the highest levels of MHV production are attained [92–94]. It is not yet clear how MHV can attain maximal levels of viral replication prior to cell death. In certain systems, apoptosis has been shown to follow cell cycle arrest [81, 82], but in other systems cell cycle progression is required for the induction of apoptosis [95]. It is possible that MHV-dependent inhibition of the cell cycle slows the activation of apoptotic pathways in MHV-infected cells to allow for maximum viral replication prior to apoptosis of the infected cells. A third proposed reason for why MHV may cause cell cycle arrest is that this might facilitate efficient MHV assembly [85]. Assembly of MHV occurs in the intermediate compartment between the endoplasmic reticulum (ER) and the Golgi apparatus and requires proper intracellular membrane structures [96, 97]. Since most membrane trafficking steps are disrupted during the M phase [98, 99], MHV-mediated host cell cycle arrest may lead to efficient MHV assembly [85]. A fourth proposed reason for why MHV causes cell cycle arrest is that this may be beneficial for cap-dependent translation of MHV mRNAs [85]. Due to the impaired function of the cap-binding protein, cap-dependent translation is reduced during the M phase [100]. All the mRNAs of MHV are capped at the 5′ end, and the translation of all MHV proteins, except the E protein, is cap-dependent [101]. Lastly, MHV-induced cell cycle arrest may have an important significance for MHV-induced pathogenesis [85]. Since noncycling cells are less susceptible to being killed by cytotoxic T cells [102], MHV-infected cells arrested in the G0/G1 phase may be less likely to be killed by cytotoxic T cells [85].
3.3 Additional Viruses That Regulate the G0/G1 Transition
The Zta protein of the EBV, a member of the Herpesvirus family, can induce a G0/G1 arrest [4]. Some viruses can also induce cell cycle entry in resting cells by dysregulating the G0/G1 transition. For example, the myxoma virus M-T5 protein can promote the transition of myxoma virus infected cells out of the G0 phase [103]. The HBV, a member of the Hepadnavirus family, has also been shown to induce an exit of cells from G0 into the G1 phase [104]; HBV regulation of the cell cycle will be described below.
4 Viruses That Regulate the G1 and S Phases of the Cell Cycle
4.1 Hepatitis C Virus Modulates the G1/S Checkpoint
HCV, a member of the Flavivirus family, is a small, enveloped virus with a positive-sense, single-stranded RNA genome. The HCV genome encodes a large polyprotein that is co- and posttranslationally processed to produce the mature structural core, E1 and E2 and nonstructural NS2, NS3, NS4A, NS4B, NS5A, and NS5B proteins [105, 106]. HCV infections are a global health problem that affects approximately 170 million people worldwide [107]. HCV is hepatotropic and is one of the major causes of chronic hepatitis, cirrhosis, and primary liver cancer, hepatocellular carcinoma (HCC) in humans [108]. Currently, there is no effective vaccine against HCV infection, and the standard treatment, consisting of a combination of pegylated interferon-α and a nucleoside analogue, ribavirin, produces a sustained virological response in only 50 % of the patients infected with genotype 1 and 80 % of the patients infected with genotypes 2 and 3 [109, 110]. The use of pegylated interferon-α and ribavirin has various side effects such as hematological complications. There are many new therapies for HCV infection in clinical development including Direct-Acting Antiviral Agents (DAA) and Host-Targeting Antiviral Agents. Two DAAs, the protease inhibitors boceprevir and telaprevir, which are both reversible covalent inhibitors of the HCV NS3-NS4A serine protease, have been approved for HCV treatment. Additionally, other DAAs, which are in phase III studies, include an NS5A inhibitor, NS5B polymerase inhibitors, and noncovalent NS3-NS4A protease inhibitors. Additionally, certain host-targeting antiviral agents, including inhibitors of cyclophilin A and microRNA (miR)122, have advanced to phase 2 or 3 clinical trials. The approval of boceprevir and telaprevir has led to the use of a triple therapy for HCV genotype 1 infection. A triple therapy regimen usually consists of one of these two protease inhibitors in combination with pegylated interferon and ribavirin. Although the triple therapy regimens are usually more effective than a combination of pegylated interferon and ribavirin without a protease inhibitor, they are associated with various side effects, and the combination of pegylated interferon and ribavirin remains the recommended treatment for HCV genotypes 2, 3, 4, 5, and 6 infection [109–111].
HCV proteins have been shown to both promote and inhibit cell cycle progression, and it is likely that the effects of HCV on the cell cycle are influenced by the experimental system used to test HCV effects on these cellular processes. The results of several studies have suggested that one or more HCV proteins can modulate cell cycle regulatory genes to affect the G1/S checkpoint in HCV-infected cells (Table 1) [112–127]. The HCV viral core protein, which forms the viral capsid, is thought to play a vital role in the development of HCV-associated HCC [11]. The results of a recent study demonstrated that the HCV core protein decreased p21 expression in human hepatoma cells. An HCV core-induced increase in the level of miR-345 was found to suppress endogenous p21 expression by targeting the 3′ untranslated region (UTR) of the p21 mRNA [117]. Decreases in the levels of p21 would lead to accelerated cell cycle progression, and increased p21 expression is frequently observed in human cancers [128]. Thus it is possible that the HCV core protein-induced decrease in p21 expression may contribute to HCV-induced HCC. These results are in agreement with previous studies that have shown a pro-proliferative effect of the core protein; HCV core induced S-phase progression in various cell systems and growth conditions [22, 122, 124, 125]. For example, HCV core protein expression increased the fraction of HepG2 cells, a human hepatoblastoma cell line, in the S phase by increasing the stability of the c-myc oncoprotein [122]. Additionally, the HCV core protein, expressed alone or in the context of HCV replication, promoted cell proliferation, DNA synthesis, and cell cycle progression in Huh-7 cells, a human hepatoma cell line [22]. In this system, HCV core activated the Wnt-β catenin signaling pathway, which was shown to be a major mediator of HCV core-induced cell proliferation. Results from another study in HepG2 cells also showed that the HCV core protein stimulated cell growth by decreasing the levels of the CDK inhibitor, p16 via induction of hypermethylation of the p16 transcription promoter [113]. The HCV NS3, NS5A, and NS5B proteins have also been shown to promote cell growth [126, 127]. The results of one study in NIH3T3 cells showed that NS3 mediated a p53-dependent transcriptional repression of p21 [129]. In murine fibroblasts and HepG2 cells, the NS5A protein repressed transcription of p21 and increased expression of Proliferating Cell Nuclear Antigen (PCNA), which is expressed during the S phase and required for DNA replication [112]. pRb expression was also shown to be decreased in HCV-infected hepatoma cells; decreased levels of pRb were caused by NS5B-dependent ubiquitination of pRb and subsequent proteasome-mediated degradation of pRb. Loss of pRb function in HCV-infected cells could promote hepatocyte proliferation and contribute to the development of HCC [127]. Overall, the results of these various studies suggest that HCV proteins can promote cell proliferation by affecting the cellular functions or levels of cell cycle regulatory proteins.
In contrast to the studies described above, which demonstrate a pro-proliferative effect of the HCV proteins, the results of another study demonstrated that NS2 can inhibit cell proliferation and induce cell cycle arrest in the S phase (Table 1). The induction of S-phase arrest in NS2-expressing cells was associated with decreased cyclin A expression [115]. The results of a different study also showed that the HCV RNA-dependent RNA polymerase, NS5B, delayed S-phase progression by interacting with the CDK-interacting protein (CINP) [118]. In fact, this study led to the identification of CINP and provides an example of how analyzing viral regulation of the cell cycle may help identify novel cell cycle regulatory proteins. Similar to the NS2- and NS5B-mediated cell cycle arrest, and in contrast to studies outlined above, the results of additional studies have suggested that the HCV core protein can impair the G1 to S phase transition through various mechanisms, including induction of p21 expression and stabilization of the CDK inhibitor, p27 (Table 1) [130–132]. Interestingly, Nguyen et al. demonstrated that the HCV core protein modulates p21 expression levels in a biphasic manner [130]. The 21-kDa, immature form, of the HCV core protein can be proteolytically processed to a 19-kDa, mature form [133]. Nguyen et al. analyzed the role of the HCV core protein in cell cycle modulation by using a HepG2-derived cell line, where the expression of the HCV core protein was under the control of an inducible promoter. At early time points after induction of core protein expression, both the 21-kDa and 19-kDa forms were equally abundant in HCV core-expressing cells; however, at later time points, the 19-kDa form accumulated and became the dominant species. The 21-kDa form of the core protein was associated with an increased expression of p21 and a concomitant decrease in CDK2 activity. These changes in p21 and CDK2 activity led to a decrease in cellular proliferation. However, accumulation of the 19-kDa form caused a decline in p21 levels. These results suggest that the HCV core protein-dependent regulation of p21 expression might depend on the early presence of the immature form of the core protein or the later expression of the mature form of the core protein during an HCV infection and may provide an explanation for the conflicting observations in various studies that have analyzed the regulation of p21 by the HCV core protein. It is possible that some studies only analyzed the effect of the 19-kDa, mature form, of the core protein or that the processing kinetics of the HCV core protein may differ in the individual studies, leading to varying effects of the core protein on p21 expression [130]. Biphasic effects on cell cycle regulatory molecules have also been demonstrated for the human cytomegalovirus (HCMV), a member of the Herpesvirus family; cyclin A expression was repressed early after HCMV infection but induced at later stages of the viral infection [134]. Overall, various studies suggest that the HCV core protein modulates cell cycle regulatory proteins and plays a vital role in HCV pathogenesis; however, the exact effects of the HCV core protein on hepatocyte cell cycle modulation remain incompletely defined.
The studies described above predominantly focused on effects of HCV proteins that were expressed in isolation or outside of the context of an authentic HCV infection. The impact of expressing the entire HCV genome on the cell cycle has also been analyzed (Table 1); however, because of the lack of an efficient HCV infection system, the effects of an HCV infection on the host cell cycle remain unclear. To begin to clarify these effects, in one study a Cre recombinase/loxP conditional system for the expression of the full-length HCV genome was generated in HepG2 cells, thus enabling the creation of a system that at least partially mimicked persistently HCV-infected hepatocytes. The results of this study showed that cells expressing the full-length HCV RNA activated the CDK-pRb-E2F pathway more effectively than observed when individual HCV proteins were expressed [114]. Full genome HCV RNA expression also enhanced anchorage-independent growth of HepG2 cells, whereas HepG2 cells only expressing HCV structural, nonstructural, or even all viral proteins showed no significant changes in anchorage-independent growth; this observation might suggest that the viral RNA itself somehow affects the cell cycle [114]. The tumor suppressor, pRb, is frequently inactivated in HCC, and HCV-mediated regulation of the CDK-pRb-E2F pathway may be one of the mechanisms responsible for the high incidence of HCC in HCV-infected patients [114, 135]. Finally, the results of a different study indicated that an HCV infection is associated with a delay in cell cycle progression. HCV-infected Huh-7.5 cells, a subline of Huh-7 hepatoma cells that can support HCV replication, showed significantly fewer cells in the S phase as compared to mock-infected cells. Further, results from gene expression analysis suggested that HCV-mediated apoptosis of Huh-7.5 cells might be a result of perturbations in cell cycle progression [119]. Interestingly, a G1 arrest was also observed in patient hepatocytes during a chronic HCV infection [121, 136, 137]. The G1 arrest was associated with increased p21 expression, which correlated with the severity of fibrosis [136]. These in vivo results suggest that the delayed cell cycle progression observed in HCV-infected Huh-7.5 cells may be physiologically relevant.
Although the studies described above provide some indications of HCV full genome effects on the cell cycle, few studies have analyzed the effects of the cell cycle status on HCV replication or the effect of the replicating virus on the cell cycle during an authentic HCV infection. Therefore, the significance of HCV-induced cell cycle arrest or proliferation for HCV replication and HCV-associated disease remains unclear. In addition, the paucity of authentic HCV replication systems, and the consequential study of HCV replication in systems that may not accurately reflect all aspects of an authentic HCV infection, has sometimes generated seemingly discrepant observations of HCV effects on the cell cycle. Although direct confirmation is lacking, various possible effects of cell cycle regulation on HCV replication have been proposed. For example, the biphasic effect of the HCV core protein on the cell cycle may be important for HCV replication, and it is possible that HCV-mediated cell cycle arrest protects cells from apoptosis during the initial stages of an HCV infection. Alternatively, during early stages of HCV infection, the immature form of the HCV core protein may regulate the expression of proteins that are required for repressing the immune response and thus help infected cells evade immune defenses [130]. Some studies have linked the effect of the cell cycle status on the translational activity of the HCV internal ribosome entry site (IRES), which mediates cap-independent translation of the HCV RNA and is located at the 5′ end of the HCV genome. The HCV IRES-dependent translation efficiency in Huh-7 cells was highest during the G0 and G1 phases of the cell cycle but was decreased during the S phase and dramatically reduced during the G2/M phase. Therefore, it is possible that HCV proteins modulate cell cycle regulatory proteins to induce a cell cycle arrest to allow efficient HCV translation and replication [138]. It has also been proposed that HCV-mediated cell cycle arrest limits the regenerative response of the liver to ongoing injury and contributes to the progression of liver disease [121, 136, 137]. In contrast, some studies have demonstrated a positive impact of cell proliferation on HCV replication. The results of one study showed that the translational activity of the HCV IRES was greatest in cells that are actively dividing [139]. In accordance with this, findings from another study suggested that HCV replication is highly dependent on cellular proliferation, and HCV RNA synthesis was strongly enhanced in the S phase. Surprisingly, and in contrast to previous studies, the same study also showed that HCV protein expression and genome replication did not affect the cell cycle status of Huh-7 cells [120].
In summary, numerous studies have analyzed the effect of HCV infection on the cell cycle status of hepatocytes. However, most of these studies were conducted with overexpression of a single HCV protein, and the results of these studies have sometimes identified contradictory effects on cell cycle regulatory proteins. Studies involving a single HCV protein may not accurately represent the expression levels of that HCV protein in HCV-infected livers and cannot analyze the consequence of interactions between different HCV proteins that could influence the cell cycle during an HCV infection. Moreover, most HCV studies that analyzed the impact of HCV proteins on the cell cycle were conducted in immortalized or transformed cell lines. Although challenging, future studies in primary hepatocytes may help delineate the exact effects of HCV on the cell cycle during an authentic HCV infection [140]. These types of studies should also consider that HCV has several genotypes and that different disease outcomes have been reported in patients infected with different HCV genotypes [141]. Therefore, it is possible that different genotypes of HCV will have different effects on the cell cycle, which might account for some of the contradictory observations that have been reported. Recently, a genetically humanized mouse model that expresses human CD81 and human occludin and can be infected with HCV was generated. This is the first mouse model where the entire HCV life cycle can be studied and provides new opportunities to understand the in vivo consequences of an HCV infection for hepatocyte genome replication and the cell cycle [142].
4.2 Small DNA Tumor Viruses Drive Cells into the S Phase
The small DNA tumor viruses are a group of double-stranded DNA viruses; representative examples of these viruses are papillomaviruses such as the HPV, Adenoviruses (Ad), and polyomaviruses such as the Simian virus 40 (SV40) [2, 143]. HPV infections are associated with the development of cervical, anal, and neck cancers [144]. Although SV40 and adenoviruses have not been linked to human cancers, they can immortalize and transform cells in culture [145]. The small DNA tumor viruses are dependent on the host cell DNA replication machinery for the replication of the viral genomes. These viruses typically infect differentiated, quiescent cells, which may not be an ideal environment for viral replication because the host cell DNA replication machinery is only available during the S phase [146, 147]. The small DNA tumor viruses do not encode a DNA polymerase or other enzymes that are involved in DNA synthesis [4]. Moreover, since quiescent cells have low levels of deoxynucleotides, the environment of quiescent cells may not be conducive to viral DNA synthesis. Thus, it is thought that small DNA viruses must induce S-phase entry of infected cells in order to create an environment that is favorable for viral replication. HPV, Ad, and SV40 have evolved strategies to promote unscheduled entry of infected cells into the S phase [6, 146, 147] (Fig. 3). Entry into S phase allows these viruses to use host enzymatic activities and cellular DNA precursors for their own DNA replication. Consequently, these viruses encode proteins that can affect cell cycle control mechanisms. For example, the small DNA tumor viruses encode proteins that can inhibit p53 and the Rb family members [6, 18, 19, 148–150] (Fig. 3). Inhibition of p53 and Rb family members by these virally encoded proteins induces the cells to enter S phase. Inhibition of p53 and Rb by the small DNA tumor virus proteins is also required for the cell transformation that is associated with HPV, Ad, or SV40 infections [6, 18, 145, 150, 151].

Regulation of the early phases of the cell cycle by small DNA tumor viruses and the herpesviruses. Small DNA tumor viruses (Human papillomavirus (HPV), Simian virus 40 (SV40), and Adenoviruses (Ad)) and herpesviruses (Kaposi’s sarcoma-associated herpesvirus (KSHV), Epstein–Barr virus (EBV), Herpes simplex virus 1 (HSV-1), and Human cytomegalovirus (HCMV)) regulate the transition from G1 to S. The mechanism used by these viruses to regulate the early phases of the cell cycle is depicted. See text and references for details
4.2.1 Small DNA Tumor Virus Oncoproteins Inhibit the Retinoblastoma Family Proteins
Transforming oncoproteins of the small DNA tumor viruses include E1A from adenovirus, E7 from HPV, and large T antigen (LTag) from SV40 [145]; these oncoproteins bind to and inactivate Rb family members, thus abrogating the need for phosphorylation by the G1 CDKs, CDK4 and CDK6 [3] (Fig. 3). The Rb family of proteins, also referred to as the pocket protein family, consists of the three proteins pRb, p107, and p130. These proteins negatively regulate the transition from the G1 to S phase [21, 152]. E1A, E7, and LTag oncoproteins contain an LXCXE (Leu-X-Cys-X-Glu, where X represents any amino acid) motif, which facilitates interaction with all three members of the Rb family. The LXCXE motif interacts with a site on Rb that is referred to as the pocket region of Rb [6, 7]. The binding of E1A, E7, or the LTag to the pocket region of Rb leads to the displacement of its cellular binding partners, HDAC and E2F. The steric disruption of the E2F-Rb complexes allows the release of the S-phase transcription factor, E2F [6, 7, 146, 153–164]. In addition to disrupting the interaction of Rb with E2F, E1A and LTag can inhibit pocket protein function by inducing posttranslational modifications [146]. The results of various studies indicate that the binding of E7 to all three Rb proteins induces their degradation by the ubiquitin-proteasome pathway [7, 146, 165, 166]. Overall, the transforming oncoproteins of the small DNA tumor viruses can inactivate Rb family members and cause unscheduled progression into the S phase. Inactivation of the Rb family by E1A, E7, and LTag would lead to the induction of transcription of E2F responsive genes, which include the E2F-controlled cell cycle and DNA synthesis genes, and help establish a favorable environment for viral replication [6, 146].
4.2.2 Small DNA Tumor Virus Oncoproteins Inhibit the p53 Tumor Suppressor Pathway
The p53 tumor suppressor pathway can be activated as a response to various cellular stresses, including DNA damage. The activation of p53 either leads to the induction of cell cycle arrest to allow time for the cell to repair any DNA damage or initiates apoptosis if the DNA damage is too extensive or cannot be repaired. Infection with many different viruses has been linked to activation of p53; extensive viral DNA replication can trigger a DNA damage response that activates p53. Since the induction of cell cycle arrest or apoptosis could prevent new virus production, many viruses, including the small DNA tumor viruses, have evolved mechanisms to inactivate the p53 tumor suppressor pathway [167]. The SV40 LTag, Ad E1B, and HPV E6 oncoproteins have been shown to bind to p53 [6, 167] (Fig. 3). LTag can directly bind and inactivate p53 [168–170]. In fact, p53 was first identified as an interaction partner of the LTag and then later shown to have an important tumor suppressor activity [171, 172]. E1B and E6 can facilitate the ubiquitination and proteasome-mediated degradation of p53 via recruitment of other cellular factors that regulate this process. E1B, in conjunction with the adenovirus protein E4-ORF6, assembles into an ubiquitin ligase complex together with cellular proteins involved in ubiquitination (Cullin 5 and Elongins B/C), to target p53 for degradation [173, 174]. E6 stimulates the degradation of p53 by recruiting the cellular ubiquitin ligase E6AP-100K [175]. Overall, small DNA tumor viruses, which usually infect quiescent cells, encode proteins that bypass restriction points in the cell cycle in order to activate the host cell replication machinery and induce cell proliferation. SV40, Ad, and HPV thus create a favorable environment for viral DNA replication.
4.3 Herpesvirus Regulation of the G1 and S Phases
Whereas small DNA tumor viruses have evolved mechanisms to activate the transcription of cellular genes that generate deoxynucleotide pools for DNA replication and rely on cellular DNA polymerases, Herpesviruses encode many of these genes in their viral genomes [4, 5, 7, 148]. Members of the Herpesviruses family are enveloped viruses that contain a large, double-stranded DNA genome that typically encodes 100–200 genes. Expression of Herpesvirus genes are temporally regulated during an infection and can be classified as immediate early, early, or late genes, reflecting their relative time of expression following infection of a cell. The Herpesvirus family is subdivided into the α-, β-, and γ-herpesviruses to distinguish various biological properties including host range and speed of replication. Within an infected cell, Herpesviruses can exist in a lytic state, where most genes are expressed and the virus is actively replicating, or in a latent state where a subset of genes are expressed and the virus is not generating infectious progeny. Herpes simplex virus 1 (HSV-1) is representative of α-herpesviruses, HCMV is representative of β-herpesviruses, and EBV and KSHVs are representative of γ-herpesviruses [2, 176–179]. Several research groups have analyzed the effects of HSV-1, HCMV, EBV, and KSHV on cell cycle regulatory pathways [4]. Here, we will describe HSV-1, HCMV, EBV, and KSHV-dependent modulation of the host cell cycle as examples of how members of the Herpesvirus family regulate the cell cycle and how this affects viral replication, cell physiology, and the development and progression of some Herpesvirus-associated diseases.
4.3.1 Kaposi’s Sarcoma-Associated Herpesvirus Regulates the G1/S Checkpoint
KSHV, also referred to as human herpesvirus 8 (HHV8), is the most recently identified human oncogenic virus. KSHV is the infectious cause of Kaposi sarcoma (KS) and two lymphoproliferative disorders that are frequently found in individuals with acquired immune deficiency syndrome (AIDS); the lymphoproliferative disorders include primary effusion lymphomas (PEL) and multicentric Castleman’s disease. KS is a common cancer in HIV-1 infected, untreated individuals. Although originally linked to diminished CD4 T cell levels, even HIV-infected individuals receiving anti-HIV therapy have a higher incidence of KS than is observed in the general population. Almost 20 years after the discovery of KSHV, palliative treatments for KS exist, but none are curative. Additionally, there is no vaccine against KSHV. Substantial advances have been made in understanding the pathobiology of KSHV, and potential targets for the treatment of KS have been suggested [180–183]. The KSHV genome encodes a large number of cellular orthologues that affect the cell cycle, DNA synthesis, and apoptotic pathways in KSHV-infected cells [180–182]. KSHV primarily infects endothelial and B cells [184, 185]. During the latent phase of a KSHV infection, viral gene expression is restricted to a subset of viral genes, and gene products are thought to avoid the host antiviral immune response and provide a proliferative advantage to the KSHV-infected cells. KSHV replication and transcriptional programs are fully activated upon induction of the lytic phase, where the virus progeny is produced, packaged, and released from the host cells [183, 186]. KSHV genes have been classified into three major categories: class 1 genes that are constitutively expressed, class II genes that are expressed during latency, but are upregulated during lytic replication, and class III genes that are only present during the lytic phase of a KSHV infection [186]. Most tumor cells in PEL or KS only express KSHV latent proteins, and only a small percentage of the tumor cells express lytic proteins [183].
KSHV expresses various proteins that can modulate the cell cycle of infected cells (Fig. 3); these KSHV-encoded proteins deregulate cell cycle checkpoints, promote cell cycle progression, and are thought to contribute to KSHV-mediated oncogenesis by functioning as growth factor receptors, signal transduction proteins, transcription factors, and cell cycle regulators [187].
The KSHV homologue of cellular cyclin D is known as the viral cyclin (v-cyclin). v-cyclin is expressed from the major latency locus of the KSHV genome and is used by KSHV to hijack cell cycle control mechanisms (Fig. 3) [183, 188]. Similar to the cellular D-type cyclins, v-cyclin interacts with and activates CDK4 and CDK6 kinases; the primary target of v-cyclin is CDK6 [189]. The v-cyclin-CDK6 complex phosphorylates pRb in vitro and in vivo [189]. The interaction of v-cyclin with CDK6 causes S-phase entry and DNA replication of KSHV-infected cells [183, 188, 190, 191]. The v-cyclin-CDK6 complex can also phosphorylate other factors that are involved in the G1/S phase transition of the cell cycle. v-cyclin-CDK6 substrates include CDK2, histone H1, p27, Cdc6, and origin-recognition complex-1 [183, 191–194]. Unlike the cellular cyclin D-CDK6 complex, which usually requires CDK6 phosphorylation by a CAK for full activation, the complex of v-cyclin with CDK6 is fully active in the unphosphorylated form [195, 196]. Importantly, v-cyclin is resistant to the action of the CDK inhibitors p21, p27, and p16 [195]. v-cyclin-CDK6 complexes can phosphorylate and inactivate p21 and p27, which favors activation of the cellular cyclin-CDK2 complexes and promotes cell cycle progression [183, 197]. Although, the results of various studies demonstrate that v-cyclin can promote S-phase entry [192, 195, 198], in primary cells, v-cyclin has been shown to induce a p53-dependent growth arrest and to sensitize cells to apoptotic signals [199, 200]. The results of studies in v-cyclin-expressing transgenic mice showed that the ability of v-cyclin to promote cell survival and tumor formation was only apparent in the absence of p53 [200, 201]. Overall, these studies confirm a role of v-cyclin in regulating the cell cycle but suggest that the exact effect of v-cyclin might be influenced by experimental conditions [199].
The latency-associated nuclear antigen (LANA1) is encoded by the major latency locus of the KSHV genome. LANA1 has no homologue in the human genome [183]. LANA1 binds to the pocket region of pRb and inhibits pRB function, thereby disrupting normal G1/S checkpoint control mechanisms (Fig. 3) [202]. Similar to the oncogenic proteins of many other tumor viruses, LANA1 can inactivate the tumor suppressor, p53, highlighting the importance of evading tumor suppressor checkpoints in viral-induced oncogenesis (Fig. 3) [183].
KSHV also possesses a number of other proteins that regulate mitogenic signaling pathways to affect the cell cycle control machinery. One such KSHV mitogenic signaling protein is the KSHV G protein-coupled receptor (vGPCR). vGPCR is expressed during early phases of KSHV lytic replication [188]. This viral chemokine receptor is homologous to the human chemokine receptors CXCR1 and CXCR2 [187]. vGPCR has been shown to activate the mitogen-activated protein kinase (MAP Kinase) and AKT signal transduction pathways, which increases the expression of angiogenic factors, such as vascular endothelial growth factor (VEGF), and can contribute to cell transformation processes [107, 188, 203]. Interestingly, KSHV encodes several DNA synthesis enzymes, including thymidine kinase, dihydrofolate reductase, thymidylate synthetase, and ribonucleotide reductase. In contrast to the cellular homologues of these KSHV genes, expression of KSHV thymidine kinase, dihydrofolate reductase, thymidylate synthetase, and ribonucleotide reductase is not under the control of the S-phase transcription factor, E2F. Consequently, expression of these KSHV DNA synthesis enzymes may enable KSHV replication even when cells are not in the S phase and when pRb is active [188, 204].
All of the KSHV proteins mentioned above can modulate host cell cycle checkpoints to prevent G1/S arrest. However, the KSHV K-bZIP protein, also referred to as the replication-associated protein (RAP), can have opposite effects on cell cycle regulation (Fig. 3) [188]. K-bZIP is expressed during lytic KSHV replication and belongs to the basic region-leucine zipper family of transcription factors [188, 205]. K-bZIP causes cell cycle arrest by inducing expression of the CDK inhibitor, p21, and the CCAAT/enhancer binding protein-α [206–208]. The results of one study also demonstrated that K-bZIP directly interacts with cyclin A-CDK2 complexes and promotes G1 arrest during early phases of KSHV lytic replication [209]. The reasons for this G1 arrest are not clear, but it is possible that the KSHV-induced arrest prevents premature apoptosis during the lytic phases of replication. It is also possible that lytic-phase KSHV DNA synthesis enzymes generate a quasi-S-phase state during the cell cycle arrest, thus enabling KSHV DNA replication. Importantly, the effects of K-bZIP are apparent during lytic replication, whereas v-cyclin and LANA1 effects are typically observed during KSHV latency [188].
Overall, the results of various studies suggest that latent and lytic phases of a KSHV infection may have different effects on the host cell cycle [188]. However, it is important to note that most studies of the effect of KSHV proteins on the cell cycle were conducted when these proteins were expressed individually and not in the context of KSHV replication, and caution should be exercised when attempting to extrapolate the results of these studies to effects in KSHV-infected cells. It may be important to determine the effects of a particular KSHV protein on the host cell cycle in the context of the actual KSHV life cycle, where multiple KSHV proteins would be acting in concert [188]. Moreover, a more comprehensive understanding of the interplay of viral and cellular factors in KSHV-infected cells will shed light on the mechanism underlying KSHV-induced tumorigenesis and may enable the development-targeted therapeutic agents [180–183].
4.3.2 Epstein–Barr Virus Regulates the Early Phases of the Cell Cycle
EBV, also referred to as human herpes virus 4 (HHV4), is the causative agent of the self-limiting, lympho-proliferative disease, infectious mononucleosis. EBV infection has also been linked to the development of Burkitt’s lymphoma, Hodgkin’s lymphoma, and nasopharyngeal carcinoma [210]. EBV preferentially infects B cells, and EBV-encoded latent genes can induce B-cell transformation in vitro [211].
Latent EBV infection has been associated with cellular proliferation (Fig. 3). The role of EBV latent proteins, such as Latent Membrane Protein 1 (LMP-1) and Epstein–Barr Nuclear Antigen 3C (EBNA-3C), in inducing cell cycle progression has been well characterized. LMP-1 induces the expression of cyclin D1 and phosphorylation of pRb [7, 212, 213]. EBNA-3C has also been implicated in inhibition of the pRb pathway and can bind to pRb in vitro [7]. EBNA-3C functions in a manner similar to SV40 LTag, Ad E1A, and HPV E7; EBNA-3C binds to the pocket region of pRb and inactivates its cell cycle inhibitory function [214]. EBNA-3C can also stabilize cyclin D by inhibiting its ubiquitination and subsequent degradation [215]. Viral nucleotide biosynthetic enzymes are not expressed during latency, which causes EBV to be reliant on the E2F-induced cellular gene expression for the very low level of EBV genome replication that is observed during latency. EBV might also activate the pRb-E2F pathway to facilitate cell cycle progression and division to expand the pool of latently infected cells. Unlike EBV lytically infected cells, latent EBV infection allows the long-term persistence of infected cells that can avoid the host antiviral immune response. Thus, the proliferation of latently infected EBV cells would lead to an increase of the infected B-cell population [7].
In order to study the EBV lytic replication cycle, latently EBV-infected cells are typically exposed to agents that induce a switch from latent to lytic EBV replication. EBV lytic replication has been shown to be associated with a cell cycle arrest (Fig. 3). When latently infected cells are treated with agents that induce the lytic phase of EBV replication, the EBV-infected cells stop dividing and arrest at the G0/G1 phase [4, 216–218]. The EBV immediate-early transactivator, Zta, also referred to as the lytic switch transactivator, can induce a G0/G1 arrest [218]. Zta acts at multiple distinct control points in the cell cycle regulatory machinery to mediate cell cycle arrest, including Zta induction of the CDK inhibitors p21 and p27. Expression of Zta has been shown to induce arrest in the G0 and G1 phases; however, it also induces the expression of certain S-phase genes [4, 7, 18, 217, 218]. Additionally, another EBV-encoded lytic transactivator, Rta, is thought to have a cell cycle activation function (Fig. 3). Rta can induce the expression of E2F, which would favor cell cycle progression [219]. Thus, despite the ability of EBV to cause cell cycle arrest during a lytic infection, it has also been shown to stimulate certain cell cycle activation pathways [7]. During a lytic infection, EBV inactivates pRb and expresses many EBV-encoded nucleotide biosynthetic enzymes (Fig. 3). Therefore, both cellular and viral nucleotide biosynthetic enzymes are present during an EBV lytic infection. Since EBV encodes many nucleotide biosynthetic enzymes during a lytic infection, it is possible that EBV is relatively resistant to the changes in E2F-mediated transcription of cellular nucleotide biosynthetic enzymes [7].
Overall, EBV seems to both inhibit and stimulate cell cycle progression. The inactivation of pRb in latently EBV-infected B cells may help expand the number of infected B cells. On the contrary, since the EBV genome contains many genes required for genome replication, lytic replication in a G0 state may prevent competition from the host cell machinery for the precursors for DNA synthesis [3].
4.3.3 Human Cytomegalovirus Regulation of the Cell Cycle
HCMV, also referred to as human herpesvirus 5 (HHV5), is not generally considered an oncogenic virus; however, HCMV infection has been implicated in certain malignant diseases [220, 221]. HCMV usually infects quiescent cells in vivo, and it is therefore likely that it is beneficial for HCMV to modulate the host cell cycle to maximize viral DNA replication [222]. Some studies have shown that HCMV infection can stimulate cellular DNA synthesis; however, most of these studies were conducted in cell lines that were not permissive to HCMV replication [222–224]. The infection of quiescent fibroblasts with HCMV leads to a reentry into the cell cycle, progression through the G1 phase, and an arrest at the G1/S border (Fig. 3) [7, 222, 225–228]. Although these observations suggested that HCMV-infected cells are arrested at the G1/S border, it is important to note that these cells exhibited characteristics of early S-phase entry, including hyperphosphorylation of pRb and increased E2F transcriptional activity [222, 226, 227]. Further, infection of cycling cells with HCMV also leads to the induction of a G1/S arrest [226, 228]. It is possible that the G1/S arrest leads to an unrestricted access to the precursors of viral replication while preventing host cell DNA synthesis [227]. During lytic infection, both cell cycle arrest, mediated by the tegument protein UL69 [229] and the immediate early IE2 protein [230], and stimulatory effects, mediated by the HCMV kinase pUL97 [231], the tegument protein pp71 [232–234], and IE2 [235–237], have been observed. Since the HCMV genome does not encode nucleotide biosynthetic enzymes, it is possible that HCMV depends on cellular E2F transcriptional targets for efficiency of lytic replication. Cell cycle effects during latent HCMV infections have not been examined [7]. Thus, HCMV expresses several proteins that allow it to modulate the cell cycle towards an S-phase like environment.
4.3.4 Herpes Simplex Virus 1 Regulation of the Cell Cycle
HSV-1, also known as Human herpesvirus 1 (HHV1), is the main cause of herpes infections that occur on the mouth and lips, including cold sores and fever blisters [238]. Similar to other Herpesviruses, HSV-1 can establish both a latent and lytic infection and can modulate the cell cycle of infected cells [4, 7]. HSV-1 cell cycle effects during latent infections remain unknown. During a lytic HSV-1 infection, HSV-1 does not stimulate the production of cellular nucleotide biosynthetic enzymes but instead induces cell cycle arrest and relies on viral nucleotide biosynthetic enzymes (Fig. 3) [7]. The results of various studies indicate that HSV-1-infected cells accumulate in the G1 phase [239–241]. Cells that are synchronized in the G0 phase and then simultaneously subjected to both serum treatment and HSV-1 infection fail to enter S phase [239–242]. HSV-1 infection also leads to an accumulation of E2F factors that are complexed with pRb and blocks cellular DNA synthesis [240, 242]. Overall, during lytic phases of HSV-1 infection, HSV-1 induces changes in cell cycle regulatory controls that are consistent with the cells arresting in the G1 phase.
The HSV-1-encoded immediate-early transcription factor, infected cell protein 0 (ICP0), can arrest cell cycle progression [243, 244]. Infected cell protein 27 (ICP27), another immediate early HSV-1 protein, has also been implicated in HSV-mediated growth arrest; ICP27 prevented phosphorylation of pRb in HSV-1-infected cells [239]. Interestingly, although pRb is hypophosphorylated in HSV-1 infected cells, the activity of CDKs that are responsible for the phosphorylation of pRb seems to be essential for HSV-1 replication, and the activity of CDKs is required for the expression of HSV-1 genes [245, 246]. Although not completely clear, it is thought that CDKs stimulate HSV-1 replication by modulating RNA Polymerase II function [247]. Further, it is possible that CDKs may enhance the ability of ICP0 to activate transcription [248]. Surprisingly, although CDK activity seems to be required for expression of HSV-1 genes, the results of two studies demonstrated that HSV-1 infection suppresses cyclin-CDK function [239, 241]. One group showed that HSV-1 infection prevents the induction of cyclin D1 and cyclin D3 following the addition of serum to serum-starved cells [239]. Additionally, results from another group showed that the infection of quiescent cells suppressed serum-induced cyclin D-CDK4/6 and cyclin E-CDK2 activity and also led to a loss of cyclin E levels [241]. However, in contrast to these studies, results of a different study showed that HSV-1 infection of serum-starved cells resulted in no change in CDK4 activity, an induction of cyclin A expression, and a transient induction of CDK2 activity [249]. These seemingly discrepant observations could be explained by a difference in the experimental systems used in these studies. The first two groups infected quiescent cells in the presence of newly replaced serum, whereas the later group infected quiescent cells in the presence of spent, not freshly replaced, medium. Thus, while the first two studies addressed the ability of HSV-1 to prevent serum-stimulated induction of cyclin-CDK function, the latter group determined whether HSV-1 infection could activate cyclin-CDK function above the uninduced, background levels [4]. Overall, the results of these various studies suggest that HSV-1 infection may partly suppress the serum-stimulated induction of cyclin-CDK activity but probably not the basal uninduced levels. Therefore, HSV-1 infection may not completely arrest cell cycle progression, and it is possible that HSV-1 regulates cyclin-CDK activity to levels that helps support efficient HSV-1 genome replication [4].
4.3.5 Conclusions from Studies That Have Analyzed the Effect of EBV, HCMV, HSV-1 Lytic Replication on the Cell Cycle
Most of the studies that have analyzed the effect of Herpesviruses on the cell cycle have focused on lytic replication. Interestingly, similar to the small DNA tumor viruses, the human Herpesviruses can modulate pRb activity during infection [7]. Due to their restricted genome size, the small DNA tumor viruses do not encode their own DNA polymerase or other accessory factors that are required for viral DNA replication. In contrast to the small DNA tumor viruses, Herpesviruses encode a DNA polymerase and some accessory factors involved in nucleotide generation [4, 7]. During lytic replication, Herpesviruses must generate large amounts of DNA. Thus, Herpesviruses have a significant requirement for nucleotide biosynthesis, metabolic, and nucleotide polymerization enzymes. Herpesviruses can either rely on their own viral machinery for the nucleotide biosynthetic enzymes or activate cellular pathways, which leads to the accumulation of these enzymes. Since Herpesviruses encode for their own DNA polymerase, it is possible that a subset of the Herpesviruses that rely on cellular nucleotide biosynthetic enzymes modulate the pRb-E2F pathway to induce the expression of these enzymes [7]. Herpesviruses might also modulate the pRb-E2F pathway in order to synchronize infected cells in the particular cell cycle state that leads to efficient replication of the viral DNA genome [7].
Results of various studies indicate that during lytic replication, EBV, HCMV, and HSV-1 induce growth arrest [4]. Synchronization in the G1 phase is an early step in the lytic infection of these viruses [4]. These Herpesvirus-encoded growth-arrest genes are components of the infecting virion and/or immediate early genes. It appears that these viruses employ very early viral products to induce a cell cycle block, which may help ensure that the cells are arrested in G0 for EBV and G1 for HSV and HCMV before viral genome replication begins. This enables the virus to ensure that cellular DNA synthesis is blocked before the virus engages in DNA replication, which may limit competition for resources between the cellular and virus DNA replication machinery [4]. Herpesviruses also employ an additional strategy to ensure that the cell is arrested at the appropriate phase before viral replication is initiated. The immediate early gene expression of these viruses has been shown to be regulated by the cell cycle; these genes are expressed immediately before the checkpoint where the respective protein has been shown to function [4]. For example, the promoter for the Zta genes of EBV is activated by G0 growth arrest signals [216, 217]. Furthermore, immediate-early HCMV gene expression was found to occur only when the infected cells progressed to the G1 phase [4]. Therefore, the viral cell cycle regulatory factors are activated at the appropriate point of the cell cycle where they function to induce growth arrest. Finally, HSV-1, HCMV, and EBV also encode immediate early proteins that affect the expression and function of p53. Interestingly, in addition to causing cell cycle arrest, Herpesviruses can also stimulate certain cell cycle activation signals [4]. The exact role of the cell cycle promotion signals during Herpesvirus replication is unclear. Overall, Herpesviruses appear to have evolved highly sophisticated mechanisms to regulate the cell cycle so as to favor viral replication.
4.4 Hepatitis B Virus Regulation of the Early Phases of the Cell Cycle
4.4.1 Hepatitis B Virus Replication and the Cell Cycle
The human HBV is a prototype member of the Hepadnavirus family of viruses. Hepadnaviruses are enveloped DNA viruses that predominately infect hepatocytes in their respective hosts [250]. Worldwide, there are over 350 million cases of chronic HBV infections; chronic HBV infection is the most common cause of HCC [250, 251]. Despite the availability of an HBV vaccine, HBV-related diseases remain a major worldwide health problem [252]. Although the association between chronic HBV infections and HCC is clear, there are still gaps in our understanding of how a chronic HBV infection can cause HCC.
HBV replication has been linked to modulation of cell cycle progression, and the status of cell proliferation pathways can also affect HBV replication in certain experimental systems [253]. Expression of the HBV genome in Huh7 cells caused these cells to progress through the G1 phase but inhibited entry into the S phase; similar results were seen in HepG2.215 cells, human hepatoblastoma cells that contain an integrated HBV genome and replicating HBV [254]. Additionally, the results of another study in HepG2.2.15 cells also showed decreased proliferation of HepG2.2.15 cells as compared to HepG2 cells. This study demonstrated that HBV modulates the expression levels of certain cell cycle regulatory proteins, which leads to a G1-phase arrest [255]. Alternatively, another study that examined the effect of HBV replication in Huh7 cells and primary marmoset hepatocytes demonstrated that in the context of replicating HBV, these cells stall in the G2 phase [256]. Overall, it appears that HBV regulates cell cycle progression; however, the exact effects of HBV infection on cell cycle may be influenced by the specific characteristics of the cell type used for the study [257].
A number of studies have analyzed the impact of the cell cycle phase on HBV replication. The levels of HBV DNA replication were found to vary in HepG2.2.15 cells depending on the phase of the cell cycle. HBV DNA levels were increased when HepG2.2.15 cells were arrested in either G1 or G2, whereas cell entry into the S phase increased the levels of cellular DNA synthesis but decreased the levels of HBV replication [258, 259]. Importantly, these results were confirmed in vivo; analysis in liver specimens from HBV-infected patients showed that hepatocytes expressing the S-phase-specific marker, PCNA, contained little or no HBV-specific DNA. Similarly, most hepatocytes that contained HBV DNA were found to be negative for PCNA [259]. Overall, these results suggest that HBV replication is decreased in actively proliferating cells and is inversely correlated with cellular DNA synthesis [257, 259]. Contrary to the studies that showed that HBV replication is regulated by the cell cycle status, one group demonstrated that HBV replication is independent of the cell cycle phase in HBV-transgenic mice [260]. However, HBV-transgenic mice do not completely mimic all aspects of an authentic HBV infection; thus, it is unclear whether results from studies conducted in HBV-transgenic mice accurately reflect all the mechanisms that can regulate HBV replication [261]. Cumulatively, the results of most studies suggest that the status of the cell cycle can influence HBV replication.
4.4.2 HBx Regulation of the Cell Cycle
HBx is a multifunctional protein that is encoded by the smallest open reading frame of the HBV genome [11]. Studies of HBV replication in some cell culture systems and in various in vivo mouse models of HBV replication demonstrated that HBx has an essential role during HBV replication [11, 257, 262, 263]. HBx can modulate cytosolic calcium levels, regulate cellular signal transduction and transcription pathways, and affect numerous cellular processes such as apoptosis and cell cycle progression [262, 263]. HBx effects have sometimes varied depending on the model system and the method of HBx expression used in a particular study [11, 263]. Thus, while many functions have been attributed to HBx, these could reflect cell type-specific consequences of a limited number of upstream initiating events that are controlled by a small number of primary HBx activities [257]. This highlights the importance of analyzing HBx activities in biologically relevant systems, such as cultured primary hepatocytes, both when HBx is expressed alone and in the context of HBV replication.
We will first describe the impact of HBx expression on cell cycle progression in immortalized or transformed cell lines. The effects of HBx expression on cell proliferation pathways in cultured primary hepatocytes will be discussed in the next section. The results of studies in immortalized or transformed cells have shown that HBx can induce cells to enter the cell cycle, enter the cell cycle but stall in the S phase, or progress more rapidly through the cell cycle [264–272]. The reported variations in HBx effects may be attributed to the use of different cell lines, varying methods of HBx expression, and the experimental conditions of the study [257]. HBx expression can cause cells in the G0 phase to exit G0 but stall at the G1/S boundary; this could be interpreted as induction of cell cycle progression beyond the G0 phase or inhibition of cell progression into the S phase. Therefore, some results that seem discrepant may actually represent varying interpretations of the same data by different researchers [257, 262]. A seminal study by the Andrisani group has provided support for the notion that HBx can have different effects on the cell cycle depending on specific cellular characteristics [268]. In these studies, two HBx-expressing cell lines were derived from the same parental AML12 liver cell line; AML12 cells are immortalized mouse hepatocytes [273]. One of the HBx-expressing cell lines displayed features consistent with that of a differentiated hepatocyte, whereas the other HBx-expressing cell line was more dedifferentiated [274, 275]. The dedifferentiated cell line displayed HBx-dependent cell cycle entry but paused early in S phase [268]. In contrast, HBx expression in the differentiated hepatocytes caused the cells to progress rapidly through the cell cycle; differentiated hepatocytes displayed HBx-dependent G1-, S-, and G2/M-phase progression [268]. Overall, the results of these studies suggest that HBx can modulate cell proliferation pathways in immortalized or transformed cells.
HBx can modulate the levels and activities of the positive regulators of the cell cycle [257]. HBx can increase the levels of cyclin D1, cyclin E, and cyclin A; activate the endogenous cyclin A promoter; promote the formation of cyclin A-CDK2 complexes; and enhance CDK1 and CDK2 activity in various immortalized and transformed cells [264, 266, 270, 276, 277]. HBx can also affect the negative cell cycle regulators, p16, p21, and p27 [257]. Results from a study in HBV-associated HCC liver sections demonstrated that the liver sections that contained high levels of methylated p16 promoters also had high expression levels of HBx. These results indicate that the expression of HBx correlates with the methylation status of the p16 promoter [278]. Similarly, HBx induced hypermethylation of the p16 promoter and downregulation of p16 protein levels in HepG2 cells [276]. Studies were also conducted in liver tissue samples from HBV-associated HCCs and corresponding HBV-infected noncancerous liver sections. HBx expression in HBV-infected noncancerous tissues correlated positively with DNA methyltransferase 1 (DNMT1) and negatively with p16 protein expression. However, in the HBV-associated HCC tissues, HBx expression still correlated positively with DNMT1 but did not correlate with the hypermethylation of the p16 promoter or with p16 protein expression. Thus, the results of this study suggest that HBx-mediated hypermethylation of p16 may play a role in the early stages of HBV-related HCC [279]. HBx has been shown to lead to both upregulation and downregulation of the Cip/Kip family members; the precise impact of HBx expression on the members of the Cip/Kip family seems to vary in different cellular contexts [257]. HBx increased p21 levels in NIH3T3 cells, a mouse embryonic fibroblast cell line, when p53 was present but did not increase the level of p21 when p53 was knocked down [280]. However, in a different study, HBx was found to increase p21 levels in Hep3B cells, a p53 mutant HCC cell line [281]. In Huh7 cells, HBx expression increased proteasomal degradation of p27 [270]. Interestingly, the results of a study in Chinese hamster ovary cells showed that the level of HBx expression influenced its effects on p21 and p27. Low levels of HBx expression resulted in an increased activity of the p21 and p27 promoters. On the contrary, when HBx was expressed at high levels, there was an inhibition of the activity of the p21 and p27 promoters [282]. This study suggests that the observed effects of HBx on CKIs can be influenced by the experimental conditions.
4.4.3 HBx Regulation of Cell Proliferation in Primary Hepatocytes
Most of the studies described above were conducted in immortalized or transformed cell lines and when HBx was overexpressed in the absence of other HBV proteins, which could contribute to the varying HBx effects that were observed. Since cellular signaling pathways that control normal cell cycle progression are usually altered in established cell lines, the effects of HBx in these cells could reflect functions that are valid in a specific cellular context but are not present in normal hepatocytes, which are the site of an authentic HBV infection. Recent studies in cultured primary hepatocytes have analyzed the effect of HBx expression, both when HBx is expressed alone and in the context of HBV replication, on hepatocyte cell cycle regulatory pathways (Fig. 4). HBx decreased the expression level of both p15 and p16 in cultured primary rat hepatocytes. Additionally, HBx increased the expression of p21 and p27. Thus, HBx expression decreased the levels of the CDK inhibitors that maintain the quiescent status of hepatocytes but increased the levels of the CDK inhibitors that prevent cell cycle progression past the late G1 phase [104]. Similar results were apparent in primary mouse hepatocytes; HBx increased the expression of both p21 and p27 and decreased cellular DNA synthesis [283]. An increase in cyclin D1 and cyclin E expression was also observed in HBx-expressing cultured primary rat hepatocytes. However, HBx expression did not induce a change in the levels of S-phase activating proteins, including cyclin A and PCNA, indicating that HBx expression in normal hepatocytes does not induce entry into the S phase [104]. Importantly, similar effects were observed in primary rat hepatocytes when HBx was expressed in the context of the HBV genome and in the presence of other HBV proteins [104]. Further, these effects of HBx were also confirmed in cultured primary human hepatocytes [284]. Although HBx upregulated CDK4 activity in primary rat hepatocytes, the increase in cyclin E expression levels was not associated with an increase in CDK2 activity [104]. Overall, the results of these studies suggest that HBx induces quiescent hepatocytes to exit G0 but stall in the G1 phase.

Modulation of cell proliferation pathways by HBV (HBx) and HTLV-1. The effect of HBV (HBx) on cell proliferation pathways in primary hepatocytes is depicted. Human T-cell lymphotropic virus type I (HTLV-1) encodes proteins, such as Tax and p30, which regulate cell cycle progression. See text and references for details
HBx-mediated exit of hepatocytes from G0 but subsequent HBx-induced arrest of hepatocytes at the G1/S phase border was shown to be critical for HBV replication in cultured primary rat hepatocytes [104]. The entry of quiescent hepatocytes into the G1 phase was necessary for the activation of the HBV polymerase [285]. The inhibition of S-phase progression was proposed to be important for HBV replication because the stalling of the cell cycle at the G1/S border might prevent competition between the host cell DNA replication machinery and the HBV replication machinery for available deoxynucleotide triphosphates (dNTPs). Since the levels of dNTPs in quiescent cells are low [286], it is possible that HBx modulation of the cell cycle may lead to an increase in the levels of cellular dNTPs that are available to the HBV polymerase. Interestingly, HBx upregulated the expression of active ribonucleotide reductase in cultured primary rat hepatocytes [285]. Cumulatively, these observations in primary hepatocytes suggest that HBx modulation of cell proliferation is essential for HBV replication. Alterations in cell cycle proteins and their regulatory mechanisms have been linked to cancer development [287], and HBx-induced changes in normal proliferation pathways of quiescent hepatocytes may facilitate HBV replication while ultimately proving detrimental to normal hepatocyte physiology and contributing to processes that influence the development of HBV-associated liver transformation [285].
4.4.4 HBx and Liver Regeneration
Several groups have used HBx-transgenic mouse models to analyze the effect of HBx expression on liver regeneration. However, because of differences in mouse strains and experimental protocols and because hepatocyte proliferation was analyzed at different times after a partial hepatectomy (PH), it is difficult to compare the results from the different groups [11]. Two of the studies suggested that HBx expression inhibits liver regeneration [288, 289], whereas the results of another study suggested that HBx did not affect total hepatocyte division but caused a subpopulation of HBx-expressing hepatocytes to enter the cell cycle prematurely [290]. These studies used mouse models in which HBx expression was controlled by the human antithrombin III gene promoter [288], the mouse albumin gene promoter [289], or the human α1-antitrypsin regulatory region [290]. Although these promoters function in hepatocytes, because the endogenous HBV HBx transcription promoter was not used to drive HBx expression, variations in the level of HBx expression that may not completely mimic normal levels of HBx and may vary between these different promoters could have influenced the HBx-mediated effect on hepatocyte proliferation. Recently, a novel HBx transgenic mouse model was generated; in these mice, HBx expression was under the control of endogenous HBx viral regulatory elements. In these mice, HBx caused delayed cell cycle progression and liver regeneration that was linked to HBx-induced IL-6 overexpression [291]. Thus, the results of this study were consistent with the previously described antiproliferative effects of HBx expression on liver regeneration. Although this system could be argued to more accurately reflect endogenous HBx levels during an HBV infection, unfortunately mRNA splice sites, which are not present in the HBV genome, and a foreign, non-HBx mRNA 3′ noncoding region were included, which likely affected HBx expression levels. Therefore, this system also does not completely mimic HBx expression during an authentic HBV infection. Cumulatively, the studies in HBx transgenic mice have demonstrated that HBx can regulate hepatocyte proliferation pathways; however, the exact impact seems to vary in different mouse models. Determining the impact of HBx expression on liver regeneration will likely provide a more accurate understanding of the effects of HBx on hepatocyte proliferation pathways in vivo. The impact of HBx on liver regeneration could be an important HBx activity that influences the development of HBV-associated HCC.
4.5 Human T-Cell Lymphotropic Virus Type I Regulation of Cell Cycle Progression
Human T-cell lymphotropic virus type I (HTLV-1) is a human oncogenic Retrovirus. Retroviruses are enveloped viruses containing a single-stranded positive-sense RNA genome; two copies of the genome are contained within each virion [292, 293]. HTLV-1 is the causative agent of Adult T-cell Leukemia (ATL), which is an aggressive malignancy of CD4+ T lymphocytes [294–296]. HTLV-1 infects an estimated 15–20 million people worldwide [293], and ATL can occur in approximately 2–5 % of the HTLV-1-infected individuals [297, 298]. HTLV-1 has also been implicated as the causative agent of tropical spastic paraparesis/HTLV-associated myelopathy (TSP/HAM) [293]. HTLV-1 is associated with malignancies that are characterized by excessive proliferation of T cells [299].
The HTLV-1 transactivator, regulatory protein, Tax is both necessary and sufficient for cell transformation and is considered to be a viral oncoprotein. Tax is a potent activator of HTLV-1 and cellular gene expression [297, 300]. The results of various studies suggest that Tax can prevent programmed cell death and increase the proliferation of HTLV-1-infected cells [297, 300]. Tax expression in cells can accelerate progression through the G1 phase and induce defects in the G1/S checkpoint, S phase, G2/M checkpoint, and the M phase [297, 300]. The oncogenic potential of Tax is thought to depend on its ability to modulate the expression levels of genes involved in cell proliferation pathways as well as the interaction of Tax with cell cycle regulatory proteins [300]. HTLV-1-transformed cells display genomic instability. Tax can inhibit cellular DNA repair pathways and override cell cycle checkpoints. These effects are thought to contribute to genomic instability and ultimately lead to Tax-mediated cellular transformation [300]. It is not clear if Tax directly inhibits DNA repair or if Tax inhibition of cell cycle checkpoints allows HTLV-1-infected cells to replicate damaged DNA and undergo mitosis before the damaged or altered DNA is repaired [300].
Cells expressing Tax have an accelerated progression through the G1 phase [301, 302] (Fig. 4). Several different mechanisms have been proposed to explain Tax-mediated disruption of G1 phase regulatory mechanisms and accelerated progression into the S phase [300]. Tax expression can activate transcription of cyclin E and cyclin D2 mRNAs [300, 302]. Tax also directly interacts with CDK4 and CDK6 and stabilizes cyclin D-CDK4 complexes [297, 303]. The results of one study indicated that Tax can stimulate CDK4 activity, and this activity correlated with the direct binding of Tax to CDK4. The cyclin D2-CDK4-Tax complex phosphorylated pRb in vitro, and the amount of phosphorylated pRb correlated with the degree of Tax protein binding to CDK4. Additionally, the cyclin D2-CDK4-Tax complexes were resistant to repression by the CDK inhibitor, p21 [303]. Tax can also stimulate proteasomal degradation of pRb, which would affect cell cycle progression by promoting passage through the G1/S checkpoint [304]. Further, Tax can activate the transcription of CDK4 and CDK2 [305] and repress the transcription of the CDK inhibitors, p18 and p19 [305, 306]. Tax can directly bind to p16 and prevent it from binding and inhibiting CDK6 [307]. Finally, Tax can inhibit p53 activation [308, 309]. Overall, the results of various reports suggest that the HTLV-1 Tax protein can disrupt normal cell cycle controls.
In contrast to the studies described above, the results of other studies have suggested that infection with HTLV-1 and the expression of Tax may not be sufficient to induce cell proliferation; the accumulation of certain genetic defects, such as those induced by IL2 or somatic mutations that inactivate the CDK inhibitors p21 and p27, may be necessary to override cell cycle checkpoints and stimulate cell proliferation [310–312]. Results of one study showed that HeLa cells infected with HTLV-1 or transduced with Tax arrested in the G1 phase; HTLV-1 infected cells, similar to the cells transduced with Tax, expressed high levels of p21 and p27 [310]. On the contrary, HOS (human osteosarcoma lineage) cells continued to proliferate after HTLV-1 infection or Tax expression; however, these cells demonstrated a reduced growth rate and exhibited mitotic aberrations. Constitutive activation of the P13K/Akt pathway in HOS cells leads to a reduction in the expression of p21 and p27, which allows HTLV-1 and Tax-induced G1 arrest to be reverted. Similar to Tax effects in HeLa cells, HTLV-1 infection or Tax expression also caused human SupT1 T cells to arrest in the G1 phase [310]. The results of this study suggest that an HTLV-1 infection usually leads to a Tax-mediated G1 arrest. Alternatively, T cells containing somatic mutations that inactivate the CDK inhibitors, p21 and p27, may proliferate after an HTLV-1 infection [310]. It is thought that in the context of an HTLV-1 infection, Tax promotes cell proliferation; oligoclonal expansion of infected T cells can lead to the onset of ATL. However, this cannot completely explain the long clinical latency of ATL following an HTLV-1 infection. The results of this study suggest that the oncogenic potentials of Tax could be revealed only when HTLV-1 infects or reactivates from T cells whose p21 and p27 function and/or expression has been lost [310]. The HTLV-1 accessory protein, p30, has also been shown to interact with cyclin E, reduce the function of cyclin E-CDK2 complexes, and delay the cell cycle before entry into S phase (Fig. 4) [311]. p30 also binds to the mRNA encoding the Tax/Rex proteins to prevent its nuclear export. Since Tax and Rex are positive regulators of viral gene expression, their inhibition by p30 leads to a decrease in virus expression [313]. This is thought to be beneficial for the establishment of a latent and persistent infection [311]. Since HTLV-1 is a highly immunogenic virus and has low genetic variability, a controlled and reduced expression of the viral proteins could be essential for viral maintenance in the course of a natural infection. The different effects of Tax and p30 on cell cycle regulation may be reflected in their different effects on HTLV-1 replication; Tax is a positive regulator of HTLV-1 replication, whereas p30 is a negative regulator [311, 314]. It is possible that rapid proliferation of cells is required at certain stages of HTLV-1 replication; however, uncontrolled proliferation of infected cells may lead to expression of viral proteins [311]. Therefore, HTLV-1 seems to have also evolved strategies that prevent rapid division of the infected cells [311]. Finally, the results of additional studies suggest that HTLV-1-encoded small proteins, such as p30 and p12, help the infected cells to evade immune defenses and prevent elimination of infected cells by host immune cells [311, 315–321]. Overall, a number of studies have analyzed the effects of various HTLV-1 proteins, particularly Tax, on the host cell cycle. However, the exact effects of these viral proteins on the cell cycle are not yet completely defined, and future studies should focus on understanding the effects of HTLV-1 proteins on the cell cycle in the context of an HTLV-1 infection and in primary T cells.
5 Viral Regulation of the G2/M Checkpoint
Some DNA and RNA viruses can induce cell cycle arrest at the G2/M phase [8]. In this section, we summarize strategies used by viruses to elicit a G2/M arrest and the potential advantages of a G2/M arrest for viral replication.
5.1 Viral Strategies for Inducing G2/M Arrest
5.1.1 Inactivation of CDK1
A number of viruses encode proteins that inhibit the activity of the cyclin B1-CDK1 complex [8] (Fig. 5). Examples include the ORF20 gene of Murine gamma herpesvirus 68 virus (MHV68), the Agnoprotein of JC human polyomavirus, ICP0 of HSV-1, E4 proteins of the HPV1, and the ς1s protein of serotype 3 reovirus, a member of the Reovirus family; members of the reovirus family are non-enveloped and contain a segmented, double-stranded RNA genome [2, 8, 322–327]. The activity of the mitosis-promoting cyclin B1-CDK1 complex can be negatively regulated by phosphorylation of CDK1 [44]. Expression of MHV68 ORF20 can induce a G2/M arrest. ORF20 expression increased CDK1 phosphorylation; the ORF20-mediated G2 arrest was a result of inactivation of the cyclin B-CDK1 complex [323]. Cells expressing Agnoprotein, which is encoded by the human neurotropic JC virus, accumulate at the G2/M phase. Agnoprotein-expressing cells showed a decrease in the expression levels and the activities of cyclins A and B. Further, Agnoprotein also stimulated p21 promoter activity, and cells continuously expressing Agnoprotein showed higher expression levels of p21 [322]. ICP0, a multifunctional HSV-1 immediate early gene product, can also induce a G2/M arrest. ICP0-induced G2/M arrest was shown to require ATM and Chk2 and correlated with phosphorylation of Cdc25C on serine 216 [324]. Phosphorylation of Cdc25C on Ser216 inactivates Cdc25C. Cdc25C is required for activation of CDK1, and inactivation of Cdc25C is an important event in establishment of the G2/M checkpoint [42, 44]. HPV1 E4 also elicits a G2/M arrest, and cells that expressed two E4 proteins (E4-17K/16K) contained inactive CDK1 complexes. During the infectious cycle of HPV1, a full length E1^E4 protein (E4-17K) is present, along with other smaller E4 polypeptides, including E4-16K, which arise by sequential cleavage of residues from the N-terminus of E4-17K. The inactivation of CDK1 was shown to be the result of an inhibitory phosphorylation on residue Tyr15 of CDK1, and the cells were found to contain elevated levels of Wee1. The kinase Wee1 inhibits CDK1 [42, 44]; interestingly, the depletion of Wee1 in cells co-expressing E4-17K and E4-16K alleviated the G2/M arrest [325]. Serotype 3 reoviruses-induced G2/M arrest was shown to require the viral S1 gene-encoded ς1s nonstructural protein. Serotype 3 reovirus infection caused a significant reduction in CDK1 activity and was associated with an increase in the inhibitory phosphorylation of CDK1. The ς1s protein was required for the inhibitory phosphorylation of CDK1 [326]. The Human Herpesvirus 6A (HHV-6A) can also induce a G2/M arrest. HHV-6A infected cells had a decrease in the activity of the cyclin B1-CDK1 complex [328]. The inactivation of the cyclin B1-CDK1 complex was associated with an increase in the inhibitory phosphorylation of CDK1, which was a result of elevated Wee1 expression and inactivation of Cdc25C. Moreover, HHV-6A infected cells had increased expression of p21; this elevated p21 expression was p53-dependent [328]. p21 can bind to the cyclin B1-CDK1 complex, inhibit its activity, and prevent G2/M transition. Finally, HHV-6 infection activated the DNA damage checkpoint kinases Chk2 and Chk1. Thus, HHV-6A infection induces a G2/M arrest by reducing cyclin B1-CDK1 activity through various regulatory mechanisms [328]. SV40 and EBV have also been reported to induce a G2/M arrest [329]. SV40 infection prevents activation of the cyclin B1-CDK1 complex, and this has been linked to maintenance of CDK1 phosphorylation [329]. A recent study showed that the EBV LMP-1 induces a G2/M-phase arrest; 14-3-3 sigma and Reprimo were found to be upregulated in LMP-1 expressing cells [330]. 14-3-3 sigma and Reprimo are p53-regulated inhibitors of G2/M progression [331, 332]. Expression of 14-3-3 sigma can result in an inhibition of the activity of various CDKs, including CDK1 [333]. Further, p53 expression results in increased mRNA levels of Reprimo and leads to the induction of a G2/M arrest. In arrested cells, Reprimo can inhibit the activity of CDK1 [331]. Taken together, the various studies described here demonstrate that many viruses affect the activity of the mitosis-promoting kinase complex, cyclin B-CDK1, in order to induce a G2/M arrest.

Examples of viruses that regulate the G2/M checkpoint. Various strategies by which viruses can induce a G2/M arrest (Inactivation of the cyclin B-CDK1 complex, Cytoplasmic retention of cyclin B-CDK1 complexes, and Inhibition of mitotic exit) are depicted. The Vpr protein of human immunodeficiency virus type 1 (HIV-1) induces a G2/M arrest; some of the mechanisms by which Vpr induces a G2/M arrest are shown. See text and references for details
5.1.2 Cytoplasmic Retention of Cyclin B1-CDK1 Complexes
Interestingly, even in the presence of active cyclin B1-CDK1 complexes, mitosis can be inhibited, provided these mitosis-promoting kinase complexes are prevented from accumulating in the nucleus [8] (Fig. 5). The viral E1^E4 protein of HPV16 uses a novel mechanism to induce G2 arrest. E1^E4 does not inhibit the kinase activity of the cyclin B1-CDK1 complex. Instead, E1^E4 sequesters the cyclin B1-CDK1 complexes on the cytokeratin network, which prevents the accumulation of active cyclin B1-CDK1 complexes in the nucleus and thus inhibits entry into mitosis [334]. A mutant of the E1^E4 protein of HPV16 that did not bind or colocalize with cyclin B1 failed to induce a G2 arrest [334]. Additionally, in vivo studies lend further significance to these in vitro observations; HPV-16 induced lesions showed cyclin B1-CDK1 activity on the cytokeratin filament network of the E1^E4-expressing cells [334]. The G2 arrest induced by the parvovirus B19 NS1 protein appears to use a mechanism that is similar to the HPV 16 E1^E4 protein-dependent regulation of cyclin B1-CDK1 localization. B19 virus-infected cells have enhanced activity of the cyclin B1-CDK1 complex; however, B19 infection causes an accumulation of cyclin B1 in the cytoplasm, thereby resulting in a G2/M arrest [335].
5.1.3 Inhibition of Mitotic Exit
Some viruses induce a G2/M arrest by allowing the cells to enter but not exit mitosis [8, 336] (Fig. 5). Examples include the effects of apoptotin protein from chicken anemia virus (CAV), E4orf4 from adenovirus, high-risk HPV E2 proteins, pUL97 and pUL21A from HCMV, ICP0 from HSV-1, and EC27 from baculovirus [54, 55, 337–344]. The CAV protein Apoptotin associates with subunit 1 of the anaphase-promoting complex/cyclosome (APC10) and induces a G2/M arrest by inhibiting the function of APC10. Expression of Apoptotin caused disruption of the APC10 complex and stabilization of APC substrates [337]. The adenovirus E4orf4 can also elicit a cell cycle block at the G2/M phase. E4orf4 alters the activity of APC to either activate or inhibit the APC; E4orf4 regulates APC in a PP2A-dependent manner [54, 55]. The detailed mechanisms that underlie E4orf regulation of APC are unknown, and it has not yet been shown whether this regulation occurs during adenovirus infection [336]. The HPV E2 proteins from high-risk but not low-risk HPV strains induce a G2/M block, which is independent of E6 and E7. E2-expressing cells that escaped the mitotic block displayed genomic instability. E2 proteins from high-risk HPV strains can also bind directly to Cdh1 and Cdc20, which are APC activators, delocalize Cdh1 to insoluble cytoplasmic aggregates, and cause the accumulation of APC substrates like cyclin B. These results suggest that the high-risk HPV E2 proteins may contribute to the oncogenic potential of HPV by inducing genomic instability [338]. The HCMV viral protein kinase pUL97 also induces Cdh1 phosphorylation during HCMV infection, which prevents the binding of Cdh1 to APC [339]. Further, during HCMV infection, HCMV pUL21A interacts with APC and targets APC4 and APC5, which are two bridge subunits of APC, for proteasomal degradation and thus leads to disruption of APC [340]. Taken together, these studies suggest that HCMV uses several mechanisms to ensure that the APC is inactivated [16, 336]. HSV-1 uses a different strategy to prevent mitotic exit. HSV-1 ICP0 induces the degradation of the kinetochore proteins, centromeric protein A (CENP-A) and CENP-C, and therefore, causes kinetochore structural defects and mitotic delay [341, 342]. Finally, the baculovirus EC27 protein is thought to act as a nondegradable cyclin B1-CDK1 analogue; however, the inhibition of mitotic exit by EC27 is not well understood [343, 344]. Additional descriptions of how viruses regulate APC can be found in a series of recently published comprehensive reviews regarding this topic [53, 336]. Overall, the results of studies described here demonstrate that many viruses can induce a G2/M arrest by interfering with mitotic progression.
5.1.4 Human Immunodeficiency Virus Type 1 Viral Protein R Induces a G2/M Arrest
HIV is a lentivirus, and a member of Retrovirus family. HIV infections can cause acquired immunodeficiency syndrome (AIDS) [345]. Two types of HIV have been characterized, HIV-1 and HIV-2; HIV-1 has higher virulence and infectivity and is the causative agent of the majority of HIV infections globally [345–347]. HIV-1 viral protein R (Vpr) is a virion-associated multifunctional accessory protein that affects multiple stages of the HIV-1 life cycle [348]. Various studies have highlighted the importance of Vpr for viral replication and pathogenesis in vivo. Vpr can activate the HIV-1 long terminal repeat (LTR) promoter and can induce a G2 arrest and apoptosis [345, 348–350].
The results of several studies have shown that HIV-1 Vpr can inhibit cell proliferation by arresting HIV-1-infected cells in the G2/M phase [348, 351] (Fig. 5). Vpr induces a G2 arrest through Tyr15 hyperphosphorylation of human CDK1 [352, 353], the CDK which regulates the entry into mitosis in all eukaryotic cells [44]. Vpr can directly bind and inhibit the phosphatase activity of Cdc25 [354]. Vpr can also stabilize and promote the kinase activity of Wee1 [355, 356]. Vpr-mediated activation of Wee1 and inhibition of Cdc25 promote phosphorylation of CDK1 during induction of G2 arrest [356–358]. The results of recent studies support the involvement of the ubiquitin proteasome system in the Vpr-induced G2 arrest [359–362]. In these studies, Vpr promoted the coordination of a E3 ubiquitin ligase complex comprised of Cullin 4A, damaged DNA-binding protein 1 (DDB1), and Vpr-binding protein (VprBP) [359–362]; this ubiquitin ligase complex ubiquitinates a specific substrate that promotes G2/M transition and thus leads to its degradation [16, 348]. Interestingly, the inhibition of polyubiquitination or the suppression of proteasome-mediated degradation alleviated the Vpr-induced G2 arrest [359, 362]. The cellular substrates that are specifically targeted by the Vpr-mediated ubiquitin proteasome system for induction of a G2/M arrest are not yet known. Identifying these substrates would lend further insight into the G2/M regulation by Vpr [16, 348].
A number of studies have analyzed the cause underlying the Vpr-induced G2 arrest, and cell cycle checkpoint proteins have been shown to be involved. Vpr and the eukaryotic DNA damage or the DNA replication checkpoint controls induce G2 arrest through the inhibitory phosphorylation of CDK1 that is regulated by Wee1 or Cdc25, and it was thought that Vpr might induce G2 arrest through the DNA damage or replication checkpoint pathways [348, 351]. However, studies in human cells showed that Vpr does not induce a G2 arrest through the DNA damage checkpoint pathway [348, 351]. Two observations which support this conclusion are that Vpr can induce G2 arrest in cells from patients with ataxia telangiectasia, which is a disorder caused by a defect in the ATM gene [358] and that Vpr expression does not increase gene mutation frequencies [363]. Instead, activation of human ATR plays a major role in the Vpr-induced G2 arrest through Ser345 phosphorylation-dependent activation of Chk1 [364–367]. Overall, the results of these studies suggest that the Vpr-induced G2 arrest may be similar to the activation of the DNA replication checkpoint rather than the DNA damage checkpoint control [348, 351]. The results of additional studies have shown that Vpr can also induce genomic instability, formation of micronuclei, and aneuploidy, which could be sensed as replication stresses that would lead to the activation of the DNA replication checkpoint [351]. Interestingly, the results of a recent study have led to the proposal of a novel mechanism underlying Vpr-induction of a G2/M arrest; the results of this study showed that the Vpr-induced G2 arrest occurs through an S-phase dependent mechanism. Although Vpr is well known to induce a G2/M arrest, the initiating event occurred in the S phase. Vpr-induced Chk1 Ser345 phosphorylation occurred in the S phase, and Vpr-expressing cells completed the S phase but arrested at the G2/M boundary. The results of this study also showed that the DNA licensing factor Cdt1 was responsible for Vpr-mediated phosphorylation of Chk1 at Ser345 and for the G2 arrest induced by Vpr. This suggests that the Vpr-induced Chk1 phosphorylation and G2/M-phase arrest may be triggered during the onset of DNA replication [365]. Finally, PP2A is also involved in the Vpr-mediated G2 arrest; okadaic acid, which is a specific inhibitor of PP2A, blocks the Vpr-induced G2/M arrest in both fission yeast and human cells [352, 368]. PP2A is a regulator of Cdc25C and G2/M checkpoint activation. Other viruses, including adenoviruses and HTLV-1, can also modulate the activity of PP2A [54, 369]. Both adenovirus E4orf4 and HTLV-1 Tax can induce a G2/M arrest [54, 370–372], and modulation of PP2A may be one of the strategies used by many viruses to induce a G2 arrest.
The results of studies with Vpr provide important examples of how viral proteins can manipulate cellular pathways at various points in order to promote efficient viral replication. The suppression of cell proliferation and G2 arrest induced by Vpr is thought to suppress human immune function by inhibiting T-cell clonal expansion [373]. Further, a Vpr-induced G2/M arrest is thought to provide an optimal cellular environment to achieve maximum levels of HIV-1 replication. The expression of HIV RNA is optimal in the G2 phase, and the ability of Vpr to manipulate the cell cycle and keep the cells in the G2 phase leads to an indirect increase in HIV LTR expression. Finally, Vpr expression leads to increased HIV-1 production, which correlated with increased LTR promoter activity in the G2 phase of HIV-1 infected cells. Overall, HIV-Vpr maximizes viral production in vivo by delaying cells in the G2 phase, where the HIV LTR is most active [349].
5.1.5 Advantages of G2/M Arrest During Virus Life Cycle
Induction of G2/M arrest by various viruses is thought to help establish a pseudo-S-phase state that may be more favorable for viral replication [8]. In this pseudo-S phase, although cellular DNA replication is complete, the cellular environment is such that the substrates and the machinery for DNA replication are available. This extends the amount of time available to DNA viruses for replication of their genomes. For some DNA viruses, this continuous replicative state can lead to an increase in viral genome copy number [8]. The levels of some viral proteins also increase in the G2/M phase [8]. For example, the results of one study demonstrated that infection with the Coronavirus, Infectious Bronchitis Virus (IBV), caused an accumulation of infected cells in the G2/M phase. Interestingly, when the effects of the cell cycle perturbations on viral replication were examined, the IBV-infected, synchronized G2/M cells showed increased viral protein expression as compared to cells in the G0 phase or the asynchronously replicating cells [9]. Some RNA viruses, such as HIV, are more transcriptionally active in G2 [349], and an increased level of transcription during G2/M may lead to production of more viral genomes [8]. Additionally, some viruses can utilize the different phases of the cell cycle to modulate protein expression via utilization of IRES-mediated translation; the IRES of HIV is upregulated in G2/M while that of HCV appears to be downregulated [8, 374, 375]. Apart from the effects on transcription and translation of viral proteins, a G2/M arrest may also impact virion assembly and release [8]. It has been proposed that the enveloped RNA viruses arrest the cell cycle prior to mitosis so as to maintain an intact intracellular organization [1]. This is beneficial to the viruses whose assembly occurs in the Golgi apparatus and ER [376]. For example, coronaviruses, such as IBV, utilize golgi and ER structures for their protein processing and assembly [377–380]. Finally, it is important to note that virus-induced G2/M arrest has been mostly studied in immortalized or transformed cell lines. Since cellular signaling pathways that regulate the cell cycle are usually altered in immortalized or transformed cell lines, the actual effect of the viral protein might be confounded in these systems [8]. Additionally, for many viruses, the consequence of a G2/M arrest for viral replication remains incompletely understood and requires further investigation, especially in systems that more accurately mimic sites of an authentic infection.
5.2 Viral Inhibition of the G2/M Checkpoint
Although all the studies described above focus on how viruses can initiate a G2 arrest, some viral proteins can also abrogate the G2/M checkpoint; examples of these viral proteins include HTLV1-Tax and EBV-EBNA3C. HTLV-1 Tax causes a G2 arrest in certain cell systems [372]; however, in some cell systems, Tax can interact with Chk1, impair Chk1 kinase activity, and inactivate Chk1-mediated phosphorylation-dependent degradation of Cdc25C, resulting in the inhibition of the gamma-irradiation-induced G2 arrest [381]. Further, EBV-EBNA3C can release a G2/M block by manipulating Chk2 signaling [382, 383].
5.3 Viral Activation of the APC
HTLV-1 Tax and HBV-HBx can activate APC [336]. HTLV-1 binds and activates APC during the S phase [384]; however, the mechanism by which it activates APC remains unknown. Additionally, the impact of HTLV-1 Tax protein on APC in the context of infection is not known [336]. It is possible that the premature activation of APC by Tax may lead to chromosome instability and contribute to the tumorigenic ability of HTLV-1 [336]. HBx has been shown to activate APC through its interaction with BubR1, which is a component of the spindle assembly checkpoint (SAC). The binding of HBx to BubR1 prevents the interaction of the APC coactivator, Cdc20 with BubR1 and therefore, induces the release of Cdc20 from the SAC. Release of Cdc20 from the SAC allows Cdc20 to associate with the core APC subunits and causes premature activation of the APC [385]. However, studies that investigated the effect of HBx on APC were conducted when HBx was expressed in Chang cells and HeLa cells, which are human cervical cancer cells, and when HBx was expressed alone and not in the context of viral replication. Therefore, the effect of HBx on APC in context of HBV replication and in primary hepatocytes warrants further investigation.
5.3.1 Benefits of Targeting APC
Inhibition of APC could be advantageous for viral replication [336]. APC can cause the ubiquitination and degradation of multiple proteins, and inhibition of APC would lead to the stabilization of its various substrates including securin, cyclin B1, thymidine kinase (TK), and ribonucleotide reductase M2 [48, 50, 336, 386]. HCMV is the only Herpesvirus that has been found to modulate the activity of APC. HCMV does not encode its own TK and RRM2 enzymes [387], which are important for nucleotide biosynthesis. It is possible that HCMV inhibits APC to prevent the degradation of these enzymes, which would allow the production of nucleotides that can be used for viral DNA replication [336]. Additionally, several HCMV proteins contain a consensus APC recognition signal, which is commonly observed in APC substrates [339]. It is possible that APC may limit viral replication by degrading these viral proteins, and inhibition of APC could enhance the stability of these viral proteins [336]. Future studies should focus on determining the impact of viral proteins on APC in the context of infection. Additionally, for certain viruses, APC has been shown to limit viral replication; therefore, it will be important to determine whether APC regulates replication by acting on viral or cellular substrates [336].
6 Conclusions and Perspectives
Many viruses have developed strategies to alter cell cycle regulatory mechanisms. Viruses often encode proteins that modify cell cycle progression by affecting the expression levels and activities of cell cycle regulatory proteins. Manipulation of the host cell cycle by viruses is thought to promote a favorable cellular environment for viral replication; however, subversion of the host cell cycle by viruses can often pose detrimental consequences to host cell physiology and contribute to viral-induced diseases.
The study of viruses, such as the small DNA tumor viruses, has led to many fundamental discoveries that have expanded our understanding of the dynamic regulation of the cell cycle. Particularly, the studies of small DNA tumor viruses have been extremely valuable in understanding the role of p53 and pRb in cell cycle control. For many viruses, such as the small DNA viruses and some Herpesviruses, the effects of infections with the viruses on the cell cycle are well understood. However, there are still many unanswered questions in regard to the exact outcomes of viral-induced cell cycle progression and arrest during infections with viruses such as HBV, HCV, HIV, and HTLV-1. Many studies that have analyzed the effects of these viruses on the host cell cycle have, by necessity, been conducted in immortalized or transformed cells. Since signaling pathways that control normal cell cycle progression are usually altered in immortalized or transformed cell lines, the effects of the viral protein, although valid in that specific cellular context, may not necessarily be similar during the course of a natural infection in normal cells. Effects of the viral proteins may be influenced by factors that are present in the transformed or immortalized cells as compared to normal cells as well as structural alteration, as described below, that might regulate these viral proteins in specific cellular systems. Future studies should focus on understanding the effects of viral proteins on the host cell cycle in systems that closely resemble a natural infection [8]. Moreover, many studies that have analyzed the effect of a particular viral protein on the host cell cycle were conducted in systems in which individual viral proteins were overexpressed, and often out of the context of the entire viral genome. These types of systems may not accurately reflect the expression level of the viral protein during an actual infection. Therefore, it will be important in future studies to analyze the effects of viral proteins on the cell cycle when these proteins are expressed in the context of viral replication, and in the presence of other viral proteins.
As described in this chapter, the multifunctionality of many viral proteins, such as those encoded by HBV, HCV, HIV, and HTLV-I, can often lead to context-dependent activities and an array of seemingly contradictory effects on cellular signaling pathways. Many viral proteins seem to have adopted a multifaceted approach to manipulate the host cell cycle. Because viruses have limits to their genome size and must encode many functions in a limited number of proteins, it is not surprising that many viral proteins are multifunctional. To this end, many viruses encode regulatory proteins that can each modulate multiple cellular factors, including those that regulate the cell cycle, to promote viral replication. Very recently, the results of two studies have provided insights into the multifunctionality of viral proteins. The results of one study showed that the viral matrix protein 40 (VP40) of the Ebola virus can rearrange into different structural assemblies. The highly plastic, unmodified, wild-type VP40 polypeptide assembled into distinct structures including a dimeric precursor, a hexameric structural component, and a nonstructural RNA-binding ring structure. Each of these distinct structures was shown to have unique and critical functions in the Ebola virus life cycle; the butterfly-shaped VP40 dimer was essential for cellular trafficking, the hexameric structural component was essential for matrix assembly, and viral budding and the RNA-binding structure had a critical role in regulating viral transcription [388]. Ebola virus encodes just 7 genes, and the ability of its protein, VP40, to adopt a different shape for a different function provides one possible explanation for how the virus can accomplish a multiple-step life cycle even though it encodes a small number of genes. The physical plasticity possessed by VP40 that enables it to arrange into distinct structures demonstrates how a structural rearrangement can allow the product of a single viral gene to accomplish a number of essential functions [388]. A second study analyzed the multifunctionality of the Adenovirus E1A protein. Multiple cellular proteins can interact with the E1A protein, and this was linked to dynamic changes in the intrinsically disordered portions of E1A that expand the repertoire of cellular proteins that can bind to E1A [389]. Many viruses have a small genome, and encoding proteins that form distinct structures that function at different stages of the virus life cycle would help these viruses to accomplish a large number of diverse functions with a small number of genes. Thus, it is possible that, like Ebola Virus and Adenoviruses, other viruses also encode proteins that undergo structural transformations, which help the viral proteins to perform different functions [388]. Consequently, future studies should continue to analyze the context-dependent effect of viral proteins on cell cycle progression as these may identify novel therapeutic targets for inhibiting viral replication in various cells as well as strategies for modulating cell cycle effects that contribute to diseases such as cancers. Moreover, these types of studies might also provide insights into the context-dependent effects of many viral proteins.
Finally, alterations in normal cell cycle regulatory mechanisms can lead to the development of many human cancers, and viral infections have been linked to a significant proportion of human cancers worldwide [12, 15]. Thus, a more comprehensive understanding of the mechanisms underlying virus-mediated alterations of the host cell cycle would help provide an in-depth understanding of virus-induced oncogenesis. The results of future studies that address the effects of virus-encoded proteins on the host cell cycle in authentic systems and in the context of viral replication may help generate new therapies that target viral proteins so as to inhibit viral replication and the development of virus-associated cancers. Moreover, these future studies could help identify novel cell cycle regulators and provide insights into many processes that influence cell transformation [16, 17]. Cell cycle regulatory proteins that are targeted by viruses also offer potential targets for antiviral and anticancer therapies; drugs that target cellular proteins instead of viral proteins may help limit the development of drug resistance in these viruses and thus limit both their replication and their ability to cause diseases such as cancer.
References
Nascimento R, Costa H, Parkhouse RM (2012) Virus manipulation of cell cycle. Protoplasma 249(3):519–528
Condit RC (2013) Principles of virology. In: Knipe DM, Howley PM (eds) Fields virology, vol 1, 6th edn. Lippincot Williams and Wilkins, Philadelphia, PA, pp 21–51
Swanton C, Jones N (2001) Strategies in subversion: de-regulation of the mammalian cell cycle by viral gene products. Int J Exp Pathol 82(1):3–13
Flemington EK (2001) Herpesvirus lytic replication and the cell cycle: arresting new developments. J Virol 75(10):4475–4481
Emmett SR, Dove B, Mahoney L, Wurm T, Hiscox JA (2005) The cell cycle and virus infection. Methods Mol Biol 296:197–218
Lavia P, Mileo AM, Giordano A, Paggi MG (2003) Emerging roles of DNA tumor viruses in cell proliferation: new insights into genomic instability. Oncogene 22(42):6508–6516
Hume AJ, Kalejta RF (2009) Regulation of the retinoblastoma proteins by the human herpesviruses. Cell Div 4:1
Davy C, Doorbar J (2007) G2/M cell cycle arrest in the life cycle of viruses. Virology 368(2):219–226
Dove B, Brooks G, Bicknell K, Wurm T, Hiscox JA (2006) Cell cycle perturbations induced by infection with the coronavirus infectious bronchitis virus and their effect on virus replication. J Virol 80(8):4147–4156
He Y, Xu K, Keiner B, Zhou J, Czudai V, Li T, Chen Z, Liu J, Klenk HD, Shu YL, Sun B (2010) Influenza A virus replication induces cell cycle arrest in G0/G1 phase. J Virol 84(24):12832–12840
Bouchard MJ, Navas-Martin S (2011) Hepatitis B and C virus hepatocarcinogenesis: lessons learned and future challenges. Cancer Lett 305(2):123–143
Dayaram T, Marriott SJ (2008) Effect of transforming viruses on molecular mechanisms associated with cancer. J Cell Physiol 216(2):309–314
Saha A, Kaul R, Murakami M, Robertson ES (2010) Tumor viruses and cancer biology: modulating signaling pathways for therapeutic intervention. Cancer Biol Ther 10(10):961–978
McLaughlin-Drubin ME, Munger K (2008) Viruses associated with human cancer. Biochim Biophys Acta 1782(3):127–150
Hanahan D, Weinberg RA (2000) The hallmarks of cancer. Cell 100(1):57–70
Chaurushiya MS, Weitzman MD (2009) Viral manipulation of DNA repair and cell cycle checkpoints. DNA Repair (Amst) 8(9):1166–1176
Howley PM, Livingston DM (2009) Small DNA tumor viruses: large contributors to biomedical sciences. Virology 384(2):256–259
Op De Beeck A, Caillet-Fauquet P (1997) Viruses and the cell cycle. In: Meijer L, Guidet S, Philippe M (eds) Progress in cell cycle research, vol 3. Plenum Press, New York, NY, pp 1–19
Vousden KH (1995) Regulation of the cell cycle by viral oncoproteins. Semin Cancer Biol 6(2):109–116
Vermeulen K, Van Bockstaele DR, Berneman ZN (2003) The cell cycle: a review of regulation, deregulation and therapeutic targets in cancer. Cell Prolif 36(3):131–149
Harper JV, Brooks G (2005) The mammalian cell cycle: an overview. Methods Mol Biol 296:113–153
Cooper GM (2000) The cell cycle. In: Cooper GM (ed) The cell: a molecular approach, 2nd edn. Sinauer Associates, Sunderland, MA, http://www.ncbi.nlm.nih.gov/books/NBK9839
Cook SJ, Balmanno K, Garner A, Millar T, Taverner C, Todd D (2000) Regulation of cell cycle re-entry by growth, survival and stress signalling pathways. Biochem Soc Trans 28(2):233–240
Amon A (1999) The spindle checkpoint. Curr Opin Genet Dev 9(1):69–75
Sherr CJ (1993) Mammalian G1 cyclins. Cell 73(6):1059–1065
Sherr CJ (1994) G1 phase progression: cycling on cue. Cell 79(4):551–555
Assoian RK, Zhu X (1997) Cell anchorage and the cytoskeleton as partners in growth factor dependent cell cycle progression. Curr Opin Cell Biol 9(1):93–98
Ohtsubo M, Theodoras AM, Schumacher J, Roberts JM, Pagano M (1995) Human cyclin E, a nuclear protein essential for the G1-to-S phase transition. Mol Cell Biol 15(5):2612–2624
Girard F, Strausfeld U, Fernandez A, Lamb NJ (1991) Cyclin A is required for the onset of DNA replication in mammalian fibroblasts. Cell 67(6):1169–1179
Walker DH, Maller JL (1991) Role for cyclin A in the dependence of mitosis on completion of DNA replication. Nature 354(6351):314–317
Lew DJ, Kornbluth S (1996) Regulatory roles of cyclin dependent kinase phosphorylation in cell cycle control. Curr Opin Cell Biol 8(6):795–804
Pines J, Hunter T (1991) Cyclin-dependent kinases: a new cell cycle motif? Trends Cell Biol 1(5):117–121
Morgan DO (1997) Cyclin-dependent kinases: engines, clocks, and microprocessors. Annu Rev Cell Dev Biol 13:261–291
Brehm A, Miska EA, McCance DJ, Reid JL, Bannister AJ, Kouzarides T (1998) Retinoblastoma protein recruits histone deacetylase to repress transcription. Nature 391(6667):597–601
Kato J, Matsushime H, Hiebert SW, Ewen ME, Sherr CJ (1993) Direct binding of cyclin D to the retinoblastoma gene product (pRb) and pRb phosphorylation by the cyclin D-dependent kinase CDK4. Genes Dev 7(3):331–342
Buchkovich K, Duffy LA, Harlow E (1989) The retinoblastoma protein is phosphorylated during specific phases of the cell cycle. Cell 58(6):1097–1105
Bracken AP, Ciro M, Cocito A, Helin K (2004) E2F target genes: unraveling the biology. Trends Biochem Sci 29(8):409–417
Hinds PW, Mittnacht S, Dulic V, Arnold A, Reed SI, Weinberg RA (1992) Regulation of retinoblastoma protein functions by ectopic expression of human cyclins. Cell 70(6):993–1006
Montagnoli A, Fiore F, Eytan E, Carrano AC, Draetta GF, Hershko A, Pagano M (1999) Ubiquitination of p27 is regulated by Cdk-dependent phosphorylation and trimeric complex formation. Genes Dev 13(9):1181–1189
Roberts JM, Koff A, Polyak K, Firpo E, Collins S, Ohtsubo M, Massague J (1994) Cyclins, Cdks, and cyclin kinase inhibitors. Cold Spring Harb Symp Quant Biol 59:31–38
Sherr CJ, Roberts JM (1999) CDK inhibitors: positive and negative regulators of G1-phase progression. Genes Dev 13(12):1501–1512
Alberts B, Jhonson A, Lewis J, Raff M, Roberts K, Walter P (2008) The cell cycle. In: Alberts B, Jhonson A, Lewis J, Raff M, Roberts K, Walter P (eds) Molecular biology of the cell, 5th edn. GS, New York, NY, pp 1053–1092
Kolupaeva V, Janssens V (2013) PP1 and PP2A phosphatases—cooperating partners in modulating retinoblastoma protein activation. FEBS J 280(2):627–643
Stark GR, Taylor WR (2006) Control of the G2/M transition. Mol Biotechnol 32(3):227–248
Branzei D, Foiani M (2008) Regulation of DNA repair throughout the cell cycle. Nat Rev Mol Cell Biol 9(4):297–308
Lobrich M, Jeggo PA (2007) The impact of a negligent G2/M checkpoint on genomic instability and cancer induction. Nat Rev Cancer 7(11):861–869
Porter LA, Donoghue DJ (2003) Cyclin B1 and CDK1: nuclear localization and upstream regulators. Prog Cell Cycle Res 5:335–347
Manchado E, Eguren M, Malumbres M (2010) The anaphase-promoting complex/cyclosome (APC/C): cell-cycle-dependent and -independent functions. Biochem Soc Trans 38(Pt 1):65–71
Barford D (2011) Structure, function and mechanism of the anaphase promoting complex (APC/C). Q Rev Biophys 44(2):153–190
Page AM, Hieter P (1999) The anaphase-promoting complex: new subunits and regulators. Annu Rev Biochem 68:583–609
Peters JM (2006) The anaphase promoting complex/cyclosome: a machine designed to destroy. Nat Rev Mol Cell Biol 7(9):644–656
Acquaviva C, Herzog F, Kraft C, Pines J (2004) The anaphase promoting complex/cyclosome is recruited to centromeres by the spindle assembly checkpoint. Nat Cell Biol 6(9):892–898
Mo M, Shahar S, Fleming SB, Mercer AA (2012) How viruses affect the cell cycle through manipulation of the APC/C. Trends Microbiol 20(9):440–448
Kornitzer D, Sharf R, Kleinberger T (2001) Adenovirus E4orf4 protein induces PP2A-dependent growth arrest in Saccharomyces cerevisiae and interacts with the anaphase-promoting complex/cyclosome. J Cell Biol 154(2):331–344
Mui MZ, Roopchand DE, Gentry MS, Hallberg RL, Vogel J, Branton PE (2010) Adenovirus protein E4orf4 induces premature APCCdc20 activation in Saccharomyces cerevisiae by a protein phosphatase 2A-dependent mechanism. J Virol 84(9):4798–4809
Smits VA, Medema RH (2001) Checking out the G(2)/M transition. Biochim Biophys Acta 1519(1–2):1–12
Lukas J, Lukas C, Bartek J (2004) Mammalian cell cycle checkpoints: signalling pathways and their organization in space and time. DNA Repair (Amst) 3(8–9):997–1007
Sancar A, Lindsey-Boltz LA, Unsal-Kacmaz K, Linn S (2004) Molecular mechanisms of mammalian DNA repair and the DNA damage checkpoints. Annu Rev Biochem 73:39–85
Rumschlag-Booms E, Rong L (2013) Influenza a virus entry: implications in virulence and future therapeutics. Adv Virol 2013:121924
La Gruta NL, Kedzierska K, Stambas J, Doherty PC (2007) A question of self-preservation: immunopathology in influenza virus infection. Immunol Cell Biol 85(2):85–92
Webster RG, Bean WJ, Gorman OT, Chambers TM, Kawaoka Y (1992) Evolution and ecology of influenza A viruses. Microbiol Rev 56(1):152–179
Simonsen L, Clarke MJ, Williamson GD, Stroup DF, Arden NH, Schonberger LB (1997) The impact of influenza epidemics on mortality: introducing a severity index. Am J Public Health 87(12):1944–1950
Morens DM, Fauci AS (2007) The 1918 influenza pandemic: insights for the 21st century. J Infect Dis 195(7):1018–1028
Samji T (2009) Influenza A: understanding the viral life cycle. Yale J Biol Med 82(4):153–159
Palese P, Shaw ML (2007) Orthomyxoviridae: the viruses and their replication. In: Knipe DM, Howley PM (eds) Fields virology, 5th edn. Lippincot Williams and Wilkins, Philadelphia, PA, pp 1647–1690
Couch RB (1996) Orthomyxoviruses. In: Baron S (ed) Medical microbiology, 4th edn. University of Texas medical branch at Galveston, Galveston, TX, http://www.ncbi.nlm.nih.gov/books/NBK8611/
Zhirnov OP, Klenk HD (2007) Control of apoptosis in influenza virus-infected cells by up-regulation of Akt and p53 signaling. Apoptosis 12(8):1419–1432
Jiang W, Wang Q, Chen S, Gao S, Song L, Liu P, Huang W (2013) Influenza A virus NS1 induces G0/G1 cell cycle arrest by inhibiting the expression and activity of RhoA protein. J Virol 87(6):3039–3052
Turpin E, Luke K, Jones J, Tumpey T, Konan K, Schultz-Cherry S (2005) Influenza virus infection increases p53 activity: role of p53 in cell death and viral replication. J Virol 79(14):8802–8811
Terrier O, Josset L, Textoris J, Marcel V, Cartet G, Ferraris O, N’Guyen C, Lina B, Diaz JJ, Bourdon JC, Rosa-Calatrava M (2011) Cellular transcriptional profiling in human lung epithelial cells infected by different subtypes of influenza A viruses reveals an overall down-regulation of the host p53 pathway. Virol J 8:285
Hale BG, Randall RE, Ortin J, Jackson D (2008) The multifunctional NS1 protein of influenza A viruses. J Gen Virol 89(Pt 10):2359–2376
Zhang S, Tang Q, Xu F, Xue Y, Zhen Z, Deng Y, Liu M, Chen J, Liu S, Qiu M, Liao Z, Li Z, Luo D, Shi F, Zheng Y, Bi F (2009) RhoA regulates G1-S progression of gastric cancer cells by modulation of multiple INK4 family tumor suppressors. Mol Cancer Res 7(4):570–580
Weber JD, Hu W, Jefcoat SC Jr, Raben DM, Baldassare JJ (1997) Ras-stimulated extracellular signal-related kinase 1 and RhoA activities coordinate platelet-derived growth factor-induced G1 progression through the independent regulation of cyclin D1 and p27. J Biol Chem 272(52):32966–32971
Li H, Ung CY, Ma XH, Li BW, Low BC, Cao ZW, Chen YZ (2009) Simulation of crosstalk between small GTPase RhoA and EGFR-ERK signaling pathway via MEKK1. Bioinformatics 25(3):358–364
Croft DR, Olson MF (2006) The Rho GTPase effector ROCK regulates cyclin A, cyclin D1, and p27Kip1 levels by distinct mechanisms. Mol Cell Biol 26(12):4612–4627
Engelhardt OG, Fodor E (2006) Functional association between viral and cellular transcription during influenza virus infection. Rev Med Virol 16(5):329–345
Yonaha M, Chibazakura T, Kitajima S, Yasukochi Y (1995) Cell cycle-dependent regulation of RNA polymerase II basal transcription activity. Nucleic Acids Res 23(20):4050–4054
Garfinkel MS, Katze MG (1992) Translational control by influenza virus. Selective and cap-dependent translation of viral mRNAs in infected cells. J Biol Chem 267(13):9383–9390
Park YW, Wilusz J, Katze MG (1999) Regulation of eukaryotic protein synthesis: selective influenza viral mRNA translation is mediated by the cellular RNA-binding protein GRSF-1. Proc Natl Acad Sci U S A 96(12):6694–6699
Pyronnet S, Dostie J, Sonenberg N (2001) Suppression of cap-dependent translation in mitosis. Genes Dev 15(16):2083–2093
Stewart SA, Poon B, Jowett JB, Xie Y, Chen IS (1999) Lentiviral delivery of HIV-1 Vpr protein induces apoptosis in transformed cells. Proc Natl Acad Sci U S A 96(21):12039–12043
Gozlan J, Lathey JL, Spector SA (1998) Human immunodeficiency virus type 1 induction mediated by genistein is linked to cell cycle arrest in G2. J Virol 72(10):8174–8180
Yuan X, Shan Y, Zhao Z, Chen J, Cong Y (2005) G0/G1 arrest and apoptosis induced by SARS-CoV 3b protein in transfected cells. Virol J 2:66
Yuan X, Wu J, Shan Y, Yao Z, Dong B, Chen B, Zhao Z, Wang S, Chen J, Cong Y (2006) SARS coronavirus 7a protein blocks cell cycle progression at G0/G1 phase via the cyclin D3/pRb pathway. Virology 346(1):74–85
Chen CJ, Makino S (2004) Murine coronavirus replication induces cell cycle arrest in G0/G1 phase. J Virol 78(11):5658–5669
Chen CJ, Sugiyama K, Kubo H, Huang C, Makino S (2004) Murine coronavirus nonstructural protein p28 arrests cell cycle in G0/G1 phase. J Virol 78(19):10410–10419
Weiss SR, Navas-Martin S (2005) Coronavirus pathogenesis and the emerging pathogen severe acute respiratory syndrome coronavirus. Microbiol Mol Biol Rev 69(4):635–664
Marra MA, Jones SJ, Astell CR, Holt RA, Brooks-Wilson A, Butterfield YS, Khattra J, Asano JK, Barber SA, Chan SY, Cloutier A, Coughlin SM, Freeman D, Girn N, Griffith OL, Leach SR, Mayo M, McDonald H, Montgomery SB, Pandoh PK, Petrescu AS, Robertson AG, Schein JE, Siddiqui A, Smailus DE, Stott JM, Yang GS, Plummer F, Andonov A, Artsob H, Bastien N, Bernard K, Booth TF, Bowness D, Czub M, Drebot M, Fernando L, Flick R, Garbutt M, Gray M, Grolla A, Jones S, Feldmann H, Meyers A, Kabani A, Li Y, Normand S, Stroher U, Tipples GA, Tyler S, Vogrig R, Ward D, Watson B, Brunham RC, Krajden M, Petric M, Skowronski DM, Upton C, Roper RL (2003) The Genome sequence of the SARS-associated coronavirus. Science 300(5624):1399–1404
Rota PA, Oberste MS, Monroe SS, Nix WA, Campagnoli R, Icenogle JP, Penaranda S, Bankamp B, Maher K, Chen MH, Tong S, Tamin A, Lowe L, Frace M, DeRisi JL, Chen Q, Wang D, Erdman DD, Peret TC, Burns C, Ksiazek TG, Rollin PE, Sanchez A, Liffick S, Holloway B, Limor J, McCaustland K, Olsen-Rasmussen M, Fouchier R, Gunther S, Osterhaus AD, Drosten C, Pallansch MA, Anderson LJ, Bellini WJ (2003) Characterization of a novel coronavirus associated with severe acute respiratory syndrome. Science 300(5624):1394–1399
Compton SR, Barthold SW, Smith AL (1993) The cellular and molecular pathogenesis of coronaviruses. Lab Anim Sci 43(1):15–28
Wege H, Siddell S, ter Meulen V (1982) The biology and pathogenesis of coronaviruses. Curr Top Microbiol Immunol 99:165–200
An S, Chen CJ, Yu X, Leibowitz JL, Makino S (1999) Induction of apoptosis in murine coronavirus-infected cultured cells and demonstration of E protein as an apoptosis inducer. J Virol 73(9):7853–7859
Chen CJ, Makino S (2002) Murine coronavirus-induced apoptosis in 17Cl-1 cells involves a mitochondria-mediated pathway and its downstream caspase-8 activation and bid cleavage. Virology 302(2):321–332
Belyavsky M, Belyavskaya E, Levy GA, Leibowitz JL (1998) Coronavirus MHV-3-induced apoptosis in macrophages. Virology 250(1):41–49
Zhu L, Anasetti C (1995) Cell cycle control of apoptosis in human leukemic T cells. J Immunol 154(1):192–200
Klumperman J, Locker JK, Meijer A, Horzinek MC, Geuze HJ, Rottier PJ (1994) Coronavirus M proteins accumulate in the Golgi complex beyond the site of virion budding. J Virol 68(10):6523–6534
Tooze J, Tooze S, Warren G (1984) Replication of coronavirus MHV-A59 in sac- cells: determination of the first site of budding of progeny virions. Eur J Cell Biol 33(2):281–293
Lowe M, Nakamura N, Warren G (1998) Golgi division and membrane traffic. Trends Cell Biol 8(1):40–44
Warren G (1993) Membrane partitioning during cell division. Annu Rev Biochem 62:323–348
Bonneau AM, Sonenberg N (1987) Involvement of the 24-kDa cap-binding protein in regulation of protein synthesis in mitosis. J Biol Chem 262(23):11134–11139
Thiel V, Siddell SG (1994) Internal ribosome entry in the coding region of murine hepatitis virus mRNA 5. J Gen Virol 75(Pt 11):3041–3046
Nishioka WK, Welsh RM (1994) Susceptibility to cytotoxic T lymphocyte-induced apoptosis is a function of the proliferative status of the target. J Exp Med 179(2):769–774
Johnston JB, Wang G, Barrett JW, Nazarian SH, Colwill K, Moran M, McFadden G (2005) Myxoma virus M-T5 protects infected cells from the stress of cell cycle arrest through its interaction with host cell cullin-1. J Virol 79(16):10750–10763
Gearhart TL, Bouchard MJ (2010) The hepatitis B virus X protein modulates hepatocyte proliferation pathways to stimulate viral replication. J Virol 84(6):2675–2686
Ashfaq UA, Javed T, Rehman S, Nawaz Z, Riazuddin S (2011) An overview of HCV molecular biology, replication and immune responses. Virol J 8:161
Lindenbach BD, Rice CM (2005) Unravelling hepatitis C virus replication from genome to function. Nature 436(7053):933–938
Sy T, Jamal MM (2006) Epidemiology of hepatitis C virus (HCV) infection. Int J Med Sci 3(2):41–46
Rosen HR (2011) Clinical practice. Chronic hepatitis C infection. N Engl J Med 364(25):2429–2438
Liang TJ, Ghany MG (2013) Current and future therapies for hepatitis C virus infection. N Engl J Med 368(20):1907–1917
Munir S, Saleem S, Idrees M, Tariq A, Butt S, Rauff B, Hussain A, Badar S, Naudhani M, Fatima Z, Ali M, Ali L, Akram M, Aftab M, Khubaib B, Awan Z (2010) Hepatitis C treatment: current and future perspectives. Virol J 7:296
Ray S, Bailey J, Thomas D (2013) Hepatitis C virus. In: Knipe DM, Howley PM (eds) Fields virology, 6th edn. Lippincot Williams and Wilkins, Philadelphia, PA, pp 795–824
Ghosh AK, Steele R, Meyer K, Ray R, Ray RB (1999) Hepatitis C virus NS5A protein modulates cell cycle regulatory genes and promotes cell growth. J Gen Virol 80(Pt 5):1179–1183
Park SH, Lim JS, Lim SY, Tiwari I, Jang KL (2011) Hepatitis C virus Core protein stimulates cell growth by down-regulating p16 expression via DNA methylation. Cancer Lett 310(1):61–68
Tsukiyama-Kohara K, Tone S, Maruyama I, Inoue K, Katsume A, Nuriya H, Ohmori H, Ohkawa J, Taira K, Hoshikawa Y, Shibasaki F, Reth M, Minatogawa Y, Kohara M (2004) Activation of the CKI-CDK-Rb-E2F pathway in full genome hepatitis C virus-expressing cells. J Biol Chem 279(15):14531–14541
Yang XJ, Liu J, Ye L, Liao QJ, Wu JG, Gao JR, She YL, Wu ZH, Ye LB (2006) HCV NS2 protein inhibits cell proliferation and induces cell cycle arrest in the S-phase in mammalian cells through down-regulation of cyclin A expression. Virus Res 121(2):134–143
Bittar C, Shrivastava S, Bhanja Chowdhury J, Rahal P, Ray RB (2013) Hepatitis C virus NS2 protein inhibits DNA damage pathway by sequestering p53 to the cytoplasm. PLoS One 8(4):e62581
Shiu TY, Huang SM, Shih YL, Chu HC, Chang WK, Hsieh TY (2013) Hepatitis C virus core protein down-regulates p21(Waf1/Cip1) and inhibits curcumin-induced apoptosis through microRNA-345 targeting in human hepatoma cells. PLoS One 8(4):e61089
Wang Y, Wang Y, Xu Y, Tong W, Pan T, Li J, Sun S, Shao J, Ding H, Toyoda T, Yuan Z (2011) Hepatitis C virus NS5B protein delays s phase progression in human hepatocyte-derived cells by relocalizing cyclin-dependent kinase 2-interacting protein (CINP). J Biol Chem 286(30):26603–26615
Walters KA, Syder AJ, Lederer SL, Diamond DL, Paeper B, Rice CM, Katze MG (2009) Genomic analysis reveals a potential role for cell cycle perturbation in HCV-mediated apoptosis of cultured hepatocytes. PLoS Pathog 5(1):e1000269
Scholle F, Li K, Bodola F, Ikeda M, Luxon BA, Lemon SM (2004) Virus-host cell interactions during hepatitis C virus RNA replication: impact of polyprotein expression on the cellular transcriptome and cell cycle association with viral RNA synthesis. J Virol 78(3):1513–1524
Sarfraz S, Hamid S, Siddiqui A, Hussain S, Pervez S, Alexander G (2008) Altered expression of cell cycle and apoptotic proteins in chronic hepatitis C virus infection. BMC Microbiol 8:133
Ruggieri A, Murdolo M, Harada T, Miyamura T, Rapicetta M (2004) Cell cycle perturbation in a human hepatoblastoma cell line constitutively expressing hepatitis C virus core protein. Arch Virol 149(1):61–74
Chen H, Pei R, Chen X (2013) Different response of two highly permissive cell lines upon HCV infection. Virol Sin 28(4):202–208
Cho JW, Baek WK, Suh SI, Yang SH, Chang J, Sung YC, Suh MH (2001) Hepatitis C virus core protein promotes cell proliferation through the upregulation of cyclin E expression levels. Liver 21(2):137–142
Honda M, Kaneko S, Shimazaki T, Matsushita E, Kobayashi K, Ping LH, Zhang HC, Lemon SM (2000) Hepatitis C virus core protein induces apoptosis and impairs cell-cycle regulation in stably transformed Chinese hamster ovary cells. Hepatology 31(6):1351–1359
Banerjee A, Ray RB, Ray R (2010) Oncogenic potential of hepatitis C virus proteins. Viruses 2(9):2108–2133
Munakata T, Liang Y, Kim S, McGivern DR, Huibregtse J, Nomoto A, Lemon SM (2007) Hepatitis C virus induces E6AP-dependent degradation of the retinoblastoma protein. PLoS Pathog 3(9):1335–1347
Abbas T, Dutta A (2009) p21 in cancer: intricate networks and multiple activities. Nat Rev Cancer 9(6):400–414
Kwun HJ, Jung EY, Ahn JY, Lee MN, Jang KL (2001) p53-dependent transcriptional repression of p21(waf1) by hepatitis C virus NS3. J Gen Virol 82(Pt 9):2235–2241
Nguyen H, Mudryj M, Guadalupe M, Dandekar S (2003) Hepatitis C virus core protein expression leads to biphasic regulation of the p21 cdk inhibitor and modulation of hepatocyte cell cycle. Virology 312(1):245–253
Yao ZQ, Eisen-Vandervelde A, Ray S, Hahn YS (2003) HCV core/gC1qR interaction arrests T cell cycle progression through stabilization of the cell cycle inhibitor p27Kip1. Virology 314(1):271–282
Otsuka M, Kato N, Lan K, Yoshida H, Kato J, Goto T, Shiratori Y, Omata M (2000) Hepatitis C virus core protein enhances p53 function through augmentation of DNA binding affinity and transcriptional ability. J Biol Chem 275(44):34122–34130
Lo SY, Masiarz F, Hwang SB, Lai MM, Ou JH (1995) Differential subcellular localization of hepatitis C virus core gene products. Virology 213(2):455–461
Salvant BS, Fortunato EA, Spector DH (1998) Cell cycle dysregulation by human cytomegalovirus: influence of the cell cycle phase at the time of infection and effects on cyclin transcription. J Virol 72(5):3729–3741
Burkhart DL, Sage J (2008) Cellular mechanisms of tumour suppression by the retinoblastoma gene. Nat Rev Cancer 8(9):671–682
Marshall A, Rushbrook S, Davies SE, Morris LS, Scott IS, Vowler SL, Coleman N, Alexander G (2005) Relation between hepatocyte G1 arrest, impaired hepatic regeneration, and fibrosis in chronic hepatitis C virus infection. Gastroenterology 128(1):33–42
Marshall A, Rushbrook S, Morris LS, Scott IS, Vowler SL, Davies SE, Coleman N, Alexander G (2005) Hepatocyte expression of minichromosome maintenance protein-2 predicts fibrosis progression after transplantation for chronic hepatitis C virus: a pilot study. Liver Transpl 11(4):427–433
Fehr C, Conrad KD, Niepmann M (2012) Differential stimulation of hepatitis C virus RNA translation by microRNA-122 in different cell cycle phases. Cell Cycle 11(2):277–285
Honda M, Kaneko S, Matsushita E, Kobayashi K, Abell GA, Lemon SM (2000) Cell cycle regulation of hepatitis C virus internal ribosomal entry site-directed translation. Gastroenterology 118(1):152–162
Grana X, Reddy EP (1995) Cell cycle control in mammalian cells: role of cyclins, cyclin dependent kinases (CDKs), growth suppressor genes and cyclin-dependent kinase inhibitors (CKIs). Oncogene 11(2):211–219
Mohd Hanafiah K, Groeger J, Flaxman AD, Wiersma ST (2013) Global epidemiology of hepatitis C virus infection: new estimates of age-specific antibody to HCV seroprevalence. Hepatology 57(4):1333–1342
Dorner M, Horwitz JA, Donovan BM, Labitt RN, Budell WC, Friling T, Vogt A, Catanese MT, Satoh T, Kawai T, Akira S, Law M, Rice CM, Ploss A (2013) Completion of the entire hepatitis C virus life cycle in genetically humanized mice. Nature 501(7466):237–241
Gaston KL (2012) Small DNA tumor viruses, 4th edn. Caister Academic Press, Bristol, UK
Gatza ML, Chandhasin C, Ducu RI, Marriott SJ (2005) Impact of transforming viruses on cellular mutagenesis, genome stability, and cellular transformation. Environ Mol Mutagen 45(2–3):304–325
Flint SJ, Enquist LW, Racaniello VR, Skalka AM (2009) Transformation and oncogenesis. In: Flint SJ, Enquist LW, Racaniello VR, Skalka AM (eds) Principles of virology, vol II, 3rd edn. ASM Press, New York, NY, pp 201–248
Helt AM, Galloway DA (2003) Mechanisms by which DNA tumor virus oncoproteins target the Rb family of pocket proteins. Carcinogenesis 24(2):159–169
Moran E (1993) DNA tumor virus transforming proteins and the cell cycle. Curr Opin Genet Dev 3(1):63–70
Nevins JR (1994) Cell cycle targets of the DNA tumor viruses. Curr Opin Genet Dev 4(1):130–134
Sherr CJ, McCormick F (2002) The RB and p53 pathways in cancer. Cancer Cell 2(2):103–112
Polager S, Ginsberg D (2009) p53 and E2f: partners in life and death. Nat Rev Cancer 9(10):738–748
Harbour JW, Dean DC (2000) The Rb/E2F pathway: expanding roles and emerging paradigms. Genes Dev 14(19):2393–2409
Henley SA, Dick FA (2012) The retinoblastoma family of proteins and their regulatory functions in the mammalian cell division cycle. Cell Div 7(1):10
Raychaudhuri P, Bagchi S, Devoto SH, Kraus VB, Moran E, Nevins JR (1991) Domains of the adenovirus E1A protein required for oncogenic activity are also required for dissociation of E2F transcription factor complexes. Genes Dev 5(7):1200–1211
Liu X, Marmorstein R (2007) Structure of the retinoblastoma protein bound to adenovirus E1A reveals the molecular basis for viral oncoprotein inactivation of a tumor suppressor. Genes Dev 21(21):2711–2716
Ikeda MA, Nevins JR (1993) Identification of distinct roles for separate E1A domains in disruption of E2F complexes. Mol Cell Biol 13(11):7029–7035
DeCaprio JA, Ludlow JW, Figge J, Shew JY, Huang CM, Lee WH, Marsilio E, Paucha E, Livingston DM (1988) SV40 large tumor antigen forms a specific complex with the product of the retinoblastoma susceptibility gene. Cell 54(2):275–283
Zalvide J, DeCaprio JA, Stubdal H (2001) Binding of SV40 large T antigen to the retinoblastoma susceptibility gene product and related proteins. Methods Mol Biol 165:213–218
Zalvide J, DeCaprio JA (1995) Role of pRb-related proteins in simian virus 40 large-T-antigen-mediated transformation. Mol Cell Biol 15(10):5800–5810
Zalvide J, Stubdal H, DeCaprio JA (1998) The J domain of simian virus 40 large T antigen is required to functionally inactivate RB family proteins. Mol Cell Biol 18(3):1408–1415
Sullivan CS, Cantalupo P, Pipas JM (2000) The molecular chaperone activity of simian virus 40 large T antigen is required to disrupt Rb-E2F family complexes by an ATP-dependent mechanism. Mol Cell Biol 20(17):6233–6243
Stubdal H, Zalvide J, DeCaprio JA (1996) Simian virus 40 large T antigen alters the phosphorylation state of the RB-related proteins p130 and p107. J Virol 70(5):2781–2788
Pagano M, Durst M, Joswig S, Draetta G, Jansen-Durr P (1992) Binding of the human E2F transcription factor to the retinoblastoma protein but not to cyclin A is abolished in HPV-16-immortalized cells. Oncogene 7(9):1681–1686
Chellappan S, Kraus VB, Kroger B, Munger K, Howley PM, Phelps WC, Nevins JR (1992) Adenovirus E1A, simian virus 40 tumor antigen, and human papillomavirus E7 protein share the capacity to disrupt the interaction between transcription factor E2F and the retinoblastoma gene product. Proc Natl Acad Sci U S A 89(10):4549–4553
Wu EW, Clemens KE, Heck DV, Munger K (1993) The human papillomavirus E7 oncoprotein and the cellular transcription factor E2F bind to separate sites on the retinoblastoma tumor suppressor protein. J Virol 67(4):2402–2407
Boyer SN, Wazer DE, Band V (1996) E7 protein of human papilloma virus-16 induces degradation of retinoblastoma protein through the ubiquitin-proteasome pathway. Cancer Res 56(20):4620–4624
Jones DL, Munger K (1997) Analysis of the p53-mediated G1 growth arrest pathway in cells expressing the human papillomavirus type 16 E7 oncoprotein. J Virol 71(4):2905–2912
O’Shea CC, Fried M (2005) Modulation of the ARF-p53 pathway by the small DNA tumor viruses. Cell Cycle 4(3):449–452
Jiang D, Srinivasan A, Lozano G, Robbins PD (1993) SV40 T antigen abrogates p53-mediated transcriptional activity. Oncogene 8(10):2805–2812
Segawa K, Minowa A, Sugasawa K, Takano T, Hanaoka F (1993) Abrogation of p53-mediated transactivation by SV40 large T antigen. Oncogene 8(3):543–548
Mietz JA, Unger T, Huibregtse JM, Howley PM (1992) The transcriptional transactivation function of wild-type p53 is inhibited by SV40 large T-antigen and by HPV-16 E6 oncoprotein. EMBO J 11(13):5013–5020
Lane DP, Crawford LV (1979) T antigen is bound to a host protein in SV40-transformed cells. Nature 278(5701):261–263
Linzer DI, Levine AJ (1979) Characterization of a 54K dalton cellular SV40 tumor antigen present in SV40-transformed cells and uninfected embryonal carcinoma cells. Cell 17(1):43–52
Harada JN, Shevchenko A, Shevchenko A, Pallas DC, Berk AJ (2002) Analysis of the adenovirus E1B-55K-anchored proteome reveals its link to ubiquitination machinery. J Virol 76(18):9194–9206
Querido E, Blanchette P, Yan Q, Kamura T, Morrison M, Boivin D, Kaelin WG, Conaway RC, Conaway JW, Branton PE (2001) Degradation of p53 by adenovirus E4orf6 and E1B55K proteins occurs via a novel mechanism involving a Cullin-containing complex. Genes Dev 15(23):3104–3117
Scheffner M, Huibregtse JM, Vierstra RD, Howley PM (1993) The HPV-16 E6 and E6-AP complex functions as a ubiquitin-protein ligase in the ubiquitination of p53. Cell 75(3):495–505
Pellet PE, Roizman B (2013) Herpesviridae. In: Knipe DM, Howley PM (eds) Fields virology, 6th edn. Lippincot Williams and Wilkins, Philadelphia, PA, pp 1802–1820
Sandri-Goldin RM (2006) Alpha Herpesviruses: molecular and cellular biology. Caister Academic Press, Bristol, UK
Flint SJ, Enquist LW, Racaniello VR, Skalka AM Patterns of infection. In: Flint SJ, Enquist LW, Racaniello VR, Skalka AM (eds) Principles of virology vol Volume II: Pathogenesis and control, vol 2, 3rd edn. ASM Press, New York, NY, pp 134–163
McGeoch DJ, Rixon FJ, Davison AJ (2006) Topics in herpesvirus genomics and evolution. Virus Res 117(1):90–104
Dittmer DP, Damania B (2013) Kaposi sarcoma associated herpesvirus pathogenesis (KSHV)-an update. Curr Opin Virol 3(3):238–244
Mesri EA, Cesarman E, Boshoff C (2010) Kaposi’s sarcoma and its associated herpesvirus. Nat Rev Cancer 10(10):707–719
Cai Q, Verma SC, Lu J, Robertson ES (2010) Molecular biology of Kaposi’s sarcoma-associated herpesvirus and related oncogenesis. Adv Virus Res 78:87–142
Jarviluoma A, Ojala PM (2006) Cell signaling pathways engaged by KSHV. Biochim Biophys Acta 1766(1):140–158
Cesarman E, Chang Y, Moore PS, Said JW, Knowles DM (1995) Kaposi’s sarcoma-associated herpesvirus-like DNA sequences in AIDS-related body-cavity-based lymphomas. N Engl J Med 332(18):1186–1191
Renne R, Blackbourn D, Whitby D, Levy J, Ganem D (1998) Limited transmission of Kaposi’s sarcoma-associated herpesvirus in cultured cells. J Virol 72(6):5182–5188
Sarid R, Flore O, Bohenzky RA, Chang Y, Moore PS (1998) Transcription mapping of the Kaposi’s sarcoma-associated herpesvirus (human herpesvirus 8) genome in a body cavity-based lymphoma cell line (BC-1). J Virol 72(2):1005–1012
Direkze S, Laman H (2004) Regulation of growth signalling and cell cycle by Kaposi’s sarcoma-associated herpesvirus genes. Int J Exp Pathol 85(6):305–319
Moore PS (2007) KSHV manipulation of the cell cycle and apoptosis. In: Arvin A, Campadelli-Fiume G, Mocarski E et al (eds) Human herpesviruses: biology, therapy, and immunoprophylaxis. Cambridge University Press, Cambridge
Chang Y, Moore PS, Talbot SJ, Boshoff CH, Zarkowska T, Godden K, Paterson H, Weiss RA, Mittnacht S (1996) Cyclin encoded by KS herpesvirus. Nature 382(6590):410
Li M, Lee H, Yoon DW, Albrecht JC, Fleckenstein B, Neipel F, Jung JU (1997) Kaposi’s sarcoma-associated herpesvirus encodes a functional cyclin. J Virol 71(3):1984–1991
Laman H, Coverley D, Krude T, Laskey R, Jones N (2001) Viral cyclin-cyclin-dependent kinase 6 complexes initiate nuclear DNA replication. Mol Cell Biol 21(2):624–635
Ellis M, Chew YP, Fallis L, Freddersdorf S, Boshoff C, Weiss RA, Lu X, Mittnacht S (1999) Degradation of p27(Kip) cdk inhibitor triggered by Kaposi’s sarcoma virus cyclin-cdk6 complex. EMBO J 18(3):644–653
Godden-Kent D, Talbot SJ, Boshoff C, Chang Y, Moore P, Weiss RA, Mittnacht S (1997) The cyclin encoded by Kaposi’s sarcoma-associated herpesvirus stimulates cdk6 to phosphorylate the retinoblastoma protein and histone H1. J Virol 71(6):4193–4198
Laman H, Peters G, Jones N (2001) Cyclin-mediated export of human Orc1. Exp Cell Res 271(2):230–237
Swanton C, Mann DJ, Fleckenstein B, Neipel F, Peters G, Jones N (1997) Herpes viral cyclin/Cdk6 complexes evade inhibition by CDK inhibitor proteins. Nature 390(6656):184–187
Kaldis P, Ojala PM, Tong L, Makela TP, Solomon MJ (2001) CAK-independent activation of CDK6 by a viral cyclin. Mol Biol Cell 12(12):3987–3999
Sarek G, Jarviluoma A, Ojala PM (2006) KSHV viral cyclin inactivates p27KIP1 through Ser10 and Thr187 phosphorylation in proliferating primary effusion lymphomas. Blood 107(2):725–732
Mann DJ, Child ES, Swanton C, Laman H, Jones N (1999) Modulation of p27(Kip1) levels by the cyclin encoded by Kaposi’s sarcoma-associated herpesvirus. EMBO J 18(3):654–663
Ojala PM, Tiainen M, Salven P, Veikkola T, Castanos-Velez E, Sarid R, Biberfeld P, Makela TP (1999) Kaposi’s sarcoma-associated herpesvirus-encoded v-cyclin triggers apoptosis in cells with high levels of cyclin-dependent kinase 6. Cancer Res 59(19):4984–4989
Verschuren EW, Klefstrom J, Evan GI, Jones N (2002) The oncogenic potential of Kaposi’s sarcoma-associated herpesvirus cyclin is exposed by p53 loss in vitro and in vivo. Cancer Cell 2(3):229–241
Verschuren EW, Hodgson JG, Gray JW, Kogan S, Jones N, Evan GI (2004) The role of p53 in suppression of KSHV cyclin-induced lymphomagenesis. Cancer Res 64(2):581–589
Radkov SA, Kellam P, Boshoff C (2000) The latent nuclear antigen of Kaposi sarcoma-associated herpesvirus targets the retinoblastoma-E2F pathway and with the oncogene Hras transforms primary rat cells. Nat Med 6(10):1121–1127
Cannon M, Philpott NJ, Cesarman E (2003) The Kaposi’s sarcoma-associated herpesvirus G protein-coupled receptor has broad signaling effects in primary effusion lymphoma cells. J Virol 77(1):57–67
Russo JJ, Bohenzky RA, Chien MC, Chen J, Yan M, Maddalena D, Parry JP, Peruzzi D, Edelman IS, Chang Y, Moore PS (1996) Nucleotide sequence of the Kaposi sarcoma-associated herpesvirus (HHV8). Proc Natl Acad Sci U S A 93(25):14862–14867
Seaman WT, Ye D, Wang RX, Hale EE, Weisse M, Quinlivan EB (1999) Gene expression from the ORF50/K8 region of Kaposi’s sarcoma-associated herpesvirus. Virology 263(2):436–449
Wang SE, Wu FY, Yu Y, Hayward GS (2003) CCAAT/enhancer-binding protein-alpha is induced during the early stages of Kaposi’s sarcoma-associated herpesvirus (KSHV) lytic cycle reactivation and together with the KSHV replication and transcription activator (RTA) cooperatively stimulates the viral RTA, MTA, and PAN promoters. J Virol 77(17):9590–9612
Wang SE, Wu FY, Fujimuro M, Zong J, Hayward SD, Hayward GS (2003) Role of CCAAT/enhancer-binding protein alpha (C/EBPalpha) in activation of the Kaposi’s sarcoma-associated herpesvirus (KSHV) lytic-cycle replication-associated protein (RAP) promoter in cooperation with the KSHV replication and transcription activator (RTA) and RAP. J Virol 77(1):600–623
Wu FY, Wang SE, Tang QQ, Fujimuro M, Chiou CJ, Zheng Q, Chen H, Hayward SD, Lane MD, Hayward GS (2003) Cell cycle arrest by Kaposi’s sarcoma-associated herpesvirus replication-associated protein is mediated at both the transcriptional and posttranslational levels by binding to CCAAT/enhancer-binding protein alpha and p21(CIP-1). J Virol 77(16):8893–8914
Izumiya Y, Lin SF, Ellison TJ, Levy AM, Mayeur GL, Izumiya C, Kung HJ (2003) Cell cycle regulation by Kaposi’s sarcoma-associated herpesvirus K-bZIP: direct interaction with cyclin-CDK2 and induction of G1 growth arrest. J Virol 77(17):9652–9661
Maeda E, Akahane M, Kiryu S, Kato N, Yoshikawa T, Hayashi N, Aoki S, Minami M, Uozaki H, Fukayama M, Ohtomo K (2009) Spectrum of Epstein–Barr virus-related diseases: a pictorial review. Jpn J Radiol 27(1):4–19
Young LS, Rickinson AB (2004) Epstein–Barr virus: 40 years on. Nat Rev Cancer 4(10):757–768
Arvanitakis L, Yaseen N, Sharma S (1995) Latent membrane protein-1 induces cyclin D2 expression, pRb hyperphosphorylation, and loss of TGF-beta 1-mediated growth inhibition in EBV-positive B cells. J Immunol 155(3):1047–1056
Dirmeier U, Hoffmann R, Kilger E, Schultheiss U, Briseno C, Gires O, Kieser A, Eick D, Sugden B, Hammerschmidt W (2005) Latent membrane protein 1 of Epstein–Barr virus coordinately regulates proliferation with control of apoptosis. Oncogene 24(10):1711–1717
Parker GA, Crook T, Bain M, Sara EA, Farrell PJ, Allday MJ (1996) Epstein–Barr virus nuclear antigen (EBNA)3C is an immortalizing oncoprotein with similar properties to adenovirus E1A and papillomavirus E7. Oncogene 13(12):2541–2549
Saha A, Halder S, Upadhyay SK, Lu J, Kumar P, Murakami M, Cai Q, Robertson ES (2011) Epstein–Barr virus nuclear antigen 3C facilitates G1-S transition by stabilizing and enhancing the function of cyclin D1. PLoS Pathog 7(2):e1001275
Fukuda M, Satoh TA, Takanashi M, Hirai K, Ohnishi E, Sairenji T (2000) Inhibition of cell growth and Epstein–Barr virus reactivation by CD40 stimulation in Epstein–Barr virus-transformed B cells. Viral Immunol 13(2):215–229
Rodriguez A, Armstrong M, Dwyer D, Flemington E (1999) Genetic dissection of cell growth arrest functions mediated by the Epstein–Barr virus lytic gene product, Zta. J Virol 73(11):9029–9038
Cayrol C, Flemington EK (1996) The Epstein–Barr virus bZIP transcription factor Zta causes G0/G1 cell cycle arrest through induction of cyclin-dependent kinase inhibitors. EMBO J 15(11):2748–2759
Swenson JJ, Mauser AE, Kaufmann WK, Kenney SC (1999) The Epstein–Barr virus protein BRLF1 activates S phase entry through E2F1 induction. J Virol 73(8):6540–6550
Michaelis M, Doerr HW, Cinatl J (2009) The story of human cytomegalovirus and cancer: increasing evidence and open questions. Neoplasia 11(1):1–9
Huang ES, Johnson RA (2000) Human cytomegalovirus—no longer just a DNA virus. Nat Med 6(8):863–864
Castillo JP, Kowalik TF (2004) HCMV infection: modulating the cell cycle and cell death. Int Rev Immunol 23(1–2):113–139
Albrecht T, Nachtigal M, St Jeor SC, Rapp F (1976) Induction of cellular DNA synthesis and increased mitotic activity in syrian hamster embryo cells abortively infected with human cytomegalovirus. J Gen Virol 30(2):167–177
Gonczol E, Plotkin SA (1984) Cells infected with human cytomegalovirus release a factor(s) that stimulates cell DNA synthesis. J Gen Virol 65(Pt 10):1833–1837
Sinclair J, Baillie J, Bryant L, Caswell R (2000) Human cytomegalovirus mediates cell cycle progression through G(1) into early S phase in terminally differentiated cells. J Gen Virol 81(Pt 6):1553–1565
Dittmer D, Mocarski ES (1997) Human cytomegalovirus infection inhibits G1/S transition. J Virol 71(2):1629–1634
Bresnahan WA, Boldogh I, Thompson EA, Albrecht T (1996) Human cytomegalovirus inhibits cellular DNA synthesis and arrests productively infected cells in late G1. Virology 224(1):150–160
Lu M, Shenk T (1996) Human cytomegalovirus infection inhibits cell cycle progression at multiple points, including the transition from G1 to S. J Virol 70(12):8850–8857
Lu M, Shenk T (1999) Human cytomegalovirus UL69 protein induces cells to accumulate in G1 phase of the cell cycle. J Virol 73(1):676–683
Wiebusch L, Hagemeier C (1999) Human cytomegalovirus 86-kilodalton IE2 protein blocks cell cycle progression in G(1). J Virol 73(11):9274–9283
Hume AJ, Finkel JS, Kamil JP, Coen DM, Culbertson MR, Kalejta RF (2008) Phosphorylation of retinoblastoma protein by viral protein with cyclin-dependent kinase function. Science 320(5877):797–799
Kalejta RF, Bechtel JT, Shenk T (2003) Human cytomegalovirus pp 71 stimulates cell cycle progression by inducing the proteasome-dependent degradation of the retinoblastoma family of tumor suppressors. Mol Cell Biol 23(6):1885–1895
Kalejta RF, Shenk T (2003) Proteasome-dependent, ubiquitin-independent degradation of the Rb family of tumor suppressors by the human cytomegalovirus pp 71 protein. Proc Natl Acad Sci U S A 100(6):3263–3268
Kalejta RF, Shenk T (2003) The human cytomegalovirus UL82 gene product (pp 71) accelerates progression through the G1 phase of the cell cycle. J Virol 77(6):3451–3459
Hagemeier C, Caswell R, Hayhurst G, Sinclair J, Kouzarides T (1994) Functional interaction between the HCMV IE2 transactivator and the retinoblastoma protein. EMBO J 13(12):2897–2903
Fortunato EA, Sommer MH, Yoder K, Spector DH (1997) Identification of domains within the human cytomegalovirus major immediate-early 86-kilodalton protein and the retinoblastoma protein required for physical and functional interaction with each other. J Virol 71(11):8176–8185
Bresnahan WA, Albrecht T, Thompson EA (1998) The cyclin E promoter is activated by human cytomegalovirus 86-kDa immediate early protein. J Biol Chem 273(34):22075–22082
Chayavichitsilp P, Buckwalter JV, Krakowski AC, Friedlander SF (2009) Herpes simplex. Pediatr Rev 30(4):119–129, quiz 130
Song B, Liu JJ, Yeh KC, Knipe DM (2000) Herpes simplex virus infection blocks events in the G1 phase of the cell cycle. Virology 267(2):326–334
de Bruyn Kops A, Knipe DM (1988) Formation of DNA replication structures in herpes virus-infected cells requires a viral DNA binding protein. Cell 55(5):857–868
Ehmann GL, McLean TI, Bachenheimer SL (2000) Herpes simplex virus type 1 infection imposes a G(1)/S block in asynchronously growing cells and prevents G(1) entry in quiescent cells. Virology 267(2):335–349
Olgiate J, Ehmann GL, Vidyarthi S, Hilton MJ, Bachenheimer SL (1999) Herpes simplex virus induces intracellular redistribution of E2F4 and accumulation of E2F pocket protein complexes. Virology 258(2):257–270
Hobbs WE 2nd, DeLuca NA (1999) Perturbation of cell cycle progression and cellular gene expression as a function of herpes simplex virus ICP0. J Virol 73(10):8245–8255
Lomonte P, Everett RD (1999) Herpes simplex virus type 1 immediate-early protein Vmw110 inhibits progression of cells through mitosis and from G(1) into S phase of the cell cycle. J Virol 73(11):9456–9467
Diwan P, Lacasse JJ, Schang LM (2004) Roscovitine inhibits activation of promoters in herpes simplex virus type 1 genomes independently of promoter-specific factors. J Virol 78(17):9352–9365
Schang LM, Phillips J, Schaffer PA (1998) Requirement for cellular cyclin-dependent kinases in herpes simplex virus replication and transcription. J Virol 72(7):5626–5637
Durand LO, Roizman B (2008) Role of cdk9 in the optimization of expression of the genes regulated by ICP22 of herpes simplex virus 1. J Virol 82(21):10591–10599
Davido DJ, Von Zagorski WF, Maul GG, Schaffer PA (2003) The differential requirement for cyclin-dependent kinase activities distinguishes two functions of herpes simplex virus type 1 ICP0. J Virol 77(23):12603–12616
Hossain A, Holt T, Ciacci-Zanella J, Jones C (1997) Analysis of cyclin-dependent kinase activity after herpes simplex virus type 2 infection. J Gen Virol 78(Pt 12):3341–3348
Seeger C, Mason WS, Zoulim F (2007) Hepadnaviruses. In: Knipe DM, Howley PM (eds) Fields virology, 5th edn. Lippincot Williams and Wilkins, Philadelphia, PA, pp 2977–3029
Block TM, Mehta AS, Fimmel CJ, Jordan R (2003) Molecular viral oncology of hepatocellular carcinoma. Oncogene 22(33):5093–5107
Nguyen VT, Law MG, Dore GJ (2009) Hepatitis B-related hepatocellular carcinoma: epidemiological characteristics and disease burden. J Viral Hepat 16(7):453–463
Madden CR, Slagle BL (2001) Stimulation of cellular proliferation by hepatitis B virus X protein. Dis Markers 17(3):153–157
Friedrich B, Wollersheim M, Brandenburg B, Foerste R, Will H, Hildt E (2005) Induction of anti-proliferative mechanisms in hepatitis B virus producing cells. J Hepatol 43(4):696–703
Wang T, Zhao R, Wu Y, Kong D, Zhang L, Wu D, Li C, Zhang C, Yu Z, Jin X (2011) Hepatitis B virus induces G1 phase arrest by regulating cell cycle genes in HepG2.2.15 cells. Virol J 8:231
Chin R, Earnest-Silveira L, Koeberlein B, Franz S, Zentgraf H, Dong X, Gowans E, Bock CT, Torresi J (2007) Modulation of MAPK pathways and cell cycle by replicating hepatitis B virus: factors contributing to hepatocarcinogenesis. J Hepatol 47(3):325–337
Casciano J, Bagga S, Yang B, Bouchard M (2012) Modulation of cell proliferation pathways by the hepatitis B virus X protein: a potential contributor to the development of hepatocellular carcinoma. In: Lau JWY (ed) Hepatocellular carcinoma-basic research. InTech, Rijeka, Croatia, pp 103–152
Huang YQ, Wang LW, Yan SN, Gong ZJ (2004) Effects of cell cycle on telomerase activity and on hepatitis B virus replication in HepG2 2.2.15 cells. Hepatobiliary Pancreat Dis Int 3(4):543–547
Ozer A, Khaoustov VI, Mearns M, Lewis DE, Genta RM, Darlington GJ, Yoffe B (1996) Effect of hepatocyte proliferation and cellular DNA synthesis on hepatitis B virus replication. Gastroenterology 110(5):1519–1528
Guidotti LG, Matzke B, Chisari FV (1997) Hepatitis B virus replication is cell cycle independent during liver regeneration in transgenic mice. J Virol 71(6):4804–4808
Guidotti LG, Matzke B, Schaller H, Chisari FV (1995) High-level hepatitis B virus replication in transgenic mice. J Virol 69(10):6158–6169
Bouchard MJ, Schneider RJ (2004) The enigmatic X gene of hepatitis B virus. J Virol 78(23):12725–12734
Benhenda S, Cougot D, Buendia MA, Neuveut C (2009) Hepatitis B virus X protein molecular functions and its role in virus life cycle and pathogenesis. Adv Cancer Res 103:75–109
Bouchard M, Giannakopoulos S, Wang EH, Tanese N, Schneider RJ (2001) Hepatitis B virus HBx protein activation of cyclin A-cyclin-dependent kinase 2 complexes and G1 transit via a Src kinase pathway. J Virol 75(9):4247–4257
Benn J, Schneider RJ (1994) Hepatitis B virus HBx protein activates Ras-GTP complex formation and establishes a Ras, Raf, MAP kinase signaling cascade. Proc Natl Acad Sci U S A 91(22):10350–10354
Benn J, Schneider RJ (1995) Hepatitis B virus HBx protein deregulates cell cycle checkpoint controls. Proc Natl Acad Sci U S A 92(24):11215–11219
Koike K, Moriya K, Yotsuyanagi H, Iino S, Kurokawa K (1994) Induction of cell cycle progression by hepatitis B virus HBx gene expression in quiescent mouse fibroblasts. J Clin Invest 94(1):44–49
Lee S, Tarn C, Wang WH, Chen S, Hullinger RL, Andrisani OM (2002) Hepatitis B virus X protein differentially regulates cell cycle progression in X-transforming versus nontransforming hepatocyte (AML12) cell lines. J Biol Chem 277(10):8730–8740
Chen HY, Tang NH, Lin N, Chen ZX, Wang XZ (2008) Hepatitis B virus X protein induces apoptosis and cell cycle deregulation through interfering with DNA repair and checkpoint responses. Hepatol Res 38(2):174–182
Mukherji A, Janbandhu VC, Kumar V (2007) HBx-dependent cell cycle deregulation involves interaction with cyclin E/A-cdk2 complex and destabilization of p27Kip1. Biochem J 401(1):247–256
Singh AK, Swarnalatha M, Kumar V (2011) c-ETS1 facilitates G1/S-phase transition by up-regulating cyclin E and CDK2 genes and cooperates with hepatitis B virus X protein for their deregulation. J Biol Chem 286(25):21961–21970
Zhang JL, Zhao WG, Wu KL, Wang K, Zhang X, Gu CF, Li Y, Zhu Y, Wu JG (2005) Human hepatitis B virus X protein promotes cell proliferation and inhibits cell apoptosis through interacting with a serine protease Hepsin. Arch Virol 150(4):721–741
Wu JC, Merlino G, Cveklova K, Mosinger B Jr, Fausto N (1994) Autonomous growth in serum-free medium and production of hepatocellular carcinomas by differentiated hepatocyte lines that overexpress transforming growth factor alpha 1. Cancer Res 54(22):5964–5973
Tarn C, Bilodeau ML, Hullinger RL, Andrisani OM (1999) Differential immediate early gene expression in conditional hepatitis B virus pX-transforming versus nontransforming hepatocyte cell lines. J Biol Chem 274(4):2327–2336
Wu JC, Merlino G, Fausto N (1994) Establishment and characterization of differentiated, nontransformed hepatocyte cell lines derived from mice transgenic for transforming growth factor alpha. Proc Natl Acad Sci U S A 91(2):674–678
Jung JK, Arora P, Pagano JS, Jang KL (2007) Expression of DNA methyltransferase 1 is activated by hepatitis B virus X protein via a regulatory circuit involving the p16INK4a-cyclin D1-CDK 4/6-pRb-E2F1 pathway. Cancer Res 67(12):5771–5778
Park SG, Chung C, Kang H, Kim JY, Jung G (2006) Up-regulation of cyclin D1 by HBx is mediated by NF-kappaB2/BCL3 complex through kappaB site of cyclin D1 promoter. J Biol Chem 281(42):31770–31777
Zhu R, Li BZ, Li H, Ling YQ, Hu XQ, Zhai WR, Zhu HG (2007) Association of p16INK4A hypermethylation with hepatitis B virus X protein expression in the early stage of HBV-associated hepatocarcinogenesis. Pathol Int 57(6):328–336
Zhu YZ, Zhu R, Fan J, Pan Q, Li H, Chen Q, Zhu HG (2010) Hepatitis B virus X protein induces hypermethylation of p16(INK4A) promoter via DNA methyltransferases in the early stage of HBV-associated hepatocarcinogenesis. J Viral Hepat 17(2):98–107
Ahn JY, Jung EY, Kwun HJ, Lee CW, Sung YC, Jang KL (2002) Dual effects of hepatitis B virus X protein on the regulation of cell-cycle control depending on the status of cellular p53. J Gen Virol 83(Pt 11):2765–2772
Park US, Park SK, Lee YI, Park JG, Lee YI (2000) Hepatitis B virus-X protein upregulates the expression of p21waf1/cip1 and prolongs G1 → S transition via a p53-independent pathway in human hepatoma cells. Oncogene 19(30):3384–3394
Leach JK, Qiao L, Fang Y, Han SL, Gilfor D, Fisher PB, Grant S, Hylemon PB, Peterson D, Dent P (2003) Regulation of p21 and p27 expression by the hepatitis B virus X protein and the alternate initiation site X proteins, AUG2 and AUG3. J Gastroenterol Hepatol 18(4):376–385
Qiao L, Leach K, McKinstry R, Gilfor D, Yacoub A, Park JS, Grant S, Hylemon PB, Fisher PB, Dent P (2001) Hepatitis B virus X protein increases expression of p21(Cip-1/WAF1/MDA6) and p27(Kip-1) in primary mouse hepatocytes, leading to reduced cell cycle progression. Hepatology 34(5):906–917
Gearhart TL, Bouchard MJ (2011) The hepatitis B virus HBx protein modulates cell cycle regulatory proteins in cultured primary human hepatocytes. Virus Res 155(1):363–367
Gearhart TL, Bouchard MJ (2010) Replication of the hepatitis B virus requires a calcium-dependent HBx-induced G1 phase arrest of hepatocytes. Virology 407(1):14–25
Yamashita M, Emerman M (2006) Retroviral infection of non-dividing cells: old and new perspectives. Virology 344(1):88–93
Ford HL, Parade AB (1999) Cancer and the cell cycle. J Cell Biochem 32–33(suppl): 166–172
Tralhao JG, Roudier J, Morosan S, Giannini C, Tu H, Goulenok C, Carnot F, Zavala F, Joulin V, Kremsdorf D, Brechot C (2002) Paracrine in vivo inhibitory effects of hepatitis B virus X protein (HBx) on liver cell proliferation: an alternative mechanism of HBx-related pathogenesis. Proc Natl Acad Sci U S A 99(10):6991–6996
Wu BK, Li CC, Chen HJ, Chang JL, Jeng KS, Chou CK, Hsu MT, Tsai TF (2006) Blocking of G1/S transition and cell death in the regenerating liver of Hepatitis B virus X protein transgenic mice. Biochem Biophys Res Commun 340(3):916–928
Hodgson AJ, Keasler VV, Slagle BL (2008) Premature cell cycle entry induced by hepatitis B virus regulatory HBx protein during compensatory liver regeneration. Cancer Res 68(24):10341–10348
Quetier I, Brezillon N, Duriez M, Massinet H, Giang E, Ahodantin J, Lamant C, Brunelle MN, Soussan P, Kremsdorf D (2013) Hepatitis B virus HBx protein impairs liver regeneration through enhanced expression of IL-6 in transgenic mice. J Hepatol 59(2):285–291
Mahieux R, Gessain A (2007) Adult T-cell leukemia/lymphoma and HTLV-1. Curr Hematol Malig Rep 2(4):257–264
Jadoul M, Poignet JL, Geddes C, Locatelli F, Medin C, Krajewska M, Barril G, Scheuermann E, Sonkodi S, Goubau P, HCV Collaborative Group (2004) The changing epidemiology of hepatitis C virus (HCV) infection in haemodialysis: European multicentre study. Nephrol Dial Transplant 19(4):904–909
Matsuoka M, Jeang KT (2007) Human T-cell leukaemia virus type 1 (HTLV-1) infectivity and cellular transformation. Nat Rev Cancer 7(4):270–280
Hinuma Y, Nagata K, Hanaoka M, Nakai M, Matsumoto T, Kinoshita KI, Shirakawa S, Miyoshi I (1981) Adult T-cell leukemia: antigen in an ATL cell line and detection of antibodies to the antigen in human sera. Proc Natl Acad Sci U S A 78(10):6476–6480
Yoshida M, Seiki M, Yamaguchi K, Takatsuki K (1984) Monoclonal integration of human T-cell leukemia provirus in all primary tumors of adult T-cell leukemia suggests causative role of human T-cell leukemia virus in the disease. Proc Natl Acad Sci U S A 81(8):2534–2537
Currer R, Van Duyne R, Jaworski E, Guendel I, Sampey G, Das R, Narayanan A, Kashanchi F (2012) HTLV tax: a fascinating multifunctional co-regulator of viral and cellular pathways. Front Microbiol 3:406
Goncalves DU, Proietti FA, Ribas JG, Araujo MG, Pinheiro SR, Guedes AC, Carneiro-Proietti AB (2010) Epidemiology, treatment, and prevention of human T-cell leukemia virus type 1-associated diseases. Clin Microbiol Rev 23(3):577–589
Franchini G, Nicot C, Johnson JM (2003) Seizing of T cells by human T-cell leukemia/lymphoma virus type 1. Adv Cancer Res 89:69–132
Marriot SJ, Semmes OJ (2005) Impact of HTLV-1 Tax on cell cycle progression and the cellular DNA damage repair response. Oncogene 24(39):5986–5995
Lemoine FJ, Marriott SJ (2001) Accelerated G(1) phase progression induced by the human T cell leukemia virus type I (HTLV-I) Tax oncoprotein. J Biol Chem 276(34):31851–31857
Ohtani K, Iwanaga R, Arai M, Huang Y, Matsumura Y, Nakamura M (2000) Cell type-specific E2F activation and cell cycle progression induced by the oncogene product Tax of human T-cell leukemia virus type I. J Biol Chem 275(15):11154–11163
Haller K, Wu Y, Derow E, Schmitt I, Jeang KT, Grassmann R (2002) Physical interaction of human T-cell leukemia virus type 1 Tax with cyclin-dependent kinase 4 stimulates the phosphorylation of retinoblastoma protein. Mol Cell Biol 22(10):3327–3338
Kehn K, Fuente Cde L, Strouss K, Berro R, Jiang H, Brady J, Mahieux R, Pumfery A, Bottazzi ME, Kashanchi F (2005) The HTLV-I Tax oncoprotein targets the retinoblastoma protein for proteasomal degradation. Oncogene 24(4):525–540
Iwanaga R, Ohtani K, Hayashi T, Nakamura M (2001) Molecular mechanism of cell cycle progression induced by the oncogene product Tax of human T-cell leukemia virus type I. Oncogene 20(17):2055–2067
Suzuki T, Narita T, Uchida-Toita M, Yoshida M (1999) Down-regulation of the INK4 family of cyclin-dependent kinase inhibitors by tax protein of HTLV-1 through two distinct mechanisms. Virology 259(2):384–391
Low KG, Dorner LF, Fernando DB, Grossman J, Jeang KT, Comb MJ (1997) Human T-cell leukemia virus type 1 Tax releases cell cycle arrest induced by p16INK4a. J Virol 71(3):1956–1962
Mulloy JC, Kislyakova T, Cereseto A, Casareto L, LoMonico A, Fullen J, Lorenzi MV, Cara A, Nicot C, Giam C, Franchini G (1998) Human T-cell lymphotropic/leukemia virus type 1 Tax abrogates p53-induced cell cycle arrest and apoptosis through its CREB/ATF functional domain. J Virol 72(11):8852–8860
Pise-Masison CA, Choi KS, Radonovich M, Dittmer J, Kim SJ, Brady JN (1998) Inhibition of p53 transactivation function by the human T-cell lymphotropic virus type 1 Tax protein. J Virol 72(2):1165–1170
Liu M, Yang L, Zhang L, Liu B, Merling R, Xia Z, Giam CZ (2008) Human T-cell leukemia virus type 1 infection leads to arrest in the G1 phase of the cell cycle. J Virol 82(17):8442–8455
Baydoun HH, Pancewicz J, Bai X, Nicot C (2010) HTLV-I p30 inhibits multiple S phase entry checkpoints, decreases cyclin E-CDK2 interactions and delays cell cycle progression. Mol Cancer 9:302
Bellon M, Baydoun HH, Yao Y, Nicot C (2010) HTLV-I Tax-dependent and -independent events associated with immortalization of human primary T lymphocytes. Blood 115(12):2441–2448
Nicot C, Dundr M, Johnson JM, Fullen JR, Alonzo N, Fukumoto R, Princler GL, Derse D, Misteli T, Franchini G (2004) HTLV-1-encoded p30II is a post-transcriptional negative regulator of viral replication. Nat Med 10(2):197–201
Bai XT, Baydoun HH, Nicot C (2010) HTLV-I p30: a versatile protein modulating virus replication and pathogenesis. Mol Aspects Med 31(5):344–349
Taylor JM, Brown M, Nejmeddine M, Kim KJ, Ratner L, Lairmore M, Nicot C (2009) Novel role for interleukin-2 receptor-Jak signaling in retrovirus transmission. J Virol 83(22):11467–11476
Fukumoto R, Dundr M, Nicot C, Adams A, Valeri VW, Samelson LE, Franchini G (2007) Inhibition of T-cell receptor signal transduction and viral expression by the linker for activation of T cells-interacting p12(I) protein of human T-cell leukemia/lymphoma virus type 1. J Virol 81(17):9088–9099
Albrecht B, Lairmore MD (2002) Critical role of human T-lymphotropic virus type 1 accessory proteins in viral replication and pathogenesis. Microbiol Mol Biol Rev 66(3):396–406, table of contents
Johnson JM, Nicot C, Fullen J, Ciminale V, Casareto L, Mulloy JC, Jacobson S, Franchini G (2001) Free major histocompatibility complex class I heavy chain is preferentially targeted for degradation by human T-cell leukemia/lymphotropic virus type 1 p12(I) protein. J Virol 75(13):6086–6094
Fukumoto R, Andresen V, Bialuk I, Cecchinato V, Walser JC, Valeri VW, Nauroth JM, Gessain A, Nicot C, Franchini G (2009) In vivo genetic mutations define predominant functions of the human T-cell leukemia/lymphoma virus p12I protein. Blood 113(16):3726–3734
Silic-Benussi M, Cavallari I, Zorzan T, Rossi E, Hiraragi H, Rosato A, Horie K, Saggioro D, Lairmore MD, Willems L, Chieco-Bianchi L, D’Agostino DM, Ciminale V (2004) Suppression of tumor growth and cell proliferation by p13II, a mitochondrial protein of human T cell leukemia virus type 1. Proc Natl Acad Sci U S A 101(17):6629–6634
Kim SJ, Ding W, Albrecht B, Green PL, Lairmore MD (2003) A conserved calcineurin-binding motif in human T lymphotropic virus type 1 p12I functions to modulate nuclear factor of activated T cell activation. J Biol Chem 278(18):15550–15557
Darbinyan A, Darbinian N, Safak M, Radhakrishnan S, Giordano A, Khalili K (2002) Evidence for dysregulation of cell cycle by human polyomavirus, JCV, late auxiliary protein. Oncogene 21(36):5574–5581
Nascimento R, Parkhouse RM (2007) Murine gammaherpesvirus 68 ORF20 induces cell-cycle arrest in G2 by inhibiting the Cdc2-cyclin B complex. J Gen Virol 88(Pt 5):1446–1453
Li H, Baskaran R, Krisky DM, Bein K, Grandi P, Cohen JB, Glorioso JC (2008) Chk2 is required for HSV-1 ICP0-mediated G2/M arrest and enhancement of virus growth. Virology 375(1):13–23
Knight GL, Turnell AS, Roberts S (2006) Role for Wee1 in inhibition of G2-to-M transition through the cooperation of distinct human papillomavirus type 1 E4 proteins. J Virol 80(15):7416–7426
Poggioli GJ, Dermody TS, Tyler KL (2001) Reovirus-induced sigma1s-dependent G(2)/M phase cell cycle arrest is associated with inhibition of p34(cdc2). J Virol 75(16):7429–7434
Patton JT (2008) Segmented double-stranded RNA viruses: structural and molecular biology. Caister Academic Press, Bristol, UK
Li L, Gu B, Zhou F, Chi J, Wang F, Peng G, Xie F, Qing J, Feng D, Lu S, Yao K (2011) Human herpesvirus 6 suppresses T cell proliferation through induction of cell cycle arrest in infected cells in the G2/M phase. J Virol 85(13):6774–6783
Scarano FJ, Laffin JA, Lehman JM, Friedrich TD (1994) Simian virus 40 prevents activation of M-phase-promoting factor during lytic infection. J Virol 68(4):2355–2361
Yeo KS, Mohidin TB, Ng CC (2012) Epstein–Barr virus-encoded latent membrane protein-1 upregulates 14-3-3sigma and Reprimo to confer G(2)/M phase cell cycle arrest. C R Biol 335(12):713–721
Ohki R, Nemoto J, Murasawa H, Oda E, Inazawa J, Tanaka N, Taniguchi T (2000) Reprimo, a new candidate mediator of the p53-mediated cell cycle arrest at the G2 phase. J Biol Chem 275(30):22627–22630
Mhawech P (2005) 14-3-3 proteins—an update. Cell Res 15(4):228–236
Laronga C, Yang HY, Neal C, Lee MH (2000) Association of the cyclin-dependent kinases and 14-3-3 sigma negatively regulates cell cycle progression. J Biol Chem 275(30):23106–23112
Davy CE, Jackson DJ, Raj K, Peh WL, Southern SA, Das P, Sorathia R, Laskey P, Middleton K, Nakahara T, Wang Q, Masterson PJ, Lambert PF, Cuthill S, Millar JB, Doorbar J (2005) Human papillomavirus type 16 E1 E4-induced G2 arrest is associated with cytoplasmic retention of active Cdk1/cyclin B1 complexes. J Virol 79(7):3998–4011
Morita E, Tada K, Chisaka H, Asao H, Sato H, Yaegashi N, Sugamura K (2001) Human parvovirus B19 induces cell cycle arrest at G(2) phase with accumulation of mitotic cyclins. J Virol 75(16):7555–7563
Fehr AR, Yu D (2013) Control the host cell cycle: viral regulation of the anaphase-promoting complex. J Virol 87(16):8818–8825
Teodoro JG, Heilman DW, Parker AE, Green MR (2004) The viral protein Apoptin associates with the anaphase-promoting complex to induce G2/M arrest and apoptosis in the absence of p53. Genes Dev 18(16):1952–1957
Bellanger S, Blachon S, Mechali F, Bonne-Andrea C, Thierry F (2005) High-risk but not low-risk HPV E2 proteins bind to the APC activators Cdh1 and Cdc20 and cause genomic instability. Cell Cycle 4(11):1608–1615
Tran K, Kamil JP, Coen DM, Spector DH (2010) Inactivation and disassembly of the anaphase-promoting complex during human cytomegalovirus infection is associated with degradation of the APC5 and APC4 subunits and does not require UL97-mediated phosphorylation of Cdh1. J Virol 84(20):10832–10843
Fehr AR, Gualberto NC, Savaryn JP, Terhune SS, Yu D (2012) Proteasome-dependent disruption of the E3 ubiquitin ligase anaphase-promoting complex by HCMV protein pUL21a. PLoS Pathog 8(7):e1002789
Everett RD, Earnshaw WC, Findlay J, Lomonte P (1999) Specific destruction of kinetochore protein CENP-C and disruption of cell division by herpes simplex virus immediate-early protein Vmw110. EMBO J 18(6):1526–1538
Lomonte P, Sullivan KF, Everett RD (2001) Degradation of nucleosome-associated centromeric histone H3-like protein CENP-A induced by herpes simplex virus type 1 protein ICP0. J Biol Chem 276(8):5829–5835
Belyavskyi M, Braunagel SC, Summers MD (1998) The structural protein ODV-EC27 of Autographa californica nucleopolyhedrovirus is a multifunctional viral cyclin. Proc Natl Acad Sci U S A 95(19):11205–11210
Ikeda M, Kobayashi M (1999) Cell-cycle perturbation in Sf9 cells infected with Autographa californica nucleopolyhedrovirus. Virology 258(1):176–188
Freed EO, Martin MA (2013) Human immunodeficiency viruses: replication. In: Knipe DM, Howley PM (eds) Fields virology, 6th edn. Lippincot Williams and Wilkins, Philadelphia, PA, pp 1502–1560
Gilbert PB, McKeague IW, Eisen G, Mullins C, Gueye NA, Mboup S, Kanki PJ (2003) Comparison of HIV-1 and HIV-2 infectivity from a prospective cohort study in Senegal. Stat Med 22(4):573–593
Sharp PM, Hahn BH (2011) Origins of HIV and the AIDS pandemic. Cold Spring Harb Perspect Med 1(1):a006841
Zhao RY, Li G, Bukrinsky MI (2011) Vpr-host interactions during HIV-1 viral life cycle. J Neuroimmune Pharmacol 6(2):216–229
Goh WC, Rogel ME, Kinsey CM, Michael SF, Fultz PN, Nowak MA, Hahn BH, Emerman M (1998) HIV-1 Vpr increases viral expression by manipulation of the cell cycle: a mechanism for selection of Vpr in vivo. Nat Med 4(1):65–71
Kogan M, Rappaport J (2011) HIV-1 accessory protein Vpr: relevance in the pathogenesis of HIV and potential for therapeutic intervention. Retrovirology 8:25
Zhao RY, Elder RT (2005) Viral infections and cell cycle G2/M regulation. Cell Res 15(3):143–149
Re F, Braaten D, Franke EK, Luban J (1995) Human immunodeficiency virus type 1 Vpr arrests the cell cycle in G2 by inhibiting the activation of p34cdc2-cyclin B. J Virol 69(11):6859–6864
He J, Choe S, Walker R, Di Marzio P, Morgan DO, Landau NR (1995) Human immunodeficiency virus type 1 viral protein R (Vpr) arrests cells in the G2 phase of the cell cycle by inhibiting p34cdc2 activity. J Virol 69(11):6705–6711
Goh WC, Manel N, Emerman M (2004) The human immunodeficiency virus Vpr protein binds Cdc25C: implications for G2 arrest. Virology 318(1):337–349
Kamata M, Watanabe N, Nagaoka Y, Chen IS (2008) Human immunodeficiency virus type 1 Vpr binds to the N lobe of the Wee1 kinase domain and enhances kinase activity for CDC2. J Virol 82(12):5672–5682
Yuan H, Kamata M, Xie YM, Chen IS (2004) Increased levels of Wee-1 kinase in G(2) are necessary for Vpr- and gamma irradiation-induced G(2) arrest. J Virol 78(15):8183–8190
Elder RT, Yu M, Chen M, Zhu X, Yanagida M, Zhao Y (2001) HIV-1 Vpr induces cell cycle G2 arrest in fission yeast (Schizosaccharomyces pombe) through a pathway involving regulatory and catalytic subunits of PP2A and acting on both Wee1 and Cdc25. Virology 287(2):359–370
Bartz SR, Rogel ME, Emerman M (1996) Human immunodeficiency virus type 1 cell cycle control: Vpr is cytostatic and mediates G2 accumulation by a mechanism which differs from DNA damage checkpoint control. J Virol 70(4):2324–2331
DeHart JL, Zimmerman ES, Ardon O, Monteiro-Filho CM, Arganaraz ER, Planelles V (2007) HIV-1 Vpr activates the G2 checkpoint through manipulation of the ubiquitin proteasome system. Virol J 4:57
Belzile JP, Duisit G, Rougeau N, Mercier J, Finzi A, Cohen EA (2007) HIV-1 Vpr-mediated G2 arrest involves the DDB1-CUL4AVPRBP E3 ubiquitin ligase. PLoS Pathog 3(7):e85
Schrofelbauer B, Hakata Y, Landau NR (2007) HIV-1 Vpr function is mediated by interaction with the damage-specific DNA-binding protein DDB1. Proc Natl Acad Sci U S A 104(10):4130–4135
Tan L, Ehrlich E, Yu XF (2007) DDB1 and Cul4A are required for human immunodeficiency virus type 1 Vpr-induced G2 arrest. J Virol 81(19):10822–10830
Mansky LM (1996) The mutation rate of human immunodeficiency virus type 1 is influenced by the vpr gene. Virology 222(2):391–400
Roshal M, Kim B, Zhu Y, Nghiem P, Planelles V (2003) Activation of the ATR-mediated DNA damage response by the HIV-1 viral protein R. J Biol Chem 278(28):25879–25886
Li G, Park HU, Liang D, Zhao RY (2010) Cell cycle G2/M arrest through an S phase-dependent mechanism by HIV-1 viral protein R. Retrovirology 7:59
Li G, Elder RT, Qin K, Park HU, Liang D, Zhao RY (2007) Phosphatase type 2A-dependent and -independent pathways for ATR phosphorylation of Chk1. J Biol Chem 282(10):7287–7298
Zimmerman ES, Chen J, Andersen JL, Ardon O, Dehart JL, Blackett J, Choudhary SK, Camerini D, Nghiem P, Planelles V (2004) Human immunodeficiency virus type 1 Vpr-mediated G2 arrest requires Rad17 and Hus1 and induces nuclear BRCA1 and gamma-H2AX focus formation. Mol Cell Biol 24(21):9286–9294
Zhao Y, Cao J, O’Gorman MR, Yu M, Yogev R (1996) Effect of human immunodeficiency virus type 1 protein R (vpr) gene expression on basic cellular function of fission yeast Schizosaccharomyces pombe. J Virol 70(9):5821–5826
Fu DX, Kuo YL, Liu BY, Jeang KT, Giam CZ (2003) Human T-lymphotropic virus type I tax activates I-kappa B kinase by inhibiting I-kappa B kinase-associated serine/threonine protein phosphatase 2A. J Biol Chem 278(3):1487–1493
Roopchand DE, Lee JM, Shahinian S, Paquette D, Bussey H, Branton PE (2001) Toxicity of human adenovirus E4orf4 protein in Saccharomyces cerevisiae results from interactions with the Cdc55 regulatory B subunit of PP2A. Oncogene 20(38):5279–5290
Shtrichman R, Sharf R, Barr H, Dobner T, Kleinberger T (1999) Induction of apoptosis by adenovirus E4orf4 protein is specific to transformed cells and requires an interaction with protein phosphatase 2A. Proc Natl Acad Sci U S A 96(18):10080–10085
Haoudi A, Daniels RC, Wong E, Kupfer G, Semmes OJ (2003) Human T-cell leukemia virus-I tax oncoprotein functionally targets a subnuclear complex involved in cellular DNA damage-response. J Biol Chem 278(39):37736–37744
Poon B, Grovit-Ferbas K, Stewart SA, Chen IS (1998) Cell cycle arrest by Vpr in HIV-1 virions and insensitivity to antiretroviral agents. Science 281(5374):266–269
Venkatesan A, Sharma R, Dasgupta A (2003) Cell cycle regulation of hepatitis C and encephalomyocarditis virus internal ribosome entry site-mediated translation in human embryonic kidney 293 cells. Virus Res 94(2):85–95
Brasey A, Lopez-Lastra M, Ohlmann T, Beerens N, Berkhout B, Darlix JL, Sonenberg N (2003) The leader of human immunodeficiency virus type 1 genomic RNA harbors an internal ribosome entry segment that is active during the G2/M phase of the cell cycle. J Virol 77(7):3939–3949
Lin GY, Lamb RA (2000) The paramyxovirus simian virus 5 V protein slows progression of the cell cycle. J Virol 74(19):9152–9166
Alonso-Caplen FV, Matsuoka Y, Wilcox GE, Compans RW (1984) Replication and morphogenesis of avian coronavirus in Vero cells and their inhibition by monensin. Virus Res 1(2):153–167
Lim KP, Liu DX (2001) The missing link in coronavirus assembly. Retention of the avian coronavirus infectious bronchitis virus envelope protein in the pre-Golgi compartments and physical interaction between the envelope and membrane proteins. J Biol Chem 276(20):17515–17523
Lontok E, Corse E, Machamer CE (2004) Intracellular targeting signals contribute to localization of coronavirus spike proteins near the virus assembly site. J Virol 78(11):5913–5922
Machamer CE, Mentone SA, Rose JK, Farquhar MG (1990) The E1 glycoprotein of an avian coronavirus is targeted to the cis Golgi complex. Proc Natl Acad Sci U S A 87(18):6944–6948
Park HU, Jeong JH, Chung JH, Brady JN (2004) Human T-cell leukemia virus type 1 Tax interacts with Chk1 and attenuates DNA-damage induced G2 arrest mediated by Chk1. Oncogene 23(29):4966–4974
Krauer KG, Burgess A, Buck M, Flanagan J, Sculley TB, Gabrielli B (2004) The EBNA-3 gene family proteins disrupt the G2/M checkpoint. Oncogene 23(7):1342–1353
Choudhuri T, Verma SC, Lan K, Murakami M, Robertson ES (2007) The ATM/ATR signaling effector Chk2 is targeted by Epstein–Barr virus nuclear antigen 3C to release the G2/M cell cycle block. J Virol 81(12):6718–6730
Liu B, Hong S, Tang Z, Yu H, Giam CZ (2005) HTLV-I Tax directly binds the Cdc20-associated anaphase-promoting complex and activates it ahead of schedule. Proc Natl Acad Sci U S A 102(1):63–68
Kim S, Park SY, Yong H, Famulski JK, Chae S, Lee JH, Kang CM, Saya H, Chan GK, Cho H (2008) HBV X protein targets hBubR1, which induces dysregulation of the mitotic checkpoint. Oncogene 27(24):3457–3464
Chabes AL, Pfleger CM, Kirschner MW, Thelander L (2003) Mouse ribonucleotide reductase R2 protein: a new target for anaphase-promoting complex-Cdh1-mediated proteolysis. Proc Natl Acad Sci U S A 100(7):3925–3929
Wiebusch L, Bach M, Uecker R, Hagemeier C (2005) Human cytomegalovirus inactivates the G0/G1-APC/C ubiquitin ligase by Cdh1 dissociation. Cell Cycle 4(10):1435–1439
Bornholdt ZA, Noda T, Abelson DM, Halfmann P, Wood MR, Kawaoka Y, Saphire EO (2013) Structural rearrangement of Ebola Virus VP40 begets multiple functions in the virus life cycle. Cell 154(4):763–774
Ferreon AC, Ferreon JC, Wright PE, Deniz AA (2013) Modulation of allostery by protein intrinsic disorder. Nature 498(7454):390–394
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2014 Springer Science+Business Media New York
About this protocol
Cite this protocol
Bagga, S., Bouchard, M.J. (2014). Cell Cycle Regulation During Viral Infection. In: Noguchi, E., Gadaleta, M. (eds) Cell Cycle Control. Methods in Molecular Biology, vol 1170. Humana Press, New York, NY. https://doi.org/10.1007/978-1-4939-0888-2_10
Download citation
DOI: https://doi.org/10.1007/978-1-4939-0888-2_10
Published:
Publisher Name: Humana Press, New York, NY
Print ISBN: 978-1-4939-0887-5
Online ISBN: 978-1-4939-0888-2
eBook Packages: Springer Protocols