
Abstract
Human papillomaviruses (HPV) are a family of small DNA tumor viruses with a size of ~52–55. The family consists of ~200 different genotypes; many of the types cause benign warts or papilloma, while a small fraction of oncogenic or “high-risk” types can cause invasive cervical cancer or other tumors. HPV infects keratinocytes in the basal layer of stratified squamous epithelia and replicates in the nucleus of infected keratinocytes along with keratinocyte differentiation. The viral genome in size of ~7.9 kb encodes six early, non-structural regulatory proteins (E1, E2, E4, E5, E6, and E7) and two late structural proteins (L1 and L2). E6 and E7 are two oncoproteins responsible for the viral oncogenesis of high-risk HPVs, including HPV16, 18, 31, 33, 35, 39, 45, 51, 52, 56, 58, 59, 68, 73, and 82. L1 is a major structural component of viral capsid and its self-assembly in vitro into a viral-like particle (VLP) provides the basis of prophylactic vaccines against infections of several HPV types. In addition to cervical cancer, high-risk HPVs are associated with the development of various anogenital cancers and certain head and neck cancers.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others


Keywords
- Cervical Cancer
- Invasive Cervical Cancer
- Recurrent Respiratory Papillomatosis
- Cervical Intraepithelial Neoplasia
- Minor Capsid Protein
These keywords were added by machine and not by the authors. This process is experimental and the keywords may be updated as the learning algorithm improves.
1 A Brief History and Classification of Human Papillomaviruses
Human papillomaviruses (HPV) are a family of small DNA tumor viruses in size of ~52–55 nm in diameter measured initially under electron microscope from skin papillomas by Joseph Melnick at Yale in 1950 (Strauss et al. 1950). Since discovery of the first two genotypes of HPV, HPV1 and HPV2, in 1977 (Orth et al. 1977) and first completion of HPV1a genome sequence in 1982 (Danos et al. 1982), the family has grown to consist of ~200 different genotypes, with 185 genotypes being completely sequenced and deposited in GenBank (www.pave.niaid.nih.gov).
Papillomaviruses were initially classified as a subfamily of the family Papovaviridae in 1962 (Melnick 1962), but reclassified in 2002 as an independent family, Papillomaviridae, in the seventh Report of the International Committee on Taxonomy of Viruses (ICTV). The family Papillomaviridae currently contains at least 29 genera and human papillomaviruses are classified into five (alpha, beta, gamma, mu, and nu) genera (Fig. 7.1). The classification of papillomaviruses depends on the most conserved L1 ORF by genotyping. Different genera share less than 60 % nucleotide sequence identity in the L1 ORF. A new type of papillomavirus is given when its DNA sequence of L1 ORF differs by more than 10 % from the closest known HPV type. Otherwise, a subtype indicates the difference between 2 and 10 % and a variant less than 2 %. HPV types are numbered in the order by their discovery. HPVs in the alpha genus cause mucosal and a fraction of cutaneous HPV lesions, while HPVs in the beta, gamma, mu, and nu genera cause cutaneous lesions (de Villiers et al. 2004; Bernard et al. 2010).

Classification of papillomaviruses, adapted with permission from Nature (Crow 2012)
HPVs are also grouped clinically as high-risk (oncogenic) types, which are frequently associated with invasive cervical cancer, and low-risk (non-oncogenic) types, which are found mainly in genital warts. Epidemiologic studies of HPV types associated with cervical cancer indicate that 15 HPV types (types 16, 18, 31, 33, 35, 39, 45, 51, 52, 56, 58, 59, 68, 73, and 82) are high-risk, and 12 HPV types (types 6, 11, 40, 42, 43, 44, 54, 61, 70, 72, 81, and 89) are low-risk. Among the high-risk HPVs, HPV16 and HPV18 are the principal causes of cervical cancer, with a combined, worldwide relative contribution of ~70 % of invasive cervical cancer (Munoz et al. 2003; de Sanjose et al. 2010).
2 General Properties of Human Papillomaviruses
2.1 HPV Virions and Genome Structure
A mature HPV particle or virion contains a double-stranded, circular genome in size of ~7.9 kb covered by an icosahedral shell or capsid. Unlike many other viruses, HPV particles do not have an envelope or a lipid membrane outside of its capsid and thus is resistant to ether treatment. An HPV capsid is composed of 72 pentamers, of which 60 are hexavalent and 12 are pentavalent (Fig. 7.2a). These pentamers are made up by two viral structural proteins, L1 and L2. Viral L1 (~55 kDa) is the major capsid protein and represents approximately 80 % of the total viral protein. Viral L2 (~70 kDa) is a minor capsid protein. Each viral pentamer is comprised of five copies of L1 attached by one L2 from the inside of the pentamer (Bishop et al. 2007; Chen et al. 2000). This is particularly true for the pentamers on the 12 vertices of icosahedral capsid shell. HPV virions are extremely stable and tolerate high temperature, low pH, proteases, and desiccation.

HPV genome structure and virion particles. (a) HPV11 VLP visualization of cryoEM reconstruction (http://nramm.scripps.edu/?p=1042). (b) HPV genome in a circular episomal form, with relative positions of viral early (PE) and late (PL) promoters and early (AE) and late (AL) polyadenylation signals
The HPV genome can be divided into three major regions: early, late, and a long control (LCR), also called the non-coding region (NCR). The three regions are separated by two polyadenylation (pA) sites: early pA (AE) and late pA (AL) sites (Fig. 7.2b). The early region encodes six ORFs (E1, E2, E1^E4, E5, E6, and E7) and this region of HPV16, HPV18, and HPV-31 also encodes an E8^E2. The late region lies downstream of the early region and encodes L1 and L2 ORFs. The LCR region (~850 bp) has no protein-coding function, but bears the origin of DNA replication and transcription factor binding sites important for viral RNA transcription (Zheng and Baker 2006).
2.2 HPV Life Cycle
HPV depends on keratinocyte differentiation for completion of its life cycle (Fig. 7.3). HPV infection is cell type-specific and requires access of virion particles to human basal cells on the basal lamina through micro-wounding induced by scratching, sexual intercourse, etc. Initial attachment of HPV virions to the host cells takes place by the interaction of viral major capsid protein L1 with the host receptor, heparan sulfate proteoglycan (Dasgupta et al. 2011). This interaction leads to a conformational change and exposes viral minor capsid protein L2 for furin/proprotein convertase digestion (Richards et al. 2006). Subsequently, exposed L2 (amino acid (aa) 108–120) interacts with the S100A10 subunit of annexin A2, a secondary cell receptor (Woodham et al. 2012; Dziduszko and Ozbun 2013), followed by viral entry of the infected cells by endocytosis. L1 dissociation from L2-viral genome, which is facilitated by cyclophilin B (Bienkowska-Haba et al. 2012), results in viral particle uncoating and leaves L2-associated viral genome in the endosome to interact with the retromer complex to be transported to the nucleus via the trans-Golgi network (Lipovsky et al. 2013). Once in the nucleus, L2 localizes the viral genome to PML bodies for transcription of early genes and establishment of infection (Day et al. 2004).

Keratinocyte differentiation-dependent HPV life cycle. (a) Infection of the cervical basal keratinocytes by viruses (circles, top left) is initiated through a microtrauma usually during sexual intercourse. The panel is modified with permission (Jia et al. 2009). (b) Expression of viral E6 and E7 is initiated in the infected basal cells (orange color on the top left for MCM7 as a surrogate for viral E6 and E7). Viral DNA replication takes place in the spinous and granular keratinocytes under intermediate or high differentiation (navy blue color on the top right for viral DNA). However, viral L1 and L2 (red color for L1 in the bottom) become detectable only in the granular and cornified keratinocytes under terminal differentiation. This panel is used with permission (Zheng and Wang 2011)
HPV DNA replication requires the A+T-rich origin of DNA replication in the LCR and two viral DNA-binding proteins, E1 as a DNA helicase and E2 as an accessory factor to E1. It occurs bidirectionally in the nuclear replication foci (Swindle et al. 1999) during two different stages of the viral life cycle. The initial DNA replication takes place in an average of once per cell cycle in the G2 phase in the infected basal cells that have already completed S phase. This replication allows the cells to maintain approximately 50–100 copies per cell. DNA repair plays a role in promotion of HPV DNA amplification (Moody and Laimins 2009; Hoskins et al. 2012; Reinson et al. 2013). The interaction of E1 with a cellular protein p80/UAF1 is required for efficient maintenance of the viral episome in undifferentiated keratinocytes (Lehoux et al. 2012; Cote-Martin et al. 2008). E2 binds to the viral genome and tethers it to mitotic chromosomes through mitosis by interaction with Brd4 on mitotic chromosomes to ensure replicated viral episomes to the nuclei of both daughter cells (You et al. 2004; Abbate et al. 2006; Wang et al. 2013).
Viral vegetative DNA replication differs from its initial replication and takes place in highly differentiated, upper layer spinous keratinocytes no longer undergoing cellular DNA synthesis. This stage of viral DNA replication is robust and presumably undertaken by a rolling circle replication mechanism (Flores and Lambert 1997; Kusumoto-Matsuo et al. 2011; Geimanen et al. 2011), leading to amplify from a low, stable copy number to several thousands of the viral genome copies per cell to be packaged into progeny virions. Viral DNA amplification is required for HPV late gene expression. Virions assemble into paracrystalline arrays in the granular layer and egress from the cornified layers of the epithelium. DNA repair also play a role in promotion of HPV DNA amplification (Moody and Laimins 2009; Hoskins et al. 2012; Reinson et al. 2013).
2.3 HPV Genome Expression
HPV transcribes its RNA from one strand of its genome in one direction and starts from two major promoters named by their transcription start site position in the virus genome. A viral early promoter upstream of E6 ORF and a viral late promoter resided in E7 ORF are responsible for the expression of viral early and late ORFs, respectively. The viral early promoter P97 in HPV-16, P99 in HPV31, and P55/P102 (previously named as P105) in HPV18 are three most studied early promoters. Their activities are tightly controlled by cis-elements in the LCR. These cis-elements, including consensus E2-binding sites, interact with cellular transcription factors and the viral transactivator/repressor E2 protein and regulate the transcription from each viral early promoter in undifferentiated keratinocytes. The resulting early primary transcripts (pre-mRNAs) carry all early ORFs, which span three exons and two introns and undergo alternative RNA splicing and polyadenylation using an early pA signal. Viral late promoter P670 in HPV16, P811 in HPV18 (Wang et al. 2011a), and P742 in HPV31 are three studied late promoters. Their activities can be induced only in differentiated keratinocytes during vegetative virus replication. Because a viral late promoter is positioned in the E7 coding region (Fig. 7.4), transcription from the late promoter has to bypass the early pA site to allow expression of the late region. As a result, a true late pre-mRNA is a chimeric transcript of the early and late regions, with the early region in its 5′ half and the late region in its 3′ half. This late pre-mRNA can be processed either into an early region transcript which is cleaved and polyadenylated at the early pA site (transcript K) or a late region transcript which is cleaved and polyadenylated at the late pA site (transcript L–O) (Fig. 7.4). Alternative RNA splicing of HPV early and late transcripts produces various species and sizes of mRNA transcripts with multiple coding potentials (Fig. 7.4 transcripts A–O). Thus, any given HPV transcript, no matter whether it is an early or late transcript, could be bicistronic, tricistronic, or even polycistronic and contains two or more ORFs. Conversely, a particular ORF can be a part of multiple mRNA species. Thus, to determine which transcript encodes which viral protein has been a challenge and we know very little about which protein is translated from which transcript. Because E6, E1, and L2 ORFs span over an intron region, the expression of these three proteins requires the retention of its corresponding intron. RNA splicing promotes expression of E7 from viral early transcripts in high-risk HPVs and E4 from viral late transcripts (Tang et al. 2006; Doorbar et al. 1990).

Genome structure and transcription map of HPV18 (Wang et al. 2011a). The bracket line in the middle of the panel represents a linear form of the virus genome for better presentation of head-to-tail junctions, promoters (arrows), early (AE) and late (AL) pA sites, and mapped cleavage sites (CS). The ORFs (open boxes) are diagramed above the bracket, and the numbers above each ORF (E6, E7, E1, E2, E5, L2, and L1) are the positions of the first nucleotide (nt) of the start codon and the last nt of the stop codon assigned to the HPV18 genome. The E1^E4 and E8^E2 ORFs span two exons with the nt positions indicated. Because the first AUG of E1^E4 and E8^E2 is positioned in the first exon, formation of an intact E1^E4 or E8^E2 ORF requires RNA splicing (dashed lines). LCR indicates a long control region. Below the bracket line are the RNA species derived from alternative promoter usage and alternative RNA splicing. Exons (heavy lines) and introns (thin lines) are illustrated for each species of RNA, with the mapped splice site positions being numbered by nt positions in the virus genome. Coding potentials for each RNA species are shown on the right. Adapted with permission (Wang et al. 2011a)
2.4 Viral Proteins
HPV genome encodes eight major viral proteins: E1, E2, E4, E5, E6, E7, L1, and L2. L1 and L2 are viral structure proteins for virus capsid formation. The other six viral proteins are non-structural and are responsible for virus replication and pathogenesis. Viral E6 and E7 are two viral oncoproteins responsible for cell transformation and tumorigenesis.
2.4.1 E1
E1 protein (~68 kDa, 649 aa residues) is a site-specific DNA-binding protein for the viral origin of DNA replication. Although relatively little is known about how E1 protein is produced in HPV-infected tissues, E1 is biochemically a well-studied protein. E1 can be subdivided into three functional regions: a C-terminal helicase/ATPase domain, a central origin-binding domain, and an N-terminal regulatory region. E1 serves as a DNA helicase and opens the DNA duplex to initiate viral DNA replication. E1 by itself has low affinity for the viral origin of replication, which contains specific, palindromic E1 binding sites. However, binding of E2 protein to specific sites adjacent to the E1 binding sites helps recruit E1 in a cooperative manner. When loaded onto the origin of DNA replication, the E1/E2 protein complex recruits host replication factors such as topoisomerase I, DNA polymerase alpha/primase, replication protein A, and Brd4 to initiate viral DNA replication (Wang et al. 2013; McBride 2008). E1 is dispensable for maintenance replication, but is essential for initial and productive replication of HPV16 DNA (Egawa et al. 2012). Overexpression of E1 blocks S-phase progression and triggers an ATM-dependent DNA damage response in viral DNA replication foci during the initial viral DNA amplification (Reinson et al. 2013; Sakakibara et al. 2011; Fradet-Turcotte et al. 2011).
2.4.2 E2
E2 protein (~42 kDa, 365 aa residues) is a viral transcription factor in addition to its role in viral DNA replication. E2 contains two defined functional domains. The N-terminal domain is crucial for transcriptional activation, whereas the C-terminal domain possesses the DNA/RNA binding and dimerization properties of the protein. These two domains are linked by a hinge region. E2 interacts with E1 and enhances the binding affinity of E1 to the replication origin (Abbate et al. 2004). E2 functions as a transcriptional activator or repressor to regulate viral early promoter activity through consensus E2-binding sites (Androphy et al. 1987; Hawley-Nelson et al. 1988; Sousa et al. 1990; Romanczuk et al. 1990), upstream of the viral early promoter. However, E2’s transcriptional repression occurs only in cells harboring integrated, but not episomal HPV16 DNA (Bechtold et al. 2003). E2 binding to the viral genome is important for tethering viral genome to mitotic chromosomes throughout mitosis (McBride 2008). E2 also plays roles in apoptosis (Parish et al. 2006; Blachon et al. 2005), ubiquitination, and intracellular trafficking (Muller et al. 2012). E2 binds RNA and interacts with RNA processing factors to regulate RNA splicing (Bodaghi et al. 2009; Lai et al. 1999) and RNA polyadenylation (Johansson et al. 2012).
2.4.3 E4
E4 (~10 kDa, 92 aa residues) is the most abundantly expressed HPV late protein and accumulates in the cytoplasm of differentiating cells in the upper epithelial layers (Griffin et al. 2012). E4 is expressed as an E1^E4 protein in which the N-terminal 5 aa residues are derived from the E1 ORF spliced to the E4 ORF. HPV16 E4 expression and cleavage of its N-terminal 17 amino residues by calpain lead E4 to multimerization, formation of amyloid-like fibers, and disruption of the normal dynamics of the cytokeratin networks (Doorbar et al. 1991; Khan et al. 2011). E4 mediates cell cycle arrest in G2 by sequestering Cdk1/cyclin B1 onto the cytokeratin network to prevent the accumulation of active Cdk1/cyclin B1 complexes in the nucleus (Davy et al. 2002; Nakahara et al. 2002). The E1^E4 protein of HPV16 might also regulate gene expression at the post-transcriptional level by interacting with a DEAD-box containing RNA helicase (Doorbar et al. 2000) and SR protein kinase 1 (SRPK1) (Bell et al. 2007).
2.4.4 E5
E5 (~10 kDa, 83 aa residues) is a small hydrophobic and oligomeric channel-forming membrane protein, which is localized in the endoplasmic reticulum (ER) and the nuclear envelope. Assembled hexameric E5 channels in membranous environments have a defined luminal diameter and stoichiometry (Wetherill et al. 2012). The N-terminus of E5 is restricted to the ER lumen, while its C terminus is exposed to the cytoplasm to mediate interactions with cytoplasmic and ER proteins (Krawczyk et al. 2010). The C terminus of E5 also induces koilocytosis, structural cellular changes that are characteristic of papillomavirus infection (Krawczyk et al. 2008). HPV16 E5 is an oncoprotein. High levels of E5 expression in the mouse skin in the presence of persistently provided exogenous estrogen induce epithelial hyperproliferation, resulting in spontaneous tumor formation and increased dysplastic disease in the cervical epithelium (Maufort et al. 2010). E5 induces anchorage-independent growth of murine fibroblasts, enhances the immortalization of human primary keritonocytes by E6 and E7, and causes cell–cell fusion (Ganguly 2012). E5 activates epidermal growth factor receptor (EGFR) signaling pathways and enhances mitogen-activated protein kinase (MAPK) activity (Zhang et al. 2005; Straight et al. 1993; Kim et al. 2006), but attenuates the TGFbeta1/Smad signaling (French et al. 2013) and down-regulates the expression of MHC class I (Campo et al. 2010).
2.4.5 E6
E6 (~18 kDa, 151 aa residues) is a nuclear oncoprotein that interacts with hundreds of cellular proteins (White et al. 2012a). E6 inactivates cellular p53 family proteins (p53, p63, and p73) essential for cell cycle control, DNA repair, and cell adhesion (Ben et al. 2011; Moody and Laimins 2010). The complex of E6 and E6-associated protein (E6-AP) functions as an ubiquitin-protein ligase in the ubiquitination of p53 (Huibregtse et al. 1993). E6 contains two hypothetical zinc fingers involved in zinc binding and three nuclear localization signals (NLS) (Tao et al. 2003) as well as a PDZ-binding site in the N-terminus (Kiyono et al. 1997; Lee et al. 1997). E6 dimerizes through its N-terminal domain and this self-association promotes the polyubiquitination of p53 by E6AP (Zanier et al. 2012). The basic-hydrophobic pocket of E6, which composes of two zinc domains and a linker helix, interacts with an acidic LxxLL motif of host proteins to exercise E6 transformation and degradation activities (Zanier et al. 2013). Besides its ability to immortalize and transform cells, E6 regulates gene expression at transcriptional and post-transcriptional levels (Desaintes et al. 1992; Klingelhutz et al. 1996) through interaction with other transcription factors/coactivators (Patel et al. 1999; Ronco et al. 1998; Kumar et al. 2002; Veldman et al. 2001, 2003; Thomas and Chiang 2005) and splicing factors, as well as its direct interaction with DNA and RNA (Bodaghi et al. 2009; Imai et al. 1989; Ristriani et al. 2000, 2001; Nomine et al. 2003). High-risk E6 enhances telomerase activity and activates several signal pathways including Akt, Wnt, Notch, and mTOC1 (Rampias et al. 2010; Lichtig et al. 2010; Weijzen et al. 2003; Spangle and Munger 2010, 2013), but inhibits apoptosis, keratinocyte differentiation, and interferon response (Ronco et al. 1998; Jackson et al. 2000; DeFilippis et al. 2003; Li et al. 1999). High-risk E6 regulates expression of a subset of cellular miRNAs (Zheng and Wang 2011; Wang et al. 2009, 2014). In E6 transgenic mice, E6 oncoprotein, in the absence of E7, synergizes with estrogen to induce cervical cancer after 9 months (Shai et al. 2007, 2010).
2.4.6 E7
E7 (~16 kDa, 98 aa residues) is a nuclear oncoprotein. The N-terminus of E7 contains conserved regions (CR) that have sequence similarity to other viruses. These conserved regions have sequence similarity to a portion of CR1 and the entire CR2 in adenovirus E1A and SV40 T antigen. E7 contributes to the binding and degradation of pRB and its related pocket proteins p107 and p130 through cullin 2 ubiquitin ligase complex associated with ZER1 (McLaughlin-Drubin and Munger 2009; Huh et al. 2007; White et al. 2012b). A conserved Leu-X-Cys-X-Glu (LXCXE) motif in the CR2 is sufficient for the association of E7 protein with pRB (Munger et al. 1989; Lee et al. 1998). A CK2 phosphorylation site is adjacent to the LXCXE motif. The C-terminal half of E7 contains a CR3 region with a zinc-binding domain and contributes to degradation of pRB and to E7 dimerization and transformation activities (Todorovic et al. 2011, 2012). E7 interacts with many cellular transcription factors/coactivators (Massimi et al. 1997; Avvakumov et al. 2003; Bernat et al. 2003; Huang and McCance 2002; Pim and Banks 2010; Zheng 2010) and participates epigenetic reprogramming (McLaughlin-Drubin et al. 2011). In addition to its cellular transformation activities, oncogenic E7 also plays a role in the viral life cycle and deregulates the cell cycle by stabilizing p21 and upregulating p16 expression. Oncogenic E7 interacts with the centrosomal regulator gamma-tubulin and induces mitotic defects and aneuploidy, leading to centrosome abnormalities and chromosomal instability (Duensing et al. 2000). High-risk E7 regulates expression of a subset of cellular miRNAs (Zheng and Wang 2011; Wang et al. 2014). In transgenic mice, the continuous expression of E7 is required for the maintenance of cervical cancers and precancerous lesions even in the presence of viral E6 (Jabbar et al. 2012; Shin et al. 2012). E7 down-regulates the expression of MHC class I (Bottley et al. 2008; Li et al. 2006) and prevents recognition of HPV-induced lesions by cytotoxic CD8 T cells.
2.4.7 L1
L1 (~55 kDa) is a major structural component of viral capsid. Both HPV16 and HPV18 L1 mRNAs initiate their translation from an initiation codon right at the splice junction of exon 2 and exon 3 of the viral late mRNA and encode a corresponding 506-aa HPV16 L1 and a 508-aa HPV18 L1 (Fig. 7.4), both of which are shorter than the one originally predicted in GenBank database (Zheng and Baker 2006; Wang et al. 2011a). L1 self-assembles into pentameric capsomers with a hollow channel at the center through a fivefold central axis when five L1 monomers come together and assemble in a symmetrical manner (Bishop et al. 2007; Chen et al. 2000). Thus, its tendency to form a pentavalent structure is directly reflected in the star-shape motif visible as a result of each capsomere. Purified capsomers can form capsids, which are stabilized by disulfide bonds between neighboring L1 molecules through their C-termini. The C-terminal half of L1 also contains activities for HPV DNA-binding and nuclear localization (Schafer et al. 2002; Nelson et al. 2002; Li et al. 1997). The N-terminal half of L1 interacts with L2. L1 capsids assembled in vitro are the basis of prophylactic vaccines against several HPV types (Schiller et al. 2012). Although most portions of L1 are well-conserved between types, the surface loops of L1 can differ substantially, probably reflecting a mechanism for evasion of neutralizing antibody responses elicited by previous papillomavirus infections.
2.4.8 L2
L2 (~70 kDa) is a minor capsid protein bearing 474 aa residues for HPV16 and 463 aa residues for HPV18. L2 exists in an oxidized state within the papillomavirus virion, with the two conserved cysteine residues forming an intramolecular disulfide bond. A single molecule of L2 interacts with an L1 pentamer via a C-terminal L1-binding domain (Finnen et al. 2003). L2 is not required for capsid formation, but participates in encapsidation of the viral genome. Up to 72 molecules of L2 can be incorporated per capsid, one beneath the axial lumen of each L1 capsomer (Buck et al. 2008). Both C- and N- terminal NLSs of L2 bind viral DNA during capsid formation and are important for nuclear localization of the DNA particle (Zhou et al. 1994; Bousarghin et al. 2003). L2 interacts with cellular proteins during the infectious entry process. After the initial binding of the virion to the cell, the N-terminus of L2 containing a consensus furin cleavage site (RxKR) is cleaved by the cellular protease, furin (Richards et al. 2006). The N-terminal L2 has a conserved transmembrane domain with three GxxxG motifs to facilitate homotypic and heterotypic interactions between transmembrane helices for vDNA translocation across the endo-/lysosomal membrane. Disruption of some of these GxxxG motifs has been shown to result in noninfectious viruses (Bronnimann et al. 2013). L2 interacts with members of T-box family, TBX2 and TBX3, and represses transcription from the long control region of HPVs (Schneider et al. 2013). A small N-terminal portion of L2 is well-conserved between different papillomavirus types. Experimental vaccines targeting these regions may offer protection against a broad range of HPV types (Jagu et al. 2013a, b).
3 HPV Infections and Transmission
HPVs can cause benign and malignant tumors in persistently infected skin and mucosal tissues anywhere in the human body. Benign tumors induced by low-risk HPVs are also called papilloma or warts, and these are in general harmless. However, when such a papilloma occurs in the larynx or upper airway, it could be life-threatening as exemplified by recurrent respiratory papillomatosis (RRP), a juvenile disease predominantly caused by HPV6 and HPV11 infection. RRP tends to recur and has the potential to spread throughout the respiratory tract (Bonagura et al. 2010). Benign tumors in the genital area are called condyloma acuminatum or genital (venereal) warts, of which around 96–100 % are caused by low-risk HPV6 and HPV11 infection. Although sexual contact as a risk factor in the development of cervical cancer was described in 1842 by Domenio Rigoni-Stern and the infectious nature of human warts was established in 1907 by Giuseppe Ciuffo’s self-inoculation experiments with a cell-free extract of common warts, a landmark breakthrough on HPV cervical infection as a cause of cervical cancer was not achieved until 1983 when HPV16 DNA was discovered in ~60 % of cervical cancer samples by Herald zur Hausen and colleagues in Germany (Durst et al. 1983). Since then, the role of various HPV genotypes in invasive cervical cancer has been extensively studied, with HPV16, 18, 31, 33, 35, 45, 52, and 58 contributing to 91 % of invasive cervical cancer, and HPV16, 18, and 45 contributing to 94 % of cervical adenocarcinomas (de Sanjose et al. 2010). Cervical cancer (Cervical Cancer Essay 00021 40/40) is the second most common cancer among women worldwide. Approximately 500,000 incident cases and 320,000 cases of attributable deaths are predicted each year. More than 80 % of cases arise in developing countries.
In addition to the cervix, high-risk HPVs also infect other anogenital areas and can lead to development of anal, vaginal, vulvar, and penile cancers. Most anal cancers (~84 %) are caused by HPV infection (Frisch et al. 1997) (Anal Cancer Essay 00022 10/10). High-risk HPV infection of oropharynx (tonsils and the back of the tongue) may lead to the development of oropharyngeal cancer (Other HPV-associated Cancer Essay 00234 262/262). Oropharyngeal cancer is more common in men than women. The prevalence of HPV in oropharyngeal cancer is increasing from 16.3 % during 1984–1989 to 71.7 % during 2000–2004 (Chaturvedi et al. 2011; Gillison et al. 2012). Epidermodysplasia verruciformis (EV) is an extremely rare autosomal recessive genetic hereditary skin disorder associated with a high risk of skin carcinoma and is characterized by high susceptibility to HPV5 and HPV8 of the skin (Orth 1986). The role of HPVs in cutaneous squamous cell carcinoma (SCC) is elusive (Iannacone et al. 2013).
HPV infection is transmitted through skin abrasions (skin warts), by sexual intercourse, during passage through an infected birth canal (juvenile RRP), and probably in other ways. Women with multiple sex partners or a history of prostitution have an increased risk of infection with HPVs and an increased risk of cervical cancer. Women with history of cervical cancer (or pre-cancer) also have an increased risk of anal cancer (Anal Cancer Essay 00022 10/10). Receptive anal intercourse increases the risk of anal cancer in both men and women. Oral HPV infection is related to oral sex behavior. Most HPV infections are transient and have either no viremia or only a minimal viremic phase. Thus, HPVs are not disseminated in general to other sites by blood in the course of HPV infections. However, detection of HPV DNA in human peripheral blood mononuclear cells and the findings of papillomavirus productive infection in lymphocytes, placenta, and bovine fetal tissues indicate that hematogeneous and vertical spread of HPVs should be carefully investigated (Freitas et al. 2013; Roperto et al. 2011).
4 Pathogenesis and Immune Responses of HPV Infections
After HPVs enter epithelial cells in the wound basal layer, virus infection is established by initiation of viral gene expression and then DNA replication. HPV life cycle is tightly linked to cell differentiation, and the time from infection to release virus can be approximately 3 weeks. Viral early gene expression and initial viral DNA replication occur in undifferentiated cells in the lower layers of the infected skin or cervix, while late gene expression and vegetative DNA replication occur in highly differentiated cells in the upper layers of the infected skin or cervix (Fig. 7.3). Although HPVs do not induce a lytic virus infection or cytolysis/cell death, viral early gene expression causes cell cycle interruption, apoptosis, keratin network collapse, DNA damage, genome instability, viral genome integration into host genome, and cell immortalization and transformation. The viral late gene expression leads to maturation of virus particles which appear as aggregates in the infected cell nuclei.
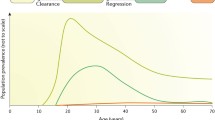
Even though most HPV infections are asymptomatic and cause no clinical problems, chronic or persistent cervical or anal HPV infection may result in histologic changes, with the infected cells displaying koilocytosis and nuclear inclusion bodies. The presence of koilocytes is a characteristic of HPV infection in all precancerous lesions and can be found in cervical smears. The period between infection and first appearance of lesions is highly variable and can range from weeks to months. HPV-induced histologic changes are classified as cervical intraepithelial neoplasias (CIN) grades 1, 2, or 3 on the basis of increasing degree of abnormality of cell growth in the cervical epithelium (Massad et al. 2013) (Cervical Cancer Essay 00021 40/40). The likelihood of spontaneous clearance versus progression to cancer in the absence of treatment varies for CIN1, CIN2, and CIN3. Histology CIN1 or cytology low-grade squamous intraepithelial lesion (LSIL) indicates abnormal cell growth that is confined to the basal 1/3 of the epithelium, usually clears spontaneously (80 % of cases) and rarely (<1 %) progresses to cancer. About 10–20 % of women with CINl progress to CIN2 and CIN3, which correspond to cytology high-grade squamous intraepithelial lesion (HSIL). CIN2 and CIN3 have a lower percentage (~40 %) of spontaneous clearance and a higher (~10 %) percentage rate of progression to cancer if not treated (Schiffman et al. 2007). CIN2 and CIN3 are distinguished by the extent of neoplasia, which is confined to the basal 2/3 of the epithelium in CIN2 or more than 2/3 in CIN3. CIN3 may involve the full thickness of the epithelium and is sometimes referred to as “cervical carcinoma in situ.” Expression of viral E6 and E7 in the infected epithelial cells inhibits cell differentiation and induces cell immortalization and transformation, resulting in disruption of virus productive life cycle and leading to viral genome integration into host genome. Although the integration of host genome is random in general, the circular HPV genome is commonly disrupted at E1 or E2 region for the integration. Thus, the degree of E1 or E2 integrity would reflect HPV genome status in cervical cancer tissues.
Because HPVs cause infection that is largely if not exclusively limited to the epithelium, they are largely shielded from the host immune response. To date, the correlates of immunity to HPV infection remain elusive. The majority of HPV infections are transient and cause no clinical problems. Most of new HPV infections clear within 1–2 years. The median duration of new infections is 6–18 months (Schiffman et al. 2007). However, the women with HIV/AIDS or immunosuppressed transplant patients take longer time to clear their HPV infection (Koshiol et al. 2006; Moscicki et al. 2004). Not all infected persons have antibodies. Approximately 60 % of women with incident HPV infections may have antibodies. The median time to seroconversion after a new infection is approximately 8 months (Ho et al. 2004). Among serum antibodies against many different viral products, the best characterized and most type-specific antibodies are those directed against conformational epitopes of the L1 protein.
HPV inhibits innate immunity and evades the host immune system. HPV infection suppresses interferon (IFN) synthesis and signaling (Ronco et al. 1998) and the expression of MHC class I in the infected cells (Bottley et al. 2008), resulting in reduced recognition of the infected cells by CD8+ T cells (Campo et al. 2010) or exclusion of CD8+ T cells from the dysplastic epithelium of HPV16-associated cervical lesions (Trimble et al. 2010). HPV-specific CD4 T cells isolated from lymph node biopsies of cervical cancer patients also suppress proliferation and cytokine (IFN-gamma, IL-2) production by responder T cells (van der Burg et al. 2007). However, T cells do play a role in preventing persistent HPV infection and inducing wart regression (Coleman et al. 1994; Nicholls et al. 2001). Healthy individuals with HPV infections display E6-specific memory T-helper cells in their blood. The majority of subjects clearing HPV16 display an HPV16 E6-specific cytotoxic T-lymphocyte (CTL) response. Failure of this response is associated with the development of cervical cancer. A significant CD8+ T-cell tumor infiltration with a higher CD8+/CD4+ and/or CD8+/regulatory T-cell ratio prevents the tumor cells from metastasizing to the tumor-draining lymph node. Moreover, T-cell defects due to a mutation in a ras homolog gene family member H (RHOH) gene and MST1, EVER1, and EVER2 deficiencies also lead to persistent EV-HPV infections (Crequer et al. 2012a, b).
5 Diagnosis of HPV Infections
5.1 Exfoliated Cell Cytology and Tissue Biopsy
Pap smear, also called a cervical smear or smear test, is a screening test to check for changes in the cervical cells from the outer opening of the cervix; it is done to identify individuals who can benefit from procedures to prevent progress to cervical cancer (Cervical Cancer Essay 00021 40/40). The test was invented by and named after Georgios Papanikolaou. Cervical lavages are performed by a colposcopist by rinsing the opening of the ectocervix with 10 mL of sterile physiologic saline for the cervical cytology. The cytological changes that can be observed include atypical squamous cells of undetermined significance (ASC-US), atypical squamous cells—cannot exclude HSIL (ASC-H), low-grade squamous intraepithelial lesion (LGSIL or LSIL), high-grade squamous intraepithelial lesion (HGSIL or HSIL), squamous cell carcinoma, and atypical glandular cells not otherwise specified (AGC or AGC-NOS) (Nayar and Solomon 2004).
Dysplasia seen on a biopsy of the cervix is grouped histologically into three categories: CIN I—mild dysplasia; CIN II—moderate to marked dysplasia; CIN III—severe dysplasia to carcinoma in situ (Massad et al. 2013; Wright et al. 2007).
5.2 Electron Microscopy
HPVs cannot be grown by conventional tissue culture methods. HPV infection can be diagnosed by electron microscopy, in which virus particles in clinical specimens can be visualized either by negative staining or thin-sectioning techniques. In thin sections of human skin warts, papillomavirus particles can be found in aggregates in an infected nucleus.
5.3 Viral Antigen Detection
HPV infection can also be diagnosed by detection of HPV proteins in infected tissues or exfoliated cells. In general, HPV L1 is detectable in cervical smears in most high-risk HPV-associated LSIL, but becomes undetectable in most of HSIL cases (Rauber et al. 2008).
Viral E2, E4, and E7 proteins have been detected from formalin-fixed paraffin-embedded cervical cancer tissues or cervical smears. Type-specific HPV E4 and E7 antibodies are particularly useful in distinguishing HPV type-specific infection of HPV16, HPV18, HPV58, and other genotypes by immunohistochemistry, ELISA, or Western blot (Griffin et al. 2012; Lidqvist et al. 2012; Xue et al. 2010; Ehehalt et al. 2012).
5.4 Viral DNA and RNA detection
Qualitative or semi-quantitative DNA or RNA tests for diagnosis of high-risk HPV infection have been developed that are based on the sequence conservation and variation of viral L1 coding regions. To date, four FDA-approved HPV DNA tests, one FDA-approved RNA test, and two Europe-approved HPV DNA tests are commercially available. These include (1) Hybrid Capture 2 (HC2) HPV DNA test (Digene Corp., Gaithersburg, MD), (2) Cervista HPV HR test (Hologic, Bedford, MA), (3) Cervista HPV 16/18 test (Hologic, Bedford, MA), (4) Cobas 4800 HPV test (Roche, Pleasanton, CA), (5) Aptima HPV assay (Gen-Probe, San Diego, CA), (6) Linear Array HPV Genotyping Test (Roche Molecular Systems Inc, Alameda, CA), and (7) INNO-LiPA HPV Genotyping Extra (Innogenetics, Ghent, Belgium) (Poljak et al. 2012).
The HC2 HPV DNA test is an in vitro nucleic acid hybridization assay with signal amplification using microplate chemiluminescence for the qualitative detection of 13 high-risk types of HPV (16, 18, 31, 33, 35, 39, 45, 51, 52, 56, 58, 59, 68) and 5 low-risk types of HPV (6, 11, 42–44). This DNA assay was the first FDA-approved HPV DNA test and has become the standard in many countries widely used in clinical studies. The HC2 distinguishes between the low-risk and high-risk groups, with the detection limits of ~5,000 genome copies, but can’t determine the specific HPV genotype
The Cervista HPV HR test and Cervista HPV16/18 test are two HPV DNA tests for high-risk HPV infections, but have no genotyping capability. The Cervista HPV HR test is an in vitro diagnostic test for detection of 14 high-risk HPV types (16, 18, 31, 33, 35, 39, 45, 51, 52, 56, 58, 59, 66, 68) in cervical specimens. The Cervista HPV HR test uses the invader chemistry and is a signal amplification method for detecting specific nucleic acid sequences.
The Cobas 4800 high-risk HPV test uses fluorescence signal to detect nucleic acids amplified by using real-time PCR methodology and detects 14 high-risk HPV types (16, 18, 31, 33, 35, 39, 45, 51, 52, 56, 58, 59, 66, 68). The Cobas 4800 system, consisting of two separate instruments (Cobas z 480 and Cobas x 480 analyzers), integrates sample preparation, amplification and detection, and result management. It has high throughput (designed to process up to 280 samples per day) and is automated.
The Aptima HPV assay detects viral E6/E7 mRNA transcripts of 14 high-risk HPVs (16, 18, 31, 33, 35, 39, 45, 51, 52, 56, 58, 59, 66, 68), but has no genotyping capability. The Aptima HPV assay uses the Gen-Probe’s Tigris DTS System, a fully automated testing system, and is a transcription-mediated amplification-based assay. The amplicons are then detected by hybridization protection with chemiluminescent-labeled single-stranded nucleic acid probes that are complementary to the amplicons.
The Linear Array HPV Genotyping Test is used in the European Union for detection and genotyping of 37 high- and low-risk HPVs (6, 11, 16, 18, 26, 31, 33, 35, 39, 40, 42, 45, 51, 52, 53, 54, 55, 56, 58, 59, 61, 62, 64, 66, 67, 68, 69, 70, 71, 72, 73 (MM9), 81, 82 (MM4), 83 (MM7), 84 (MM8), IS39, and CP6108). The test utilizes amplification of target DNA by PCR with PGMY09/11 primers and then hybridizes the amplicons to multiple HPV genotype-specific probes fixed on a membrane strip.
INNO-LiPA HPV Genotyping Extra uses the principles of reverse line blot hybridization. The test amplifies HPV DNA with SPF10 primers at the L1 region and then hybridizes the amplicons to the probes fixed on membrane strips in sequence-specific lines. The INNO-LiPA test detects and distinguishes 28 low- and high-risk HPVs including 18 high-risk HPVs (16, 18, 26, 31, 33, 35, 39, 45, 51–53, 56, 58, 59, 66, 68, 73, 82), 7 low-risk HPVs (6, 11, 40, 43, 44, 54, 70), and 3 additional genotypes (69, 71, 74).
5.5 Serology
HPV serology is a valuable tool to study immune status prior to and after HPV vaccination. Although most HPV infections are cleared spontaneously within 2 years, the majority of infected subjects develop and maintain serum antibodies to viral L1 for up to five subsequent years. Serology is not commonly used for clinical diagnosis because more than 40 % of women do not seroconverted after HPV infection. The standard methods are VLP ELISA and pseudovirion neutralization assays (http://home.ccr.cancer.gov/lco/NeutralizationAssay.htm). Both methods have similar sensitivity and specificity in the detection of serum L1 antibodies in the majority of infected subjects. There is recent evidence that antibody to HPV16 E6 can be predictive of subsequent development of oropharyngeal or anal cancer with a high degree of specificity, and this will be an important area for future research (Kreimer et al. 2013).
5.6 Detection of Cellular Surrogates of High-risk E6 and E7 in Infected Tissues
A few cellular biomarkers of high-risk HPV E6 and E7 expression have been widely used to indicate viral oncoprotein expression in pre-cancer lesions or cancers. These include cell cycle inhibitors (p16Ink4a and p18Ink4c), MCM7 (minichromosome maintenance protein 7), cyclin E2, and a subset of miRNAs (Zheng and Wang 2011; Wang et al. 2011b, 2014; Klaes et al. 2001; Middleton et al. 2003).
6 Epidemiology and Prevention of HPV Infections
The global prevalence of HPV infection among women with normal cytology is around 11–12 %, with a higher rate in Africa, Caribbean, and South America and a lower rate in Asia, Europe and Northern America. The prevalence of any HPV is about 26.8 % in females aged 14–59 and 44.8 % among women aged 20–24 years in the United States (Dunne et al. 2007). The majority of HPV infections are transient and asymptomatic; 70 % of new HPV infections clear within 1 year, and approximately 90 % clear within 2 years. The median duration of new infections is 8 months. Up to 50 % of young women with one type of HPV infection may acquire another type of HPV infection within a few years. Although co-infections with multiple high-risk HPVs may lead to an increased risk of CIN2/3, persistent infection with any high-risk HPV is the most important risk factor for cervical lesions and cervical cancer. The risk of cancer development varies by HPV type, with HPV16 being more oncogenic than other high-risk HPV types. Factors associated with cervical cancer (Cervical Cancer Essay 00021 40/40) also include an increased number of life-time sexual partners, increased age, other sexually transmitted infections, immune suppression, and other host factors. There are also epidemiology reports of smoking being associated with increased cervical cancer, although it isn’t clear if this may be due to association with other risk factor (Haverkos et al. 2003). The time between initial HPV infection and development of cervical cancer is usually decades. Many aspects of the natural history of HPV remain to be understood, including the role and duration of naturally acquired immunity after HPV infection.
Because HPV is the most common sexually transmitted infectious agent, condom use and circumcision are two common practices to prevent sexual partners from HPV transmission. Pap smear screening is widely used in developed world to check for changes in the cervical cells from the outer opening of the cervix and to prevent progress to cervical cancer by treatment of these lesions. While likely, it is not known if screening for and treatment of anal HSIL will similarly prevent anal cancer; the United States National Cancer Institute has recently initiated a large randomized clinical trial to study this issue. Two FDA-licensed prophylactic HPV vaccines, Gardasil from Merck (USA) and Cervarix from GlaxoSmithKline (UK), are very safe and effective in preventing new or persistent HPV infections and reducing HPV infection-induced cervical, vulvar, vaginal, anal, and penile lesions. Their use will be an important public health measure to prevent HPV-associated cancers (Schiller et al. 2012). Both vaccines are comprised of viral DNA-free, L1 VLP. Cervarix contains L1 VLP from HPV16 and 18 produced in insect cells, while Gardasil is comprised of L1 VLP from HPV6, 11, 16 and 18 produced in yeast. Because it contains L1 VLP from HPV6 and HPV11, Gardasil immunization also provides protection against genital warts (Donovan et al. 2011). In the presence of adjuvant aluminates, both the quadrivalent Gardasil and the bivalent Cervarix are highly immunogenic and excellent HPV type-specific protection is provided to girls and boys at age 11 or 12 years who receive all three vaccine doses (0, 1, and 6 month). Catch-up vaccination with either one of HPV vaccines is approved by the US FDA for young women through age 26 or young men through age 21 (Cervical Cancer Essay 00021 40/40).
Due to the limited inclusion of HPV types, the current vaccines do not provide cross-immune protection against other high-risk mucosal HPVs. However, there is evidence to suggest that HPV L2 might work as a pan-HPV vaccine against different HPVs. A small N-terminal portion of L2 is well-conserved among different HPVs, and experimental vaccines targeting these conserved domains offer protection against a broad range of HPV types (Jagu et al. 2013b).
Most of therapeutic vaccine approaches target high-risk HPV E6, E7 or both to control disease progression in preexisting HPV infections and lesions. To date, these studies have provided only a gleam of success. An experimental HPV16 E6 and E7 synthetic long peptide vaccine can increase the number and activity of HPV16-specific CD4+ and CD8+ T cells in patients with vulvar intraepithelial neoplasia or cervical cancer (Welters et al. 2008; Kenter et al. 2009). HPV VLP entry into NK cells triggers cytotoxic activity and cytokine secretion (Renoux et al. 2011), although VLP does not activate Langerhans cells (Fausch et al. 2002). A therapeutic HPV16/18 DNA vaccine with optimized E6 and E7 codons stimulate high titers of anti-E6/E7 antibodies and high levels of cellular immunity by both CD8+ and CD4+ T cells (Bagarazzi et al. 2012).
7 Conclusion
HPVs have been well recognized as a group of small DNA tumor viruses that are highly transmissible through sexual or close contact. In the past decades, HPV infection has been found in association with development of various human cancers, including cervical, anogenital, oropharyngeal, and even some skin cancers. Although there is more to be learned about the basic biology of HPV, the translational research on HPVs has remarkably advanced our understanding of HPV epidemiology, disease progression, clinical diagnosis, and cervical cancer prevention. Today, L1-based HPV vaccination has become a general practice and has been mandated in many countries to protect girls and boys from HPV infections and HPV-related precancerous lesions and cancer.
References
Abbate EA, Berger JM, Botchan MR. The X-ray structure of the papillomavirus helicase in complex with its molecular matchmaker E2. Genes Dev. 2004;18:1981–96.
Abbate EA, Voitenleitner C, Botchan MR. Structure of the papillomavirus DNA-tethering complex E2:Brd4 and a peptide that ablates HPV chromosomal association. Mol Cell. 2006;24: 877–89.
Androphy EJ, Lowy DR, Schiller JT. Bovine papillomavirus E2 trans-activating gene product binds to specific sites in papillomavirus DNA. Nature. 1987;325:70–3.
Avvakumov N, Torchia J, Mymryk JS. Interaction of the HPV E7 proteins with the pCAF acetyltransferase. Oncogene. 2003;22:3833–41.
Bagarazzi ML et al. Immunotherapy against HPV16/18 generates potent TH1 and cytotoxic cellular immune responses. Sci Transl Med. 2012;4:155ra138.
Bechtold V, Beard P, Raj K. Human papillomavirus type 16 E2 protein has no effect on transcription from episomal viral DNA. J Virol. 2003;77:2021–8.
Bell I, Martin A, Roberts S. The E1circumflexE4 protein of human papillomavirus interacts with the serine-arginine-specific protein kinase SRPK1. J Virol. 2007;81:5437–48.
Ben KY et al. The human papillomavirus E6 oncogene represses a cell adhesion pathway and disrupts focal adhesion through degradation of TAp63beta upon transformation. PLoS Pathog. 2011;7:e1002256.
Bernard HU, Burk RD, Chen Z, van Doorslaer K, zur Hausen H, de Villiers EM. Classification of papillomaviruses (PVs) based on 189 PV types and proposal of taxonomic amendments. Virology. 2010;401:70–9.
Bernat A, Avvakumov N, Mymryk JS, Banks L. Interaction between the HPV E7 oncoprotein and the transcriptional coactivator p300. Oncogene. 2003;22:7871–81.
Bienkowska-Haba M, Williams C, Kim SM, Garcea RL, Sapp M. Cyclophilins facilitate dissociation of the human papillomavirus type 16 capsid protein L1 from the L2/DNA complex following virus entry. J Virol. 2012;86:9875–87.
Bishop B et al. Crystal structures of four types of human papillomavirus L1 capsid proteins: understanding the specificity of neutralizing monoclonal antibodies. J Biol Chem. 2007;282:31803–11.
Blachon S, Bellanger S, Demeret C, Thierry F. Nucleo-cytoplasmic shuttling of high risk human Papillomavirus E2 proteins induces apoptosis. J Biol Chem. 2005;280:36088–98.
Bodaghi S, Jia R, Zheng ZM. Human papillomavirus type 16 E2 and E6 are RNA-binding proteins and inhibit in vitro splicing of pre-mRNAs with suboptimal splice sites. Virology. 2009;386(1):32–43.
Bonagura VR et al. Recurrent respiratory papillomatosis: a complex defect in immune responsiveness to human papillomavirus-6 and -11. APMIS. 2010;118:455–70.
Bottley G, Watherston OG, Hiew YL, Norrild B, Cook GP, Blair GE. High-risk human papillomavirus E7 expression reduces cell-surface MHC class I molecules and increases susceptibility to natural killer cells. Oncogene. 2008;27:1794–9.
Bousarghin L, Touze A, Combita-Rojas AL, Coursaget P. Positively charged sequences of human papillomavirus type 16 capsid proteins are sufficient to mediate gene transfer into target cells via the heparan sulfate receptor. J Gen Virol. 2003;84:157–64.
Bronnimann MP, Chapman JA, Park CK, Campos SK. A transmembrane domain and GxxxG motifs within L2 are essential for papillomavirus infection. J Virol. 2013;87:464–73.
Buck CB et al. Arrangement of L2 within the papillomavirus capsid. J Virol. 2008;82:5190–7.
Campo MS et al. HPV-16 E5 down-regulates expression of surface HLA class I and reduces recognition by CD8 T cells. Virology. 2010;407:137–42.
Chaturvedi AK et al. Human papillomavirus and rising oropharyngeal cancer incidence in the United States. J Clin Oncol. 2011;29:4294–301.
Chen XS, Garcea RL, Goldberg I, Casini G, Harrison SC. Structure of small virus-like particles assembled from the L1 protein of human papillomavirus 16. Mol Cell. 2000;5:557–67.
Coleman N et al. Immunological events in regressing genital warts. Am J Clin Pathol. 1994;102:768–74.
Cote-Martin A et al. Human papillomavirus E1 helicase interacts with the WD repeat protein p80 to promote maintenance of the viral genome in keratinocytes. J Virol. 2008;82:1271–83.
Crequer A et al. Inherited MST1 deficiency underlies susceptibility to EV-HPV infections. PLoS One. 2012a;7:e44010.
Crequer A et al. Human RHOH deficiency causes T cell defects and susceptibility to EV-HPV infections. J Clin Invest. 2012b;122:3239–47.
Crow JM. HPV: the global burden. Nature. 2012;488:S2–3.
Danos O, Katinka M, Yaniv M. Human papillomavirus 1a complete DNA sequence: a novel type of genome organization among papovaviridae. EMBO J. 1982;1:231–6.
Dasgupta J et al. Structural basis of oligosaccharide receptor recognition by human papillomavirus. J Biol Chem. 2011;286:2617–24.
Davy CE et al. Identification of a G(2) arrest domain in the E1 wedge E4 protein of human papillomavirus type 16. J Virol. 2002;76:9806–18.
Day PM, Baker CC, Lowy DR, Schiller JT. Establishment of papillomavirus infection is enhanced by promyelocytic leukemia protein (PML) expression. Proc Natl Acad Sci U S A. 2004;101:14252–7.
de Sanjose S et al. Human papillomavirus genotype attribution in invasive cervical cancer: a retrospective cross-sectional worldwide study. Lancet Oncol. 2010;11:1048–56.
de Villiers EM, Fauquet C, Broker TR, Bernard HU, zur Hausen H. Classification of papillomaviruses. Virology. 2004;324:17–27.
DeFilippis RA, Goodwin EC, Wu L, DiMaio D. Endogenous human papillomavirus E6 and E7 proteins differentially regulate proliferation, senescence, and apoptosis in HeLa cervical carcinoma cells. J Virol. 2003;77:1551–63.
Desaintes C, Hallez S, Van Alphen P, Burny A. Transcriptional activation of several heterologous promoters by the E6 protein of human papillomavirus type 16. J Virol. 1992;66:325–33.
Donovan B et al. Quadrivalent human papillomavirus vaccination and trends in genital warts in Australia: analysis of national sentinel surveillance data. Lancet Infect Dis. 2011;11:39–44.
Doorbar J et al. Detection of novel splicing patterns in a HPV16-containing keratinocyte cell line. Virology. 1990;178:254–62.
Doorbar J, Ely S, Sterling J, McLean C, Crawford L. Specific interaction between HPV-16 E1-E4 and cytokeratins results in collapse of the epithelial cell intermediate filament network. Nature. 1991;352:824–7.
Doorbar J et al. The E1E4 protein of human papillomavirus type 16 associates with a putative RNA helicase through sequences in its C terminus. J Virol. 2000;74:10081–95.
Duensing S et al. The human papillomavirus type 16 E6 and E7 oncoproteins cooperate to induce mitotic defects and genomic instability by uncoupling centrosome duplication from the cell division cycle. Proc Natl Acad Sci U S A. 2000;97:10002–7.
Dunne EF et al. Prevalence of HPV infection among females in the United States. JAMA. 2007;297:813–9.
Durst M, Gissmann L, Ikenberg H, zur Hausen H. A papillomavirus DNA from a cervical carcinoma and its prevalence in cancer biopsy samples from different geographic regions. Proc Natl Acad Sci U S A. 1983;80:3812–5.
Dziduszko A, Ozbun MA. Annexin A2 and S100A10 regulate human papillomavirus type 16 entry and intracellular trafficking in human keratinocytes. J Virol. 2013;87:7502–15.
Egawa N et al. The E1 protein of human papillomavirus type 16 is dispensable for maintenance replication of the viral genome. J Virol. 2012;86:3276–83.
Ehehalt D et al. Detection of human papillomavirus type 18 E7 oncoprotein in cervical smears: a feasibility study. J Clin Microbiol. 2012;50:246–57.
Fausch SC, Da Silva DM, Rudolf MP, Kast WM. Human papillomavirus virus-like particles do not activate Langerhans cells: a possible immune escape mechanism used by human papillomaviruses. J Immunol. 2002;169:3242–9.
Finnen RL, Erickson KD, Chen XS, Garcea RL. Interactions between papillomavirus L1 and L2 capsid proteins. J Virol. 2003;77:4818–26.
Flores ER, Lambert PF. Evidence for a switch in the mode of human papillomavirus type 16 DNA replication during the viral life cycle. J Virol. 1997;71:7167–79.
Fradet-Turcotte A, Bergeron-Labrecque F, Moody CA, Lehoux M, Laimins LA, Archambault J. Nuclear accumulation of the papillomavirus E1 helicase blocks S-phase progression and triggers an ATM-dependent DNA damage response. J Virol. 2011;85:8996–9012.
Freitas AC, Mariz FC, Silva MA, Jesus AL. Human papillomavirus vertical transmission: review of current data. Clin Infect Dis. 2013;56:1451–6.
French D et al. Expression of HPV16 E5 down-modulates the TGFbeta signaling pathway. Mol Cancer. 2013;12:38.
Frisch M et al. Sexually transmitted infection as a cause of anal cancer. N Engl J Med. 1997;337:1350–8.
Ganguly N. Human papillomavirus-16 E5 protein: oncogenic role and therapeutic value. Cell Oncol (Dordr). 2012;35:67–76.
Geimanen J et al. Development of a cellular assay system to study the genome replication of high- and low-risk mucosal and cutaneous human papillomaviruses. J Virol. 2011;85: 3315–29.
Gillison ML et al. Human papillomavirus and diseases of the upper airway: head and neck cancer and respiratory papillomatosis. Vaccine. 2012;30 Suppl 5:F34–54.
Griffin H et al. E4 antibodies facilitate detection and type-assignment of active HPV infection in cervical disease. PLoS One. 2012;7:e49974.
Haverkos HW, Soon G, Steckley SL, Pickworth W. Cigarette smoking and cervical cancer: Part I: a meta-analysis. Biomed Pharmacother. 2003;57:67–77.
Hawley-Nelson P, Androphy EJ, Lowy DR, Schiller JT. The specific DNA recognition sequence of the bovine papillomavirus E2 protein is an E2-dependent enhancer. EMBO J. 1988;7:525–31.
Ho GY, Studentsov YY, Bierman R, Burk RD. Natural history of human papillomavirus type 16 virus-like particle antibodies in young women. Cancer Epidemiol Biomarkers Prev. 2004;13:110–6.
Hoskins EE et al. The fanconi anemia pathway limits human papillomavirus replication. J Virol. 2012;86:8131–8.
Huang SM, McCance DJ. Down regulation of the interleukin-8 promoter by human papillomavirus type 16 E6 and E7 through effects on CREB binding protein/p300 and P/CAF. J Virol. 2002;76:8710–21.
Huh K et al. Human papillomavirus type 16 E7 oncoprotein associates with the cullin 2 ubiquitin ligase complex, which contributes to degradation of the retinoblastoma tumor suppressor. J Virol. 2007;81:9737–47.
Huibregtse JM, Scheffner M, Howley PM. Cloning and expression of the cDNA for E6-AP, a protein that mediates the interaction of the human papillomavirus E6 oncoprotein with p53. Mol Cell Biol. 1993;13:775–84.
Iannacone MR et al. Case-control study of cutaneous human papillomavirus infection in Basal cell carcinoma of the skin. J Invest Dermatol. 2013;133:1512–20.
Imai Y, Tsunokawa Y, Sugimura T, Terada M. Purification and DNA-binding properties of human papillomavirus type 16 E6 protein expressed in Escherichia coli. Biochem Biophys Res Commun. 1989;164:1402–10.
Jabbar SF et al. Cervical cancers require the continuous expression of the human papillomavirus type 16 E7 oncoprotein even in the presence of the viral E6 oncoprotein. Cancer Res. 2012;72:4008–16.
Jackson S, Harwood C, Thomas M, Banks L, Storey A. Role of Bak in UV-induced apoptosis in skin cancer and abrogation by HPV E6 proteins. Genes Dev. 2000;14:3065–73.
Jagu S et al. Optimization of multimeric human papillomavirus L2 vaccines. PLoS One. 2013a;8:e55538.
Jagu S et al. Phylogenetic considerations in designing a broadly protective multimeric L2 vaccine. J Virol. 2013b;87:6127–36.
Jia R et al. Control of the papillomavirus early-to-late switch by differentially expressed SRp20. J Virol. 2009;83:167–80.
Johansson C et al. HPV-16 E2 contributes to induction of HPV-16 late gene expression by inhibiting early polyadenylation. EMBO J. 2012;31:3212–27.
Kenter GG et al. Vaccination against HPV-16 oncoproteins for vulvar intraepithelial neoplasia. N Engl J Med. 2009;361:1838–47.
Khan J et al. Role of calpain in the formation of human papillomavirus type 16 E1^E4 amyloid fibers and reorganization of the keratin network. J Virol. 2011;85:9984–97.
Kim SH et al. Human papillomavirus 16 E5 up-regulates the expression of vascular endothelial growth factor through the activation of epidermal growth factor receptor, MEK/ ERK1,2 and PI3K/Akt. Cell Mol Life Sci. 2006;63:930–8.
Kiyono T, Hiraiwa A, Fujita M, Hayashi Y, Akiyama T, Ishibashi M. Binding of high-risk human papillomavirus E6 oncoproteins to the human homologue of the Drosophila discs large tumor suppressor protein. Proc Natl Acad Sci U S A. 1997;94:11612–6.
Klaes R et al. Overexpression of p16(INK4A) as a specific marker for dysplastic and neoplastic epithelial cells of the cervix uteri. Int J Cancer. 2001;92:276–84.
Klingelhutz AJ, Foster SA, McDougall JK. Telomerase activation by the E6 gene product of human papillomavirus type 16. Nature. 1996;380:79–82.
Koshiol JE et al. Time to clearance of human papillomavirus infection by type and human immunodeficiency virus serostatus. Int J Cancer. 2006;119:1623–9.
Krawczyk E et al. Koilocytosis: a cooperative interaction between the human papillomavirus E5 and E6 oncoproteins. Am J Pathol. 2008;173:682–8.
Krawczyk E, Suprynowicz FA, Sudarshan SR, Schlegel R. Membrane orientation of the human papillomavirus type 16 E5 oncoprotein. J Virol. 2010;84:1696–703.
Kreimer AR et al. Evaluation of human papillomavirus antibodies and risk of subsequent head and neck cancer. J Clin Oncol. 2013;31:2708–15.
Kumar A et al. Human papillomavirus oncoprotein E6 inactivates the transcriptional coactivator human ADA3. Mol Cell Biol. 2002;22:5801–12.
Kusumoto-Matsuo R, Kanda T, Kukimoto I. Rolling circle replication of human papillomavirus type 16 DNA in epithelial cell extracts. Genes Cells. 2011;16:23–33.
Lai MC, Teh BH, Tarn WY. A human papillomavirus E2 transcriptional activator. The interactions with cellular splicing factors and potential function in pre-mRNA processing. J Biol Chem. 1999;274:11832–41.
Lee SS, Weiss RS, Javier RT. Binding of human virus oncoproteins to hDlg/SAP97, a mammalian homolog of the Drosophila discs large tumor suppressor protein. Proc Natl Acad Sci U S A. 1997;94:6670–5.
Lee JO, Russo AA, Pavletich NP. Structure of the retinoblastoma tumour-suppressor pocket domain bound to a peptide from HPV E7. Nature. 1998;391:859–65.
Lehoux M, Fradet-Turcotte A, Lussier-Price M, Omichinski JG, Archambault J. Inhibition of human papillomavirus DNA replication by an E1-derived p80/UAF1-binding peptide. J Virol. 2012;86:3486–500.
Li M, Cripe TP, Estes PA, Lyon MK, Rose RC, Garcea RL. Expression of the human papillomavirus type 11 L1 capsid protein in Escherichia coli: characterization of protein domains involved in DNA binding and capsid assembly. J Virol. 1997;71:2988–95.
Li S et al. The human papilloma virus (HPV)-18 E6 oncoprotein physically associates with Tyk2 and impairs Jak-STAT activation by interferon-alpha. Oncogene. 1999;18:5727–37.
Li H, Ou X, Xiong J, Wang T. HPV16E7 mediates HADC chromatin repression and downregulation of MHC class I genes in HPV16 tumorigenic cells through interaction with an MHC class I promoter. Biochem Biophys Res Commun. 2006;349:1315–21.
Lichtig H et al. HPV16 E6 augments Wnt signaling in an E6AP-dependent manner. Virology. 2010;396:47–58.
Lidqvist M et al. Detection of human papillomavirus oncoprotein E7 in liquid-based cytology. J Gen Virol. 2012;93:356–63.
Lipovsky A et al. Genome-wide siRNA screen identifies the retromer as a cellular entry factor for human papillomavirus. Proc Natl Acad Sci U S A. 2013;110:7452–7.
Massad LS et al. 2012 updated consensus guidelines for the management of abnormal cervical cancer screening tests and cancer precursors. Obstet Gynecol. 2013;121:829–46.
Massimi P, Pim D, Banks L. Human papillomavirus type 16 E7 binds to the conserved carboxy-terminal region of the TATA box binding protein and this contributes to E7 transforming activity. J Gen Virol. 1997;78(Pt 10):2607–13.
Maufort JP, Shai A, Pitot HC, Lambert PF. A role for HPV16 E5 in cervical carcinogenesis. Cancer Res. 2010;70:2924–31.
McBride AA. Replication and partitioning of papillomavirus genomes. Adv Virus Res. 2008;72: 155–205.
McLaughlin-Drubin ME, Munger K. The human papillomavirus E7 oncoprotein. Virology. 2009;384:335–44.
McLaughlin-Drubin ME, Crum CP, Munger K. Human papillomavirus E7 oncoprotein induces KDM6A and KDM6B histone demethylase expression and causes epigenetic reprogramming. Proc Natl Acad Sci U S A. 2011;108:2130–5.
Melnick JL. Papova virus group. Science. 1962;135:1128–30.
Middleton K et al. Organization of human papillomavirus productive cycle during neoplastic progression provides a basis for selection of diagnostic markers. J Virol. 2003;77:10186–201.
Moody CA, Laimins LA. Human papillomaviruses activate the ATM DNA damage pathway for viral genome amplification upon differentiation. PLoS Pathog. 2009;5:e1000605.
Moody CA, Laimins LA. Human papillomavirus oncoproteins: pathways to transformation. Nat Rev Cancer. 2010;10:550–60.
Moscicki AB, Ellenberg JH, Farhat S, Xu J. Persistence of human papillomavirus infection in HIV-infected and -uninfected adolescent girls: risk factors and differences, by phylogenetic type. J Infect Dis. 2004;190:37–45.
Muller M et al. Large scale genotype comparison of human papillomavirus E2-host interaction networks provides new insights for e2 molecular functions. PLoS Pathog. 2012;8:e1002761.
Munger K, Werness BA, Dyson N, Phelps WC, Harlow E, Howley PM. Complex formation of human papillomavirus E7 proteins with the retinoblastoma tumor suppressor gene product. EMBO J. 1989;8:4099–105.
Munoz N et al. Epidemiologic classification of human papillomavirus types associated with cervical cancer. N Engl J Med. 2003;348:518–27.
Nakahara T, Nishimura A, Tanaka M, Ueno T, Ishimoto A, Sakai H. Modulation of the cell division cycle by human papillomavirus type 18 E4. J Virol. 2002;76:10914–20.
Nayar R, Solomon D. Second edition of ‘The Bethesda System for reporting cervical cytology’ - atlas, website, and Bethesda interobserver reproducibility project. Cytojournal. 2004;1:4.
Nelson LM, Rose RC, Moroianu J. Nuclear import strategies of high risk HPV16 L1 major capsid protein. J Biol Chem. 2002;277:23958–64.
Nicholls PK et al. Regression of canine oral papillomas is associated with infiltration of CD4+ and CD8+ lymphocytes. Virology. 2001;283:31–9.
Nomine Y et al. Domain substructure of HPV E6 oncoprotein: biophysical characterization of the E6 C-terminal DNA-binding domain. Biochemistry. 2003;42:4909–17.
Orth G. Epidermodysplasia verruciformis: a model for understanding the oncogenicity of human papillomaviruses. Ciba Found Symp. 1986;120:157–74.
Orth G, Favre M, Croissant O. Characterization of a new type of human papillomavirus that causes skin warts. J Virol. 1977;24:108–20.
Parish JL et al. E2 proteins from high- and low-risk human papillomavirus types differ in their ability to bind p53 and induce apoptotic cell death. J Virol. 2006;80:4580–90.
Patel D, Huang SM, Baglia LA, McCance DJ. The E6 protein of human papillomavirus type 16 binds to and inhibits co- activation by CBP and p300. EMBO J. 1999;18:5061–72.
Pim D, Banks L. Interaction of viral oncoproteins with cellular target molecules: infection with high-risk vs low-risk human papillomaviruses. APMIS. 2010;118:471–93.
Poljak M, Cuzick J, Kocjan BJ, Iftner T, Dillner J, Arbyn M. Nucleic acid tests for the detection of alpha human papillomaviruses. Vaccine. 2012;30 Suppl 5:F100–6.
Rampias T et al. Activation of Wnt signaling pathway by human papillomavirus E6 and E7 oncogenes in HPV16-positive oropharyngeal squamous carcinoma cells. Mol Cancer Res. 2010;8: 433–43.
Rauber D, Mehlhorn G, Fasching PA, Beckmann MW, Ackermann S. Prognostic significance of the detection of human papilloma virus L1 protein in smears of mild to moderate cervical intraepithelial lesions. Eur J Obstet Gynecol Reprod Biol. 2008;140:258–62.
Reinson T et al. Engagement of the ATR-dependent DNA damage response at the human papillomavirus 18 replication centers during the initial amplification. J Virol. 2013;87:951–64.
Renoux VM et al. Human papillomavirus entry into NK cells requires CD16 expression and triggers cytotoxic activity and cytokine secretion. Eur J Immunol. 2011;41:3240–52.
Richards RM, Lowy DR, Schiller JT, Day PM. Cleavage of the papillomavirus minor capsid protein, L2, at a furin consensus site is necessary for infection. Proc Natl Acad Sci U S A. 2006;103: 1522–7.
Ristriani T et al. HPV oncoprotein E6 is a structure-dependent DNA-binding protein that recognizes four-way junctions. J Mol Biol. 2000;296:1189–203.
Ristriani T, Nomine Y, Masson M, Weiss E, Trave G. Specific recognition of four-way DNA junctions by the C-terminal zinc-binding domain of HPV oncoprotein E6. J Mol Biol. 2001;305: 729–39.
Romanczuk H, Thierry F, Howley PM. Mutational analysis of cis elements involved in E2 modulation of human papillomavirus type 16 P97 and type 18 P105 promoters. J Virol. 1990;64:2849–59.
Ronco LV, Karpova AY, Vidal M, Howley PM. Human papillomavirus 16 E6 oncoprotein binds to interferon regulatory factor-3 and inhibits its transcriptional activity. Genes Dev. 1998;12: 2061–72.
Roperto S et al. PBMCs are additional sites of productive infection of bovine papillomavirus type 2. J Gen Virol. 2011;92:1787–94.
Sakakibara N, Mitra R, McBride AA. The papillomavirus E1 helicase activates a cellular DNA damage response in viral replication foci. J Virol. 2011;85:8981–95.
Schafer F, Florin L, Sapp M. DNA binding of L1 is required for human papillomavirus morphogenesis in vivo. Virology. 2002;295:172–81.
Schiffman M, Castle PE, Jeronimo J, Rodriguez AC, Wacholder S. Human papillomavirus and cervical cancer. Lancet. 2007;370:890–907.
Schiller JT, Castellsague X, Garland SM. A review of clinical trials of human papillomavirus prophylactic vaccines. Vaccine. 2012;30 Suppl 5:F123–38.
Schneider MA et al. The transcription factors TBX2 and TBX3 interact with human papillomavirus 16 (HPV16) L2 and repress the long control region of HPVs. J Virol. 2013;87:4461–74.
Shai A, Brake T, Somoza C, Lambert PF. The human papillomavirus E6 oncogene dysregulates the cell cycle and contributes to cervical carcinogenesis through two independent activities. Cancer Res. 2007;67:1626–35.
Shai A, Pitot HC, Lambert PF. E6-associated protein is required for human papillomavirus type 16 E6 to cause cervical cancer in mice. Cancer Res. 2010;70:5064–73.
Shin MK, Sage J, Lambert PF. Inactivating all three rb family pocket proteins is insufficient to initiate cervical cancer. Cancer Res. 2012;72:5418–27.
Sousa R, Dostatni N, Yaniv M. Control of papillomavirus gene expression. Biochim Biophys Acta. 1990;1032:19–37.
Spangle JM, Munger K. The human papillomavirus type 16 E6 oncoprotein activates mTORC1 signaling and increases protein synthesis. J Virol. 2010;84:9398–407.
Spangle JM, Munger K. The HPV16 E6 oncoprotein causes prolonged receptor protein tyrosine kinase signaling and enhances internalization of phosphorylated receptor species. PLoS Pathog. 2013;9:e1003237.
Straight SW, Hinkle PM, Jewers RJ, McCance DJ. The E5 oncoprotein of human papillomavirus type 16 transforms fibroblasts and effects the downregulation of the epidermal growth factor receptor in keratinocytes. J Virol. 1993;67:4521–32.
Strauss MJ, Bunting H, Melnick JL. Virus-like particles and inclusion bodies in skin papillomas. J Invest Dermatol. 1950;15:433–44.
Swindle CS, Zou N, Van Tine BA, Shaw GM, Engler JA, Chow LT. Human papillomavirus DNA replication compartments in a transient DNA replication system. J Virol. 1999;73:1001–9.
Tang S, Tao M, McCoy Jr JP, Zheng ZM. The E7 oncoprotein is translated from spliced E6*I transcripts in high-risk human papillomavirus type 16- or type 18-positive cervical cancer cell lines via translation reinitiation. J Virol. 2006;80:4249–63.
Tao M, Kruhlak M, Xia S, Androphy E, Zheng ZM. Signals that dictate nuclear localization of human papillomavirus type 16 oncoprotein E6 in living cells. J Virol. 2003;77:13232–47.
Thomas MC, Chiang CM. E6 oncoprotein represses p53-dependent gene activation via inhibition of protein acetylation independently of inducing p53 degradation. Mol Cell. 2005;17:251–64.
Todorovic B, Massimi P, Hung K, Shaw GS, Banks L, Mymryk JS. Systematic analysis of the amino acid residues of human papillomavirus type 16 E7 conserved region 3 involved in dimerization and transformation. J Virol. 2011;85:10048–57.
Todorovic B et al. Conserved region 3 of human papillomavirus 16 E7 contributes to deregulation of the retinoblastoma tumor suppressor. J Virol. 2012;86:13313–23.
Trimble CL et al. Human papillomavirus 16-associated cervical intraepithelial neoplasia in humans excludes CD8 T cells from dysplastic epithelium. J Immunol. 2010;185:7107–14.
van der Burg SH et al. Association of cervical cancer with the presence of CD4+ regulatory T cells specific for human papillomavirus antigens. Proc Natl Acad Sci U S A. 2007;104:12087–92.
Veldman T, Horikawa I, Barrett JC, Schlegel R. Transcriptional activation of the telomerase hTERT gene by human papillomavirus type 16 E6 oncoprotein. J Virol. 2001;75:4467–72.
Veldman T, Liu X, Yuan H, Schlegel R. Human papillomavirus E6 and Myc proteins associate in vivo and bind to and cooperatively activate the telomerase reverse transcriptase promoter. Proc Natl Acad Sci U S A. 2003;100:8211–6.
Wang X et al. Oncogenic HPV infection interrupts the expression of tumor-suppressive miR-34a through viral oncoprotein E6. RNA. 2009;15:637–47.
Wang X, Meyers C, Wang HK, Chow LT, Zheng ZM. Construction of a full transcription map of human papillomavirus type 18 during productive viral infection. J Virol. 2011a;85:8080–92.
Wang X, Meyers C, Guo M, Zheng ZM. Upregulation of p18Ink4c expression by oncogenic HPV E6 via p53-miR-34a pathway. Int J Cancer. 2011b;129:1362–72.
Wang X, Helfer CM, Pancholi N, Bradner JE, You J. Recruitment of Brd4 to the human papillomavirus type 16 DNA replication complex is essential for replication of viral DNA. J Virol. 2013;87:3871–84.
Wang X et al. miRNAs are biomarkers of oncogenic HPV infections. Proc Natl Acad Sci U S A. 2014;111(11):4262–7.
Weijzen S, Zlobin A, Braid M, Miele L, Kast WM. HPV16 E6 and E7 oncoproteins regulate Notch-1 expression and cooperate to induce transformation. J Cell Physiol. 2003;194:356–62.
Welters MJ et al. Induction of tumor-specific CD4+ and CD8+ T-cell immunity in cervical cancer patients by a human papillomavirus type 16 E6 and E7 long peptides vaccine. Clin Cancer Res. 2008;14:178–87.
Wetherill LF et al. High-risk human papillomavirus E5 oncoprotein displays channel-forming activity sensitive to small-molecule inhibitors. J Virol. 2012;86:5341–51.
White EA, Kramer RE, Tan MJ, Hayes SD, Harper JW, Howley PM. Comprehensive analysis of host cellular interactions with human papillomavirus E6 proteins identifies new E6 binding partners and reflects viral diversity. J Virol. 2012a;86:13174–86.
White EA et al. Systematic identification of interactions between host cell proteins and E7 oncoproteins from diverse human papillomaviruses. Proc Natl Acad Sci U S A. 2012b;109:E260–7.
Woodham AW et al. The S100A10 subunit of the annexin A2 heterotetramer facilitates L2-mediated human papillomavirus infection. PLoS One. 2012;7:e43519.
Wright Jr TC, Massad LS, Dunton CJ, Spitzer M, Wilkinson EJ, Solomon D. 2006 consensus guidelines for the management of women with cervical intraepithelial neoplasia or adenocarcinoma in situ. J Low Genit Tract Dis. 2007;11:223–39.
Xue Y et al. HPV16 E2 is an immediate early marker of viral infection, preceding E7 expression in precursor structures of cervical carcinoma. Cancer Res. 2010;70:5316–25.
You J, Croyle JL, Nishimura A, Ozato K, Howley PM. Interaction of the bovine papillomavirus E2 protein with Brd4 tethers the viral DNA to host mitotic chromosomes. Cell. 2004;117:349–60.
Zanier K et al. Solution structure analysis of the HPV16 E6 oncoprotein reveals a self-association mechanism required for E6-mediated degradation of p53. Structure. 2012;20:604–17.
Zanier K et al. Structural basis for hijacking of cellular LxxLL motifs by papillomavirus E6 oncoproteins. Science. 2013;339:694–8.
Zhang B, Srirangam A, Potter DA, Roman A. HPV16 E5 protein disrupts the c-Cbl-EGFR interaction and EGFR ubiquitination in human foreskin keratinocytes. Oncogene. 2005;24:2585–8.
Zheng ZM. Viral oncogenes, noncoding RNAs, and RNA splicing in human tumor viruses. Int J Biol Sci. 2010;6:730–55.
Zheng ZM, Baker CC. Papillomavirus genome structure, expression, and post-transcriptional regulation. Front Biosci. 2006;11:2286–302.
Zheng ZM, Wang X. Regulation of cellular miRNA expression by human papillomaviruses. Biochim Biophys Acta. 2011;1809:668–77.
Zhou J, Sun XY, Louis K, Frazer IH. Interaction of human papillomavirus (HPV) type 16 capsid proteins with HPV DNA requires an intact L2 N-terminal sequence. J Virol. 1994;68:619–25.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2014 Springer Science+Business Media New York
About this chapter
Cite this chapter
Zheng, ZM. (2014). Human Papillomaviruses. In: Yarchoan, R. (eds) Cancers in People with HIV and AIDS. Springer, New York, NY. https://doi.org/10.1007/978-1-4939-0859-2_7
Download citation
DOI: https://doi.org/10.1007/978-1-4939-0859-2_7
Published:
Publisher Name: Springer, New York, NY
Print ISBN: 978-1-4939-0858-5
Online ISBN: 978-1-4939-0859-2
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)