
Abstract
Hepatitis B virus causes chronic infection in about 300 million of people worldwide and results in about one million deaths a year, because of virus-induced end-stage liver disease and liver cancer. Mechanisms from acute hepatitis B virus (HBV) infection progress into chronicity, repeated bouts of inflammations were reviewed, with emphasis on the unusual modest innate immune responses against the virus. Chronic inflammation paves the way for malignant transformation of the infected hepatocytes. Recent studies in human and animal liver cancer models identify major genetic alteration pathways leading to carcinogenesis. A better understanding of HBV persistence and carcinogenesis will provide new insights for more effective interventions to control chronic hepatitis B and its sequelae in the future.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others


Keywords
These keywords were added by machine and not by the authors. This process is experimental and the keywords may be updated as the learning algorithm improves.
Virus Structure and Genetics
Hepatitis B virus (HBV) is the prototypic member of the family Hepadnaviridae, which includes a group of hepatotropic viruses with different species tropism, sharing remarkable similarities in the genome organization and the unique replication strategy through reverse transcription of an RNA pregenome [1]. Based on the sequence similarity and host ranges, the Hepadnavirus family is divided into the genera orthohepadnavirus of mammals and avihepadnavirus of birds. Human HBV can be further grouped into eight genotypes, which are based on the more than 8 % difference of genome sequence [2].
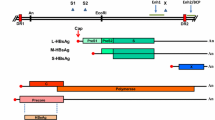
HBV is a small enveloped DNA virus with the genome size of 3.2 kb. The virion of HBV, Dane particle, is approximately 42 nm in diameter. It is composed of an internal icosahedral nucleocapsid containing a DNA genome, and an outer lipid bilayer membrane made of three forms of envelope proteins, including large (L), middle (M) and small (S) proteins. The S protein is abundant in HBV virions and in two subviral spheres and filamentous structures lacking viral nucleic acids. The latter two forms outnumber HBV virions in around 100–10,000-fold excess and are the major sources of HBsAg in the blood. The excessive HBsAg has been suggested to counteract the host neutralizing antibody against HBV infection. The HBV nucleocapsid is assembled by the dimers of core protein (HBcAg). HBcAg consists of the N-terminal self-assembly domain and C-terminal arginine-rich domain, which harbors three phosphorylation sites critical for packaging pregenome/polymerase complex and DNA synthesis [3].
The genome organization of HBV is complex, featured by the circular partially double-stranded DNA, also termed relaxed-circular DNA (rcDNA), and overlapping open reading frames (ORFs). All the ORFs are in the same direction, defining the minus and plus strands of DNA. The minus strand of rcDNA spans the entire length of 3.2 kb genome with a redundant 5′-flap attached with a viral polymerase. An RNA primer is attached at the 5′ end of the plus strand DNA. The 3′ extension of the plus strand is incomplete and only approximately two thirds of the genome, leaving an open gap with variable length between its 5′ and 3′ ends.
HBV contains four ORFs encoding core/precore, polymerase, envelope, and X proteins. Upon initiation of the viral replication cycle, HBV rcDNA needs to be converted into covalently closed circular DNA (cccDNA) in nuclei, serving as a transcriptional template of viral RNAs. Under a physiological condition, cccDNA is most likely associated with histones and other proteins to form a viral minichromosome [4, 5]. Four unidirectional RNAs, precore/pregenomic, preS1, preS2/S, and X, with 3.5, 2.4, 2.1, and 0.7 kb in length, respectively, are transcribed under the regulation of four distinct promoters and two enhancers. Besides, epigenetic regulation, including histone modification and DNA methylation, has been suggested to play a role in the control of viral transcription [6–8]. All the four viral transcripts terminate at the same polyadenylation signal, so they possess an identical 3′ end sequence but with variable 5′ transcriptional start sites. There exist two different 3.5 kb greater-than-genome transcripts, precore and pregenomic transcripts, which are generated through the heterogeneous transcription start sites [9]. The precore protein is translated from the precore mRNA, beginning upstream of the start codon of the precore gene, and is further processed to generate secretory hepatitis B e antigen (HBeAg), which may have immunoregulatory functions [10]. The pregenomic RNA, starting downstream of the start codon of the precore gene, is not only the template for viral replication through the reverse transcription [11], but also the mRNA for translation of both core and polymerase proteins.
The three envelope proteins, L, M, and S proteins, are translated from their own start sites in preS1 (L protein) and preS2/S (M and S proteins) mRNAs, respectively, and share the same C-terminal amino acids, called the S domain. As a consequence, compared to the S protein, the L protein contains extra preS1 and preS2 domains, and the M protein contains an extra preS2 domain. The remaining 0.7 kb transcript is responsible for the translation of multifunctional X protein, which interacts with various cellular factors to regulate host and viral transcription, cell-cycle progression, DNA-damage repair, and apoptosis [12, 13]. There are a few spliced viral RNAs whose functions remain elusive.
Cellular Targets of Infection
HBV is a hepatotropic virus. In the liver, hepatocytes are the main cell type, comprising 60–70 % of the liver cell mass. Other cells include bile ductal epithelial cells, stellate cells (Ito cells), sinusoidal endothelial cells, and Kupffer cells. Both hepatocytes and bile ductal epithelial cells originate from the common progenitor cells that can differentiate to hepatocytes and ductal epithelial cells. Among these cells, hepatocytes are the primary targets of HBV infection [14]. The hepatic tropism of HBV is probably due to the limited distribution of the receptor sodium taurocholate cotransporting polypeptide (NTCP) on hepatocytes and hepatocyte-enriched transcription factors.
Previous studies investigated ducks infected by duck hepatitis B virus (DHBV) have demonstrated that DHBV could infect and replicate in cells other than hepatocytes. The replicative intermediates and cccDNA can be found not only in hepatocytes, but also in bile ductal epithelial cells, acinar cells of the pancreas, proximal tubular epithelial cells of the kidney, and the spleen in chronically infected ducks [15–17]. Similarly, several human studies reported that, in addition to hepatocytes, HBV can replicate in bile duct epithelial cells, and in the extrahepatic sites, including pancreas [18], spleen [19], peripheral blood mononuclear cells (PBMCs) [20–22], and bone marrow cells [23]. Of note, these studies could not unambiguously demonstrate that the replicative intermediates and cccDNA of HBV existed in these cells; hence it remains unclear whether HBV is just passively absorbed [24] or indeed actively replicates in these non-hepatocyte cells.
So far, hepatocytes are the only cells that are definitely shown to replicate HBV. Hepatocyte regeneration occurs in response to killing of the infected hepatocytes by antiviral cytotoxic T cells. Hepatocyte replacement primarily results from division of the remaining hepatocytes, but in chronic HBV infection, progenitor cells may also contribute to hepatocyte replacement by proliferation and differentiation into hepatocytes [14]. During liver injury, stellate cells respond by producing collagen fibers [25]. In the persistent liver injury due to chronic viral infection, continuous deposition of collagen fibers from stellate cells leads to hepatic fibrosis, and eventually evolves to cirrhosis.
Viral Latency and Replication
Entry of HBV into the infected hepatocytes is generally believed to undergo the receptor-mediated endocytosis [26]. After entry, the naked nucleocapsid is released into the cytoplasm and the viral genome is translocated to the nuclei. The intracytosolic transportation of viral genome is facilitated by the viral capsid interacting with cellular microtubules. The subsequent translocation through the nuclear pore complex is mediated by the nuclear transport machinery importin-α and importin-β [27, 28]. In the nuclei of the infected hepatocytes, rcDNA is converted into cccDNA.
The conversion from rcDNA to cccDNA is complicated. Comparison of the structure difference between rcDNA and cccDNA revealed various critical steps required for the cccDNA formation, including the fill-in of the 3′-gap in plus, removal of the viral polymerase covalently linked to the 5′ end of minus strand, removal of the redundant flap at the 5′ end of minus strand, removal of the RNA primer at the 5′ end of plus strand, and ligation of the resulting nicked plus and minus strands. Both viral polymerase and DNA repair machinery may contribute to this process [29, 30]. However, the detailed mechanisms and the host factors involved in this process remain unclear. The study of HBV cccDNA is also hampered by the fact that HBV cannot efficiently form cccDNA in vitro, particularly in cell lines. Therefore, most of the information regarding the HBV replication comes from the studies on the replication of its kinship DHBV, which is a good model system to investigate the cccDNA formation because of its high efficiency in formation of cccDNA in both human and avian cell lines [29].
HBV cccDNA is the template for viral transcription. Aforementioned, the 3.5 kb greater-than-genome pgRNA not only serves as the mRNA for synthesis of core and polymerase proteins, but also is used as a template for reverse transcription. Although HBV shares the same replication strategy with the retrovirus, there exist several distinct features between HBV and retroviruses [1]. Retrovirus forms a double-stranded linear pre-integration complex and subsequently integrates to the host genome, whereas HBV generates rcDNA within the viral nucleocapsid in the cytoplasm of infected hepatocytes and forms episomal cccDNA in the nuclei. Mature nucleocapsids are then either directed to the secretory pathway in endoplasmic reticulum (ER) for envelopment or are redirected towards the nucleus to establish a cccDNA pool. The latter is a pathway termed intracellular recycling [29, 31].
HBV cccDNA exhibits remarkable stability. Even after years of recovery from acute HBV infection, persistent intrahepatic cccDNA can be still detected even in the presence of active antiviral immunity [32]. Additionally, HBV reactivation can be found in the patients with prior resolved hepatitis B receiving anti-lymphoma therapy [33], indicating the transcription-competent HBV DNA can hide in a latent reservoir of host even under the control of effective antiviral immunity for a very long time. This indicates that the existence of intrahepatic cccDNA is responsible for the persistence of replication-competent HBV in the liver. The persistent cccDNA also forms a major barrier to eradication of HBV by nucleos(t)ide analogues (NA). Although HBV replication can be effectively suppressed by NA treatment, HBV cccDNA can last very long under this condition because NAs only block the reverse transcription process, but fail to destroy existing cccDNA. Clearance of HBV cccDNA likely requires to eliminate all the infected hepatocytes by lysis of them, although some noncytolytic mechanisms may also contribute to the clearance of HBV cccDNA [34, 35]. Taken together, HBV cccDNA is responsible for viral latency and failure of NAs in eradicating HBV. Future therapeutic strategy aiming to cure chronic HBV should target intrahepatic cccDNA.
Immune Response
HBV is a non-cytopathic virus and the associated liver damage is thought to be immune-mediated [35, 36]. The outcome of acute HBV infection is the consequence of a complex interaction between virus and host immune responses. HBV infection in the majority of immunocompetent adults results in a self-limited and transient hepatitis, whereas in 5 % of the infected adults and more than 90 % of neonates with perinatal infection become chronically infected. Cellular immune response to HBV infection contributes to both viral clearance and liver injury [37]. In contrast to acute HBV infection which exhibits a robust and wide-spectrum HBV-specific T cell immunity, chronic HBV infection is characterized with an inefficient (oligoclonal) T cell response unable to clear HBV from the liver [35, 36, 38]. In a portion of patients with persistent HBV infection, the sustained weak T cell immunity causes recurrent liver damage without clearance of viruses, and eventually leads to cirrhosis and hepatocellular carcinoma (HCC) [35].
Control of viral infection often requires coordination of innate and adaptive immunity. The innate immune system detects the invasion of pathogens by recognition of the pathogen-associated molecular pattern (PAMP) through the pattern recognition receptors (PRRs). Three groups of PRRs have been identified so far, including the Toll-like receptor (TLR), RIG-I-like receptor (RLR) and NOD-like receptor (NLR). Appropriate activation of innate immunity is usually necessary for induction of robust adaptive immunity against the invading pathogens [39]. However, the role of innate immunity in acute HBV infection has been debated. HBV infection exhibits a long incubation period, around 1–6 months, before the appearance of symptoms. In the experimental HBV infection in chimpanzees and natural infection in humans, during the incubation period, HBV remains undetectable or at a very low level until it undergoes exponential growth, usually 8 weeks after infection [40, 41]. By analyzing the serial expression profiles of intrahepatic genes following acute HBV infection in experimental chimpanzees, the genes of conventional innate immunity could not be detected in early and expansion phase of acute HBV infection, suggesting the stealth nature of acute HBV infection [42]. Similarly, studies of the innate immune response in acutely HBV-infected patients revealed that type I interferon (IFN) could be barely detectable in the early phase of HBV infection [43]. Therefore, the innate immunity seems to be silent in the initial stage of HBV infection. Consistently, only mild or no symptoms are noticed in the early period of acute HBV infection. Of note, the stealth nature of acute HBV infection cannot be totally attributable to the tolerogenic liver microenvironment because the innate immune response could be readily detected following acute infection of HCV, which is another hepatotropic virus and often leads to persistent infection [44].
Nevertheless, this long-assumed notion that HBV is a stealth virus has been challenged recently. Using an in vitro HBV recombinant baculovirus infection system with HepG2 and HepaRG cells, it was shown that HBV infection could activate strong type I IFN response [45]. Besides, it was also shown that, upon infection of primary human liver cells, HBV is recognized by nonparenchymal cells, mainly Kupffer cells [46], although they are not infected. This recognition leads to release of IL-6, which subsequently suppresses HBV replication by inhibition of the activity of HNF-1 and HNF-4, two liver-enriched transcription factors critical for HBV transcription. Furthermore, it was found that the woodchuck hepatitis virus (WHV) infection activates production of proinflammatory cytokines, IFN-γ and IL-12, and both NK and NKT cell responses within a few hours after inoculation with a liver-pathogenic dose of virus, although the induction of adaptive immune response is delayed 4–5 weeks later [47]. Altogether, these studies suggest that innate immunity could be activated by HBV infection. Therefore, it raises the questions about how the host innate immune system recognizes HBV infection, whether or which PRRs are required for HBV recognition, and also how HBV attenuates the innate immune response. Interestingly, a recent study reported that the assembled nucleocapsid, but not free core antigen, controls HBV clearance in an immune-competent mouse model of HBV persistence, suggesting the potential role of assembled viral nucleocapsid in dictating the viral clearance [48]. Besides, it is found that NKT cells could sense HBV infection-induced alternations of self-lipid through CD1d and thus contribute to control of HBV infection [49]. Moreover, HBV virions and proteins, such as HBsAg and HBeAg, can suppress the response elicited by TLRs of parenchymal and nonparenchymal liver cells [50]. These data further support the importance of innate immunity in the control of HBV infection, may be independent of IFN-α, and suggest the sensing mechanisms of host for recognition of HBV infection.
In contrast to the controversial role of innate immunity in the control of HBV infection, adaptive immunity is well known to control viral clearance. Adaptive immunity can be strongly induced following HBV infection, because natural infection of HBV in adults can cause robust neutralizing antibody and T cell immunity. After the start of exponential increase following the initial incubation phase of HBV replication, HBV-specific CD4 and CD8 T cell responses are timely activated and become readily detectable [41, 51]. The important role of CD8 T cell immunity in the control of HBV infection has been documented [52]. It has been also observed in chimpanzees [40] and in humans [41] that maximal reduction in HBV levels occurs before significant destruction of infected hepatocytes. Additionally, the antiviral CD8 T cell response appears when HBV viral load starts to decline. Therefore, in addition to the cytolytic effects of cellular immunity, noncytolytic inhibition of HBV replication by innate and cellular immunity should also contribute to the viral clearance [53].
In chronic HBV infection, the immune response of patients fails to clear virus. Chronic HBV infection exhibits several defects of immune responses, which are currently under intensive investigation and are the potential targets for developing the therapeutic strategy to eradicate HBV infection. Analysis of the spectrum and magnitude of cellular immunity against HBV reveals the distinct profiles of T cell immunity between resolved and persistent HBV infection [38]. The reasons causing the defects of cellular immunity are multifactorial, probably due to the tolerogenic environment of the liver [54], the effects of regulatory T cells (Tregs) [55, 56], and the persistent exposure to a high viral load. These combined mechanisms can result in either deletion of HBV-specific cellular immunity or its functional exhaustion [57]. Exhausted T cells are characteristic by the upregulation of negative co-stimulatory molecules or dysregulation of the co-stimulatory pathway, like PD-1/PD-L1 [38, 58, 59].
Perinatal transmission is the primary route of HBV infection in Asia. As mentioned above, the infection of newborn results in chronic infection in more than 90 %. This probably results from the immaturity of the hepatic immune system, although the detailed mechanisms causing this phenomenon remain unclear [60]. Three distinct virological and immunological phases, including immune tolerance, immune clearance, and inactive residual phases, are often noted in chronic hepatitis B (CHB) patients, particularly in perinatally acquired subjects [61]. The immune tolerance phase is characterized with positivity of HBeAg, high viremia, and no or only minimal hepatic damage, indicating that infected hepatocytes are not attacked by the host immune system. The transition from the immune tolerance phase to clearance phase often occurs in patients aged between 20 and 40 years old. So far, the mechanisms triggering the loss of immune tolerance are also unknown. During the phase of immune clearance, most patients experience decline of viremia and fluctuation of hepatitis activity and finally enter the inactive residual phase, which is accompanied by loss of HBeAg, a low viral replication and the remission of hepatitis, leading to a better clinical outcome. Of note, although most patients with chronic HBV infection become inactive carriers, a portion of patients have the sustained weak T cell immune response that causes recurrent liver damage, and results in persistent hepatic necroinflammation, eventually leading to liver cirrhosis and HCC.
HBeAg seroconversion, defined as loss of HBeAg and appearance of anti-HBe antibody, is thus a virological landmark that usually signifies remission of both viral replication and hepatitis activity, and partial recovery of cellular immunity against HBV. Additionally, during HBeAg seroconversion, mutant strains that decrease or even abolish production of HBeAg are preferentially selected [61]. Despite its important role in the natural history of HBV infection, the biological function of HBeAg is still unclear because it is not required for HBV assembly, replication, or infection [10]. Clinical observation suggests that serum HBeAg may have an immunomodulatory function in immune tolerance phase, whereas cytosolic HBeAg may serve as a target for the inflammatory immune response in the immune clearance phase [10]. Although interesting, this speculation has not been unambiguously demonstrated by experiments.
In conclusion, the coordinated response of innate and adaptive immunity plays a role in the control of acute HBV infection. Cellular immunity against HBV infection contributes to both viral clearance and liver damage. Persistent necroinflammation caused by sustained weak T cell response in CHB patients often leads to liver cirrhosis and even HCC. Understanding the immunobiology and immunopathogenesis of HBV infection should facilitate the development of novel therapeutic strategies for CHB.
Viral Oncogenes and Cellular Transformation
The incidence of HCC ranks fifth for cancers worldwide and causes about half-a-million deaths every year [62]. More than two-thirds of HCC occur in Asia because of the high prevalence of chronic HBV infection, which is usually an ultimate outcome of chronic hepatitis caused by persistent HBV viral infection [63, 64]. Vaccination of newborns against HBV has been initiated worldwide and has effectively reduced persistent HBV infections from 15 % in the prevaccination era to 1 % in the postvaccination era [65]. However, vaccination cannot control infection in adults who are already chronically infected. There are about 350 million HBsAg carriers in the world [62].
Fortunately, orally administered NAs have been developed to control the progression of liver disease in CHB patients [66, 67]. Current NAs (Lamivudine, Adefovir Entecavir, Telbivudine, Tenofovir, etc.) inhibit viral replication by blocking the HBV reverse transcriptase activity. Such treatments can only block the viral replication and subsequent infections, but cannot eradicate the existing virus genome (cccDNA) and cannot reduce the viral transcription and protein translation. There are still abundant viral proteins with carcinogenic activities in the HBV-infected hepatocytes, which continuously contribute to the carcinogenic process in CHB patients despite receiving antiviral therapies.
HBV can encode oncogenic viral proteins that directly contribute to carcinogenesis, including HBx and deletion mutants of pre-S proteins [68, 69]. HBx is a nonstructural viral protein that has multifunctional regulator modulating gene transcription, cell responses to genotoxic stress, protein degradation, apoptosis, and several signaling pathways [13, 63]. Its role in malignant transformation of hepatocytes has been demonstrated in the liver-specific HBx transgenic mouse models [70, 71]. Several possible carcinogenic mechanisms for HBx have been reported, with the major one coming from its effects in antagonizing the p53-dependent antitumor functions, including transcriptional activation and apoptosis [72–74]. In addition, HBx has the ability to stimulate several cytoplasmic signaling pathways to promote cell proliferation and anti-apoptosis activity, such as Ras/MEK/MAPK, JNK/JAK/STAT, and PI3K/Akt/GSK-3 [13, 74]. The c-Src kinase as the upstream key switch for HBx to turn on these kinase signaling cascades has been reported [13, 74]. It has been shown that HBx can trigger the release of Ca2+ ions from the ER and mitochondria, which in turn activates the Ca2+-responsive Pyk2 kinase and leads to c-Src activation [75]. In addition, HBx can also affect a variety of cellular transcriptional factors, either directly or indirectly. It can directly interact with components of the basal transcription machinery, such as ribosome-binding protein 5 and TATA-binding protein, as well as the transcriptional activators of CREB/ATF and NF-κB [13, 74]. It is noteworthy that an increasing list of HBx-responsive transcription factors have been identified, including NF-κB, NFAT, AP-1, and androgen receptor (AR) [13, 68, 76]. With the evidence from both cell culture-based assay and the hydrodynamic injection-based mouse models, these HBx-modulated cellular events could also stimulate HBV viral transcription and replication, which also leads to an increased risk of HBV-associated HCC [13, 77–79].
In addition to HBx, frequent deletions at the pre-S region of HBV have been identified in progressive liver diseases and were associated with a higher risk of HCC in CHB patients [69, 80]. A recent report showed that the mice carrying transgenes of HBV with a replication-competent pre-S2 deletion mutant develop liver cancers, including HCC, at the end of 2-year follow-up, supporting the carcinogenic potential of this mutant [81]. The truncated HBsAg accumulates in the ER, which can induce ER stress and oxidative DNA damage. Its significance in contributing to hepatocarcinogenesis awaits further clarification [81–83].
Aside from specific viral oncogenic proteins, several viral genetic variations were found to be associated with the carcinogenic process. These variations are mainly due to the lack of proofreading function of the HBV reverse transcriptase. Among them, the viral genotype attracts much attention, which is based on overall genome sequence divergence [84, 85]. Currently, at least eight HBV genotypes (A–H) have been designated, showing distinct geographical distribution [86]. The HBV viral genotype was shown to affect the clinical outcome. In Asia, where genotypes B and C are endemically dominant, a higher prevalence of genotype C was identified in patients with severe liver diseases, liver cirrhosis, and HCC [87]. Likewise, in Western countries, where HBV genotypes A and D are dominant, genotype D was found to be associated with more severe liver disease and a higher incidence of HCC than genotype A [87]. Intriguingly, in longitudinal cohort studies, the risk of HBV-related HCC was shown to be further significantly increased when viral load was included in the analysis in combination with the worse genotypes [88, 89].
Some other specific virus genetic variations (or mutations) were also demonstrated to correlate with the risk of HCC. For example, the A1762T/G1764A basal core promoter mutant was shown to be an HBV titer-independent risk factor for HCC, whereas the precore G1896A mutant confers a protective effect [89]. Presumably, the specific virus genetic variants associated with the risk of HCC are those selected during the chronic inflammation processes. They might have differential contributions to the inflammation and tumorigenesis in hepatocytes, such as enhancing cell proliferation activity or suppressing apoptosis activity, although the underlying molecular mechanisms in hepatocarcinogenesis for these genetic variations have not yet been identified.
Host Cofactors of HBV-Related Pathogenesis
As the virus-related carcinogenic process is an interplay between the hepatitis virus and the host hepatocytes, host factors are also involved in pathogenesis. Since tumor-associated inflammatory response has recently been added as another carcinogenic factor, at the earliest stages of neoplastic progression, which is critical for fostering the incipient neoplasias to acquire other hallmark capabilities into full-blown cancers [90]. This can also be applied to HBV-induced hepatocarcinogenesis. HBV causes decades of chronic inflammation in the liver; the persistent necrosis and regeneration predispose the accumulation of carcinogenic events in hepatocytes. During inflammation, Kupffer cells and other immune cells are recruited to the liver. They will then release the proinflammatory cytokines and chemokines, such as TNF-α, IL-1β, IL-6, CXCL8, and CXCR4 [91–95], which then stimulate the aberrant proliferation of hepatocytes through activating the transcriptional activators of NF-κB, STAT3, and hypoxia-inducible factor 1α. The process not only stimulates the tumorigenic activity of hepatocytes but also leads to the production of some more inflammatory mediators, which further recruit and activate immune cells in the liver tissues [91–95]. Such an amplification loop establishes a cancer-prone inflammatory microenvironment in the liver. Evidence from the genetic manipulation of mice has confirmed the central role of NF-κB in tipping the interplay between target liver cells and immune cells in favor of tumorigenic processes [95–97].
The long-term persistent inflammation induces the continuous cycles of excess hepatocyte death and regeneration. During the process, not only the viral variants associated with HCC are selected, the hepatocytes with aberrations conferring growth or survival advantages are also selected for clonal expansion at the stage of liver cirrhosis, resulting in the monoclonal characteristics in cirrhotic nodules which can be regarded precancerous [98, 99]. When such lesions gain further genetic aberrations conferring other capabilities of tumor cells for the resulting HCC, such as invasiveness, limitless replication, or angiogenesis, they become malignant [90]. Currently, integration of the increasing data derived from the genome-wide analyses of HCCs has delineated several signaling pathways involved in hepatocarcinogenesis [100–102]. The two major pathways are the activation of Wnt-signaling pathway in (~70 % of HCC, with frequent mutations of β-catenin and Axin-1) [103–105], and the dysregulation of cell-cycle G1/S transition in (~80 % of HCC, with aberrations of p53, INK4a, ARF, Rb, and cyclin D1) [106, 107]. Some other pathways have also been documented, including the activation of the insulin-like growth factor (IGF) signaling pathway [108–110], the Ras/MAPK signaling pathway, the mTOR pathway [111], the c-met pathway [101], the JAK/STAT pathway [112], the hedgehog pathway [113], and the telomerase reverse transcriptase (TERT) activation [102].
Among these pathways, the occurrence of p53 and β-catenin mutations shows intriguing viral-etiology-associated mutation patterns. Most p53 mutations belong to the missense somatic mutations, which occur more frequently in HBV-related HCCs [114]. A hotspot p53 mutation at codon 249, with a selective arginine-to-serine substitution (caused by G to T transversion), was identified to be strongly associated with exposure to aflatoxin B1 in combination with chronic HBV infection [115]. The possibility therefore exists that p53 might play an important role as the checkpoint for the HBV-specific carcinogenic mechanisms. In contrast to p53, oncogenic β-catenin mutations were scarcely identified in HBV-related HCC [114] and were identified mainly in hepatitis-related HCCs [116]. Most β-catenin mutations in HCC occur predominantly at the N-terminus as the dominant Wnt-signaling activation mutants, which help β-catenin escape the degradation regulated by GSK-3β/APC/Axin complex [117]. According to the finding that HBx can activate the β-catenin signaling in HCC cells through inhibiting the c-Src/GSK-3β-dependent signaling pathway [118, 119], it raised the possibility that HBV infection confers a mechanism to activate β-catenin signaling and bypass the need for the activating mutations. More recently, the results from our liver-specific β-catenin KO mouse model further supported that HBx can synergistically collaborate with β-catenin in stimulating the carcinogenic process from hepatic progenitor cells to HCC in this mouse model [120].
In addition to genetic and gene expression aberrations, microRNAs (miRNAs) have recently been reported as important host factors of carcinogenic process associated with HBV [121, 122]. Several miRNAs were identified with the ability to regulate HBV mRNA levels, either by direct binding to the viral transcripts or by targeting to cellular factors regulating the viral transcription. For example, miR-155 and miR-372 can decrease the transcription of HBV through targeting C/EBP-β and PRKACB, respectively [123, 124]. In contrast, miR-1 can enhance the activity of the HBV core promoter by augmenting a positive transcription factor of farnesoid X receptor alpha (FXRA) [125]. Our recent study also pointed out miR-18a can repress the estrogen receptor alpha (ERα) [126], which in turn represses the transcription of HBV through blocking the essential transcription factor of HNF4α binding to the HBV enhancer I [127]. And several miRNAs were recently identified with potential to directly bind to the HBV transcripts and regulate their expression, such as miR-210, miR-199a, and miR-125a [128–130]. In addition to affect the viral transcription, several miRNAs were reported involved in regulating the replication of HBV, mainly by indirectly affecting the key host factors. For example, miR-122 restricts HBV replication through regulating Heme oxygenase-1 (HO-1) gene [131]; and miR-1 and miR-152 repress the expression of HDAC4 and DNMT1 to regulate the HBV replication epigenetically on viral cccDNA [125, 132]. Intriguingly, the host miRNAs can in turn also be regulated by the specific HBV proteins, such as HBx, with the functional significance still awaiting to be investigated [133]. Finally, by comparing the miRNA expression profiles of paired HCC liver tissues, numerous miRNAs showed differential expression patterns in HCC with distinct viral etiology [134–137]. For example, the aberrant changes of miR-96, miR-602, miR-26, and miR-18a seem to occur preferentially in the HBV-related HCC [122, 126, 138]. Their roles in HBV-related hepatocarcinogenesis warrants further investigation.
The unique gender difference issue for HBV-related HCC again has attracted lots of attention recently. The male preference of HCC is more evident in HBV-related than HCV-related HCC [62], with the male to female ratio of 3–7:1 for HBV-related HCC and 1.5–3:1 for HCV-related HCC [139, 140]. Epidemiological studies highly suggested the involvement of androgen and estrogen axes in regulating this gender difference specific in HBV-related HCC. For example, the elevated serum testosterone levels and the genetic polymorphisms for increased androgen activities were significantly associated with increased risk of HCC in male HBsAg carriers [141, 142], suggesting the androgen axis as a tumor promoter function in HBV-related male HCC. In contrast, the estrogen axis was considered as a tumor protector in female HCC. Early oophorectomy was associated an increased risk; whereas postmenopausal hormone replacement therapy was associated with a lower risk of female HBV-related HCC [143].
Our recent studies demonstrated that HBV is actually the virus responsive for the sex hormones in hepatocytes. Both the androgen and estrogen pathways are involved in regulating the transcription of HBV, to control the major HCC risk factor of viral titers in HBV-infected patients, and thus provides possible mechanisms for their involvement in the gender difference of HCC. First, for the androgen part of study, we have identified that the ligand-stimulated AR could increase the transcription of HBV mRNAs through its direct binding to the two androgen-responsive element (ARE) motifs in the enhancer I region of HBV genome [144]. Moreover, the HBx in turn increases the transcriptional activity of ligand bound AR through enhancing the dimerization and the activity of AR N-terminal transactivation domain, by the two key kinase switches of c-Src and GSK-3 [76, 145]. The results indicated a potential positive feedback loop between ligand-stimulated AR and HBx protein, which persistently increases the risk of HCC in male HBV carriers. The finding has been confirmed in the HBV transgenic mouse models [146, 147].
For the estrogen part of study, we first found that HBV titers were increased in female HBV transgenic mice after ovariectomy and decreased in male mice supplemented with estrogen [127]. Next, in the HepG2 hepatoma cells, we found that upregulation of ERα can reduce the HBV transcription, through a specific region within enhancer I of HBV. To further dissect the mechanism, we found that ERα represses the transcription of HBV mediated by squelching the liver-enriched transcription factor HNF4α, which is an essential positive factor for the active transcription of HBV in hepatocytes. This mechanism might account for the lower viral load and reduced incidence of liver cancer in HBV-infected women [127]. Moreover, we have also identified a novel miR-18a elevation-mediated mechanism for suppressing the expression of ERα in more than 60 % of female HCC [126]. This mechanism not only attributes to the decreased activity of estrogen pathway, but also points out a potential mechanism for indirectly regulating the transcription of HBV.
Therefore, the gender disparity in HCC can actually be attributed to both androgen and estrogen sex hormones, targeting to regulate the transcription of HBV. Using the DEN-induced HCC mouse model, which also showed a gender difference of HCC, it has been demonstrated that estrogens can protect hepatocytes from malignant transformation via downregulation of the secretion of IL-6 from Kupffer cells [148]. Of note, an intriguing HNF4α-miRNA inflammatory feedback circuit has been identified, which is critical in this DEN-induced hepatocarcinogenesis model [149]. Since IL-6 and STAT3 are also the key members in this circuit, the involvement of this circuit in regulation of the estrogen function on HBV is worthy to be studied. Using the same animal model, Foxa1 and Foxa2 as the key factors regulating the transcription of both the AR and ER alpha target genes in hepatocytes have recently been identified [150]. The sexually dimorphic HCC is completely reversed in Foxa1- and Foxa2-deficient mice in this mouse model. The involvement of Foxa1/2 in regulating the AR and ER alpha effects on the transcription of HBV is also an intriguing issue to be addressed accordingly.
Abbreviations
- AR:
-
Androgen receptor
- ARE:
-
Androgen-responsive element
- cccDNA:
-
Covalently closed circular DNA
- CHB:
-
Chronic hepatitis B
- DHBV:
-
Duck hepatitis B Virus
- ER:
-
Endoplasmic reticulum
- ERα:
-
Estrogen receptor alpha
- FXRA:
-
Farnesoid X receptor alpha
- HBcAg:
-
Hepatitis B core antigen
- HBeAg:
-
Hepatitis B e antigen
- HBV:
-
Hepatitis B virus
- HCC:
-
Hepatocellular carcinoma
- HO-1:
-
Heme oxygenase-1
- IFN:
-
Interferon
- IGF:
-
Insulin-like growth factor
- miRNA:
-
microRNA
- NA:
-
Nucleos(t)ide analogue
- NLR:
-
NOD-like receptor
- NTCP:
-
Sodium taurocholate cotransporting polypeptide
- ORFs:
-
Open reading frames
- PAMP:
-
Pathogen-associated molecular pattern
- PBMC:
-
Peripheral blood mononuclear cell
- PRR:
-
Pattern recognition receptor
- rcDNA:
-
Relaxed-circular DNA
- RLR:
-
RIG-I-like receptor
- TERT:
-
Telomerase reverse transcriptase
- TLR:
-
Toll-like receptor
- Treg:
-
Regulatory T cell
- WHV:
-
Woodchuck hepatitis virus
References
Nassal M. Hepatitis B, viruses: reverse transcription a different way. Virus Res. 2008;134:235–49.
Schaefer S. Hepatitis B, virus taxonomy and hepatitis B virus genotypes. World J Gastroenterol. 2007;13:14–21.
Lan YT, Li J, Liao W, Ou J. Roles of the three major phosphorylation sites of hepatitis B virus core protein in viral replication. Virology. 1999;259:342–8.
Bock CT, Schranz P, Schroder CH, Zentgraf H. Hepatitis B virus genome is organized into nucleosomes in the nucleus of the infected cell. Virus Genes. 1994;8:215–29.
Newbold JE, Xin H, Tencza M, Sherman G, Dean J, Bowden S, Locarnini S. The covalently closed duplex form of the hepadnavirus genome exists in situ as a heterogeneous population of viral minichromosomes. J Virol. 1995;69:3350–7.
Pollicino T, Belloni L, Raffa G, Pediconi N, Squadrito G, Raimondo G, Levrero M. Hepatitis B virus replication is regulated by the acetylation status of hepatitis B virus cccDNA-bound H3 and H4 histones. Gastroenterology. 2006;130:823–37.
Guo Y, Li Y, Mu S, Zhang J, Yan Z. Evidence that methylation of hepatitis B virus covalently closed circular DNA in liver tissues of patients with chronic hepatitis B modulates HBV replication. J Med Virol. 2009;81:1177–83.
Levrero M, Pollicino T, Petersen J, Belloni L, Raimondo G, Dandri M. Control of cccDNA function in hepatitis B virus infection. J Hepatol. 2009;51:581–92.
Yu X, Mertz JE. Promoters for synthesis of the pre-C and pregenomic mRNAs of human hepatitis B virus are genetically distinct and differentially regulated. J Virol. 1996;70:8719–26.
Milich D, Liang TJ. Exploring the biological basis of hepatitis B e antigen in hepatitis B virus infection. Hepatology. 2003;38:1075–86.
Summers J, Mason WS. Replication of the genome of a hepatitis B-like virus by reverse transcription of an RNA intermediate. Cell. 1982;29:403–15.
Murakami S. Hepatitis B, virus X protein: a multifunctional viral regulator. J Gastroenterol. 2001;36:651–60.
Bouchard MJ, Schneider RJ. The enigmatic X gene of hepatitis B virus. J Virol. 2004;78:12725–34.
Seeger C, Mason WS. Hepatitis B virus biology. Microbiol Mol Biol Rev. 2000;64:51–68.
Halpern MS, England JM, Deery DT, Petcu DJ, Mason WS, Molnar-Kimber KL. Viral nucleic acid synthesis and antigen accumulation in pancreas and kidney of Pekin ducks infected with duck hepatitis B virus. Proc Natl Acad Sci U S A. 1983;80:4865–9.
Freiman JS, Jilbert AR, Dixon RJ, Holmes M, Gowans EJ, Burrell CJ, Wills EJ, et al. Experimental duck hepatitis B virus infection: pathology and evolution of hepatic and extrahepatic infection. Hepatology. 1988;8:507–13.
Tagawa M, Omata M, Yokosuka O, Uchiumi K, Imazeki F, Okuda K. Early events in duck hepatitis B virus infection. Sequential appearance of viral deoxyribonucleic acid in the liver, pancreas, kidney, and spleen. Gastroenterology. 1985;89:1224–9.
Shimoda T, Shikata T, Karasawa T, Tsukagoshi S, Yoshimura M, Sakurai I. Light microscopic localization of hepatitis B virus antigens in the human pancreas. Possibility of multiplication of hepatitis B virus in the human pancreas. Gastroenterology. 1981;81:998–1005.
Di Bisceglie AM, Hoofnagle JH. Hepatitis B virus replication within the human spleen. J Clin Microbiol. 1990;28:2850–2.
Romet-Lemonne JL, McLane MF, Elfassi E, Haseltine WA, Azocar J, Essex M. Hepatitis B virus infection in cultured human lymphoblastoid cells. Science. 1983;221:667–9.
Pasquinelli C, Laure F, Chatenoud L, Beaurin G, Gazengel C, Bismuth H, Degos F, et al. Hepatitis B virus DNA in mononuclear blood cells. A frequent event in hepatitis B surface antigen-positive and -negative patients with acute and chronic liver disease. J Hepatol. 1986;3:95–103.
Bouffard P, Lamelin JP, Zoulim F, Pichoud C, Trepo C. Different forms of hepatitis B virus DNA and expression of HBV antigens in peripheral blood mononuclear cells in chronic hepatitis B. J Med Virol. 1990;31:312–7.
Romet-Lemonne JL, Elfassi E, Haseltine W, Essex M. Infection of bone marrow cells by hepatitis B virus. Lancet. 1983;2:732.
Kock J, Theilmann L, Galle P, Schlicht HJ. Hepatitis B virus nucleic acids associated with human peripheral blood mononuclear cells do not originate from replicating virus. Hepatology. 1996;23:405–13.
Mathew J, Geerts A, Burt AD. Pathobiology of hepatic stellate cells. Hepatogastroenterology. 1996;43:72–91.
Glebe D, Urban S. Viral and cellular determinants involved in hepadnaviral entry. World J Gastroenterol. 2007;13:22–38.
Rabe B, Vlachou A, Pante N, Helenius A, Kann M. Nuclear import of hepatitis B virus capsids and release of the viral genome. Proc Natl Acad Sci U S A. 2003;100:9849–54.
Kann M, Schmitz A, Rabe B. Intracellular transport of hepatitis B virus. World J Gastroenterol. 2007;13:39–47.
Kock J, Rosler C, Zhang JJ, Blum HE, Nassal M, Thoma C. Generation of covalently closed circular DNA of hepatitis B viruses via intracellular recycling is regulated in a virus specific manner. PLoS Pathog. 2010;6:e1001082.
Sohn JA, Litwin S, Seeger C. Mechanism for CCC DNA synthesis in hepadnaviruses. PLoS One. 2009;4:e8093.
Tuttleman JS, Pourcel C, Summers J. Formation of the pool of covalently closed circular viral DNA in hepadnavirus-infected cells. Cell. 1986;47:451–60.
Rehermann B, Ferrari C, Pasquinelli C, Chisari FV. The hepatitis B virus persists for decades after patients’ recovery from acute viral hepatitis despite active maintenance of a cytotoxic T-lymphocyte response. Nat Med. 1996;2:1104–8.
Yeo W, Chan TC, Leung NW, Lam WY, Mo FK, Chu MT, Chan HL, et al. Hepatitis B virus reactivation in lymphoma patients with prior resolved hepatitis B undergoing anticancer therapy with or without rituximab. J Clin Oncol. 2009;27:605–11.
Dandri M, Petersen J. Hepatitis B virus cccDNA clearance: killing for curing? Hepatology. 2005;42:1453–5.
Guidotti LG, Chisari FV. Immunobiology and pathogenesis of viral hepatitis. Annu Rev Pathol. 2006;1:23–61.
Rehermann B, Nascimbeni M. Immunology of hepatitis B virus and hepatitis C virus infection. Nat Rev Immunol. 2005;5:215–29.
Maini MK, Boni C, Lee CK, Larrubia JR, Reignat S, Ogg GS, King AS, et al. The role of virus-specific CD8(+) cells in liver damage and viral control during persistent hepatitis B virus infection. J Exp Med. 2000;191:1269–80.
Boni C, Fisicaro P, Valdatta C, Amadei B, Di Vincenzo P, Giuberti T, Laccabue D, et al. Characterization of hepatitis B virus (HBV)-specific T-cell dysfunction in chronic HBV infection. J Virol. 2007;81:4215–25.
Takeuchi O, Akira S. Innate immunity to virus infection. Immunol Rev. 2009;227:75–86.
Guidotti LG, Rochford R, Chung J, Shapiro M, Purcell R, Chisari FV. Viral clearance without destruction of infected cells during acute HBV infection. Science. 1999;284:825–9.
Webster GJ, Reignat S, Maini MK, Whalley SA, Ogg GS, King A, Brown D, et al. Incubation phase of acute hepatitis B in man: dynamic of cellular immune mechanisms. Hepatology. 2000;32:1117–24.
Wieland S, Thimme R, Purcell RH, Chisari FV. Genomic analysis of the host response to hepatitis B virus infection. Proc Natl Acad Sci U S A. 2004;101:6669–74.
Dunn C, Peppa D, Khanna P, Nebbia G, Jones M, Brendish N, Lascar RM, et al. Temporal analysis of early immune responses in patients with acute hepatitis B virus infection. Gastroenterology. 2009;137:1289–300.
Bertoletti A, Ferrari C. Kinetics of the immune response during HBV and HCV infection. Hepatology. 2003;38:4–13.
Lucifora J, Durantel D, Testoni B, Hantz O, Levrero M, Zoulim F. Control of hepatitis B virus replication by innate response of HepaRG cells. Hepatology. 2010;51:63–72.
Hosel M, Quasdorff M, Wiegmann K, Webb D, Zedler U, Broxtermann M, Tedjokusumo R, et al. Not interferon, but interleukin-6 controls early gene expression in hepatitis B virus infection. Hepatology. 2009;50:1773–82.
Guy CS, Mulrooney-Cousins PM, Churchill ND, Michalak TI. Intrahepatic expression of genes affiliated with innate and adaptive immune responses immediately after invasion and during acute infection with woodchuck hepadnavirus. J Virol. 2008;82:8579–91.
Lin YJ, Wu HL, Chen DS, Chen PJ. Hepatitis B virus nucleocapsid but not free core antigen controls viral clearance in mice. J Virol. 2012;86:9266–73.
Zeissig S, Murata K, Sweet L, Publicover J, Hu Z, Kaser A, Bosse E, et al. Hepatitis B virus-induced lipid alterations contribute to natural killer T cell-dependent protective immunity. Nat Med. 2012. doi:10.1038/nm.2811 (Epub ahead of print).
Wu J, Meng Z, Jiang M, Pei R, Trippler M, Broering R, Bucchi A, et al. Hepatitis B virus suppresses toll-like receptor-mediated innate immune responses in murine parenchymal and nonparenchymal liver cells. Hepatology. 2009;49:1132–40.
Fisicaro P, Valdatta C, Boni C, Massari M, Mori C, Zerbini A, Orlandini A, et al. Early kinetics of innate and adaptive immune responses during hepatitis B virus infection. Gut. 2009;58:974–82.
Thimme R, Wieland S, Steiger C, Ghrayeb J, Reimann KA, Purcell RH, Chisari FV. CD8(+) T cells mediate viral clearance and disease pathogenesis during acute hepatitis B virus infection. J Virol. 2003;77:68–76.
Guidotti LG, Chisari FV. Noncytolytic control of viral infections by the innate and adaptive immune response. Annu Rev Immunol. 2001;19:65–91.
Crispe IN. The liver as a lymphoid organ. Annu Rev Immunol. 2009;27:147–63.
Stoop JN, van der Molen RG, Baan CC, van der Laan LJ, Kuipers EJ, Kusters JG, Janssen HL. Regulatory T cells contribute to the impaired immune response in patients with chronic hepatitis B virus infection. Hepatology. 2005;41:771–8.
Stross L, Gunther J, Gasteiger G, Asen T, Graf S, Aichler M, Esposito I, et al. Foxp3+ regulatory T cells protect the liver from immune damage and compromise virus control during acute, experimental hepatitis B virus infection. Hepatology. 2012. doi:10.1002/hep.25765 (Epub ahead of print).
Wherry EJ, Blattman JN, Murali-Krishna K, van der Most R, Ahmed R. Viral persistence alters CD8 T-cell immunodominance and tissue distribution and results in distinct stages of functional impairment. J Virol. 2003;77:4911–27.
Fisicaro P, Valdatta C, Massari M, Loggi E, Biasini E, Sacchelli L, Cavallo MC, et al. Antiviral intrahepatic T-cell responses can be restored by blocking programmed death-1 pathway in chronic hepatitis B. Gastroenterology. 2010;138:682–93, 693.e1-4.
Schurich A, Khanna P, Lopes AR, Han KJ, Peppa D, Micco L, Nebbia G, et al. Role of the coinhibitory receptor cytotoxic T lymphocyte antigen-4 on apoptosis-Prone CD8 T cells in persistent hepatitis B virus infection. Hepatology. 2011;53:1494–503.
Tan AT, Koh S, Goh V, Bertoletti A. Understanding the immunopathogenesis of chronic hepatitis B virus: an Asian prospective. J Gastroenterol Hepatol. 2008;23:833–43.
Chen DS. From hepatitis to hepatoma: lessons from type B viral hepatitis. Science. 1993;262:369–70.
Parkin DM, Bray F, Ferlay J, Pisani P. Estimating the world cancer burden: Globocan 2000. Int J Cancer. 2001;94:153–6.
Tan A, Yeh SH, Liu CJ, Cheung C, Chen PJ. Viral hepatocarcinogenesis: from infection to cancer. Liver Int. 2008;28:175–88.
Gomaa AI, Khan SA, Toledano MB, Waked I, Taylor-Robinson SD. Hepatocellular carcinoma: epidemiology, risk factors and pathogenesis. World J Gastroenterol. 2008;14:4300–8.
Kao JH, Chen DS. Global control of hepatitis B virus infection. Lancet Infect Dis. 2002;2:395–403.
Hoofnagle JH, Doo E, Liang TJ, Fleischer R, Lok AS. Management of hepatitis B: summary of a clinical research workshop. Hepatology. 2007;45:1056–75.
Ghany M, Liang TJ. Drug targets and molecular mechanisms of drug resistance in chronic hepatitis B. Gastroenterology. 2007;132:1574–85.
Neuveut C, Wei Y, Buendia MA. Mechanisms of HBV-related hepatocarcinogenesis. J Hepatol. 2010;52:594–604.
Chen BF, Liu CJ, Jow GM, Chen PJ, Kao JH, Chen DS. High prevalence and mapping of pre-S deletion in hepatitis B virus carriers with progressive liver diseases. Gastroenterology. 2006;130:1153–68.
Kim CM, Koike K, Saito I, Miyamura T, Jay G. HBx gene of hepatitis B virus induces liver cancer in transgenic mice. Nature. 1991;351:317–20.
Wu BK, Li CC, Chen HJ, Chang JL, Jeng KS, Chou CK, Hsu MT, et al. Blocking of G1/S transition and cell death in the regenerating liver of Hepatitis B virus X protein transgenic mice. Biochem Biophys Res Commun. 2006;340:916–28.
Elmore LW, Hancock AR, Chang SF, Wang XW, Chang S, Callahan CP, Geller DA, et al. Hepatitis B virus X protein and p53 tumor suppressor interactions in the modulation of apoptosis. Proc Natl Acad Sci U S A. 1997;94:14707–12.
Ogden SK, Lee KC, Barton MC. Hepatitis B viral transactivator HBx alleviates p53-mediated repression of alpha-fetoprotein gene expression. J Biol Chem. 2000;275:27806–14.
Arbuthnot P, Capovilla A, Kew M. Putative role of hepatitis B virus X protein in hepatocarcinogenesis: effects on apoptosis, DNA repair, mitogen-activated protein kinase and JAK/STAT pathways. J Gastroenterol Hepatol. 2000;15:357–68.
Bouchard MJ, Wang LH, Schneider RJ. Calcium signaling by HBx protein in hepatitis B virus DNA replication. Science. 2001;294:2376–8.
Chiu CM, Yeh SH, Chen PJ, Kuo TJ, Chang CJ, Chen PJ, Yang WJ, et al. Hepatitis B virus X protein enhances androgen receptor-responsive gene expression depending on androgen level. Proc Natl Acad Sci U S A. 2007;104:2571–8.
Lin YJ, Huang LR, Yang HC, Tzeng HT, Hsu PN, Wu HL, Chen PJ, et al. Hepatitis B virus core antigen determines viral persistence in a C57BL/6 mouse model. Proc Natl Acad Sci U S A. 2010;107:9340–5.
McClain SL, Clippinger AJ, Lizzano R, Bouchard MJ. Hepatitis B virus replication is associated with an HBx-dependent mitochondrion-regulated increase in cytosolic calcium levels. J Virol. 2007;81:12061–5.
Keasler VV, Hodgson AJ, Madden CR, Slagle BL. Enhancement of hepatitis B virus replication by the regulatory X protein in vitro and in vivo. J Virol. 2007;81:2656–62.
Yeung P, Wong DK, Lai CL, Fung J, Seto WK, Yuen MF. Association of hepatitis B virus pre-S deletions with the development of hepatocellular carcinoma in chronic hepatitis B. J Infect Dis. 2011;203:646–54.
Na B, Huang Z, Wang Q, Qi Z, Tian Y, Lu CC, Yu J, et al. Transgenic expression of entire hepatitis B virus in mice induces hepatocarcinogenesis independent of chronic liver injury. PLoS One. 2011;6:e26240. Epub 2011 Oct 12.
Hsieh YH, Su IJ, Wang HC, Chang WW, Lei HY, Lai MD, Chang WT, et al. Pre-S mutant surface antigens in chronic hepatitis B virus infection induce oxidative stress and DNA damage. Carcinogenesis. 2004;25:2023–32.
Su IJ, Wang HC, Wu HC, Huang WY. Ground glass hepatocytes contain pre-S mutants and represent preneoplastic lesions in chronic hepatitis B virus infection. J Gastroenterol Hepatol. 2008;23:1169–74.
Norder H, Courouce AM, Magnius LO. Complete genomes, phylogenetic relatedness, and structural proteins of six strains of the hepatitis B virus, four of which represent two new genotypes. Virology. 1994;198:489–503.
Okamoto H, Kurai K, Okada S, Yamamoto K, Lizuka H, Tanaka T, Fukuda S, et al. Full-length sequence of a hepatitis C virus genome having poor homology to reported isolates: comparative study of four distinct genotypes. Virology. 1992;188:331–41.
Kurbanov F, Tanaka Y, Mizokami M. Geographical and genetic diversity of the human hepatitis B virus. Hepatol Res. 2010;40:14–30.
Liu CJ, Kao JH. Genetic variability of hepatitis B virus and response to antiviral therapy. Antivir Ther. 2008;13:613–24.
Yu MW, Yeh SH, Chen PJ, Liaw YF, Lin CL, Liu CJ, Shih WL, et al. Hepatitis B virus genotype and DNA level and hepatocellular carcinoma: a prospective study in men. J Natl Cancer Inst. 2005;97:265–72.
Yang HI, Yeh SH, Chen PJ, Iloeje UH, Jen CL, Su J, Wang LY, et al. Associations between hepatitis B virus genotype and mutants and the risk of hepatocellular carcinoma. J Natl Cancer Inst. 2008;100:1134–43.
Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell. 2011;144:646–74.
Mantovani A, Allavena P, Sica A, Balkwill F. Cancer-related inflammation. Nature. 2008;454:436–44.
Karin M, Lawrence T, Nizet V. Innate immunity gone awry: linking microbial infections to chronic inflammation and cancer. Cell. 2006;124:823–35.
Gao B, Jeong WI, Tian Z. Liver: an organ with predominant innate immunity. Hepatology. 2008;47:729–36.
Lin WW, Karin M. A cytokine-mediated link between innate immunity, inflammation, and cancer. J Clin Invest. 2007;117:1175–83.
Karin M. Nuclear factor-kappaB in cancer development and progression. Nature. 2006;441:431–6.
Sun B, Karin M. NF-kappaB signaling, liver disease and hepatoprotective agents. Oncogene. 2008;27:6228–44.
Pikarsky E, Porat RM, Stein I, Abramovitch R, Amit S, Kasem S, Gutkovich-Pyest E, et al. NF-kappaB functions as a tumour promoter in inflammation-associated cancer. Nature. 2004;431:461–6.
Paradis V, Laurendeau I, Vidaud M, Bedossa P. Clonal analysis of macronodules in cirrhosis. Hepatology. 1998;28:953–8.
Yeh SH, Chen PJ, Shau WY, Chen YW, Lee PH, Chen JT, Chen DS. Chromosomal allelic imbalance evolving from liver cirrhosis to hepatocellular carcinoma. Gastroenterology. 2001;121:699–709.
Wong CM, Ng IO. Molecular pathogenesis of hepatocellular carcinoma. Liver Int. 2008;28:160–74.
Villanueva A, Newell P, Chiang DY, Friedman SL, Llovet JM. Genomics and signaling pathways in hepatocellular carcinoma. Semin Liver Dis. 2007;27:55–76.
Llovet JM, Bruix J. Molecular targeted therapies in hepatocellular carcinoma. Hepatology. 2008;48:1312–27.
de La Coste A, Romagnolo B, Billuart P, Renard CA, Buendia MA, Soubrane O, Fabre M, et al. Somatic mutations of the beta-catenin gene are frequent in mouse and human hepatocellular carcinomas. Proc Natl Acad Sci U S A. 1998;95:8847–51.
Miyoshi Y, Iwao K, Nagasawa Y, Aihara T, Sasaki Y, Imaoka S, Murata M, et al. Activation of the beta-catenin gene in primary hepatocellular carcinomas by somatic alterations involving exon 3. Cancer Res. 1998;58:2524–7.
Satoh S, Daigo Y, Furukawa Y, Kato T, Miwa N, Nishiwaki T, Kawasoe T, et al. AXIN1 mutations in hepatocellular carcinomas, and growth suppression in cancer cells by virus-mediated transfer of AXIN1. Nat Genet. 2000;24:245–50.
Tannapfel A, Busse C, Weinans L, Benicke M, Katalinic A, Geissler F, Hauss J, et al. INK4a-ARF alterations and p53 mutations in hepatocellular carcinomas. Oncogene. 2001;20:7104–9.
Malumbres M, Barbacid M. To cycle or not to cycle: a critical decision in cancer. Nat Rev Cancer. 2001;1:222–31.
Schirmacher P, Held WA, Yang D, Chisari FV, Rustum Y, Rogler CE. Reactivation of insulin-like growth factor II during hepatocarcinogenesis in transgenic mice suggests a role in malignant growth. Cancer Res. 1992;52:2549–56.
Alexia C, Fallot G, Lasfer M, Schweizer-Groyer G, Groyer A. An evaluation of the role of insulin-like growth factors (IGF) and of type-I IGF receptor signalling in hepatocarcinogenesis and in the resistance of hepatocarcinoma cells against drug-induced apoptosis. Biochem Pharmacol. 2004;68:1003–15.
Zhao H, Dupont J, Yakar S, Karas M, LeRoith D. PTEN inhibits cell proliferation and induces apoptosis by downregulating cell surface IGF-IR expression in prostate cancer cells. Oncogene. 2004;23:786–94.
Villanueva A, Chiang DY, Newell P, Peix J, Thung S, Alsinet C, Tovar V, et al. Pivotal role of mTOR signaling in hepatocellular carcinoma. Gastroenterology. 2008;135:1972–83.
Calvisi DF, Ladu S, Gorden A, Farina M, Conner EA, Lee JS, Factor VM, et al. Ubiquitous activation of Ras and Jak/Stat pathways in human HCC. Gastroenterology. 2006;130:1117–28.
Osipo C, Miele L. Hedgehog signaling in hepatocellular carcinoma: novel therapeutic strategy targeting hedgehog signaling in HCC. Cancer Biol Ther. 2006;5:238–9.
Laurent-Puig P, Legoix P, Bluteau O, Belghiti J, Franco D, Binot F, Monges G, et al. Genetic alterations associated with hepatocellular carcinomas define distinct pathways of hepatocarcinogenesis. Gastroenterology. 2001;120:1763–73.
Bressac B, Kew M, Wands J, Ozturk M. Selective G to T mutations of p53 gene in hepatocellular carcinoma from southern Africa. Nature. 1991;350:429–31.
Huang H, Fujii H, Sankila A, Mahler-Araujo BM, Matsuda M, Cathomas G, Ohgaki H. Beta-catenin mutations are frequent in human hepatocellular carcinomas associated with hepatitis C virus infection. Am J Pathol. 1999;155:1795–801.
Giles RH, van Es JH, Clevers H. Caught up in a Wnt storm: Wnt signaling in cancer. Biochim Biophys Acta. 2003;1653:1–24.
Cha MY, Kim CM, Park YM, Ryu WS. Hepatitis B virus X protein is essential for the activation of Wnt/beta-catenin signaling in hepatoma cells. Hepatology. 2004;39:1683–93.
Ding Q, Xia W, Liu JC, Yang JY, Lee DF, Xia J, Bartholomeusz G, et al. Erk associates with and primes GSK-3beta for its inactivation resulting in upregulation of beta-catenin. Mol Cell. 2005;19:159–70.
Wang EY, Yeh SH, Tsai TF, Huang HP, Jeng YM, Lin WH, Chen WC, et al. Depletion of beta-catenin from mature hepatocytes of mice promotes expansion of hepatic progenitor cells and tumor development. Proc Natl Acad Sci U S A. 2011;108:18384–9.
Liu WH, Yeh SH, Chen PJ. Role of microRNAs in hepatitis B virus replication and pathogenesis. Biochim Biophys Acta. 1809;2011:678–85.
Ladeiro Y, Couchy G, Balabaud C, Bioulac-Sage P, Pelletier L, Rebouissou S, Zucman-Rossi J. MicroRNA profiling in hepatocellular tumors is associated with clinical features and oncogene/tumor suppressor gene mutations. Hepatology. 2008;47:1955–63.
Wang B, Majumder S, Nuovo G, Kutay H, Volinia S, Patel T, Schmittgen TD, et al. Role of microRNA-155 at early stages of hepatocarcinogenesis induced by choline-deficient and amino acid-defined diet in C57BL/6 mice. Hepatology. 2009;50:1152–61.
Wang J, Liu X, Wu H, Ni P, Gu Z, Qiao Y, Chen N, et al. CREB up-regulates long non-coding RNA, HULC expression through interaction with microRNA-372 in liver cancer. Nucleic Acids Res. 2010;38:5366–83.
Zhang X, Zhang E, Ma Z, Pei R, Jiang M, Schlaak JF, Roggendorf M, et al. Modulation of hepatitis B virus replication and hepatocyte differentiation by MicroRNA-1. Hepatology. 2011;53:1476–85.
Liu WH, Yeh SH, Lu CC, Yu SL, Chen HY, Lin CY, Chen DS, et al. MicroRNA-18a prevents estrogen receptor-alpha expression, promoting proliferation of hepatocellular carcinoma cells. Gastroenterology. 2009;136:683–93.
Wang SH, Yeh SH, Lin WH, Yeh KH, Yuan Q, Xia NS, Chen DS, et al. Estrogen Receptor-alpha represses transcription of HBV genes via interaction with hepatocyte nuclear factor 4alpha. Gastroenterology. 2012;142:989–98.
Wu FL, Jin WB, Li JH, Guo AG. Targets for human encoded microRNAs in HBV genes. Virus Genes. 2011;42:157–61.
Zhang GL, Li YX, Zheng SQ, Liu M, Li X, Tang H. Suppression of hepatitis B virus replication by microRNA-199a-3p and microRNA-210. Antiviral Res. 2010;88:169–75.
Potenza N, Papa U, Mosca N, Zerbini F, Nobile V, Russo A. Human microRNA hsa-miR-125a-5p interferes with expression of hepatitis B virus surface antigen. Nucleic Acids Res. 2011;39:5157–63.
Qiu L, Fan H, Jin W, Zhao B, Wang Y, Ju Y, Chen L, et al. miR-122-induced down-regulation of HO-1 negatively affects miR-122-mediated suppression of HBV. Biochem Biophys Res Commun. 2010;398:771–7.
Huang J, Wang Y, Guo Y, Sun S. Down-regulated microRNA-152 induces aberrant DNA methylation in hepatitis B virus-related hepatocellular carcinoma by targeting DNA methyltransferase 1. Hepatology. 2010;52:60–70.
Wang Y, Lu Y, Toh ST, Sung WK, Tan P, Chow P, Chung AY, et al. Lethal-7 is down-regulated by the hepatitis B virus x protein and targets signal transducer and activator of transcription 3. J Hepatol. 2010;53:57–66.
Ji J, Shi J, Budhu A, Yu Z, Forgues M, Roessler S, Ambs S, et al. MicroRNA expression, survival, and response to interferon in liver cancer. N Engl J Med. 2009;361:1437–47.
Ura S, Honda M, Yamashita T, Ueda T, Takatori H, Nishino R, Sunakozaka H, et al. Differential microRNA expression between hepatitis B and hepatitis C leading disease progression to hepatocellular carcinoma. Hepatology. 2009;49:1098–112.
Murakami Y, Yasuda T, Saigo K, Urashima T, Toyoda H, Okanoue T, Shimotohno K. Comprehensive analysis of microRNA expression patterns in hepatocellular carcinoma and non-tumorous tissues. Oncogene. 2006;25:2537–45.
Mizuguchi Y, Mishima T, Yokomuro S, Arima Y, Kawahigashi Y, Shigehara K, Kanda T, et al. Sequencing and bioinformatics-based analyses of the microRNA transcriptome in hepatitis B-related hepatocellular carcinoma. PLoS One. 2011;6:e15304.
Yang L, Ma Z, Wang D, Zhao W, Chen L, Wang G. MicroRNA-602 regulating tumor suppressive gene RASSF1A is overexpressed in hepatitis B virus-infected liver and hepatocellular carcinoma. Cancer Biol Ther. 2010;9:803–8.
Lee CM, Lu SN, Changchien CS, Yeh CT, Hsu TT, Tang JH, Wang JH, et al. Age, gender, and local geographic variations of viral etiology of hepatocellular carcinoma in a hyperendemic area for hepatitis B virus infection. Cancer. 1999;86:1143–50.
Shiratori Y, Shiina S, Imamura M, Kato N, Kanai F, Okudaira T, Teratani T, et al. Characteristic difference of hepatocellular carcinoma between hepatitis B- and C- viral infection in Japan. Hepatology. 1995;22:1027–33.
Yu MW, Yang YC, Yang SY, Cheng SW, Liaw YF, Lin SM, Chen CJ. Hormonal markers and hepatitis B virus-related hepatocellular carcinoma risk: a nested case-control study among men. J Natl Cancer Inst. 2001;93:1644–51.
Yu MW, Cheng SW, Lin MW, Yang SY, Liaw YF, Chang HC, Hsiao TJ, et al. Androgen-receptor gene CAG repeats, plasma testosterone levels, and risk of hepatitis B-related hepatocellular carcinoma. J Natl Cancer Inst. 2000;92:2023–8.
Yu MW, Chang HC, Chang SC, Liaw YF, Lin SM, Liu CJ, Lee SD, et al. Role of reproductive factors in hepatocellular carcinoma: impact on hepatitis B- and C-related risk. Hepatology. 2003;38:1393–400.
Wang SH, Yeh SH, Lin WH, Wang HY, Chen DS, Chen PJ. Identification of androgen response elements in the enhancer I of hepatitis B virus: a mechanism for sex disparity in chronic hepatitis B. Hepatology. 2009;50:1392–402.
Yang WJ, Chang CJ, Yeh SH, Lin WH, Wang SH, Tsai TF, Chen DS, et al. Hepatitis B virus X protein enhances the transcriptional activity of the androgen receptor through c-Src and glycogen synthase kinase-3beta kinase pathways. Hepatology. 2009;49:1515–24.
Wu MH, Ma WL, Hsu CL, Chen YL, Ou JH, Ryan CK, Hung YC, et al. Androgen receptor promotes hepatitis B virus-induced hepatocarcinogenesis through modulation of hepatitis B virus RNA transcription. Sci Transl Med. 2010;2:32ra35.
Tian Y, Kuo CF, Chen WL, Ou JH. Enhancement of hepatitis B virus replication by androgen and its receptor in mice. J Virol. 2012;86:1904–10.
Naugler WE, Sakurai T, Kim S, Maeda S, Kim K, Elsharkawy AM, Karin M. Gender disparity in liver cancer due to sex differences in MyD88-dependent IL-6 production. Science. 2007;317:121–4.
Hatziapostolou M, Polytarchou C, Aggelidou E, Drakaki A, Poultsides GA, Jaeger SA, Ogata H, et al. An HNF4alpha-miRNA inflammatory feedback circuit regulates hepatocellular oncogenesis. Cell. 2011;147:1233–47.
Li Z, Tuteja G, Schug J, Kaestner KH. Foxa1 and foxa2 are essential for sexual dimorphism in liver cancer. Cell. 2012;148:72–83.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2014 Springer Science+Business Media New York
About this chapter
Cite this chapter
Yang, HC., Yeh, SH., Chen, PJ., Chen, DS. (2014). Hepatitis B Virus: Pathogenesis and Host Immune Response. In: Hudnall, S. (eds) Viruses and Human Cancer. Springer, New York, NY. https://doi.org/10.1007/978-1-4939-0870-7_6
Download citation
DOI: https://doi.org/10.1007/978-1-4939-0870-7_6
Published:
Publisher Name: Springer, New York, NY
Print ISBN: 978-1-4939-0869-1
Online ISBN: 978-1-4939-0870-7
eBook Packages: MedicineMedicine (R0)