
Abstract
Asthma is increasingly recognised as a heterogeneous disease, with multiple phenotypes that differ in severity, pathology, therapeutic response and long-term outcome. A combination of genetic, epigenetic and environmental factors are thought to contribute to the molecular diversity of the disease, with sensitisation (T-cell differentiation) dependent on the local microenvironment and the nature of the invading pathogen. A strong body of evidence exists associating numerous environmental and genetic components in asthma development, with multiple asthma genes involved independently (through the inheritance of polymorphisms) or through the interaction with the environment to increase risk. However, the inability to reproduce inheritance patterns and the dramatic increase in incidence over the last decade provides strong evidence that changes in the environment have activated a pre-existing susceptibility, including the alteration in epigenetic regulation, to play an important role in disease. The role of epigenetic regulation and modulation in the development of asthma and allergy has been widely speculated. Interestingly, factors known to be involved in disease susceptibility including genetic predisposition and exposure to environmental stimuli (in utero and post-natal) have been explored as factors involved in the mechanisms associated with the epigenome. Thus, it is proposed that modification of the epigenome in the regulation of important pathways, including those involved in asthma-associated gene expression and T-cell differentiation play a direct role in disease. In addition, current research focuses on the central role of the airway epithelium in asthma development and progression. Inherently defective in disease, the mechanisms associated with epithelial dysfunction, including the increased susceptibility to injury and the inability to activate normal repair processes are yet to be completely elucidated. Trefoil factor 2 (TFF2), previously shown to be upregulated in asthma and involved in airway epithelial restitution fails to protect the epithelium from pathogen-induced injury. By focusing on the role of epigenetic mechanisms, the epithelium and TFF2 in asthma pathogenesis, this chapter highlights their potential as targets in future therapeutic research.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others

Keywords
9.1 Introduction
The prevalence, complexity and severity of allergic disorders, including asthma continues to increase on a global scale (Pawankar et al. 2012). According to the World Health Organisation, 300 million people worldwide are afflicted with asthma, a disease that remains a heavy burden on health-care systems as these numbers steadily rise (Weinberg 2011). The dramatic increase in incidence over the last 20 years provides strong evidence that changes in the environment (in utero and post-natal pathogen exposure) have activated a pre-existing susceptibility, including the alteration in epigenetic regulation, to play an important role in disease development (Yang and Schwartz; Yuyama et al. 2002). In addition, the airway epithelium has proven to be central in its progression and pathogenesis, providing a link between the development of the disease and the clinically significant events characteristic of human asthma (Hackett and Knight 2007). By focusing on trefoil factor 2 (TFF2), a protein shown to be upregulated in asthma and involved in airway epithelial restitution, from an endogenous mechanistic aspect highlights the requirement to elucidate the role of the epithelium in asthma, and provides information into targeting the epithelium in future therapeutic research.
9.2 Asthma, Airway Remodelling and Disease Phenotypes
To date, asthma is defined as an inflammatory respiratory disease, characterised by sudden, chronic symptoms of wheezing, sputum production, variable reversible airflow limitation and airway hyperresponsiveness (AHR) (Bousquet et al. 2000). As a highly heterogeneous disease of the respiratory tract, current management strategies are inadequate in their ability to prevent the development of the disease. Anti-inflammatory corticosteroids are the most effective and therefore, most widely prescribed therapeutic agent in response to active inflammation during exacerbations (Murata and Ling). However, a subset of patients with chronic, severe disease may be classified as corticosteroid resistant, as clinical symptoms persist despite high dosage levels (Barnes and Adcock 2009).
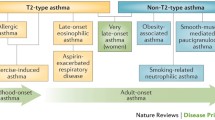
Molecular heterogeneity between human asthma patients, including differences observed in the immune response following allergen exposure, is thought to contribute to corticosteroid-resistance and the inability to establish the exact mechanisms involved in disease development and progression (Woodruff et al. 2009). Allergic asthma is characterised by the production of Th2 cells and its associated cytokines in response to allergen exposure in atopic individuals, contributing to 75–80 % of all asthmatic patients (Holgate 2008) (Fig. 9.1). Interestingly however, it has been shown that over 40 % of the Western population is classified as atopic with only approximately 7 % of those expressing atopy in the form of asthma (Beasley et al. 1989). Recent developments also suggest that a significant proportion of severe, allergic asthma cases are driven by alternative inflammatory pathways (Prescott 2006), including that driven by Th1 and Th17 cells (Fig. 9.1). Although, atopic asthma is managed well with corticosteroids generally, it has been shown that cases mediated by Th1- and Th17-immune pathways are largely corticosteroid resistant, with persistence of clinically relevant symptoms including AHR (Robins et al.; Yang et al. 2009; McKinley et al. 2008; Cui et al. 2005). These pathways appear to derive from a common naive precursor cell, whose differentiation pathway is determined by cytokine, environmental, genetic and epigenetic signals during primary antigenic stimulation and sensitisation (Bluestone et al. 1995; Abbas et al. 1996; O’Garra et al. 1998). Collectively, the inherent immune and phenotypic heterogeneity observed in human asthma highlights the importance of understanding the mechanisms involved in the development of disease, with rationale centred on establishing a common factor in disease pathogenesis for future, target-based therapeutic research. In this chapter, two pathways will be explored in detail—the role of epigenetic mechanisms in the development of asthma, and the involvement of the airway epithelium in disease progression.

The phenotypic subtypes of asthma. During early sensitisation, caused by a combination of epigenetic, genetic and environmental factors, T-cells undergo differentiation dependent on the local microenvironment and the nature of the invading pathogen. Exposure to allergens including house dust mite and pollens direct naive T-cells to differentiate along a Th2 pathway, with individuals developing atopy. This type of asthma which involves the recruitment of mast cells and eosinophils is most common, and also the most likely to be controlled with current corticosteroid treatment. Conversely, individuals’ exposure to pollutants and irritants usually in utero and post-natal development cause naive T-cells to differentiate into Th1 of Th17 cells. Recruitment of neutrophils in this immune pathway causes a non-allergic asthma phenotype, with most cases persisting into adulthood, and more severe with corticosteroid-resistance
A number of pathologically significant events are also thought to contribute to the limitations in current asthma treatment, with the inability of corticosteroids to reverse and/or prevent airway remodelling observed in both childhood and adult cases of chronic, severe disease (Holgate et al.). The structural alterations associated include epithelial goblet cell metaplasia, collagen deposition and thickening of the subepithelial lamina reticularis with increased matrix deposition, smooth muscle hyperplasia and hypertrophy, and angiogenesis (Vignola et al. 2000). The exact mechanisms that contribute to the process of airway remodelling in asthma are yet to be elucidated, with current knowledge indicating an interrelationship with inflammation and AHR. Until recently, these structural alterations have been considered to be a secondary phenomenon, developing late in disease progression as a direct consequence of persistent inflammation (Fedorov et al. 2005). Although this is observed in a subset of patients with late-onset disease, airway remodelling has also been shown to be a consistent feature of childhood asthma independent of inflammation, with no requirement for concurrent eosinophil infiltration of the airway tissue (Payne et al. 2003; Prescott 2006). In order to explain this observation, it is proposed that the alterations in airway structure are, at least in part, regulated through the ability of epithelial cells to communicate with the underlying mesenchyme to maintain and propagate remodelling and inflammatory responses throughout the airway wall (Davies 2009; Hackett 2012).
9.3 Epigenetic Mechanisms and the Development of Asthma
There is a strong body of evidence associating numerous environmental and genetic components in asthma development. Changes in the primary DNA sequence of genes involved in both immune and inflammatory pathways are either independently associated with asthma, or interact with the environment to affect disease risk. However, due to the irreproducibility of inheritance patterns and the dramatic increase in asthma incidence in a relatively short period of time, the ability of these factors to affect and be affected by epigenetic mechanisms have brought about an alternative explanation for disease development (Fig. 9.2).

Venn diagram illustrating the relationship between environmental exposures, epigenetic modulation and genetic susceptibility in the development of asthma. These factors are interrelated, with many of the environmental factors displayed also affecting the epigenome. In addition, gene expression including those genes known to be involved in asthma susceptibility, including ADAM33 is subject to epigenetic modulation. A combination of these factors is thought to cause disease development, with the exact contribution of each factor yet to be elucidated
9.3.1 Genetic Basis of Asthma: Heritability and Susceptibility
A complex heritable disease, asthma has a strong familial connection (36–79 % heritability) with a non-Mendelian pattern of inheritance and polymorphisms in more than 100 genes (Ober and Yao 2011; Vercelli 2008). The genes involved in asthma susceptibility may be categorised into four main groups: (1) genes associated with immunoregulation, (2) genes associated with Th-cell differentiation, (3) genes associated with epithelial structure and function and (4) genes associated with lung function, remodelling and disease severity (Vercelli 2008). In addition to these genes, positional cloning studies have found other gene polymorphisms involved in asthma development and susceptibility including ADAM33 and TFF2 (Zhang et al. 2012; Allen et al. 2003). Interestingly, asthma shows a parent-of-origin transmission of inheritance, with an affected mother significantly more likely to transmit the disease than an affected father (Demenais et al. 2001). Similar to the mode of inheritance observed for multiple epigenetic mechanisms, a number of known genes associated with asthma are transmitted in a parent-of-origin fashion, including the FCERIB locus (Sandford et al. 2000) and the Spink5gene (Liu et al. 2009). Yet the inability to replicate these associations between differing studies, the diverse nature of genes subject to asthma- and allergy-related polymorphisms and the finding that genetic susceptibility may explain the cause of disease in only a subset of patients, implicate other mechanisms, such as epigenetics, in the development and progression of disease.
In order to appreciate the potential importance of epigenetic modulation in the development of asthma, numerous studies have established that DNA methylation is involved in the differential expression of disease-associated genes ADAM33, ALOX12 and PTGDR (Yang et al. 2008; Morales et al. 2012; Isidoro-García et al. 2011). Polymorphisms of ADAM33, found by means of positional cloning, are strongly associated with asthma and AHR (Van Eerdewegh et al. 2002). Through the analysis of bronchial biopsy samples from human subjects, it was found that the ADAM33 gene contains a regulatory CpG island within its promoter (Yang et al. 2008). Bisulfite sequencing demonstrated that this region is subject to epigenetic (methylation) modulation, with the region hypomethylated in ADAM33-expressing fibroblasts and hypermethylated in epithelial cell which fail to express the gene. Recent advances in epigenetic research in asthma have also linked methylation modulation of ALOX12 gene expression in children with or without persistent wheeze (Morales et al. 2012). Furthermore, interplay between genetic polymorphisms and methylation levels were observed, with genetic variation influencing ALOX12 methylation. In another study, expression of the asthma susceptibility gene PTGDR is shown to be regulated by epigenetic mechanisms, with allergic asthma patients exhibiting a distinct methylation pattern (demethylation of promoter) as compared to controls (Isidoro-García et al. 2011). Assessment of the important asthma susceptibility locus 17q21 also presents the interrelationship between genetic polymorphisms and epigenetic regulation (Berlivet et al. 2012). Using lymphoblastoid cell lines, this study showed that the locus harbours three genes—ZPBP2, GSDMB and ORMDL3 which exhibit allele-specific differences in gene expression levels determined by distinct polymorphism and DNA methylation profiles in vitro. Although the exact role of ADAM33, ALOX12, PTGDR and locus 17q21 in asthma development is yet to be completely elucidated, the link between their expression regulated by DNA methylation, and their involvement in disease susceptibility provides rationale to further research the epigenetic mechanisms involved in asthma pathogenesis.
9.3.2 Gene–Environment Interactions in Asthma Development and the Role of Epigenetic Regulation
Exposure to various environmental pollutants and pathogens in utero and in post-natal (early childhood) development is thought to be a central factor in asthma pathogenesis. Besides studies involving the complex interaction between genetic susceptibility and the environment, many of the factors associated with asthma development including exposure to cigarette smoke, air pollutants and microbial allergens have been shown to be involved in epigenetic modulation.
As the most important risk factor for asthma development in children, and its known ability to alter epigenetic mechanisms, extensive research can be found involving the exposure to cigarette smoke and its role in disease pathogenesis (Lodrup Carlsen and Carlsen 2001). Direct inhalation of cigarette smoke components, including 7,12-dimethylbenz{a}anthracene (DMBA) was found to alter DNA methylation patterns in mouse lung (Phillips and Goodman 2009). Several studies have also shown that in utero cigarette smoke exposure affects DNA methylation in multiple tissues, including the placenta which resulted in CpG methylation and expression of numerous genes involved in the oxidative stress pathway in smokers, compared to levels measured in placenta from non-smokers (Suter et al. 2011). In addition to this, an association between in utero exposure to cigarette smoke and the development of asthma has been extensively researched (Xepapadaki et al. 2009; Lee et al. 2012). Genes involved in airway inflammation, such as AXL and PTPRO were found to be methylated in response to in utero cigarette smoke exposure in buccal DNA of the offspring (Breton et al. 2009). Furthermore, studies have demonstrated histone modification associations between environmental exposure and asthma risk. A significant reduction of HDAC2 expression in the airways of rats and different strains of mice following cigarette smoke exposure produced similar results to HDAC2 expression levels observed relatively in asthmatic smokers (Yang et al. 2006; Cosio et al. 2004). Adenuga et al., found that exposure to cigarette smoke to mice caused a decrease in HDAC1 and HDAC2 activity in the lung through the ability of cigarette components to induce phosphorylation and serine/threonine proteasomal degradation (Adenuga et al. 2009). Collectively, the link between epigenetic modulation with cigarette smoke exposure and asthma risk is displayed, with many of the epigenetic mechanisms observed in smokers, also observed in asthma patients.
Air pollution may also impact asthma development via pollutant-induced and pollutant-regulated epigenetic modulation. Research demonstrates that in utero exposure to airborne polycyclic aromatic hydrocarbons (PAH) correlates to asthma in offspring, caused by DNA methylation of CpG regions in the ACSL3 promoter (Perera et al. 2009). In addition, 3-year PAH exposure was associated with higher methylation of FoxP3, reduced Treg function and increased asthma severity in children with asthma (Nadeau et al. 2010). The association between in utero exposure to dichlorodiphenyldichloroethylene (DDE) and asthma in children (Sunyer et al. 2005) has been established, with levels of DDE in cord blood within the Menorca cohort inversely correlated with DNA methylation in one of the ALOX12 (previously described) CpG regions (Morales et al. 2012). Furthermore, diesel exhaust particulate (DEP) matter has been reported to induce airway inflammation and exacerbate asthma in vivo (Cao et al. 2007). This same study also indicates that DEP exposure induces COX-2 gene expression through the acetylation of histone H4, the degradation of HDAC1 and the recruitment of histone acetyltransferase (HAT) p300 in association with the COX2 promoter. Children exposed to particulate air pollution display reduced methylation in a CpG site of the NOS2 gene promoter region (Salam et al. 2012). Involved in nitric oxide synthesis, promoter methylation in NOS2 effects exhaled nitric oxide (FeNO) with higher methylation in ARG1 and ARG2 also associated with lower FeNO, and a strong correlation produced in children with asthma (Breton et al. 2011). Recent studies have also demonstrated that lower methylation of the NOS2 and IL-6 gene promoter in nasal epithelium is associated with higher FeNO in children with asthma (Baccarelli et al. 2012).
Early life bacterial and viral infections have thought to contribute to asthma development, with subsequent infections resulting in disease exacerbations following initial sensitization and exposure. In a murine Aspergillus fumigatus-sensitised murine model, exposure to A. fumigatus in conjunction with DEP resulted in methylation of several CpG sites at the Ifng promoter and demethylation at one IL-4 promoter with methylation modulation correlating significantly with changes in IgE expression and production (Liu et al. 2008). Stimulation of human CD4+ T-cells derived from asthma patients with the allergen components of house dust mite led to demethylation in several CpG sites in the IL-4 promoter and increased expression levels of IL-4 in supernatant when compared to those control non-atopic subjects (Kwon et al. 2008). Stimulation of multiple T-cell and monocytic cell lines with lipopolysaccharide enhanced IL-8 release through toll-like receptor-4, which induces histone H3 and H4 acetylation at the IL-8 promoter (Tsaprouni et al. 2007).
In addition to the research involved in establishing the role of epigenetic modulation in enhancing asthma susceptibility, environmental factors including the exposure to specific bacterial and viral infections, may protect from disease development. Exposure to farm life in early development (in utero and post-natal) significantly reduces the risk of asthma and allergy (Schaub et al. 2009; Ege et al. 2011). An abundance of bacteria and their components, including endotoxin is found in such an environment, with the risk of asthma development decreasing with exposure to high concentrations of endotoxin, and the microbial diversity of the environment (Ege et al. 2011). Furthermore, cord blood from neonates subjected to an in utero and post-natal rural environment possessed increased numbers and activity of Treg cells, FoxP3 demethylation and increased FoxP3 gene expression when compared to levels observed in non-rural neonates (Schaub et al. 2009). Another study found that when pregnant mice were exposed to farm-derived gram-negative bacterium Acinetobacter lwoffii F78, and subsequently challenged offspring with ovalbumin (OVA) to induce an allergic airways disease phenotype, supernatant from splenic mononuclear cells had significantly increased levels of IFN-γ and reduction of IL-4, IL-5 and IL-13 expression (Brand et al. 2011). In utero exposure to protective bacteria was also demonstrated to be mediated through the modifications of histone H4 at the Ifng promoter in mice, with reduced acetylation of H4 in those offspring unexposed and prevention of this reduced acetylation in those exposed. Inhibition of acetylation in H4 was found to increase airway inflammation and AHR in OVA-challenged mice through its ability to regulate IFN-γ production.
9.3.3 In Utero
Exposures including those previously mentioned have been known to modulate the epigenome and enhance susceptibility to asthma development in offspring. Further investigation of these exposures, including that of the maternal diet has been shown to be risk factors in disease pathogenesis. Hollingsworth et al. displayed in a murine model the importance of dietary methyl donors in disease, with exposure to these donors associated with increased allergic airway inflammation (Hollingsworth et al. 2008). The same study aimed to explain such a phenomenon, with airway inflammation thought to be mediated by the increased methylation of RUNX3 gene. Given the finding that RUNX3-deficient mice possess elevated levels of serum IgE and increased AHR (Fainaru et al. 2005), inhibition of RUNX3 gene expression through DNA methylation may be associated with asthma development. Further research is required to fully elucidate the role of this gene in disease.
9.3.4 Epigenetic Regulation and Modulation of the Immune System in Allergy and Asthma
As an immune-mediated disease, asthma is characterised by the ability of T-cells to differentiate mainly towards a Th2-phenotype (observed in allergic asthma), although other subtypes including Th1 and Th17 may be involved in non-allergic cases (Lloyd and Hessel 2010). As discussed previously, these cases are thought to be more severe, non-resoluting and resistant to corticosteroid treatment. Important pathways in the immune response, such as T-cell differentiation and Treg function have been shown to be regulated by epigenetic mechanisms (Jones and Chen 2006; White et al. 2006). Multiple studies have shown that differentiation of naive T-cells into mature T cells is accompanied by changes in both methylation status and chromatin structure in cytokine genes dependent on the subtype (Table 9.1) (Lovinsky-Desir et al. 2012). Maturation of naive T-cells into Th2 cells involves the modification of epigenomic structure of IL-4 and IL-13 genes, while differentiation into Th1 cells is accompanied by epigenomic changes in the structure of the IFN-γ locus (Agarwal et al. 1998).
Specifically, research suggests that Th1-cell differentiation is accompanied by the progressive demethylation of CpG sites in the Ifng promoter (White et al. 2006), in addition to methylation of a highly conserved DNase I-hypersensitive region at the 3′ end of the IL-4 locus (Lee et al. 2002). Reports also indicate that in Th1 cells, hyperacetylation of histones H3 and H4 occur at the Ifng locus via a Stat4 and T-bet-dependent mechanism (Chang et al. 2008), with no such pattern observed at the IL-4 locus (Fields et al. 2002). Chang et al. also found that inhibition of histone deacetylase (HDAC) through the T-bet-dependent removal of Sin3A-HDAC complexes, in naive T cells stimulates acquisition of H4 acetylation, Ifng transcription and ultimately, Th1 differentiation (Chang et al. 2008).
Conversely, Th2-cell differentiation is accompanied by the appearance of DNase I hypersensitive sites with demethylation around these sites within the IL-4 and IL-13 promoters (Santangelo et al. 2002), and the increased methylation of the Ifng promoter (Jones and Chen 2006). To detail the pathway involved in Th2 differentiation, Kim et al. 2010) reports that STAT6, an important gene in signalling IL-4 transcription, possesses enhanced susceptibility to DNA methylation (Kim and Lee 2011). Another study indicates that the RAD50-hypersensitive site 7 within the Th2 cytokine locus control region (LCR) is subject to demethylation in a STAT6-dependent manner and only in cells stimulated under conditions similar to that during Th2 differentiation (Kim et al. 2007). Chromatin modifications are also thought to contribute to the epigenetic mechanisms regulating differentiation of naive T-cells into Th2-cells. Acetylation of histones H3 and H4 in the IL-4 and IL-13 region (Avni et al. 2002) through GATA3 regulation is found to be important in transcription of Th2-associated genes (Fields et al. 2002). Furthermore, chromatin remodelling of the Th2 LCR gene is important in the regulation and coordination of Th2 cytokine production, airway inflammation and AHR (Koh et al.). The importance of individual HDACs in T-cell differentiation has also been established, with a loss of HDAC1 displaying an increase in airway inflammation, mucus hypersecretion and AHR (Grausenburger et al. 2010). It was also demonstrated in this study that HDAC1 is recruited to the IL-4 gene locus, indicating a role of HDAC1 in regulating IL-4 production. Recent advances have indicated that there are functionally relevant epigenetic pathways involved in T-cell subset differentiation. Allan et al. explored the role of the SUV39H1-H3K9me3-HP1α silencing pathway in the control of Th2-lineage stability (Allan et al. 2012). This pathway involves the histone methylase SUV29H1, which regulates the methylation of histone H3 on the HP1α locus, promotes transcriptional silencing of Th1 loci, and ensures naive T-cells follow a pathway towards Th2 differentiation.
A role for Th17 cells and their associated cytokines have been implicated in the immunopathology of asthma (Doe et al. 2010). Similar to that observed in Th1- and Th2-differentiation, Th17-cell differentiation is found to be regulated, at least in part, by epigenetic mechanisms. TGF-β and IL-6/IL-21 have been shown to induce Th17 cell differentiation (Mangan et al. 2006), with gene transcription regulated by RORγt and Stat5 (Ivanov et al. 2006; Laurence et al. 2007). Modulations in chromatin structure of Th17-associated genes have been established in multiple studies. In one report, methylation of histone H3K4 independent of any repressive H3K27 modifications are found at the IL17a/IL17f gene promoters (Wei et al. 2009) in Th17 differentiation. Another study observed an increase in histone H3 acetylation at the IL17a/IL17f gene promoters as a result of the Th17 lineage pathway (Akimzhanov et al. 2007). Both papers showed significant H3K27 methylation within the Ifng promoter, with an absence of H3 acetylation and H3K4 methylation also observed in Th17-cells (Wei et al. 2009; Akimzhanov et al. 2007). Mukasa et al., established that chromatin structure of the gene loci involved in Th17 cell differentiation, including IL17a/IL17f, Ifng and Rorc, are not stable, but change in response to environmental stimuli (Mukasa et al.). These authors also report that TGF-β (an important cytokine implicated in asthma pathogenesis) induces an increase in IL17a/IL17f gene expression through the enhanced methylation of histone H3K4 across the locus. The ability of the local cellular environment, in conjugation with chromatin remodelling, to regulate Th17 cell differentiation may indicate an important relationship between disease states and physiological conditions, although this requires extensively more research.
Regulatory T-cells (Tregs) are central in the maintenance of self-tolerance, with the transcription factor FoxP3 crucial for the regulation and expression of active Tregs (Ansel et al. 2003). Through the regulation of FoxP3 by epigenetic mechanisms, the investigation of Treg cell differentiation has been explored by multiple studies. In short, complete demethylation of the conserved FoxP3 promoter region provides stability to FoxP3 expression and commitment to the Treg phenotype in humans (Janson et al. 2008). Regulation of the methylation profile in FoxP3 gene is dependent on STAT3, a downstream target for IL-6 during allergic airway inflammation in a murine model following OVA sensitisation (Doganci et al. 2005). Interestingly, the FoxP3 methylation profile characterises and distinguishes natural Tregs (found in the thymus) from those induced by TGF-β (found in the periphery) (Lal et al. 2009). Specifically, this paper reports that the upstream enhancer in the FoxP3 gene is demethylated in natural Tregs (with acetylation of histone H3) and methylated in TGF-β-induced Tregs. TGF-β, a major cytokine implicated in the pathogenesis of asthma including the progression to airway remodelling, has been shown to have the ability to modulate the methylation profile of FoxP3 by decreasing methylation in the first intronic CpG region of the gene (Kim and Leonard 2007). In addition, following T-cell receptor signalling, an increased binding to cyclic-AMP response element-binding protein/activating transcription factor was observed, and in this study led to increased expression of FoxP3 gene. In the context of allergic asthma, authors used purified T-cells from the spleen and lymph nodes of mice induced to display an allergic airways disease phenotype (through the administration of OVA) (Kim et al. 2010). Results indicated that memory Th2 cells had the ability to redifferentiate into functional FoxP3+ Treg cells by TGF-β when stimulated in the presence of retinoic acid and rapamycin.
The role of epigenetics in the development of asthma and allergy has been widely speculated, with research aimed to establish its significance. Interestingly, factors known to be involved in asthma aetiology including, genetic susceptibility and exposure to environmental stimuli (in utero and post-natal), have been explored as factors involved in modulating the epigenome. Due to this, it is proposed that epigenetic modulation and regulation of important pathways, including those involved in gene expression and T-cell differentiation play a direct role in disease development. Figure 9.2 summarises the interrelationship between epigenetic mechanisms, environmental stimuli and genetic susceptibility, with all three factors understood to possess a role, at least in part, to asthma pathogenesis. However, the exact role of each factor and the precise relationship between such factors are yet to be completely established and require further investigation.
9.4 Mechanisms Involved in the Development and Progression of Asthma: The Airway Epithelium
Under normal circumstances, the airway epithelium containing ciliated columnar, mucus-secreting goblet and surfactant-secreting, progenitor Clara cells forms a highly regulated and impermeable barrier to environmental insults during respiration (Xiao et al. 2011). This barrier serves to maintain tissue homeostasis, and when compromised by environmental pathogens activate an immunological response that aims to protect the underlying lung tissue (Davies 2009). The involvement of the epithelium in asthma pathogenesis is thought to be of primary significance, and may not simply be a consequence of chronic inflammation (Hackett and Knight 2007). During disease exacerbations, an inherently impaired epithelial barrier renders the airway susceptible to infection, which in turn stimulates sensitisation and immune activation to physiologically innocuous pathogens (Xiao et al. 2011) (Fig. 9.3). The mechanisms, which cause the epithelium to be inherently defective both in its increased susceptibility to injury and inability to activate normal repair processes, are inadequately defined. It may be plausible however, that epigenetic modulation in association with exposure to environmental stimuli may render the epithelium inherently defective during in utero and post-natal development as previously described (Hammad and Lambrecht 2008). The direct and intimate involvement of the epithelium during asthma development and disease progression is thus, an attractive target for current and future research.

The central role of the epithelium in asthma development and progression. An inherently defective epithelium thought to be caused by a combination of genetic, epigenetic and environmental factors in asthma is subject to allergen and/or pollutant sensitisation in utero and/or in post-natal development. Sensitisation and subsequent exposure to these allergens induces epithelial cell activation, with these cells possessing increased susceptibility to injury. Through the activation of epithelial cells, and the direct role of injury to the immune response following pathogen invasion, these cells in addition to other inflammatory cells including fibroblasts, macrophages, mast cells and eosinophils release multiple pro-inflammatory and pro-remodelling growth factors and cytokines in order to eliminate the incriminating pathogen and initiate repair responses in an attempt to restore the epithelium to full functionality. An upregulation of repair molecules including trefoil factor 2, a peptide involved in epithelial restitution leads to an aberrant repair process. Goblet cell metaplasia is one such consequence of this aberrant response. Collectively, these events characteristic of the inherently defective epithelium in asthma are important for the propagation and maintenance of inflammation and remodelling throughout the airway wall, with a negative feedback loop perpetuating disease
Evidence to suggest that the airway epithelium is inherently defective in human asthma patients involves global gene expression profiling of the epithelium from human disease biopsy samples in comparison to that of healthy control samples (Kicic et al. 2010). The primary disruption of epithelial tight junctions allows pathogens to infiltrate the underlying airway wall, resulting in a complex interaction with immune and inflammatory cells (Holgate 2007). Despite the passage of airway epithelial cells of human asthma patients several times, and the separation of these cells from any inflammatory cells or mediators for a prolonged period, it has been demonstrated that these cells are unable to form effectively functioning tight junctions (Wan et al. 2000). Furthermore results from the same study measured a marked reduction in transepithelial resistance, indicating an increased leakiness. Additional studies suggest that cells appear to be less differentiated (Kicic et al. 2010) and adhesion proteins including E-cadherin, essential for epithelial structure and cell-to-cell communication, are downregulated within the diseased epithelium as compared to control samples (Trautmann et al. 2005). An increased susceptibility of epithelial cells to oxidant-induced damage and apoptosis (Bucchieri et al. 2002), and the abnormal expression of several pro-inflammatory transcription factors (Sampath et al. 1999) have also been reported in asthma patients compared to healthy controls. These findings support the hypothesis that fundamental alterations in the epithelial barrier function contribute to the onset and progression of asthma (Kicic et al. 2010).
Activation of epithelial cells following environmental stimuli exposure is a key event in the recognition of pathogens that coordinate the subsequent immune response. The involvement of dendritic cells in conjunction with the inherently dysfunctional epithelium is thought to be central in allergen sensitisation and presentation of allergen to circulating T-cells (Lambrecht and Hammad 2009). Isolation of dendritic cells from the airways of mice exposed to allergen and transferred to naive mice induced the production of Th2 cells which were specific to house dust mite, which was involved in the initial sensitisation process (Hammad et al.). On the other hand, depletion of dendritic cells from the airway of both naive or allergen-sensitised mice resulted in the inability to initiate the production of Th2 cells or the development of airway inflammation when these mice were exposed to the allergen in question (van Rijt et al. 2005). Upregulation of important cytokines, including thymic stromal lymphopoietin (TSLP), granulocyte-macrophage colony-stimulating factor (GM-CSF), IL-25 and IL-33, are important in the initiation of the immune response and allergen sensitisation in asthma. Mice exposed to OVA with the overexpression of GM-CSF induce spontaneous sensitisation and a Th2-related response (Stampfli et al. 1998). Furthermore, a deficiency in GM-CSF causes a failure to sensitise mice to house dust mite allergen (Cates et al. 2004), and attenuates the ability of DEP to increase allergic sensitisation (Ohta et al. 1999). Interestingly, when human epithelial cells collected from asthma patients were cultured, results indicated that these cells continuously overproduce GM-CSF (Ritz et al. 2002) independent of allergen exposure, which suggests that the production of GM-CSF may be epigenetically regulated in disease (Ritz et al. 2002). Yet once initiated and activated, the epithelial response to environmental stimuli are maintained through the downstream effects of the initial immune response, including the production of important pro-inflammatory and pro-remodelling cytokines, providing a negative feedback loop that can perpetuate damage to the epithelium, and ultimately affect disease progression and severity.
Due to the innate dysregulation and intrinsic defection of the airway epithelium both biochemically and functionary in asthma, it has been shown that these cells have an increased susceptibility to injury. Ultrastructural changes, characterised by the sloughing of columnar cells, and goblet cell metaplasia with increased mucus secretion and airway plugging occur in the epithelium in association with damage and provide evidence of dysfunctional activation in the presence of a predetermined stimuli (Hamilton et al. 2001). Selective loss of columnar cells is observed in sufferers of asthma and, when compared with that of non-asthmatic subjects, may be increased by 15–45 % (Zhou et al. 2011). An increased susceptibility to injury is thought to be caused by the predetermined capacity of epithelial cells to activate pathways involved in the immune response. Regulation and promotion of epithelial cell activation has been shown to be caused by a combination of early life exposure to allergen and downstream signalling cascades (Lambrecht and Hammad 2012). Viral infection of the airway epithelium with respiratory syncytial virus (RSV) or cigarette smoke exposure induces the upregulation of toll-like receptor 4 (TLR4) expression and promotes its localisation to the cell membrane, which in turn increases the responsiveness of epithelial cells to endotoxin (Pace et al. 2008; Monick et al. 2003). In addition, the role of the transcription factor nuclear factor-κB (NF-κB) in the expression of multiple inflammatory cytokines and the activation of the epithelium has been established, with mice deficient in the NF-κB subunits p50 or p65 displaying reduced responses to endotoxin and exposure to other allergens (Yang et al. 1998). Newer studies also exhibited the ability of constitutively activated NF-κB within epithelial cells to activate dendritic cells, cause injury to the epithelium itself and promote sensitisation to OVA in a murine model (Ather et al. 2011). Conversely, inhibition of epithelial expression of NF-κB in mice reduced the recruitment of Th2 cells in the lung and attenuated airway remodelling (Broide et al. 2005). This increased susceptibility to damage, coupled with defective epithelium activation, results in the induction of an ineffective and impaired repair phenotype (Holgate 1998).
Under normal circumstances, a damaged epithelium is able to activate a self-repair process with efficiency (Erjeflt and Persson 1997). Injury to the airway epithelium initiates a repair pathway involving the migration, proliferation and differentiation of neighbouring undamaged cells (Holgate 2008). The ultimate outcome of such a process involves resolution and return of the tissue to its normal structure and function (Davies 2009). However, it has become increasingly apparent that this normal repair process is compromised and rendered defective in the epithelium of asthma patients. Most interesting is the finding that the expression of a large number of genes involved in the epithelial repair process was reduced (Zhang et al. 2012). Additionally, airway epithelial cells have been shown to proliferate at a faster rate than control cells, with asthma samples displaying a dramatic impairment in wound healing ability (Stevens et al. 2008). In detailed studies, epithelial wounds in airway tissue from asthma patients were unable to close after 10 days, with a subset of samples found to possess a maximum wound closure of less than 70 % 30 days post-wound induction. During normal repair processes, progenitor Clara cells have been shown to proliferate and undergo phenotypic differentiation to re-establish the integrity and functional arrangement of the epithelium (Crosby and Waters 2010). However, with chronic Clara cell depletion as observed in human samples and induced allergic airway disease (AAD) in mice, the ineffectiveness of the repair process during injury is highlighted (Perl et al. 2011). Downregulation of the primary extracellular protein fibronectin observed in diseased epithelial cells demonstrates the significant impairment of repair processes in wound healing (Kicic et al. 2010) as this protein profoundly influences the survival, proliferation and differentiation of epithelial cells in physiological conditions (Zahm et al. 1991). Knocking down of fibronectin expression in healthy airway epithelial cells in vitro resulted in a significant impairment in wound healing, with addition of fibronectin to the same cells reversing the impairment, and restoring the capacity of these cells to self-repair. Furthermore, a normal response to epithelial injury includes the upregulation of receptors that drive proliferation and repair, including members of the epithelial growth factor receptor (EGFR) family (Le Cras et al. 2011). Despite the finding that expression of EGFR is markedly increased, especially in areas of columnar cell loss (Amishima et al. 1998), this upregulation does not correlate with the proliferative response of repairing cells (Puddicombe et al. 2000). Accordingly, this data demonstrates that the response to injury and the repair process that ensues is dysregulated in the epithelium of patients diagnosed with asthma.
Following environmental stimuli-induced injury, the epithelium provides the microenvironment for persistent and chronic inflammation and the irreversible structural alterations of airway remodelling (Xiao et al. 2011). The interactions and signalling observed between epithelial, mesenchymal, neural and ECM cells are necessary to initiate numerous functions in the lung, allowing an exchange of information between these elements in response to various stimuli (Evans et al. 1999). The activation of aberrant repair pathways and the production of numerous growth factors and cytokines are thought to be central to the severity, pathology, therapeutic response and long-term outcome of disease (Holgate 1998). The mechanisms, which cause the epithelium to be inherently defective both in its increased susceptibility to injury and inability to activate normal repair processes, are inadequately defined. A combination of environmental, genetic and epigenetic factors is thought to contribute, yet their exact involvement is yet to be elucidated. The difficulty in establishing the complex interaction between these factors in the development of disease has led research to focus on targeting asthma progression and the ability to attenuate and/or reverse epithelial dysfunction.
9.5 Trefoil Factor 2
TFF2 is one of the three known mammalian trefoil peptides (White 2001). Small and protease resistant, these proteins are well established as protective molecules in the gut (Franic et al. 2005). Specifically, TFF2 is produced by intestinal epithelium and promotes repair by mediating epithelial restitution and inhibiting apoptosis (Hoffmann 2005). Studies utilising methods of gene disruption to generate mice deficient in TFF2 indicate that this protein promotes gastric mucosal healing through the inhibition of acid secretion and stimulation of mucosal proliferation (White 2001). Interestingly, TFF2 is upregulated in diverse pathologic conditions of the gastrointestinal tract, including at the sites of gastric and duodenal ulceration, and Crohn’s disease (Wright 1993). Yet despite this upregulation, the dissolution of epithelial integrity is a consistent feature of these gastrointestinal conditions. In an attempt to explain the cause of such a phenomenon, research suggests that while short-term upregulation of TFF2 appears to be beneficial in epithelial repair processes, chronic upregulation may in fact contribute to pathologically significant events, including the progression to gastric cancer formation (White 2001). With some reports demonstrating an upregulation of TFF2 expression in the gastric tissue of chronically infected H. Pylori mice (Nomura et al. 2005) and in patients with H. Pylori-associated chronic gastritis and gastric cancer (Hu et al. 2003; Leung et al. 2002). Nevertheless, the mechanisms involved in the upregulation of TFF2 in numerous chronic pathological conditions and tumour formation have yet to be established in the gastrointestinal tract setting, with its functional capacity questioned as this observed upregulation is unable to prevent epithelial injury and mediate a normal repair response that re-establishes normal barrier structure and function. Further studies are required to understand this phenomenon in the gastrointestinal tract, which would further benefit research in understanding the role of TFF2 in the airway.
The role of TFF2 in the lung is less established, with its expression only more recently documented in the epithelial cells of human and animal lung (Kuperman et al. 2005; Nikolaidis et al. 2006). Studies suggest that TFF2 in the lung possesses a similar protective function to that expressed in the gut. TFF2 is found to have the capacity to promote cell migration of human epithelial cells and activation of rapid repair mechanisms in response to injurious stimuli in the lung (Oertel et al. 2001). Furthermore, TFF2 has been observed to enhance airway epithelial cell survival during restitution processes by inhibiting apoptosis and promoting angiogenesis (Hoffmann 2005). It is therefore plausible to hypothesise that TFF2 plays a key role in the initiation and/or progression of airway remodelling in asthma (Royce et al. 2011). However, the exact role of TFF2 in pathogenesis and its contribution to such structural alterations in this disease remains to be elucidated.
Previously, studies have demonstrated the rapid induction of TFF2 by mucus positive airway epithelial cells with expression not contributing to the regulation of the inflammatory response in a mouse model of acute airway inflammation (Nikolaidis et al. 2006). However recent reports suggest that TFF2 has an important role in the regulation of IL-33 at mucosal surfaces and the development of the type 2 allergic immune response in the lung following allergen exposure (Wills Karp et al. 2012). Specifically, TFF2 promotes IL-33 release from lung epithelia and alveolar macrophages initiated be an injurious event, which in turn is thought to be required for the production of major inflammatory cytokines IL-4 and IL-13, and AHR. Thus in light of these recent findings, TFF2 may have the capacity to mediate both inflammatory events and epithelial restitution processes in the lung in response to allergic insult.
Although expression of TFF2 is increased in asthma, it does not lead to an appropriate repair response and restitution of epithelium to normal functionality (Holgate 2000). Upregulation of TFF2 in asthma fails to prevent the progression of airway inflammation and epithelial remodelling changes suggesting that this expression change may be insufficient to prevent chronic epithelial injury and/or promote normal repair processes. Furthermore, the upregulation of pro-inflammatory and pro-remodelling mediators may override the potential benefits associated with increased TFF2 expression. Alternatively, there may be a failure to downregulate TFF2 expression after restitution, with the consequence that epithelial cells are inappropriately ‘held’ in a repair phenotype (Holgate 2000). Similar to research concerning the gastrointestinal tract, further studies are required to establish the exact mechanisms that upregulate TFF2 in asthma and the role of this upregulation in disease.
9.6 Novel Therapeutic Strategies in Asthma
As support for the role of epigenetic modulation in allergy and asthma continues to strengthen, the capacity to alter these changes has proven to be an attractive target for future therapeutic research. The clinical utility of HDAC inhibitors (HDACi) in oncology are relatively well characterised, with the mechanisms associated with their anticancer effects involving cell death and apoptosis, differentiation, decreased migration and invasion, and cell cycle arrest (Bolden et al. 2006). The role of HDACs in asthma has also been widely reported, with HDAC expression and activity of class I enzymes HDAC1 and HDAC2, decreased in bronchial biopsies from patients with asthma compared with normal subjects (Ito et al. 2002). In addition, decreased HDAC activity was implicated in alveolar macrophages and peripheral blood mononuclear cells in patients with asthma than in control subjects (Cosio et al. 2004). Conversely both studies report an increase in HAT activity in patients with asthma (Ito et al. 2002; Cosio et al. 2004). Interestingly, the molecular mechanisms and actions of corticosteroids involves downregulating the expression of multiple inflammatory genes by reversing the increased HAT activity observed in asthma, through the recruitment of HDAC2 (Barnes 2006). Furthermore, corticosteroid insensitivity (resistance) has been correlated with a reduction in HDAC activity in patients with asthma (Hew et al. 2006). Collectively, these findings suggest that HDACi may have a therapeutic benefit in reducing the severity of disease exacerbations through their ability to activate HDACs in order to attenuate the expression of numerous inflammatory genes.
Preliminary animal studies involving a number of general HDACi have indicated a potential for their use in asthma. In one report, mice sensitised and subsequently challenged with OVA to exhibit an AAD phenotype were administered with a representative HDACi, Trichostatin A (TSA) (Choi et al. 2005). Results indicate that TSA attenuates AHR and inflammation following methacholine administration. However, a recent study involving mice sensitised and challenged with A. fumigates disputes the finding that TSA has anti-inflammatory properties (Banerjee et al. 2012). In contrast, the ability of TSA to inhibit drug-induced bronchoconstriction was validated in this study in both the murine model and in human lung samples. Inhibition of drug-induced constriction in in vitro studies involving the administration of suberoylanilide hydroxamic acid (SAHA) to isolated guinea pig tracheal rings has also been published (Assem et al. 2008). The administration of the broad-spectrum HDACi, valproic acid was administered to AAD mice (induced through OVA sensitisation and exposure) in order to evaluate its capacity to attenuate the important characteristics of asthma (Royce et al.). Although valproic acid treatment was found not to affect inflammatory cell or infiltrate counts, administration resulted in reduced epithelial thickness and subepithelial collagen deposition when compared to vehicle-treated mice. In confirmation of previous findings, valproic acid also attenuated methacholine-induced AHR. Taken together, these results show the ability of HDACi to reverse drug-induced bronchoconstriction, with further research required to establish the ability of this class of drugs in reducing airway inflammation and remodelling changes.
The epithelium has been shown to be central in asthma pathogenesis, through its ability to activate the immune response, and propagate and maintain airway inflammation and remodelling. Through its ability to connect the molecular mechanisms involved in the development and progression of asthma pathogenesis, the epithelium is an attractive target for therapeutic research. Involved in epithelial restitution in the airway, TFF2 has been shown to be upregulated in asthma with endogenous expression levels seemingly inadequate to protect and/or reverse epithelial damage caused by invading pathogens. Interestingly, TFF2 has been shown to be important in the regulation of airway remodelling in two murine models of AAD (Royce et al.). In this study, TFF2-deficient AAD mice (AAD induced by either OVA or A. fumigates) exhibited increased goblet cell metaplasia, increased subepithelial fibrosis deposition (in both models) and increased epithelial thickness (in A. fumigates model). Thus, it is shown that TFF2 has the capacity to attenuate important epithelial and subepithelial remodelling events, characteristic of chronic, severe disease. A pilot study examining the effect of exogenous TFF2 treatment in a chronic OVA AAD model in mice support the contention that endogenous TFF2 fails to prevent epithelial injury and promote reparation (Royce et al. 2012). Despite no significant difference in the inflammatory response reported between TFF2-treated and untreated chronic AAD mice, important remodelling changes including goblet cell metaplasia, and lamina reticularis thickness was significantly attenuated with such treatment. Furthermore, bronchial epithelial apoptosis and AHR was significantly reduced following exogenous TFF2 treatment. Similar to those findings involving the administration of HDACi to animal models of AAD, further investigation is required to establish the capability of exogenous TFF2 in diminishing the inflammatory response, decreasing AHR and attenuating the structural alterations that constitute airway remodelling.
9.7 Conclusion
As prevalence, complexity and severity of asthma continues to increase on a global scale, despite advances in medicine and the understanding of disease, the importance of elucidating its development and pathogenesis is emphasised. The molecular heterogeneity of the disease is thought to be contributed to a combination of genetic, epigenetic and environmental factors. The role of genetic susceptibility and inheritance, and the exposure to various environmental pathogens in utero and in post-natal development have been widely studied in the acquisition of an asthma phenotype. Epigenetic regulation, modulated through interactions with genetic and environmental factors, has been explored in an attempt to explain the dramatic increase in incidence over the last decade. Modulation of the epigenome in the regulation of important asthma-associated pathways, including gene expression and T-cell differentiation is thought to play a direct role in conferring asthma susceptibility, development and progression. In addition, an inherently defective epithelium is thought to be of primary significance. It is hypothesised that the epithelium has the capacity to propagate and maintain remodelling and inflammatory changes, through its altered communication with the underlying mesenchyme. TFF2, known to be involved in epithelial restitution has previously been shown to be upregulated in asthma. Despite this, increased expression fails to lead to complete restoration of the epithelium to normal functionality. By understanding the limited knowledge available, we aim to establish the mechanisms involved in the pathogenesis of asthma, and more specifically determine the roles of epigenetic modulation, the epithelium and TFF2 in disease. This chapter discussed the inadequacy of current research and provided the rationale to focus on targeting these mechanisms for future therapies.
References
Abbas AK, Murphy KM, Sher A (1996) Functional diversity of helper T lymphocytes. Nature 383(6603):787–793
Adenuga D, Yao H, March TH, Seagrave J, Rahman I (2009) Histone deacetylase 2 is phosphorylated, ubiquitinated, and degraded by cigarette smoke. Am J Respir Cell Mol Biol 40:464–473
Agarwal S, Rao A (1998) Modulation of chromatin structure regulates cytokine gene expression during T cell differentiation. Immunity 9(6):765–775
Akimzhanov AM, Yang XO, Dong C (2007) Chromatin remodeling of interleukin-17 (IL-17)-IL-17F cytokine gene locus during inflammatory helper T cell differentiation. J Biol Chem 282:5969–5972
Allan RS, Zueva E, Cammas F, Schreiber HA, Masson V, Belz GT, Roche D, Maison C, Quivy J-P, Almouzni G, Amigorena S (2012) An epigenetic silencing pathway controlling T helper 2 cell lineage commitment. Nature 487:249–253
Allen M, Heinzmann A, Noguchi E, Abecasis G, Broxholme J, Ponting C, Bhattacharyya S, Tinsley J, Zhang Y, Holt R, Jones EY, Lench N, Carey A, Jones H, Dickens N, Dimon C, Nicholls R, Baker C, Xue L, Townsend E, Kabesch M, Weiland S, Carr D, Von Mutius E, Adcock I, Barnes P, Lathrop GM, Edwards M, Moffatt M, Cookson WOCM (2003) Positional cloning of a novel gene influencing asthma from chromosome 2q14. Nat Genet 35:258–263
Amishima M et al (1998) Expression of epidermal growth factor and epidermal growth factor receptor immunoreactivity in the asthmatic human airway. Am J Respir Crit Care Med 157(6):1907–1912
Ansel KM, Lee DU, Rao A (2003) An epigenetic view of helper T cell differentiation. Nat Immunol 4:616–623
Assem E-SK et al (2008) Effects of a selection of histone deacetylase inhibitors on mast cell activation and airway and colonic smooth muscle contraction. Int Immunopharmacol 8(13):1793–1801
Ather JL et al (2011) Airway epithelial NF-kappaB activation promotes allergic sensitization to an innocuous inhaled antigen. Am J Respir Cell Mol Biol 44(5):631–638
Avni O, Lee D, Macian F, Szabo S, Glimcher L, Rao A (2002) T(H) cell differentiation is accompanied by dynamic changes in histone acetylation of cytokine genes. Nat Immunol 3:643–651
Baccarelli A, Rusconi F, Bollati V, Catelan D, Accetta G, Hou L, Barbone F, Bertazzi PA, Biggeri A (2012) Nasal cell DNA methylation, inflammation, lung function and wheezing in children with asthma. Epigenomics 4:91–100
Banerjee A et al (2012) Trichostatin A abrogates airway constriction, but not inflammation, in murine and human asthma models. Am J Respir Cell Mol Biol 46(2):132–138
Barnes PJ (2006) How corticosteroids control inflammation: quintiles prize lecture 2005. Br J Pharmacol 148(3):245–254
Barnes PJ, Adcock IM (2009) Glucocorticoid resistance in inflammatory diseases. Lancet 373(9678):1905–1917
Beasley R et al (1989) Cellular events in the bronchi in mild asthma and after bronchial provocation. Am Rev Respir Dis 139(3):806–817
Berlivet S, Moussette S, Ouimet M, Verlaan D, Koka V, Al Tuwaijri A, Kwan T, Sinnett D, Pastinen T, Naumova A (2012) Interaction between genetic and epigenetic variation defines gene expression patterns at the asthma-associated locus 17q12-q21 in lymphoblastoid cell lines. Hum Genet 131:1161–1171
Bluestone JA et al (1995) TCR gamma delta cells: a specialized T-cell subset in the immune system. Annu Rev Cell Dev Biol 11:307–353
Bolden JE, Peart MJ, Johnstone RW (2006) Anticancer activities of histone deacetylase inhibitors. Nat Rev Drug Discov 5(9):769–784
Bousquet J et al (2000) Asthma: from bronchoconstriction to airways inflammation and remodeling. Am J Respir Crit Care Med 161(5):1720–1745
Brand S, Teich R, Dicke T, Harb H, Yildirim AÃ, Tost JR, Schneider-Stock R, Waterland RA, Bauer U-M, Von Mutius E, Garn H, Pfefferle PI, Renz H (2011) Epigenetic regulation in murine offspring as a novel mechanism for transmaternal asthma protection induced by microbes. J Allergy Clin Immunol 128:618–625, e7
Breton CV, Byun H-M, Wenten M, Pan F, Yang A, Gilliland FD (2009) Prenatal tobacco smoke exposure affects global and gene-specific DNA methylation. Am J Respir Crit Care Med 180:462–467
Breton CV, Byun H-M, Wang X, Salam MT, Siegmund K, Gilliland FD (2011) DNA methylation in the arginase-nitric oxide synthase pathway is associated with exhaled nitric oxide in children with asthma. Am J Respir Crit Care Med 184:191–197
Broide DH et al (2005) Allergen-induced peribronchial fibrosis and mucus production mediated by IκB kinase β-dependent genes in airway epithelium. Proc Natl Acad Sci USA 102(49):17723–17728
Bucchieri F et al (2002) Asthmatic bronchial epithelium is more susceptible to oxidant-induced apoptosis. Am J Respir Cell Mol Biol 27(2):179–185
Cao D, Bromberg PA, Samet JM (2007) COX-2 expression induced by diesel particles involves chromatin modification and degradation of HDAC1. Am J Respir Cell Mol Biol 37:232–239
Cates EC et al (2004) Intranasal exposure of mice to house dust mite elicits allergic airway inflammation via a GM-CSF-mediated mechanism. J Immunol 173(10):6384–6392
Chang S, Collins PL, Aune TM (2008) T-bet dependent removal of Sin3A-histone deacetylase complexes at the Ifng locus drives Th1 differentiation. J Immunol 181:8372–8381
Choi JH et al (2005) Trichostatin A attenuates airway inflammation in mouse asthma model. Clin Exp Allergy 35(1):89–96
Cosio BG, Mann B, Ito K, Jazrawi E, Barnes PJ, Chung KF, Adcock IM (2004) Histone acetylase and deacetylase activity in alveolar macrophages and blood mononocytes in asthma. Am J Respir Crit Care Med 170:141–147
Crosby L, Waters C (2010) Epithelial repair mechanisms in the lung. Am J Physiol Lung Cell Mol Physiol 298(6):L715–L731
Cui J et al (2005) TH1-mediated airway hyperresponsiveness independent of neutrophilic inflammation. J Allergy Clin Immunol 115(2):309–315
Davies D (2009) The role of the epithelium in airway remodeling in asthma. Proc Am Thorac Soc 6(8):678–682
Demenais F, Chaudru V, Martinez M (2001) Detection of parent-of-origin effects for atopy by model-free and model-based linkage analyses. Genet Epidemiol 21(Suppl 1):S186–S191
Doe C, Bafadhel M, Siddiqui S, Desai D, Mistry V, Rugman P, Mccormick M, Woods J, May R, Sleeman MA, Anderson IK, Brightling CE (2010) Expression of the T helper 17-associated cytokines IL-17A and IL-17F in asthma and COPD. Chest 138:1140–1147
Doganci A, Eigenbrod T, Krug N, De Sanctis GT, Hausding M, Erpenbeck VJ, Haddad E-B, Schmitt E, Bopp T, Kallen K-J, Herz U, Schmitt S, Luft C, Hecht O, Hohlfeld JM, Ito H, Nishimoto N, Yoshizaki K, Kishimoto T, Rose-John S, Renz H, Neurath MF, Galle PR, Finotto S (2005) The IL-6R alpha chain controls lung CD4+CD25+ Treg development and function during allergic airway inflammation in vivo. J Clin Invest 115:313–325
Ege MJ, Strachan DP, Cookson WO, Moffatt MF, Gut I, Lathrop M, Kabesch M, Genuneit J, Büchele G, Sozanska B, Boznanski A, Cullinan P, Horak E, Bieli C, Braun-Fahrländer C, Heederik D, Von Mutius E (2011) Gene-environment interaction for childhood asthma and exposure to farming in Central Europe. J Allergy Clin Immunol 127:138–144, e4
Erjeflt JS, Persson CG (1997) Airway epithelial repair: breathtakingly quick and multipotentially pathogenic. Thorax 52(11):1010–1012
Evans MJ et al (1999) The attenuated fibroblast sheath of the respiratory tract epithelial-mesenchymal trophic unit. Am J Respir Cell Mol Biol 21(6):655–657
Fainaru O, Shseyov D, Hantisteanu S, Groner Y (2005) Accelerated chemokine receptor 7-mediated dendritic cell migration in Runx3 knockout mice and the spontaneous development of asthma-like disease. Proc Natl Acad Sci U S A 102:10598–10603
Fedorov IA et al (2005) Epithelial stress and structural remodelling in childhood asthma. Thorax 60(5):389–394
Fields PE, Kim ST, Flavell RA (2002) Cutting edge: changes in histone acetylation at the IL-4 and IFN-gamma loci accompany Th1/Th2 differentiation. J Immunol 169:647–650
Franic TV et al (2005) Reciprocal changes in trefoil 1 and 2 expression in stomachs of mice with gastric unit hypertrophy and inflammation. J Pathol 207(1):43–52
Grausenburger R et al (2010) Conditional deletion of histone deacetylase 1 in T cells leads to enhanced airway inflammation and increased Th2 cytokine production. J Immunol 185(6):3489–3497
Hackett T-L (2012) Epithelial-mesenchymal transition in the pathophysiology of airway remodelling in asthma. Curr Opin Allergy Clin Immunol 12(1):53–59
Hackett T-L, Knight DA (2007) The role of epithelial injury and repair in the origins of asthma. Curr Opin Allergy Clin Immunol 7(1):63–68
Hamilton LM et al (2001) The bronchial epithelium in asthma–much more than a passive barrier. Monaldi Arch Chest Dis 56(1):48–54
Hammad H, Lambrecht BN (2008) Dendritic cells and epithelial cells: linking innate and adaptive immunity in asthma. Nat Rev Immunol 8(3):193–204
Hew M et al (2006) Relative corticosteroid insensitivity of peripheral blood mononuclear cells in severe asthma. Am J Respir Crit Care Med 174(2):134
Hoffmann W (2005) Trefoil factors TFF (trefoil factor family) peptide-triggered signals promoting mucosal restitution. Cell Mol Life Sci 62(24):2932–2938
Holgate ST (1998) The inflammation-repair cycle in asthma: the pivotal role of the airway epithelium. Clin Exp Allergy 28(suppl 5):97–103
Holgate ST (2000) The bronchial epithelial origins of asthma. Chem Immunol 78:62–71
Holgate S (2007) Epithelium dysfunction in asthma. J Allergy Clin Immunol 120(6):1233–1244
Holgate ST (2008) Pathogenesis of asthma. Clin Exp Allergy 38(6):872–897
Hollingsworth JW, Maruoka S, Boon K, Garantziotis S, Li Z, Tomfohr J, Bailey N, Potts EN, Whitehead G, Brass DM, Schwartz DA (2008) In utero supplementation with methyl donors enhances allergic airway disease in mice. J Clin Invest 118:3462–3469
Hu G-Y et al (2003) Expression of TFF2 and Helicobacter pylori infection in carcinogenesis of gastric mucosa. World J Gastroenterol 9(5):910–914
Isidoro-García M, Sanz C, García-Solaesa V, Pascual M, Pescador DB, Lorente F, Dávila I (2011) PTGDR gene in asthma: a functional, genetic, and epigenetic study. Allergy 66:1553–1562
Ito K et al (2002) Expression and activity of histone deacetylases in human asthmatic airways. Am J Respir Crit Care Med 166(3):392–396
Ivanov II, Mckenzie BS, Zhou L, Tadokoro CE, Lepelley A, Lafaille JJ, Cua DJ, Littman DR (2006) The orphan nuclear receptor RORgammat directs the differentiation program of proinflammatory IL-17+ T helper cells. Cell 126:1121–1133
Janson PCJ, Winerdal ME, Marits P, Thörn M, Ohlsson R, Winqvist O (2008) FOXP3 promoter demethylation reveals the committed Treg population in humans. PLoS One 3:e1612
Jones B, Chen J (2006) Inhibition of IFN-gamma transcription by site-specific methylation during T helper cell development. EMBO J 25(11):2443–2452
Kicic A et al (2010) Decreased fibronectin production significantly contributes to dysregulated repair of asthmatic epithelium. Am J Respir Crit Care Med 181(9):889–898
Kim B-S et al (2010) Conversion of Th2 memory cells into Foxp3+ regulatory T cells suppressing Th2-mediated allergic asthma. Proc Natl Acad Sci 107(19):8742–8747
Kim S-H, Lee C-E (2011) Counter-regulation mechanism of IL-4 and IFN-α signal transduction through cytosolic retention of the pY-STAT6:pY-STAT2:p48 complex. Eur J Immunol 41:461–472
Kim H-P, Leonard WJ (2007) CREB/ATF-dependent T cell receptor-induced FoxP3 gene expression: a role for DNA methylation. J Exp Med 204:1543–1551
Kim ST, Fields PE, Flavell RA (2007) Demethylation of a specific hypersensitive site in the Th2 locus control region. Proc Natl Acad Sci U S A 104:17052–17057
Koh BH, Hwang SS, Kim JY, Lee W, Kang M-J, Lee CG, Park J-W, Flavell RA, Lee GR (2010) Th2 LCR is essential for regulation of Th2 cytokine genes and for pathogenesis of allergic asthma. Proc Natl Acad Sci 107:10614–10619
Kuperman D et al (2005) Dissecting asthma using focused transgenic modeling and functional genomics. J Allergy Clin Immunol 116(2):305–311
Kwon N-H, Kim J-S, Lee J-Y, Oh M-J, Choi D-C (2008) DNA methylation and the expression of IL-4 and IFN-gamma promoter genes in patients with bronchial asthma. J Clin Immunol 28:139–146
Lal G, Zhang N, Van Der Touw W, Ding Y, Ju W, Bottinger EP, Reid SP, Levy DE, Bromberg JS (2009) Epigenetic regulation of Foxp3 expression in regulatory T cells by DNA methylation. J Immunol 182:259–273
Lambrecht BN, Hammad H (2009) Biology of lung dendritic cells at the origin of asthma. Immunity 31(3):412–424
Lambrecht BN, Hammad H (2012) The airway epithelium in asthma. Nat Med 18(5):684–692
Lane N et al (2010) Regulation in chronic obstructive pulmonary disease: the role of regulatory T-cells and Th17 cells. Clin Sci 119:75–86
Laurence A, Tato CM, Davidson TS, Kanno Y, Chen Z, Yao Z, Blank RB, Meylan FO, Siegel R, Hennighausen L, Shevach EM, O’Shea JJ (2007) Interleukin-2 signaling via STAT5 constrains T helper 17 cell generation. Immunity 26:371–381
Le Cras T et al (2011) Epithelial EGF receptor signaling mediates airway hyperreactivity and remodeling in a mouse model of chronic asthma. Am J Physiol Lung Cell Mol Physiol 300(3):L414–L421
Lee DU, Agarwal S, Rao A (2002) Th2 lineage commitment and efficient IL-4 production involves extended demethylation of the IL-4 gene. Immunity 16:649–660
Lee SL, Lam TH, Leung TH, Wong WH, Schooling M, Leung GM, Lau YL (2012) Foetal exposure to maternal passive smoking is associated with childhood asthma, allergic rhinitis, and eczema. Scientific World Journal 2012:542983
Leung W et al (2002) Expression of trefoil peptides (TFF1, TFF2, and TFF3) in gastric carcinomas, intestinal metaplasia, and non-neoplastic gastric tissues. J Pathol 197(5):582–588
Liu J, Ballaney M, Al-Alem U, Quan C, Jin X, Perera F, Chen L-C, Miller RL (2008) Combined inhaled diesel exhaust particles and allergen exposure alter methylation of T helper genes and IgE production in vivo. Toxicol Sci 102:76–81
Liu Q, Xia Y, Zhang W, Li J, Wang P, Li H, Wei C, Gong Y (2009) A functional polymorphism in the SPINK5 gene is associated with asthma in a Chinese Han population. BMC Med Genet 10:59
Lloyd CM, Hessel EM (2010) Functions of T cells in asthma: more than just TH2 cells. Nat Rev Immunol 10(12):838–848
Lodrup Carlsen KC, Carlsen K-H (2001) Effects of maternal and early tobacco exposure on the development of asthma and airway hyperreactivity. Curr Opin Allergy Clin Immunol 1:139–143
Lovinsky-Desir S, Miller RL (2012) Epigenetics, asthma, and allergic diseases: a review of the latest advancements. Curr Allergy Asthma Rep 12(3):211–220
Mangan PR, Harrington LE, O’Quinn DB, Helms WS, Bullard DC, Elson CO, Hatton RD, Wahl SM, Schoeb TR, Weaver CT (2006) Transforming growth factor-[beta] induces development of the TH17 lineage. Nature 441:231–234
McKinley L et al (2008) TH17 cells mediate steroid-resistant airway inflammation and airway hyperresponsiveness in mice. J Immunol 181(6):4089–4097
Monick MM et al (2003) Respiratory syncytial virus up-regulates TLR4 and sensitizes airway epithelial cells to endotoxin. J Biol Chem 278(52):53035–53044
Morales E, Bustamante M, Vilahur N, Escaramis G, Montfort M, De Cid R, Garcia-Esteban R, Torrent M, Estivill X, Grimalt JO, Sunyer J (2012) DNA hypomethylation at ALOX12 is associated with persistent wheezing in childhood. Am J Respir Crit Care Med 185:937–943
Mukasa R et al (2010) Epigenetic instability of cytokine and transcription factor gene loci underlies plasticity of the T helper 17 cell lineage. Immunity 32(5):616–627
Murata A, Ling PM (2012) Asthma diagnosis and management. Emerg Med Clin North Am 30(2):203–222
Nadeau K, Mcdonald-Hyman C, Noth EM, Pratt B, Hammond SK, Balmes J, Tager I (2010) Ambient air pollution impairs regulatory T-cell function in asthma. J Allergy Clin Immunol 126:845–852, e10
Nikolaidis N et al (2006) Allergen induced TFF2 is expressed by mucus-producing airway epithelial cells but is not a major regulator of inflammatory responses in the murine lung. Exp Lung Res 32(10):483–497
Nomura S et al (2005) Alterations in gastric mucosal lineages induced by acute oxyntic atrophy in wild-type and gastrin-deficient mice. Am J Physiol Gastrointest Liver Physiol 288(2):G362–G375
Ober C, Yao T-C (2011) The genetics of asthma and allergic disease: a 21st century perspective. Immunol Rev 242:10–30
Oertel M et al (2001) Trefoil factor family-peptides promote migration of human bronchial epithelial cells: synergistic effect with epidermal growth factor. Am J Respir Cell Mol Biol 25(4):418–424
O’Garra A (1998) Cytokines induce the development of functionally heterogeneous T helper cell subsets. Immunity 8(3):275–283
Ohta K et al (1999) Diesel exhaust particulate induces airway hyperresponsiveness in a murine model: essential role of GM-CSF. J Allergy Clin Immunol 104(5):1024–1030
Pace E et al (2008) Cigarette smoke increases Toll-like receptor 4 and modifies lipopolysaccharide-mediated responses in airway epithelial cells. Immunology 124(3):401–411
Pawankar R et al (2012) Allergic diseases and asthma: a major global health concern. Curr Opin Allergy Clin Immunol 12(1):39–41
Payne DNR et al (2003) Early thickening of the reticular basement membrane in children with difficult asthma. Am J Respir Crit Care Med 167(1):78–82
Perera F, Tang W-Y, Herbstman J, Tang D, Levin L, Miller R, Ho S-M (2009) Relation of DNA methylation of 5′-CpG island of ACSL3 to transplacental exposure to airborne polycyclic aromatic hydrocarbons and childhood asthma. PLoS One 4:e4488
Perl A-K, Riethmacher D, Whitsett J (2011) Conditional depletion of airway progenitor cells induces peribronchiolar fibrosis. Am J Respir Crit Care Med 183(4):511–521
Phillips JM, Goodman JI (2009) Inhalation of cigarette smoke induces regions of altered DNA methylation (RAMs) in SENCAR mouse lung. Toxicology 260:7–15
Prescott SL (2006) The development of respiratory inflammation in children. Paediatr Respir Rev 7(2):89–96
Puddicombe SM et al (2000) Involvement of the epidermal growth factor receptor in epithelial repair in asthma. FASEB J 14(10):1362–1374
Ritz SA et al (2002) On the generation of allergic airway diseases: from GM-CSF to Kyoto. Trends Immunol 23(8):396–402
Royce S et al (2009) Effect of extracellular matrix composition on airway epithelial cell and fibroblast structure: implications for airway remodeling in asthma. Ann Allergy Asthma Immunol 102(3):238–246
Royce S et al (2011) Trefoil factor 2 regulates airway remodeling in animal models of asthma. J Asthma 48(7):653–659
Royce SG et al (2013) Trefoil factor 2 reverses airway remodeling changes in allergic airways disease. Am J Respir Cell Mol Biol 48(1):135–144
Salam MT, Byun H-M, Lurmann F, Breton CV, Wang X, Eckel SP, Gilliland FD (2012) Genetic and epigenetic variations in inducible nitric oxide synthase promoter, particulate pollution, and exhaled nitric oxide levels in children. J Allergy Clin Immunol 129:232–239, e7
Sampath D et al (1999) Constitutive activation of an epithelial signal transducer and activator of transcription (STAT) pathway in asthma. J Clin Invest 103(9):1353–1361
Sandford AJ, Chagani T, Zhu S, Weir TD, Bai TR, Spinelli JJ, Fitzgerald JM, Behbehani NA, Tan WC, Paré PD (2000) Polymorphisms in the IL4, IL4RA, and FCERIB genes and asthma severity. J Allergy Clin Immunol 106:135–140
Santangelo S, Cousins DJ, Winkelmann NEE, Staynov DZ (2002) DNA methylation changes at human Th2 cytokine genes coincide with DNase I hypersensitive site formation during CD4+ T cell differentiation. J Immunol 169:1893–1903
Schaub B, Liu J, Höppler S, Schleich I, Huehn J, Olek S, Wieczorek G, Illi S, Von Mutius E (2009) Maternal farm exposure modulates neonatal immune mechanisms through regulatory T cells. J Allergy Clin Immunol 123:774–782, e5
Stämpfli MR et al (1998) GM-CSF transgene expression in the airway allows aerosolized ovalbumin to induce allergic sensitization in mice. J Clin Invest 102(9):1704
Stevens PT et al (2008) Dysregulated repair in asthmatic paediatric airway epithelial cells: the role of plasminogen activator inhibitor-1. Clin Exp Allergy 38(12):1901–1910
Sunyer J, Torrent M, Muñoz-Ortiz L, Ribas-Fitó NR et al (2005) Prenatal dichlorodiphenyldichloroethylene (DDE) and asthma in children. Environ Health Perspect 113:1787–1790
Suter M, Ma J, Harris AS, Patterson L, Brown KA, Shope C, Showalter L, Abramovici A, Aagaard-Tillery KM (2011) Maternal tobacco use modestly alters correlated epigenome-wide placental DNA methylation and gene expression. Epigenetics 6:1284–1294
Trautmann A et al (2005) Apoptosis and loss of adhesion of bronchial epithelial cells in asthma. Int Arch Allergy Immunol 138(2):142–150
Tsaprouni LG, Ito K, Adcock IM, Punchard N (2007) Suppression of lipopolysaccharide- and tumour necrosis factor-α-induced interleukin (IL)-8 expression by glucocorticoids involves changes in IL-8 promoter acetylation. Clin Exp Immunol 150:151–157
Van Eerdewegh P, Little RD, Dupuis J, Del Mastro RG, Falls K, Simon J, Torrey D, Pandit S, Mckenny J, Braunschweiger K, Walsh A, Liu Z, Hayward B, Folz C, Manning SP, Bawa A, Saracino L, Thackston M, Benchekroun Y, Capparell N, Wang M, Adair R, Feng Y, Dubois J, Fitzgerald MG, Huang H, Gibson R, Allen KM, Pedan A, Danzig MR, Umland SP, Egan RW, Cuss FM, Rorke S, Clough JB, Holloway JW, Holgate ST, Keith TP (2002) Association of the ADAM33 gene with asthma and bronchial hyperresponsiveness. Nature 418:426–430
van Rijt LS et al (2005) In vivo depletion of lung CD11c+ dendritic cells during allergen challenge abrogates the characteristic features of asthma. J Exp Med 201(6):981–991
Vercelli D (2008) Discovering susceptibility genes for asthma and allergy. Nat Rev Immunol 8:169–182
Vignola AM, Kips J, Bousquet J (2000) Tissue remodeling as a feature of persistent asthma. J Allergy Clin Immunol 105(6):1041–1053
Wan H et al (2000) Quantitative structural and biochemical analyses of tight junction dynamics following exposure of epithelial cells to house dust mite allergen Der p 1. Clin Exp Allergy 30(5):685–698
Wei G, Wei L, Zhu J, Zang C, Hu-Li J, Yao Z, Cui K, Kanno Y, Roh T-Y, Watford WT, Schones DE, Peng W, Sun H-W, Paul WE, O’shea JJ, Zhao K (2009) Global mapping of H3K4me3 and H3K27me3 reveals specificity and plasticity in lineage fate determination of differentiating CD4+ T cells. Immunity 30:155–167
Weinberg EG (2011) The WAO white book on allergy 2011-2012: review article. Curr Allergy Clin Immunol 24(3):156–157
White SR (2001) Trefoil peptides in airway epithelium: an important addition to the plethora of peptides. Am J Respir Cell Mol Biol 25(4):401–404
White GP, Hollams EM, Yerkovich ST, Bosco A, Holt BJ, Bassami MR, Kusel M, Sly PD, Holt PG (2006) CpG methylation patterns in the IFNγ promoter in naive T cells: variations during Th1 and Th2 differentiation and between atopics and non-atopics. Pediatr Allergy Immunol 17:557–564
Wills Karp M et al (2012) Trefoil factor 2 rapidly induces interleukin 33 to promote type 2 immunity during allergic asthma and hookworm infection. J Exp Med 209(3):607–622
Woodruff PG et al (2009) T-helper type 2–driven inflammation defines major subphenotypes of asthma. Am J Respir Crit Care Med 180(5):388
Wright NA (1993) Trefoil peptides and the gut. Gut 34(5):577–579
Xepapadaki P, Manios Y, Liarigkovinos T, Grammatikaki E, Douladiris N, Kortsalioudaki C, Papadopoulos NG (2009) Association of passive exposure of pregnant women to environmental tobacco smoke with asthma symptoms in children. Pediatr Allergy Immunol 20:423–429
Xiao C et al (2011) Defective epithelial barrier function in asthma. J Allergy Clin Immunol 128(3):549–556, e1
Yang IV, Schwartz DA (2011) Epigenetic control of gene expression in the lung. Am J Respir Crit Care Med 183(10):1295
Yang M, Kumar RK, Foster PS (2009) Pathogenesis of steroid-resistant airway hyperresponsiveness: interaction between IFN-gamma and TLR4/MyD88 pathways. J Immunol 182(8):5107–5115
Yang L et al (1998) Essential role of nuclear factor kB in the induction of eosinophilia in allergic airway inflammation. J Exp Med 188(9):1739–1750
Yang S-R, Chida AS, Bauter MR, Shafiq N, Seweryniak K, Maggirwar SB, Kilty I, Rahman I (2006) Cigarette smoke induces proinflammatory cytokine release by activation of NF-kappaB and posttranslational modifications of histone deacetylase in macrophages. Am J Physiol Lung Cell Mol Physiol 291:L46–L57
Yang Y, Haitchi HM, Cakebread J, Sammut D, Harvey A, Powell RM, Holloway JW, Howarth P, Holgate ST, Davies DE (2008) Epigenetic mechanisms silence a disintegrin and metalloprotease 33 expression in bronchial epithelial cells. J Allergy Clin Immunol 121:1393–1399, e14
Yuyama N et al (2002) Analysis of novel disease-related genes in bronchial asthma. Cytokine 19(6):287–296
Zahm JM, Chevillard M, Puchelle E (1991) Wound repair of human surface respiratory epithelium. Am J Respir Cell Mol Biol 5(3):242–248
Zhang Y, Moffatt M, Cookson WOC (2012) Genetic and genomic approaches to asthma: new insights for the origins. Curr Opin Pulm Med 18:6–13
Zhou C et al (2011) Epithelial apoptosis and loss in airways of children with asthma. J Asthma 48(4):358–365
Acknowledgements
The support of the Australian Institute of Nuclear Science and Engineering is acknowledged. TCK was the recipient of AINSE awards. TCK is a Future Fellow and Epigenomic Medicine Laboratory is supported by the Australian Research Council. Supported in part by the Victorian Government’s Operational Infrastructure Support Program.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2014 Springer Science+Business Media New York
About this chapter
Cite this chapter
Tortorella, S., Royce, S.G., Karagiannis, T.C. (2014). Molecular Mechanisms in the Development and Progression of Asthma: The Role of Epigenetic Regulation and the Airway Epithelium. In: Maulik, N., Karagiannis, T. (eds) Molecular mechanisms and physiology of disease. Springer, New York, NY. https://doi.org/10.1007/978-1-4939-0706-9_9
Download citation
DOI: https://doi.org/10.1007/978-1-4939-0706-9_9
Published:
Publisher Name: Springer, New York, NY
Print ISBN: 978-1-4939-0705-2
Online ISBN: 978-1-4939-0706-9
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)