
Abstract
Membrane proteins (MPs) exhibit a broad range of activities, which are crucial for cell survival. They can be pumps, channels, enzymes, scaffolds, signal transmitters, or a combination of these functions. Understanding their molecular mechanisms generally requires their extraction out of membranes and their purification. Solubilization and isolation are usually carried out using detergents, which disrupt the membrane and adsorb onto the hydrophobic surface of the transmembrane domain of MPs, keeping them water soluble. Detergents, however, tend to inactivate most MPs more or less rapidly, making their biochemical and biophysical studies challenging. Specially designed amphipathic polymers called “amphipols” (APols) have been developed with the view of improving the stability of MPs in aqueous solutions. In this chapter, the properties of APols and of the complexes they form with MPs are summarized, and a brief overview of APol applications that have been validated thus far is presented. Five experimental protocols are described in detail: (1) trapping MPs in APols, (2) measuring the amount of APol bound per MP, (3) APol-assisted folding of MPs, (4) APol-assisted production of MPs by cell-free expression, and (5) immobilizing MPs onto solid surfaces for screening purposes using functionalized APols.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others
Keywords
- Critical Micellar Concentration
- Immobilize Metal Affinity Chromatography
- Critical Aggregation Concentration
- Styrene Maleic Acid
- Detergent Removal
These keywords were added by machine and not by the authors. This process is experimental and the keywords may be updated as the learning algorithm improves.
7.1 General Introduction to the Properties and Uses of Amphipols
7.1.1 Inactivation of Membrane Proteins in the Presence of Detergents and Strategies for Membrane Proteins Stabilization
In their native environment, i.e., the membrane, membrane proteins (MPs) are stabilized by various types of physical factors provided by the membrane architecture itself, such as membrane thickness, accessibility to water, the distribution of charges, lipid asymmetry, or other properties like transmembrane gradients, viscosity, etc. However, specific molecular interactions between proteins, lipids, and cofactors play a key role in MP stability (for discussions, see, e.g., Bowie 2001; Garavito and Ferguson-Miller 2001; Popot and Engelman 2000). Detergents compete with these interactions, inducing destabilization. In other words, most detergents can be considered as being too good a solvent, breaking more interactions than would be desirable. The extent of this problem varies from one detergent to another, leading to distinguishing “weak” and “strong” detergents. It also varies considerably depending on the nature of MPs and, in particular, tends to be more severe for α-helical than for β-barrel MPs. This variability creates a bias in our understanding of the structure and function of MPs, representing an important bottleneck.
To improve the stability of detergent-solubilized MPs, several approaches can be resorted to, such as: (1) transfer to a “weak” detergent like Tween or digitonin, (2) working close to the critical micellar concentration (CMC) of the detergent in order to limit the volume of the micellar phase, (3) supplementing the micelles with lipids or cofactors, (4) working fast enough so as to collect data or form crystals before the inactivation of the protein sets in, or (5) select or engineer more stable MPs. An alternative is to replace classical detergents by bilayer-like environments such as lipid vesicles, bicelles, nanodiscs, or cubic phases, or by novel, less aggressive surfactants such as detergents with multiple, branched, or cyclic hydrophobic moieties (see, e.g., Chae et al. 2010; Hong et al. 2011; Hovers et al. 2011) or stabilizing polar heads (Matar-Merheb et al. 2011), fluorinated surfactants (Breyton et al. 2004, 2009, 2010; Chabaud et al. 1998; Popot 2010; see also Chap. 8 in this volume), amphipathic peptides (Koutsopoulos et al. 2012; Schafmeister et al. 1993; Wang et al. 2011; Zhao et al. 2006), lipopeptides (McGregor et al. 2003; Privé 2009), or polymers such as styrene maleic acid lipid particles (SMALPs; Knowles et al. 2009; Long et al. 2013; Rajesh et al. 2011), and amphipols (Popot et al. 2011; Tribet et al. 1996; Zoonens and Popot 2014).
7.1.2 General Properties of APols in Aqueous Solution
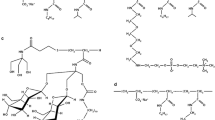
Amphipols (APols) are short and flexible amphipathic polymers, designed so as to bind to the transmembrane domain of MPs by multiple hydrophobic contact points. MP/APol complexes, as a result, should present a low k off and a small K D. They should not dissociate even at extreme dilutions and, when they do, should do so extremely slowly (see below). This would make them radically different from MP/detergent complexes, in which the protein-bound detergent molecules are in rapid equilibrium with free monomers and micelles, and dissociate upon dilution below the CMC. The first APols to have been synthesized comprise a polyacrylic acid backbone onto which octylamine and isopropylamine side chains have been randomly grafted (Tribet et al. 1996; Fig. 7.1). The most widely used APol, called A8-35, features 35 % of ungrafted carboxylic acid groups. Above pH 7, all of those are ionized (Gohon et al. 2004). Twenty-five percent of the carboxylic groups have been derivatized with octyl chains, giving A8-35 its amphipathy, and the last 40 % with isopropyl groups, so as to reduce the charge density along the polymer. The average molecular weight (MW) of A8-35 is ~ 4.3 kDa (Giusti et al. 2014b) .

Molecular structures of various amphipols: A8-35, SAPol, PC-APol, and NAPol. The groups conferring the aqueous solubility to the polymers are circled in red, the alkyl chains in blue, and the groups that modulate the charge density in green. The three types of groups are randomly distributed along the chain. For A8-35, the molar percentages of each group are x = 35 %, y = 25 %, and z = 40 %. The cartoons are reprinted with permissions from Proc Natl Acad Sci USA 93:15047–15050, copyright 1996 National Academy of Sciences, USA (Tribet et al. 1996), Biopolymers 95:811–823, copyright 2011 Wiley Periodicals, Inc. (Dahmane et al. 2011), Langmuir 23:3025–3035, copyright 2007 American Chemical Society (Diab et al. 2007b) and Langmuir 28:4625–4639, copyright 2012 American Chemical Society (Sharma et al. 2012), respectively.
The solution properties of A8-35 have been intensively studied and previously reviewed (Popot 2010; Popot et al. 2003, 2011; Zoonens and Popot 2014). A8-35 is highly soluble in water (> 200 g L−1). Its concentration can reach up to 100 g L−1 without affecting significantly the viscosity of the solution (L.J. Catoire, personal communication). In aqueous solutions, its molecules self-associate to form small, compact, hydrated particles with an average MW of ~ 40 kDa (Gohon et al. 2006) . Based on an average MW of ~ 4.3 kDa per individual molecule, A8-35 particles therefore contain an average of ~ 9–10 molecules, corresponding to ~ 80 octyl chains. The minimal concentration at which A8-35 particles start to assemble—the critical aggregation concentration (CAC)—is ~ 0.002 g L−1 (Giusti et al. 2012). Whereas the general organization of A8-35 particles and detergent micelles resemble each other, with similar sizes and the presence of a hydrophobic core and a hydrophilic surface, differences include the smaller number of molecules per A8-35 particle, a much slower rate of exchange with the solution (expected from indirect evidence, but not measured directly yet), and a higher viscosity: According to molecular dynamics (MD) calculations, the backbone of A8-35 in a particle moves ~ 10 × more slowly than the hydrophilic head group of micellar sodium dodecyl sulfate (SDS; Perlmutter et al. 2011). Upon size-exclusion chromatography (SEC), APol particles show a Stokes radius, R S, of 3.15 nm (Gohon et al. 2006) , with a highly homogeneous size distribution provided that the pH of the solution is ≥ 7 (Gohon et al. 2004, 2006) and divalent cations are absent (Picard et al. 2006). Indeed, the solubility of A8-35 particles being due to their negative charges, the protonation or complexation of carboxylate groups makes them less hydrophilic, leading them to aggregate (isolated carboxylates have a pKa of around 4.8; in A8-35, however, because of their close proximity, some of them start to protonate at or slightly below pH 7) . Multivalent cations, such as Ca2+, can bridge particles, leading, depending on the concentration of Ca2+, to the formation of small oligomers or to massive precipitation (Diab et al. 2007a; Picard et al. 2006) .
7.1.3 A Library of APols
APols with chemical structures different from that of A8-35 have been designed in order to make them insensitive to pH (Fig. 7.1) . Replacing the isopropyl groups by sulfonate groups yields sulfonated APols (SAPols), which remain water soluble even at pH 2 (Dahmane et al. 2011). Zwitterionic APols with phosphocholine polar head groups (PC-APols; Diab et al. 2007a, b; Tribet et al. 2009) and nonionic APols (NAPols) carrying sugar groups (Bazzacco et al. 2012; Sharma et al. 2012) have also been synthesized and validated. This new generation of APols widens the field of possible applications.
A8-35, even if its pH sensitivity can create limitations in some specific applications, like nuclear magnetic resonance (NMR), remains one of the easiest APols to synthesize and to label, and by far the most thoroughly studied one in terms both of its properties and of its applications. This makes it very attractive to diversify, label, and/or functionalize it, and has led to the creation of a library of A8-35 variants specially designed for specific applications (Table 7.1) . Variations around the A8-35 structure concern, for instance, the percentage of charges (A8-75), the length of the backbone (A34-35), or both (A34-75; Tribet et al. 1996). Isotopically labeled or functionalized versions of A8-35 have also been synthesized and validated. For example, A8-35 and A8-75 have been labeled with such isotopes as 14C (Tribet et al. 1997), 3H (Gohon et al. 2008), and 2H (Giusti et al. 2014b; Gohon et al. 2004, 2006). 14C- and 3H-labeled APols are useful as tracers for the detection of APols in solution, and have been used to quantify the amount of MP-bound A8-35 (Gohon et al. 2008; Tribet et al. 1997), whereas the deuterated versions have been used in NMR, analytical ultracentrifugation (AUC), and small-angle neutron scattering (SANS) experiments (see e.g., Catoire et al. 2009, 2010a; Gohon et al. 2008; Zoonens et al. 2005) .
Grafting a small percentage of a tag or label onto A8-35 does not affect its solution properties (reviewed in Le Bon et al. 2014b), while functionalizing it for specific purposes. Fluorescent APols (FAPols) bearing different fluorophores, which cover a broad range of excitation and emission wavelengths, have thus been synthesized (Fernandez et al. 2014; Giusti et al. 2012; Opačić et al. 2014; Zoonens et al. 2007). Affinity tags have also been grafted onto A8-35, such as biotin (Charvolin et al. 2009), polyhistidine (Giusti et al. 2014a), randomly distributed imidazole groups F. Giusti, unpublished data, an oligonucleotide (Le Bon et al. 2014a), or a thiamorpholine (unpublished data), yielding tagged APols nicknamed BAPol, HistAPol, ImidAPol, OligAPol and SulfidAPol, respectively (Table 7.1). Biotin has also been grafted onto a PC-APol (Basit et al. 2012) and a NAPol (Ferrandez et al. 2014). Tagged APols can be used to immobilize MPs onto solid supports (see below) .
7.1.4 General Properties of MP/APol Complexes
The activity, stability, and physical–chemical features of MP/APol complexes have been investigated in some details (for reviews, see Popot 2010; Popot et al. 2003, 2011; Zoonens and Popot 2014) . Briefly, APols adsorb exclusively onto the hydrophobic transmembrane region of MPs (Althoff et al. 2011; Cao et al. 2013; Catoire et al. 2009; Liao et al. 2013; Perlmutter et al. 2014; Zoonens et al. 2005), forming a compact layer ca. 1.5–2 nm thick (Althoff et al. 2011; Gohon et al. 2008; Perlmutter et al. 2014). Their association is very stable as long as no competing surfactant is present in the medium (Zoonens et al. 2007). On the other hand, because they are freely miscible with other surfactants , MP-bound APols can be easily displaced by detergents or other APols (Tribet et al. 1997, 2009; Zoonens et al. 2007). Similarly, MPs trapped in APols can be delivered to lipid bilayers (Nagy et al. 2001; Pocanschi et al. 2006) or three-dimensional (3D) lipid phases (Polovinkin et al. 2014) .
After trapping, MP/A8-35 complexes are almost—although not completely—as homogeneous as MP/detergent ones, and they present the same sensitivity as pure A8-35 particles to low pH and divalent cations (see, e.g., Gohon et al. 2008; Picard et al. 2006; Zoonens et al. 2007). The size of MP/APol complexes is slightly larger than that of MP/detergent ones, and, as a result, their rotational correlation time, τ c, is slightly longer (Catoire et al. 2010b). A major difference between APol-trapped and detergent-solubilized MPs is their stability, which is usually much higher in APols (Fig. 7.2). For example, the denaturation temperature of A8-35-trapped BLT1, a G protein-coupled receptor (GPCR) of leukotriene LTB4, is increased by ~ 11 °C as compared to that in Fos-choline-16 (Fig. 7.2a; Dahmane et al. 2009). Similar effects can be observed for β-barrel MPs, such as the monomeric outer membrane protein OmpA from Escherichia coli (Pocanschi et al. 2013), or a trimeric porin, MOMP, the major outer membrane protein from Chlamydia trachomatis (Fig. 7.2b; Tifrea et al. 2011) .

Comparison of membrane protein stability in detergent and after trapping in A8-35. a Thermostability curves of a GPCR, the BLT1 leukotriene receptor, in the detergent Fos-choline 16 supplemented with lipids (asolectin) in a 2:1 mass ratio (blue curve), in pure A8-35 (red), and in a mixture of A8-35 and lipids in a 5:1 mass ratio (gray). The stability was monitored by following, during storage at 4 °C, the ability of the GPCR to specifically bind LTB4. Adapted with permission from Biochemistry 48:6516–6521, copyright 2009 American Chemical Society (Dahmane et al. 2009). b Thermostability of a trimeric porin, MOMP from Chlamydia trachomatis, monitored by CD in the detergent Z3-14 (open symbols) and after trapping with A8-35 (solid symbols). Reprinted from Vaccine 29:4623–46231, copyright 2011, with permission from Elsevier (Tifrea et al. 2011). c Evolution of the ATPase activity of the calcium pump (Serca-1a) over time after removal of Ca2+ from preparations. Sarcoplasmic reticulum vesicles were solubilized in the detergent C12E8 (blue curve), then supplemented with A8-35 (magenta), and finally diluted under the CMC of the detergent, so that the protein’s environment became predominantly the APol (green; adapted from Champeil et al. 2000)
In some cases, APols may affect the activity of the MPs they bind to. No or very moderate effects have been seen on the functional and pharmacological properties of the nicotinic acetylcholine receptor (Martinez et al. 2002), bacteriorhodopsin (Bazzacco et al. 2012; Dahmane et al. 2013; Gohon et al. 2008), or GPCRs (Banères et al. 2011; Bazzacco et al. 2012; Catoire et al. 2010a; Dahmane et al. 2009; Damian et al. 2012; Rahmeh et al. 2012). APol-trapped MPs can be recognized by antibodies (Charvolin et al. 2009; Giusti et al. 2014a; Le Bon et al. 2014b), by toxins (Charvolin et al. 2009), as well as by other soluble proteins (Basit et al. 2012). However, presumably because of repulsive electrostatic interactions , A8-35 slows down the binding to GPCRs of G proteins and of arrestin, whereas NAPols do not (Bazzacco et al. 2012). The sarcoplasmic calcium pump (Ca2+-ATPase) is reversibly inhibited after trapping in APols (Champeil et al. 2000; Picard et al. 2006) . At the same time, its half-life following calcium removal increases by ~ 60 × over that in detergent solution (Fig. 7.2c). Our current working hypothesis is that both effects originate from the viscosity of the APol environment (Perlmutter et al. 2011), which would slow down the large conformational transitions of the transmembrane helix bundle that take place both during the functional cycle of the Ca2+-ATPase and at the onset of denaturation (for discussions, see Picard et al. 2006; Popot et al. 2003, 2011). Other stabilization mechanisms also come into play, among which a less efficient competition of APols, as compared to detergents, with stabilizing protein/protein and protein/lipid interactions, as well as the reduction of the hydrophobic sink that is made possible by the high affinity of APols for MPs and their very low CAC (for a discussion, see Popot et al. 2011). The extent and origin of the stabilizing effect may vary from protein to protein, and different APols stabilize MPs to different extents. It seems, for instance, that the lesser the charge density along the APol chain, the better the stabilization (see e.g., Bazzacco et al. 2012; Picard et al. 2006) .
7.1.5 Overview of APol Applications
APols were initially developed to handle MPs in aqueous solutions under less destabilizing conditions than can be achieved with detergents . This expectation has been largely vindicated (Popot 2010; Popot et al. 2011), despite a few exceptions (such as cytochrome b 6 f; see Tribet et al. 1996). Their field of applications covers essentially all of those that are classically implemented in detergent solutions, with the benefit of improved stability. In addition, a few specific uses have been validated, based on the specific properties of MP/APol complexes and the rich chemistry of APols. Table 7.2 summarizes those applications that have been validated so far (for more details, see Popot 2010; Popot et al. 2011; Zoonens and Popot 2014) . Two applications that remain problematical to date are infrared studies in the amide band absorption region—because all existing APols absorb in this region—and crystallization. Extensive tests have shown that a model MP, cytochrome bc 1, does not crystallize when trapped in pure A8-35, whereas it does in a mixture of APols and detergent (Charvolin et al. 2014). Two phenomena can probably be incriminated, electrostatic repulsion between the complexes, which is reduced upon diluting A8-35 with a nonionic detergent, and the relative heterogeneity of MP/APol complexes, probably due to imperfect relaxation of the APol belt to its free energy minimum, which disappears in MP/APol/detergent ternary complexes (Zoonens et al. 2007). While not a good medium for crystallization, APols can be used to produce, by folding them from inclusion bodies or by cell-free expression (CFE), the MPs to be crystallized. APol-trapped MPs can then be transferred to a more favorable crystallization medium, such as lipid 3D phases (Polovinkin et al. 2014) .
7.1.6 Conclusion
In summary, APols are promising new surfactants for biochemical and biophysical studies of MPs in aqueous solution, because they form with MPs small and compact water-soluble complexes while improving the stability of the MPs they interact with. The APol layer surrounding the transmembrane domain of MPs is very stable, but it can be exchanged for other surfactants. The chemistry of APols allows modifications and labeling, generating a library of molecules with different molecular structures and more than a dozen types of labeled or functionalized versions of APol A8-35, expanding the scope of applications in both basic and applied research. Among the many novel surfactants developed to replace classical detergents, APols present the advantage of being remarkably easy to use. Their implementation is facilitated by the vast corpus of works describing in detail their properties and those of MP/APol complexes, as well as procedures for most conceivable applications, and by the commercial availability of APol A8-35.
In the next section, five protocols are presented in detail, describing: (1) how to trap MPs in APols so as to improve their stability, (2) how to quantify the amount of APol bound per MP, (3) how to fold an MP to its native state using APols, (4) how to produce MPs by CFE in presence of NAPols, and finally (5) how to immobilize MPs onto a solid surface for surface plasmon resonance (SPR) measurements.
7.2 Detailed Protocols for Some Major Applications
7.2.1 Protein Trapping in APols
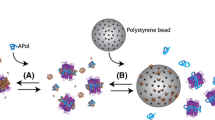
APols are not (or very weak) detergents . As a consequence, they are poorly efficient for direct extraction of MPs from biological membranes, even if this has been observed in very few cases (Popot et al. 2003) . Because of this feature, detergents must be used for solubilization, unless MPs are produced directly in the presence of APols (cf. § 7.2.4). The trapping procedure consists, then, of replacing detergents by APols in a sample of MPs, which, in general, has already been purified (Fig. 7.3). The protocol is simple, easy, fast, and requires no important biochemical optimization .

Trapping membrane proteins in amphipols. a Schematic representation of MP trapping in APols. Adapted with permission from Proc Natl Acad Sci USA 102:8893–8898, copyright 2005 National Academy of Sciences, USA (Zoonens et al. 2005). b Determination of the optimal MP/APol mass ratio by measuring the optical density at 280 nm of samples of tOmpA trapped at different mass ratios of A8-35 before and after ultracentrifugation at 60,000 × g for 20 min at 4 °C (from Zoonens 2004). c SEC profiles of tOmpA in the detergent C8E4, in a mixture of detergent and A8-35, and after trapping in A8-35. Reprinted with permission from Biochemistry 46:10392–10404, copyright 2007 American Chemical Society (Zoonens et al. 2007)
7.2.1.1 Preparation of a Stock Solution of APols
A8-35 is supplied as a white powder, which can be stored at room temperature . Note that most of APols are very stable molecules, except for NAPols and FAPols, which carry sugar groups and fluorescent probes, respectively . Storage of NAPols at − 20 °C is advisable whatever their conditioning, i.e., in powder or in solution, because sugars can be hydrolyzed. FAPols need to be protected from UV-visible light with aluminum foil. When needed, a stock solution of APols at 100 g L−1, or 10 % w/w, is prepared with MilliQ water (water purified on an A10 Advantage Millipore system):
-
Weigh some powder, for instance 20 mg, with an analytical balance in an Eppendorf tube or, if possible, in a small glass vial (note: The powder is sometimes very electrostatic and caution is required).
-
Add 180 µL of MilliQ water in order to reach a final mass of 200 mg.
-
Homogenize the solution with a vortex or by magnetic stirring. Incubate at least a couple of hours before use for a good rehydration of the lyophilized powder. The solution is then kept at 4 °C or, if need be, frozen at − 20 °C .
7.2.1.2 Determination of the Protein Concentration
The exchange of detergent for APols is carried out by supplying APols pre-solubilized in water to the sample of MPs (Fig. 7.3a) . The amount of APol to be added is calculated on the basis of the mass of MP initially present in the sample. The concentration of protein must be known, at least approximately. After purification, the concentration of protein in the detergent solution is determined by its optical density from UV-visible spectra. If the epsilon of the protein is unknown, its concentration can be assessed by colorimetric measurements such as bicinchoninic acid (BCA) assay. Alternatively, amino acid analysis after HCl hydrolysis can also be used .
7.2.1.3 Determination of the Optimal MP/APol Mass Ratio
The sole optimization to do is that of the mass ratio of APols required to keep soluble the MP well dispersed in aqueous solution after detergent removal. For that, the protein and detergent concentrations are kept unchanged while increasing concentrations of APols are tested :
-
Determine the MP/APol mass ratios to be tested. Table 7.3 gives an example, considering the concentration of MP to be 1 g L−1 and the volume of aliquots to be 500 μL for each condition.
Table 7.3 Example of conditions to be tested to determine the optimal MP/APol mass ratio -
Pipet seven aliquots of equal volume of MP present in detergent solution. Add the appropriate volume of APols calculated. Note that dilution effects of the final volume can be neglected up to 10 % of variation after adding APols. Keep aside the two control samples.
-
Mix and incubate for 15–20 min at either room temperature or 4 °C depending on the stability of the protein of interest.
When APols are supplied to the samples, they mix freely with detergent molecules in micelles and at the transmembrane surface of the protein, as shown by fluorescence and isothermal calorimetry studies (Tribet et al. 2009; Zoonens et al. 2007). This leads to the formation of MP/detergent/APol ternary complexes .
7.2.1.4 Detergent Removal
This step can be achieved in various ways . Most often, detergent removal is carried out by adsorption into polystyrene beads (Bio-Beads). Note that APols do not absorb onto Bio-Beads (Zoonens 2004; Zoonens et al. 2007). The mass of beads to be added is typically 20 × the mass of detergent present in the sample. For instance, if the concentration of detergent is 6 g L−1 in 500 μL, the amount of Bio-Beads to be added is ~ 120 mg:
-
Calculate the appropriate amount of beads according to the mass of the detergent present in each sample. Weigh the beads and add them in the five samples containing APols plus in one of the two control samples, which will become the negative control (note: Bio-Beads are usually washed out successively in ethanol and water prior usage and then stored in water. Before weighing, dry them on a tissue paper for maximal water removal). The last sample, without beads, represents the positive control .
-
Incubate the samples for 2 h under gentle shaking at either room temperature or 4 °C.
-
Let the beads sediment by gravity or proceed to a quick centrifugation. Pipet the samples using a microcapillary tip that excludes the beads and put them in new Eppendorf tubes .
Alternatively, it is possible to eliminate the detergent micelles by dilution under the CMC of the detergent. Note that this method is more suitable to detergents with a high CMC rather than detergents with a low one, such as n-dodecyl-β-d-maltoside (DDM), because even under CMC these detergents are still able to keep MPs soluble. If the dilution method is employed, dilute the five samples containing APols plus that of the negative control with a detergent-free buffer. Dilute the last sample, which becomes the positive control, with buffer containing detergent at the same concentration as initially in the sample .
Whatever the protocol used, some detergent monomers can still be present in the samples. Usually, they are not problematic, as long as the negative control shows that the monomers cannot keep the MP in solution in the absence of APols, but if need be they can be eliminated by dialysis or by several cycles of dilution/concentration using ultrafiltration devices. Note that the presence of APols in the external dialysis buffer is not required, as APols do not cross standard dialysis membranes of 12–14-kDa MW cutoff. Indeed, the MW of the particles of A8-35 is ~ 40 kDa (Gohon et al. 2006), and, because of its low CAC (0.002 g L−1; Giusti et al. 2012), there are very few free molecules in solution. Another procedure for detergent removal, albeit seldom used, is to adsorb it onto cyclodextrins (Althoff et al. 2011) .
7.2.1.5 Identification of the Best MP/APol Ratio
-
Measure the UV-visible spectrum of each sample .
-
Centrifuge the samples at 100,000 × g for 20 min (Note: The speed and duration of the centrifugation step are given for a small protein of about 30 kDa. These parameters can be adjusted if the protein of interest is bigger) .
-
Take off the supernatants and measure again their UV-visible spectra.
-
Calculate the percentage of protein kept in the supernatant for each condition. This experiment determines the minimal MP/APol mass ratio required to keep MPs soluble (Fig. 7.3b). However, to establish the minimal MP/APol mass ratio required to obtain homogeneous complexes, which is slightly higher, it is recommended to analyze the samples by SEC (Fig. 7.3c) .
The optimal MP/APol mass ratios for two model MPs of small MW like bacteriorhodopsin of H. salinarium (BR, 27 kDa) and the transmembrane domain of OmpA of E. coli (tOmpA, 19 kDa) are 1:5 and 1:4, respectively (Gohon et al. 2008; Zoonens et al. 2007). These ratios exceed by ≥ 2 × the amount of A8-35 that binds to these MPs (see next protocol). This is because APols, which have a weak dissociating power, cannot prevent protein/protein interaction if they are not present in excess in the sample. To keep MP/APol complexes homogeneously distributed, an excess of APols is thus required. There is no need, however, to increase the concentration of APols beyond the minimal concentration yielding an acceptable monodispersity because, due to the hydrophobic sink effect, this may compromise the stability of fragile MPs (Popot et al. 2011) .
7.2.2 Measuring of the Amount of Bound APols
APols specifically adsorb onto the transmembrane region of MPs (Althoff et al. 2011; Cao et al. 2013; Catoire et al. 2009; Liao et al. 2013; Zoonens et al. 2005), where they form a compact layer ca. 1.5–2 nm thick (Althoff et al. 2011; Gohon et al. 2008; Perlmutter et al. 2014) . The amount of polymers constituting the belt surrounding MPs was estimated in two studies using BR and tOmpA as model MPs. The first determination is based on extensive physical measurements carried out on BR complexed with either plain or deuterated A8-35, using primarily SANS and AUC (Gohon et al. 2008). It is thought to give a relatively accurate measurement of the amount of A8-35 bound per BR monomer, but is extremely laborintensive. The second study relied on the use of a fluorescent APol (FAPol). Under the conditions used in Zoonens et al. (2007), it yielded what is thought to be a lower limit to the amount of A8-35 bound to the tOmpA monomer (see below). We describe below first, how to express the amount of APol bound per MP, next, how to estimate a priori the amount of APol a given MP is likely to bind, and then three protocols for measuring it using FAPols .
7.2.2.1 Why is it Preferable to Express the Amount of APols Bound per MP in Mass Rather than as a Number of Molecules?
APols being highly polydisperse polymers, the size of individual molecule varies considerably and their MW can be estimated only on average. The average MW of A8-35 molecules is ~ 4.3 kDa (note: This revised mass is twice smaller than had been initially estimated; for a discussion, see Giusti et al. 2014b). However, despite the variable MW of individual APol chains, the particles they form in solution migrate upon SEC with a size distribution as narrow as that of globular proteins. SANS and AUC analyses indicate that they feature a well-defined Stokes radius (R S = 3.15 nm) and MW (40 kDa; Gohon et al. 2006). Interestingly, the size and homogeneity of A8-35 particles do not depend on the degree of heterogeneity of the molecules that constitute them. Indeed, a version of A8-35 with a restricted length polydispersity forms particles with the same apparent size and dispersity as standard A8-35 particles (F. Giusti and C. Tribet, unpublished results). The average MW of individual molecules being only a very rough estimate, the amount of APol bound per MP is much more meaningfully expressed in mass ratio rather than as a molar stoichiometry. Similarly, in the case of functionalized APols, to preserve accuracy and reliability, the number of fluorophores or tags is expressed as their number per 40-kDa APol particle, which can be used as a mass reference, rather than as their number per APol chain, which has no great significance and is inaccurate .
7.2.2.2 How to Estimate a Priori the Likely Amount of APols Bound per MP Based on Structural Data?
In the case of α-helical MPs, the most thoroughly studied MP/APol complexes are those of BR with A8-35. In the complexes, the protein/APol mass ratio is ~ 1:2, i.e., ~ 54 kDa of A8-35 per monomer of BR (27 kDa; Gohon et al. 2008). Lipids (~ 9 kDa) are also present in the complexes. In the case of β-barrel MPs, the best-characterized complexes, in terms of composition, are those of tOmpA with A8-35. The mass ratio that has been estimated is ~ 1:1.3, i.e., ~ 25 kDa of A8-35 per monomer of tOmpA (19 kDa; Zoonens et al. 2007). This value should be considered as a minimal value, however, because the conditions under which the measurements were done (see below) led to some aggregation and, very likely, to the loss of some APol. A more likely assumption of bound APol per tOmpA monomer would be ~ 45 kDa, which corresponds to the amount of APol needed, in MD calculations, to fully cover the transmembrane domain of OmpX, an eight-stranded β-barrel whose dimensions are very similar to those of tOmpA (Perlmutter et al. 2014) .
Based on these values, and assuming that the volume of the A8-35 belt surrounding an MP is roughly proportional to the perimeter of the transmembrane domain to be covered, it is possible to estimate the amount of APols interacting with any other MP. The only information needed is the dimensions of the hydrophobic domain of the protein of interest, modeled as a cylinder filled out by the transmembrane helices or delimited by the number of β-strands, whose perimeter increases roughly as the square root of its volume (α-helical bundles), or linearly with the number of β-strands. For instance, the transmembrane domain of BR is a bundle of seven α-helices. If the MP of interest has a similar organization, like, for example, a GPCR, it can be expected to bind approximately the same amount of A8-35, i.e., ~ 54 kDa (note, however, that BR/A8-35 complexes comprise ~ 9 kDa of lipids (Gohon et al. 2008), which increase the transmembrane perimeter). On the other hand, if the MP contains twice more helices than BR and features a more or less homothetic shape, the volume of its transmembrane domain doubles while the transmembrane surface increases by ~ 40 %, and one can expect in the ballpark of 75 kDa of bound A8-35. It is fair to say, however, that too few reliable measurements are available to date to gather how reliable such an approach is . The only other relatively precise estimate of bound A8-35 has been obtained with the cytochrome bc 1 dimer, which has 22 transmembrane helices. On the basis of the above calculations, one would expect it to bind ~ 96 kDa A8-35. The experimental estimate is only 49–63 kDa (Charvolin et al. 2014; Popot et al. 2003). Note also that it is not unreasonable to expect that the ionic strength may modulate the volume and mass of the belts of ionic APols because it modulates the repulsion between charged polar groups (see Popot et al. 2003). Despite these uncertainties, estimating a priori the probable mass ratio of MP to APol in complexes is useful to provide guidelines when planning trapping experiments, or when endeavoring to measure experimentally the amount of bound APol .
7.2.2.3 How to Experimentally Measure the Quantity of APols Bound per MP?
As previously mentioned, the mass of APol to add for trapping is in excess of that of APol that actually binds to the surface of the MP. After trapping, some APol remains present as free particles in the sample. Measuring the amount of bound APols can be carried out by several approaches. Whatever the method chosen, the analysis is considerably facilitated by using FAPols as tracers:
-
Prepare a stock solution of APol/FAPol mixture.
FAPols carrying various fluorescent probes have been synthesized (see Table 7.1). The choice of FAPol depends on the absorption spectrum of the protein of interest. For instance, if the protein absorbs only at 280 nm, FAPolNBD, which shows a maximum absorption at 490 nm, is suitable. On the other hand, if the protein absorbs also visible light, as BR does, another FAPol, like FAPolAlexa647, must be chosen in order to avoid overlapping between protein and FAPol absorption bands .
Pure FAPols usually absorb too much at the peak of absorbance of fluorophore, and possibly also at 280 nm, interfering with protein determination. They are better used diluted with nonfluorescent APol (A8-35). Because labeled and unlabeled APols freely and rapidly mix in salty aqueous solutions (Zoonens et al. 2007), they can be mixed from two stock solutions prepared at 100 g L−1. A convenient FAPol/A8-35 ratio is one at which the absorbance of the FAPol at its maximal absorption wavelength is, in the complexes with the MP, ~ 25 % of that of the protein at 280 nm. This ratio can be estimated a priori based on the extinction coefficients of the protein and FAPol, the estimation of the amount of APols bound per MP (cf. § 7.2.2.2), and on the MP/APol mass ratio needed for trapping. If the protein possesses many tryptophan residues, its extinction coefficient may be high enough so that no dilution of the FAPol stock solution is necessary :
-
Measure the spectral absorbance of the FAPol/A8-35 mixture (or pure FAPol if dilution with A8-35 is not necessary) and determine the relative contribution of APols at 280 nm and at the peak of absorbance (note: Even if neither APol nor FAPol absorb significantly at 280 nm, it is advisable to quantify it).
After the complexes with an MP have been formed, they must be separated from the excess of APol used at the trapping step.
Method 1: Size-exclusion chromatography (SEC). This approach is appropriate for large MPs—40 kDa or above—because MP/APol complexes and free APol particles are sufficiently resolved:
-
Wash the gel filtration column with three column volumes of running buffer. Note that, for analytic analysis of the samples, APol is usually not required in the running buffer, in contrast to detergents, which must always be present above their CMC. However, for this particular experiment, it cannot be excluded that a small amount of APol leaches from the protein as the complexes migrate into APol-free buffer. It might therefore be preferable to saturate the solution with which the column is equilibrated with “some” free APol, such as the CAC or twice the CAC, so as to prevent desorption, and to subtract the corresponding background. However, this modification to the procedure has not been carefully investigated yet. The composition of standard buffer is 20 mM Tris-HCl, 150 mM NaCl, and pH 8.0 but it can be modified provided that pH is above 7 and divalent cations are absent .
-
Inject an aliquot of FAPol/A8-35 mixture (or pure FAPol) at 10 g L−1. The elution profile is monitored at two wavelengths, 280 nm and the maximum absorption wavelength of the fluorophore, for example, 490 nm for FAPolNBD. Determine the elution volume of APol particles.
-
After trapping the protein in the FAPol/A8-35 mixture, inject an aliquot of the sample at an appropriate concentration in order to get a good signal-to-noise ratio of the elution peak. Follow the elution of MP/A8-35/FAPolNBD complexes at the two wavelengths 280 and 490 nm . If the separation from free APol particles is good, calculate the amount of bound APols per MP as follows: Integrate the peak area of MP/A8-35/FAPolNBD complexes at 280 and 490 nm in order to determine, respectively, the mass of MP and that of FAPolNBD which had co-migrated with the protein. A subtraction of the APol contribution to the absorption of the protein at 280 nm may have to be applied, based on the ratio of the surface of the peaks at 280 and 490 nm observed with the pure FAPol/A8-35 mixture or on the ratio of the optical densities at 280 and 490 nm measured from a UV-visible spectrum . The total mass of APol is then calculated taking into account the dilution of FAPol with A8-35. The ratio of APol and MP masses gives the amount of bound APols per MP (note: If the elution peaks of APol particles and MP/APol complexes overlap, use a more resolutive gel filtration column or try another separation procedure).
Method 2: Immobilized metal affinity chromatography (IMAC). The presence of a tag fused to the MP under study makes it possible to immobilize MP/APol complexes onto an affinity column and to eliminate the excess of APol particles . This procedure is particularly convenient when the protein is small and MP/APol complexes cannot be efficiently separated from free APol particles by SEC. Note that free APol particles are, however, required to keep homogeneous MP/APol complexes. Indeed, as previously mentioned, APols are poorly dissociating surfactants, and so, in the absence of free APol particles, small MP/APol oligomers start to form, which is likely to be accompanied by some desorption of the MP-bound APol (Zoonens et al. 2007). Because of this effect, the MP/APol ratio determined by this method must be taken as an estimate by default unless buffers are saturated with some free APol :
-
After trapping the MP in the FAPol/A8-35 mixture, inject the sample on an affinity resin. For instance, if the protein has a polyhistidine tag, load the sample on a Ni:NTA column. The majority of the protein (~ 80 %) will be retained on the resin (Giusti et al. 2014a).
-
Rinse the resin with equilibration buffer to wash out free FAPol/A8-35 particles. Elute the MP/FAPol/A8-35 complexes with a buffer containing imidazole. Note that, as said before, the presence of APol in equilibration and elution buffers at the CAC or twice the CAC may be advisable, so as to prevent desorption .
-
Desalt the sample to remove imidazole and measure the optical density of the sample at 280 nm and at the maximum wavelength of FAPol. The concentrations of MP and FAPol are calculated using their respective extinction coefficients. Subtract, if need be, the contribution of FAPol at 280 nm and calculate the total mass of APol if FAPol was initially mixed with A8-35 before trapping. The ratio of APol and MP masses gives the amount of bound APol per MP .
Method 3: Analytical ultracentrifugation (AUC) . The MP/APol mass ratio in complexes can be precisely determined by sedimentation velocity (SV) measurements using AUC. AUC is a priori applicable to any MP, because the density of APol particles and that of MP/APol complexes are different enough for them to separate during the centrifugation run, even if their hydrodynamic radii are not very different . For example, the sedimentation coefficients (S) of A8-35 particles (R S ≈ 3.15 nm; Gohon et al. 2006) and BR/A8-35 complexes (R S ≈ 5.0 nm; Gohon et al. 2008) are 1.6 and 3.2 S, respectively, making them easily distinguishable (Gohon et al. 2006, 2008). The specific volume of the sodium salt of A8-35, \({{\bar{v}}_{2}}\), is 0.809 L g−1, its density, \(\rho ={1}/{{{{\bar{v}}}_{2}}}\;\), 1.236 L g−1 (Gohon et al. 2004, 2006) . The MP/APol mass ratio can be determined by sophisticated AUC measurements involving the comparison of sedimentation properties of complexes formed between the protein and unlabeled or deuterated A8-35 and/or simultaneous measurements of the absorbance and refractive index of the complexes (Gohon et al. 2008). However, with the advent of FAPols, it is simpler to measure the respective absorbance of the protein and the APol in the complexes, as done above for the complexes separated by SEC or affinity chromatography:
-
After MP trapping in FAPol/A8-35 mixture, adjust the sample concentration by dilution or concentration so that the protein absorbance at 280 nm, in the AUC cell, reaches ~ 0.5 .
-
Define the parameters of the SV run, namely time and speed, according to the sedimentation coefficient of the protein under study. For instance, in the case of small MPs, like BR or tOmpA, the SV experiment is carried out at 42,000 rpm during 4 h. The migration of the particles and complexes is followed at two wavelengths, 280 nm and the maximum absorption wavelength of FAPol, and, if available, with interference optics, which gives a measure of the refractive index.
-
Measure the solvent density and viscosity.
-
Analyze the SV profiles with Sedfit or an equivalent program (for details, see Gohon et al. 2008). The distribution c(S) of sedimentation coefficients (S) shows peaks reflecting the migration of MP/APol complexes and of free APol particles during the SV run. Integrate the peak areas at 280 nm and at the second wavelength. As noted above for SEC and IMAC experiments, the contribution of APols at 280 nm may have to be subtracted from the protein signal. MP and FAPol masses can be determined from their respective extinction coefficients. Calculate the total mass of APol from the FAPol/A8-35 ratio. The ratio of APol and MP masses gives the amount of bound APol per MP .
Whereas this procedure is technologically more demanding, it presents the advantage, over that by SEC and by IMAC, that the MP/APol complexes are never separated from the free APol, which eliminates the risks of desorption and/or aggregation.
7.2.3 APols-Assisted Folding of a MP
APols have proven to be very helpful to fold MPs expressed as inclusion bodies in E. coli, like class A GPCRs or porins (Banères et al. 2011; Bazzacco et al. 2012; Dahmane et al. 2009, 2011, 2013; Pocanschi et al. 2006; see also Chap. 3 by JL Banères in this volume; Fig. 7.4) . The protocol used for α-helical MPs is derived from one initially developed to refold BR in lipids (see Popot et al. 1987, in which many useful practical details can be found). It is quite simple but requires some optimization regarding the quantity of APol to be added and the presence or absence of lipids. For variants and the effect of various modifications to this protocol, see Dahmane et al. 2013 .

Amphipol-assisted folding of membrane proteins to their native state. a Folding of BR from a denaturated state in SDS. Top, UV-visible spectra of samples before and after precipitation of SDS with KCl in the presence of A8-35 at various MP/APol mass ratios. In SDS, the sample is colored yellow because BR is denaturated in bacterio-opsin (BO) and the retinal is released in solution. After refolding, the color shifts to purple because the retinal has rebound to the protein, indicating that BO has recovered its native structure. Bottom, SEC profiles of the same three samples after folding. Reprinted with permission from Biochemistry 45:13954–13961, copyright 2006 American Chemical Society (Pocanschi et al. 2006). b Comparison of the yield of folding of four GPCRs in detergent + lipids (D + L) and in A8-35 + lipids (A + L). Adapted with permission from Biochemistry 48:6516–6521, copyright 2009 American Chemical Society (Dahmane et al. 2009). c Folding of a porin, full-length OmpA from E. coli, after 19 × dilution from 8 M urea into an A8-35 solution. The unfolded (U) and folded (F) forms of OmpA were separated by SDS-PAGE. Reprinted from Eur Biophys J 42:103–118, copyright 2013, with kind permission from Springer Science and Business Media (Pocanschi et al. 2013)
7.2.3.1 Solubilization and Purification of MPs in Denaturing Conditions
Inclusion bodies are clusters principally comprised of misfolded forms of the protein of interest, but they can also contain some DNA and other bacterial proteins . They need to be solubilized and purified in denaturing conditions. For α-helical MPs, the denaturing agent is usually SDS, whereas for β-barrel MPs it is urea. Purification is most often carried out by affinity chromatography. For instance, if the protein of interest is fused to a polyhistidine tag, purification can be carried out on Ni:NTA resin. Note that the concentration of SDS and urea tolerated by the resin is given by the supplier. Also, it is essential to work at room temperature, because both urea and SDS crystallize at low temperature:
-
Isolate the inclusion bodies by differential centrifugations and determine the concentration of protein by the BCA assay.
-
Prepare the solubilization buffer containing the appropriate denaturing agent. For example, for α-helical MPs, the buffer contains 10 mM Tris–HCl, pH 7.5, 100 mM NaH2PO4, 6 M urea, 0.8 % SDS, 10 % glycerol, 4 mM β-mercaptoethanol (adapted from protocols described in Banères et al. 2005; Damian et al. 2006). For porins, the solubilization buffer contains 10 mM borate, pH 10.0, 8 M urea, 2 mM EDTA (Pocanschi et al. 2006, 2013; note: The presence of reducing agent is required only if cystein residues are present. The solubility of urea can be increased to 10 M by heating).
-
Dissolve the inclusion bodies in the appropriate solubilization buffer at a final concentration of 10 g L−1 and incubate overnight at room temperature (note: Sonication pulses can be applied to speed up solubilization) .
-
• Centrifuge the sample for 20 min at 20,000 × g in order to remove insoluble material.
-
Proceed to the purification step. Note that for a purification of α-helical MPs on Ni:NTA resin, the buffers are: (1) equilibration buffer: 50 mM Tris–HCl, pH 8.0, 300 mM NaCl, 0.8 % SDS, 4 mM β-mercaptoethanol; (2) elution buffer: 50 mM Tris–HCl, pH 8.0, 300 mM NaCl, 0.8 % SDS, 400 mM imidazole, 4 mM β-mercaptoethanol; and (3) desalting buffer: 50 mM Tris–HCl, pH 8.0, 0.8 % SDS, 4 mM β-mercaptoethanol. For purifying β-barrel MPs, SDS in each buffer is replaced by 8 M urea.
-
Determine the concentration of protein by UV-absorbance or by the BCA assay .
7.2.3.2 Renaturation in APols
This step consists of either exchanging SDS for APols (protocol 1) or diluting urea in the presence of APols (protocol 2). The optimal MP/APol mass ratio must be determined by carrying out folding tests with variable amounts of APols.
Protocol 1 (for α-helical MPs):
-
Distribute 0.25 mg of the MP to be folded in three Eppendorf tubes. Add increasing volumes of APol—5, 12.5, 25 μL—from a stock solution at 100 g L−1 in order to obtain MP/APol mass ratios equal to 1:2, 1:5, and 1:10. Note that lipids generally help in the folding process, cf. (Dahmane et al. 2009, 2013). To test it, they can be supplied to the samples so that the APol/lipid mass ratio is 1:0.2, but this ratio can be optimized, as well as the nature of the lipids .
-
Mix and incubate the samples for 30 min at room temperature.
-
SDS is eliminated by precipitation with KCl added from a 4-M stock solution so that the final concentration of KCl in the samples is equal to 150 mM plus the concentration of SDS. For example, if the volume of the sample is 500 μL and the concentration of SDS is 0.8 % (28 mM), the final KCl concentration should be 178 mM. The volume of KCl to be added is thus 44.5 μL.
-
Incubate 30 min at room temperature under vigorous stirring.
-
Centrifuge the samples 5 min at the maximum speed of a benchtop centrifuge at 20 °C.
-
Collect the supernatant and repeat the centrifugation step.
-
Measure the optical density of samples.
Protocol 2 (for β-barrel MPs):
-
Fix, e.g., to 1 mg the mass of protein to fold. Dilute the sample by a 10× dilution factor into urea-free buffer containing 5 mg of APols, so that the final MP/APol mass ratio is 1:5. Test also ratios 1:2 and 1:10. Note that the dilution factor and speed of dilution can be optimized. If need be, incubate the samples at 40 °C for 24 hours.
-
Concentrate the samples using an ultrafiltration device and measure the optical density of supernatants.
7.2.3.3 Further Renaturation
To increase the yield of folding, urea or SDS traces can be further eliminated by a dialysis step:
-
Dialyze the sample for 24 h at room temperature using a standard dialysis membrane of 12–14-kDa MW cutoff. Note that APols are not needed in the external bath, but the presence of 150 mM KCl is required to prevent redissolution of crystallites of potassium dodecyl sulfate that may not have been totally removed by centrifugation. The volume of the external bath is ~ 500 × larger than the volume of the samples.
-
Recover the sample and centrifuge it for 5 min at the maximum speed of a benchtop centrifuge .
-
Measure the optical density of the samples.
-
If the buffer needs to be exchanged, proceed to a second dialysis for 24 h at 4 °C.
The solubility of MP is not a criterion of folding. The simplest and most direct proof that the protein adopts its native conformation is to check its activity. If the activity assay is not easy to set up, the yield of folding can be assessed by other approaches such as ligand-binding experiments using equilibrium dialysis. In that case, ligand titration can be monitored by radioactivity or changes in the intensity of emission fluorescence or absorption. If the protein is naturally colored in its native conformation due to the binding of a cofactor, such as retinal for BR, the native state can be quantified by spectral absorption changes. It is also possible to check the homogeneity and size of the protein by SEC, its secondary structure by CD , the local environment of tryptophan residues by CD and fluorescence measurements, and the melting temperature by differential scanning calorimetry or by fluorescence thermal shift. In the case of porins, the folded state of the protein can usually be assessed by SDS-PAGE, upon which, as a rule, folded and unfolded forms exhibit different electrophoretic mobilities, or by dot blot if an antibody recognizing the native state of the protein is available, or by protease digestion, etc.
7.2.4 APols-Assisted CFE of a MP
Overexpression of MPs in vivo under their native form is extremely tricky because, unlike soluble proteins, these proteins, to be functional, have to be targeted and inserted into the membrane . Because this insertion process can be inefficient and is frequently toxic, it represents a major limiting factor for protein production. One strategy to overcome this problem is to resort to CFE (Zubay 1973; see also Chap. 2 by F. Bernhard in this volume). CFE presents many attractive features, among which to do away with the toxicity issue and to allow labeling using limited amounts of isotopically labeled or unnatural amino acids (Kigawa et al. 1999). Unfortunately, probably due to their anionic character, charged APols such as A8-35 or SAPols inhibit the CFE of MPs (Park et al. 2011). Such is not the case, however, for nonionic APols (NAPols; Bazzacco et al. 2012; Fig. 7.5). Excellent results have been obtained when BR was expressed in vitro in the presence of NAPols: The majority of BR (~ 90 %) was properly folded and remained stable over several months, whereas in DDM, in spite of similar production and folding yields, it tends to precipitate rapidly (Bazzacco et al. 2012; Park et al. 2011). Because APols are so much milder than detergents, developing APol-assisted CFE of MPs appears as a very promising approach .

CFE is carried out using a commercial system (5prime) or a homemade lysate (see Chap. 2 by F. Bernhard in this volume), in which an E. coli lysate provides the machinery to drive coupled transcription and translation in the presence of a DNA template (note: Prokaryotic lysate from E. coli is commonly employed to produce both pro- and eukaryotic MPs. The protocol below was developed using this lysate, but it is certainly possible to use eukaryotic lysates such as that from wheat germ). For large-scale production, a semipermeable membrane allows for a continuous supply of substrates and the removal of inhibitory by-products, thus extending the duration of expression and the protein synthesis yield .
7.2.4.1 CFE Small-Scale Reaction
To optimize the concentration of NAPols to be used, small-scale syntheses are carried out in the presence of a concentration range of NAPols :
-
Prepare a stock solution of NAPols at 100 g L−1, or 10 % w/w, in MilliQ water as previously described in protocol 7.2.1.1.
-
Small-scale syntheses are carried out in the presence of 0.5 µg of plasmid and 3, 5, 8, and 10 g L−1 of NAPols in 50 µL of lysate. Note that the quantity of NAPol tends to be larger than is actually necessary. Indeed, because the amount of synthesized proteins is hardly predictable, it is better to add an excess of it. The reaction is performed for 6 h in a thermomixer (Eppendorf) at 700 rpm and 25 °C (note: It is possible to optimize also the temperature, Mg2+ concentration, and other parameters, as exhaustively described in the same volume by Bernhard et al.) .
-
At the end of the small-scale tests, 10 µL samples are diluted with the same volume of 10 mM Tris–HCl, pH 8.0 buffer, and then centrifuged for 10 min at 16,000 × g at room temperature to check for protein solubility by Western blot .
7.2.4.2 CFE Large-Scale Reaction
Once the concentration of NAPols to be used has been optimized, the reaction can be scaled up to 1 mL of lysate :
-
A new solution of NAPols is prepared directly in the CFE reaction buffer used to dissolve all the components of the lysate. This allows reaching the desired final volume (1 mL) in the presence of the adequate quantity of NAPol. The solution of NAPols is stirred overnight to insure complete solubilization. Note that it is recommended to add 1 % of sodium azide in the solution.
-
15 µg of plasmid, in CFE reaction buffer, is incubated for 20 h in the thermomixer in the presence of NAPols at the optimized temperature (note: It is not necessary to add NAPols in the feeding chamber) .
7.2.4.3 Sample Collection
Plasmids for CFE (pIVEX) usually contain a hexa-His fusion tag to allow detection and purification of the expressed protein. After the lysate has been collected, the protein can be purified by IMAC, either in batch or using a pre-packed column. As previously mentioned, it is not necessary to add NAPols in the purification buffers.
7.2.5 APols-Mediated Immobilization of MPs
The development of sensors carrying MPs as recognition motifs has multiple applications in diagnostics and/or fundamental understanding of molecular interactions necessary for the design of new drug-discovery strategies . The development of such biosensors requires the isolation and immobilization of MPs onto solid supports without alteration of their native conformation and function and the use of appropriate bionanotechnological platforms for high-sensitivity detection .
Surface plasmon resonance (SPR; see Rich and Myszka 2005) is a label-free technique based on the propagation of an evanescent wave along a gold-coated surface. It permits the detection of changes of refractive index in the vicinity of the surface that result from the binding of partners . This technique requires the immobilization of one of the interacting partners onto the surface. Only a few studies have been published on isolated MPs due to the difficulty to maintain them functional in solution, and because direct interaction of the proteins with the surface tends to impair their function. Specific immobilization strategies are usually a good alternative (Jonkheijm et al. 2008). Various affinity tags can be used for protein immobilization, including polyhistidine tags, which bind to Ni:NTA motifs, affinity tags recognized by antibodies, and biotin or streptags binding to streptavidin . Functionalized APols fulfill several criteria for a specific immobilization of functional MPs onto surfaces: (1) They stabilize MPs, providing a longer lifetime to the isolated protein; (2) they form with MPs permanent complexes, which do not dissociate in the absence of free APols; biosensors can thus be used in surfactant-free buffers, which simplifies their implementation and considerably limits the risk of missing weak or moderate interactions with ligands; and, finally, (3) the complexes formed between MPs and functionalized APols will bind onto the solid surface of the biosensor without the need for any genetic or chemical modification of the protein (Fig. 7.6; Charvolin et al. 2009) .

Amphipol-mediated immobilization of membrane proteins onto solid supports a Principle of MPs immobilization using functionalized APols. b Measurement of the immobilization of an MP (left) and of the interaction of another with a specific antibody (right) by SPR. Left, application to streptavidin-coated chip lanes of cytochrome bc 1 complexed either by plain A8-35 (gray) or by biotinylated A8-35 (BAPol; black). Right, application of an antibody raised against tOmpA to lanes carrying BAPol-immobilized tOmpA (black) or other MPs (other traces). Reprinted with permission from Proc Natl Acad Sci USA 106:405–410, copyright 2009 National Academy of Sciences, USA. (Charvolin et al. 2009)
The protocol described here is for MPs trapped in biotinylated A8-35 (BAPol), but it can be adapted to MPs trapped in other functionalized APols, such as HistAPol, ImidAPol, or OligAPol (cf. § 7.1.3). If the MP possesses a tag, its trapping with unfunctionalized APol, e.g., plain A8-35, will improve its stability and the immobilization of the complexes can be carried out following the same approach . This protocol is adapted for Biacore instruments (GE Healthcare), which are the most widely distributed commercial instruments. Specific conditions are indicated in the case of a Biacore X100 instrument. The signal can be optimized by adjusting experimental conditions (Karlsson and Fält 1997). For more details about data analysis, see Rich and Myszka 2005 .
7.2.5.1 Experimental Setup
MP/BAPols complexes (called ligands in the Biacore terminology) are immobilized to the Biacore sensor chip before addition of the interacting molecules, for instance, antibodies (called analyte in the Biacore terminology), to reveal specific interactions.
Choose the adequate sensor chips. The binding capacity of most of Biacore chips is improved by pre-coupling of a dextran matrix. Existing CM5 chips, which are the most widely used, are already premodified for coupling of various proteins. If the immobilization is carried out via BAPols, the SA sensor chip (CM5 chips premodified with streptavidin) is used. Note that alternatives of SA sensor chips are the use of a standard CM5 chip followed by aminocoupling of streptavidin, or the SA capture chip, which is regenerable. In the case of charge repulsion between the sample to be immobilized and the surface, it is possible to improve the detection signal by using commercial chips with shorter dextran matrices. Note that two flow cells are required for the measurements: The sample flow cell, where the total signal will result from specific and nonspecific binding onto surfaces, and the reference flow cell, used to evaluate the nonspecific binding of proteins onto surfaces.
7.2.5.2 Preparation of Samples
Note that it is important to exclude compounds influencing the refractive index of the solutions (e.g., glycerol). Moreover, all samples and buffers should be filtered on 0.22-µm filters or centrifuged 10 min at 10,000 × g:
-
Trap the protein in BAPol as described in protocol 7.2.1. The final protein concentration to fix for a good signal depends on the protein under study. For example, in the case of BR, the protein concentration should be 0.03 g L−1. Typical quantities required for the experiments are 0.06–0.1 µg of total protein .
-
Prepare the solutions of antibodies (a nonspecific antibody and a specific one for negative and positive controls, respectively), both solutions at two different concentrations (0.055 and 0.01 g L−1). Typical quantities required for the experiments are 6–10 µg of total protein. Note that these quantities are indicative and should be optimized depending on sample purity.
-
Prepare the running buffer, HBS-N, which contains 10 mM HEPES, pH 7.4, and 150 mM NaCl. Note that the dextran matrix and the APols are both negatively charged and so, the ionic strength of buffer can be optimized for an optimal signal with minimal repulsion.
-
Prepare the surface regeneration solution (1 M NaCl, 50 mM NaOH).
7.2.5.3 Measurements
The signal observed during an SPR experiment is proportional to the refractive index, which is proportional to the mass for molecules with the same refractive index. Proteins have approximately the same refractive index and the empirical conversion factor (1 RU ≈ 1 pg mm−2) can be used for estimating the mass bound to the surface (Stenberg et al. 1991) :
-
Follow the instructions of the manufacturer for conditioning the sensor surface.
-
After obtaining a stable baseline, immobilize the proteins onto the sensor surface, which becomes the sample flow cell. For this step, start a new cycle using only the second flow cell for its functionalization and inject the sample. In the case of qualitative analysis, it is recommended to try to saturate the surface with the MP/BAPol complexes for the highest possible response from antibody binding in the next step. This can be achieved by repeating consecutive injections until stabilization of the signal (note: For a flow rate of 10 µL min−1, use consecutive 60-s injections of MP/BAPol complexes at 0.03 g L−1 to saturate the surface). On the other hand, in the case of kinetic studies, it is usually necessary to limit the density of immobilized ligand to avoid diffusion-limited data, analyte depletion, and analyte rebinding.
-
Prepare the reference flow cell (the first flow cell), which can either be left unmodified, or be modified with either BAPol alone or a different MP trapped in BAPol.
-
Stop the manual run after obtaining a stable signal in both cells .
-
Measure the binding of the analyte, for instance, that of an antibody raised against the MP under study. It is recommended to use two different concentrations of antibodies (0.05 g L−1 for the first injection and 0.01 g L−1 for the second one) and one regeneration solution in each cycle. The solutions will be added according to the series of the following cycles:
-
Cycle 1: Blank cycle with running buffer
-
Cycle 2: Cycle with a nonspecific antibody to be used for negative control
-
Cycle 3: Cycle with a specific antibody raised against the MP under study
-
The sample parameters are: 30 µL min−1 flow rate, 180 s contact time, and 120 s dissociation time. The regeneration parameters are: 30 µL min−1 flow rate, 60 s contact time, and wash in running buffer.
References
Althoff T, Mills DJ, Popot J-L, Kühlbrandt W (2011) Assembly of electron transport chain components in bovine mitochondrial supercomplex I1III2IV1. EMBO J 30:4652–4664
Banères J-L, Mesnier D, Martin A, Joubert L, Dumuis A, Bockaert J (2005) Molecular characterization of a purified 5-HT4 receptor. A structural basis for drug efficacy. J Biol Chem 280:20253–20260
Banères J-L, Popot J-L, Mouillac B (2011) New advances in production and functional folding of G protein-coupled receptors. Trends Biotechnol 29:314–322
Basit H, Sharma S, Van der Heyden A, Gondran C, Breyton C, Dumy P, Winnik FM, Labbé P (2012) Amphipol mediated surface immobilization of FhuA: a platform for label-free detection of the bacteriophage protein pb5. Chem Commun 48:6037–6039
Bazzacco P, Sharma KS, Durand G, Giusti F, Ebel C, Popot J-L, Pucci B (2009) Trapping and stabilization of integral membrane proteins by hydrophobically grafted glucose-based telomers. Biomacromolecules 10:3317–3326
Bazzacco P, Billon-Denis E, Sharma KS, Catoire LJ, Mary S, Le Bon C, Point E, Banères J-L, Durand G, Zito F, Pucci B, Popot J-L (2012) Non-ionic homopolymeric amphipols: application to membrane protein folding, cell-free synthesis, and solution NMR. Biochemistry 51:1416–1430
Bechara C, Bolbach G, Bazzacco P, Sharma SK, Durand G, Popot J-L, Zito F, Sagan S (2012) MALDI mass spectrometry analysis of membrane protein/amphipol complexes. Anal Chem 84:6128–6135
Bowie JU (2001) Stabilizing membrane proteins. Curr Opin Struct Biol 11:397–402
Breyton C, Chabaud E, Chaudier Y, Pucci B, Popot J-L (2004) Hemifluorinated surfactants: a non-dissociating environment for handling membrane proteins in aqueous solutions? FEBS Lett 564:312–318
Breyton C, Gabel F, Abla M, Pierre Y, Lebaupain F, Durand G, Popot J-L, Ebel C, Pucci B (2009) Micellar and biochemical properties of (hemi)fluorinated surfactants are controlled by the size of the polar head. Biophys J 97:1077–1086
Breyton C, Pucci B, Popot J-L (2010) Amphipols and fluorinated surfactants: two alternatives to detergents for studying membrane proteins in vitro. In: Mus-Veteau I (ed) Heterologous expression of membrane proteins: methods and protocols, vol 601. The Humana Press, Totowa, pp 219–245
Cao E, Liao M, Cheng Y, Julius D (2013). TRPV1 structures in distinct conformations reveal activation mechanisms. Nature 504:113–118
Catoire LJ, Zoonens M, van Heijenoort C, Giusti F, Popot J-L, Guittet E (2009) Inter- and intramolecular contacts in a membrane protein/surfactant complex observed by heteronuclear dipole-to-dipole cross-relaxation. J Magn Res 197:91–95
Catoire LJ, Damian M, Giusti F, Martin A, van Heijenoort C, Popot J-L, Guittet E, Banères J-L (2010a) Structure of a GPCR ligand in its receptor-bound state: leukotriene B4 adopts a highly constrained conformation when associated to human BLT2. J Am Chem Soc 132:9049–9057
Catoire LJ, Zoonens M, van Heijenoort C, Giusti F, Guittet E, Popot J-L (2010b) Solution NMR mapping of water-accessible residues in the transmembrane β-barrel of OmpX. Eur Biophys J 39:623–630
Catoire LJ, Damian M, Baaden M, Guittet E, Banères J-L (2011) Electrostatically-driven fast association and perdeuteration allow detection of transferred cross-relaxation for G protein-coupled receptor ligands with equilibrium dissociation constants in the high-to-low nanomolar range. J Biomol NMR 50:191–195
Chabaud E, Barthélémy P, Mora N, Popot J-L, Pucci B (1998) Stabilization of integral membrane proteins in aqueous solution using fluorinated surfactants. Biochimie 80:515–530
Chae PS, Rasmussen SGF, Rana R, Gotfryd K, Chandra R, Goren MA, Kruse AC, Nurva S, Loland CJ, Pierre Y, Drew D, Popot J-L, Picot D, Fox BG, Guan L, Gether U, Byrne B, Kobilka BK, Gellman SH (2010) Maltose-neopentyl glycol (MNG) amphiphiles for solubilization, stabilization and crystallization of membrane proteins. Nat Methods 7:1003–1008
Champeil P, Menguy T, Tribet C, Popot J-L, le Maire M (2000) Interaction of amphipols with the sarcoplasmic reticulum Ca2+-ATPase. J Biol Chem 275:18623–18637
Charvolin D, Perez J-B, Rouvière F, Giusti F, Bazzacco P, Abdine A, Rappaport F, Martinez KL, Popot J-L (2009) The use of amphipols as universal molecular adapters to immobilize membrane proteins onto solid supports. Proc Natl Acad Sci U S A 106:405–410
Charvolin D, Picard M, Huang L-S, Berry EA, Popot J-L (2014) Solution behavior and crystallization of cytochrome bc 1 in the presence of amphipols J Membr Biol, in the press
Cvetkov TL, Huynh KW, Cohen MR, Moiseenkova-Bell VY (2011) Molecular architecture and subunit organization of TRPA1 ion channel revealed by electron microscopy. J Biol Chem 286:38168–38176
Dahmane T, Damian M, Mary S, Popot J-L, Banères J-L (2009) Amphipol-assisted in vitro folding of G protein-coupled receptors. Biochemistry 48:6516–6521
Dahmane T, Giusti F, Catoire LJ, Popot J-L (2011) Sulfonated amphipols: synthesis, properties and applications. Biopolymers 95:811–823
Dahmane T, Rappaport F, Popot J-L (2013) Amphipol-assisted folding of bacteriorhodopsin in the presence and absence of lipids. Functional consequences. Eur Biophys J 42:85–101
Damian M, Martin A, Mesnier D, Pin J-P, Banères J-L (2006) Asymmetric conformational changes in a GPCR dimer controlled by G-proteins. EMBO J 13:5693–5702
Damian M, Marie J, Leyris J-P, Fehrentz J-A, Verdié P, Martinez J, Banères J-L, Mary S (2012) High constitutive activity is an intrinsic feature of ghrelin receptor protein: a study with a functional monomeric GHS-R1a receptor reconstituted in lipid discs. J Biol Chem 287:3630–3641
Diab C, Tribet C, Gohon Y, Popot J-L, Winnik FM (2007a) Complexation of integral membrane proteins by phosphorylcholine-based amphipols. Biochim Biophys Acta 1768:2737–2747
Diab C, Winnik FM, Tribet C (2007b) Enthalpy of interaction and binding isotherms of non-ionic surfactants onto micellar amphiphilic polymers (amphipols). Langmuir 23:3025–3035
Elter S, Raschle T, Arens S, Viegas A, Gelev V, Etzkorn M, Wagner G (2014) The use of amphipols for NMR structural characterization of 7-TM proteins. J Membr Biol, in the press
Etzkorn M, Raschle T, Hagn F, Gelev V, Rice AJ, Walz T, Wagner G (2013) Cell-free expressed bacteriorhodopsin in different soluble membrane mimetics: biophysical properties and NMR accessibility. Structure 21:394–401
Feinstein HE, Tifrea D, Popot J-L, de la MLM, Cocco MJ (2014) Amphipols stabilize the Chlamydia major outer membrane protein vaccine formulation J Membr Biol, in the press
Fernandez A, Le Bon C, Baumlin N, Giusti F, Crémel G, Popot J-L, Bagnard D (2014) In vivo characterization of the biodistribution profile of amphipols J Membr Biol, in the press
Ferrandez Y, Dezi M, Bosco M, Urvoas A, Valério M, Le Bon C, Giusti F, Broutin I, Durand G, Polidori A, Popot J-L, Picard M, Minard P (2014) Amphipol-mediated screening of molecular ortheses specific for membrane protein targets J Membr Biol, in the press
Flötenmeyer M, Weiss H, Tribet C, Popot J-L, Leonard K (2007) The use of amphipathic polymers for cryo-electron microscopy of NADH: ubiquinone oxidoreductase (complex I). J Microsc 227:229–235
Garavito RM, Ferguson-Miller S (2001) Detergents as tools in membrane biochemistry. J Biol Chem 276:32403–32406
Giusti F, Popot J-L, Tribet C (2012) Well-defined critical association concentration and rapid adsorption at the air/water interface of a short amphiphilic polymer, amphipol A8-35: a study by Förster resonance energy transfer and dynamic surface tension measurements. Langmuir 28:10372–10380
Giusti F, Kessler P, Westh Hansen R, Lloret N, Le Bon C, Mourier G, Popot J-L, Martinez KL, Zoonens M (2014a) Synthesis of polyhistidine-bearing amphipols and its use for immobilization of membrane proteins. In submission
Giusti F, Rieger J, Catoire L, Qian S, Calabrese AN, Watkinson TG, Casiraghi M, Radford SE, Ashcroft AE, Popot J-L (2014b) Synthesis, characterization and applications of a perdeuterated amphipol. J Membr Biol, DOI 10.1007/s00232-014-9656-x
Gohon Y, Pavlov G, Timmins P, Tribet C, Popot J-L, Ebel C (2004) Partial specific volume and solvent interactions of amphipol A8-35. Anal Biochem 334:318–334
Gohon Y, Giusti F, Prata C, Charvolin D, Timmins P, Ebel C, Tribet C, Popot J-L (2006) Well-defined nanoparticles formed by hydrophobic assembly of a short and polydisperse random terpolymer, amphipol A8-35. Langmuir 22:1281–1290
Gohon Y, Dahmane T, Ruigrok R, Schuck P, Charvolin D, Rappaport F, Timmins P, Engelman DM, Tribet C, Popot J-L, Ebel C (2008) Bacteriorhodopsin/amphipol complexes: structural and functional properties. Biophys J 94:3523–3537
Gohon Y, Vindigni J-D, Pallier A, Wien F, Celia H, Giuliani A, Tribet C, Chardot T, Briozzo P (2011) High water solubility and fold in amphipols of proteins with large hydrophobic regions: oleosins and caleosin from seed lipid bodies. Biochim Biophys Acta 1808:706–716
Gorzelle BM, Hoffman AK, Keyes MH, Gray DN, Ray DG, Sanders CR II (2002) Amphipols can support the activity of a membrane enzyme. J Am Chem Soc 124:11594–11595
Hong W-X, Baker KA, Ma X, Stevens RC, Yeager M, Zhang Q (2011) Design, synthesis and properties of branch-chained maltoside detergents for stabilization and crystallization of integral membrane proteins: human connexin 26. Langmuir 26:8690–8696
Hovers J, Potschies M, Polidori A, Pucci B, Raynal S, Bonneté F, Serrano-Vega M, Tate C, Picot D, Pierre Y, Popot J-L, Nehmé R, Bidet M, Mus-Veteau I, Bußkamp H, Jung K-H, Marx A, Timmins PA, Welte W (2011) A class of mild surfactants that keep integral membrane proteins water-soluble for functional studies and crystallization. Mol Membr Biol 28:171–181
Jonkheijm P, Weinrich D, Schröder H, Niemeyer CM, Waldmann H (2008) Chemical strategies for generating protein biochips. Angew Chem Int Ed Engl 47:9618–9647
Karlsson R, Fält A (1997) Experimental design for kinetic analysis of protein-protein interactions with surface plasmon resonance biosensors. J Immunol Method 200:121–133
Kigawa T, Yabuki T, Yoshida Y, Tsutsui M, Ito Y, Shibata T, Yokoyama S (1999) Cell-free production and stable-isotope labeling of milligram quantities of proteins. FEBS Lett 442:15–19
Knowles TJ, Finka R, Smith C, Lin Y-P, Dafforn T, Overduin M (2009) Membrane proteins solubilized intact in lipid containing nanoparticles bounded by styrene maleic acid copolymer. J Am Chem Soc 131:7484–7485
Koutsopoulos S, Kaiser L, Eriksson HM, Zhang S (2012) Designer peptide surfactants stabilize diverse functional membrane proteins. Chem Soc Rev 41:1721–1728
Le Bon C, Della Pia EA, Giusti F, Lloret N, Zoonens M, Martinez KL, Popot J-L (2014a) Synthesis of an oligonucleotide-derivatized amphipol and its use to trap and immobilize membrane proteins Nucleic Acids Res, DOI: 10.1093/nar/gku250.
Le Bon C, Popot J-L, Giusti F (2014b) Labeling and functionalizing amphipols for biological applications J Membr Biol, DOI 10.1007/s00232-014-9655-y
Leney AC, McMorran LM, Radford SE, Ashcroft AE (2012) Amphipathic polymers enable the study of functional membrane proteins in the gas phase. Anal Chem 84:9841–9847
Liao M, Cao E, Julius D, Cheng Y (2013) Structure of the TRPV1 ion channel determined by electron cryo-microscopy. Nature 504:107–112
Long AR, O’Brien CC, Malhotra K, Schwall CT, Albert AD, Watts A, Alder NN (2013) A detergent-free strategy for the reconstitution of active enzyme complexes from native biological membranes into nanoscale discs. BMC Biotechnol 13:41. doi:10.1186/1472-6750-1113-1141
Martinez KL, Gohon Y, Corringer P-J, Tribet C, Mérola F, Changeux J-P, Popot J-L (2002) Allosteric transitions of Torpedo acetylcholine receptor in lipids, detergent and amphipols: molecular interactions vs. physical constraints. FEBS Lett 528:251–256
Matar-Merheb R, Rhimi M, Leydier A, Huché F, Galián C, Desuzinges-Mandon E, Ficheux D, Flot D, Aghajari H, Kahn R, Di Pietro A, Jault J-M, Coleman AW, Falson P (2011) Structuring detergents for extracting and stabilizing functional membrane proteins. PLoS ONE 6:e18036
McGregor C-L, Chen L, Pomroy NC, Hwang P, Go S, Chakrabartty A, Privé GG (2003) Lipopeptide detergents designed for the structural study of membrane proteins. Nat Biotechnol 21:171–176
Nagy JK, Kuhn Hoffmann A, Keyes MH, Gray DN, Oxenoid K, Sanders CR (2001) Use of amphipathic polymers to deliver a membrane protein to lipid bilayers. FEBS Lett 501:115–120
Ning Z, Hawley B, Seebun D, Figeys D (2014) APols aided protein precipitation: a rapid method for protein concentrating for proteomic analysis. J Membr Biol, in the press
Opačić M, Giusti F, Broos J, Popot J-L (2014) Amphipol A8-35 preserves the activity of detergent-sensitive mutants of Escherichia coli mannitol permease EIImtl. J Membr Biol, in the press
Park K-H, Billon-Denis E, Dahmane T, Lebaupain F, Pucci B, Breyton C, Zito F (2011) In the cauldron of cell-free synthesis of membrane proteins: playing with new surfactants. New Biotech 28:255–261
Perlmutter JD, Drasler WJ, Xie W, Gao J, Popot J-L, Sachs JN (2011) All-atom and coarse-grained molecular dynamics simulations of a membrane protein stabilizing polymer. Langmuir 27:10523–10537
Perlmutter JD, Popot J-L, Sachs JN (2014) Molecular dynamics simulations of a membrane protein/amphipol complex J Membr Biol, in the press
Picard M, Dahmane T, Garrigos M, Gauron C, Giusti F, le Maire M, Popot J-L, Champeil P (2006) Protective and inhibitory effects of various types of amphipols on the Ca2+-ATPase from sarcoplasmic reticulum: a comparative study. Biochemistry 45:1861–1869
Planchard N, Point E, Dahmane T, Giusti F, Renault M, Le Bon C, Durand G, Milon A, Guittet E, Zoonens M, Popot J-L, Catoire LJ (2014) The use of amphipols for solution NMR studies of membrane proteins: advantages and limitations as compared to other solubilizing media J Membr Biol, DOI 10.1007/s00232-014-9654-z
Pocanschi CL, Dahmane T, Gohon Y, Rappaport F, Apell H-J, Kleinschmidt JH, Popot J-L (2006) Amphipathic polymers: tools to fold integral membrane proteins to their active form. Biochemistry 45:13954–13961
Pocanschi C, Popot J-L, Kleinschmidt JH (2013) Folding and stability of outer membrane protein A (OmpA) from Escherichia coli in an amphipathic polymer, amphipol A8-35. Eur Biophys J 42:103–118
Polovinkin V, Gushchin I, Balandin T, Chervakov P, Round E, Schevchenko V, Popov A, Borshchevskiy V, Popot J-L, Gordeliy V (2014) High-resolution structure of a membrane protein transferred from amphipol to a lipidic mesophase. J Membr Biol, in the press
Popot J-L (2010) Amphipols, nanodiscs, and fluorinated surfactants: three non-conventional approaches to studying membrane proteins in aqueous solutions. Annu Rev Biochem 79:737–775
Popot J-L, Engelman DM (2000) Helical membrane protein folding, stability and evolution. Annu Rev Biochem 69:881–923
Popot J-L, Gerchman S-E, Engelman DM (1987) Refolding of bacteriorhodopsin in lipid bilayers: a thermodynamically controlled two-stage process. J Mol Biol 198:655–676
Popot J-L, Berry EA, Charvolin D, Creuzenet C, Ebel C, Engelman DM, Flötenmeyer M, Giusti F, Gohon Y, Hervé P, Hong Q, Lakey JH, Leonard K, Shuman HA, Timmins P, Warschawski DE, Zito F, Zoonens M, Pucci B, Tribet C (2003) Amphipols: polymeric surfactants for membrane biology research. Cell Mol Life Sci 60:1559–1574
Popot J-L, Althoff T, Bagnard D, Banères J-L, Bazzacco P, Billon-Denis E, Catoire LJ, Champeil P, Charvolin D, Cocco MJ, Crémel G, Dahmane T, de la MLM, Ebel C, Gabel F, Giusti F, Gohon Y, Goormaghtigh E, Guittet E, Kleinschmidt JH, Kühlbrandt W, Le Bon C, Martinez KL, Picard M, Pucci B, Rappaport F, Sachs JN, Tribet C, van Heijenoort C, Wien F, Zito F, Zoonens M (2011) Amphipols from A to Z. Annu Rev Biophys 40:379–408
Prata C, Giusti F, Gohon Y, Pucci B, Popot J-L, Tribet C (2001) Non-ionic amphiphilic polymers derived from tris(hydroxymethyl)-acrylamidomethane keep membrane proteins soluble and native in the absence of detergent. Biopolymers 56:77–84
Privé G (2009) Lipopeptide detergents for membrane protein studies. Curr Opin Struct Biol 19:1–7
Rahmeh R, Damian M, Cottet M, Orcel H, Mendre C, Durroux T, Sharma KS, Durand G, Pucci B, Trinquet E, Zwier JM, Deupi X, Bron P J-LB, Mouillac B, Granier S (2012) Structural insights into biased G protein-coupled receptor signaling revealed by fluorescence spectroscopy. Proc Natl Acad Sci U S A 109:6733–6738
Rajesh S, Knowles TJ, Overduin M (2011) Production of membrane proteins without cells or detergents. N Biotech 28:250–254
Raschle T, Hiller S, Etzkorn M, Wagner G (2010) Nonmicellar systems for solution NMR spectroscopy of membrane proteins. Curr Opin Struct Biol 20:471–479
Rich RL, Myszka DG (2005) Survey of the year 2004 commercial optical biosensor literature. J Mol Recognit 18:431–478
Schafmeister CE, Miercke LJW, Stroud RA (1993) Structure at 2.5 Å of a designed peptide that maintains solubility of membrane proteins. Science 262:734–738
Sharma KS, Durand G, Gabel F, Bazzacco P, Le Bon C, Billon-Denis E, Catoire LJ, Popot J-L, Ebel C, Pucci B (2012) Non-ionic amphiphilic homopolymers: synthesis, solution properties, and biochemical validation. Langmuir 28:4625–4639
Stenberg E, Persson B, Roos H, Urbaniczky C (1991) Quantitative determination of surface concentration of protein with surface plasmon resonance using radio-labeled proteins. J Colloid Interface Sci 143:513–526
Tifrea D, Pal S, Cocco MJ, Popot J-L, de la Maza LM (2014) Increased immuno accessibility of MOMP epitopes in a vaccine formulated with amphipols may account for the very robust protection elicited against a vaginal challenge with C. muridarum. J Immunol, in the press
Tifrea DF, Sun G, Pal S, Zardeneta G, Cocco MJ, Popot J-L, de la MLM (2011) Amphipols stabilize the Chlamydia major outer membrane protein and enhance its protective ability as a vaccine. Vaccine 29:4623–4631
Tribet C, Audebert R, Popot J-L (1996) Amphipols: polymers that keep membrane proteins soluble in aqueous solutions. Proc Natl Acad Sci U S A 93:15047–15050
Tribet C, Audebert R, Popot J-L (1997) Stabilisation of hydrophobic colloidal dispersions in water with amphiphilic polymers: application to integral membrane proteins. Langmuir 13:5570–5576
Tribet C, Mills D, Haider M, Popot J-L (1998) Scanning transmission electron microscopy study of the molecular mass of amphipol/cytochrome b 6 f complexes. Biochimie 80:475–482
Tribet C, Diab C, Dahmane T, Zoonens M, Popot J-L, Winnik FM (2009) Thermodynamic characterization of the exchange of detergents and amphipols at the surfaces of integral membrane proteins. Langmuir 25:12623–12634
Wang X, Corin K, Baaske P, Wienken CJ, Jerabek-Willemsen M, Duhr S, Braun D, Zhang S (2011) Peptide surfactants for cell-free production of functional G protein-coupled receptors. Proc Natl Acad Sci U S A 108:9049–9054
Zhao X, Nagai Y, Reeves PJ, Kiley P, Khorana HG, Zhang S (2006) Designer short peptide surfactants stabilize G protein-coupled receptor bovine rhodopsin. Proc Natl Acad Sci U S A 103:17707–17712
Zoonens M (2004) Caractérisation des complexes formés entre le domaine transmembranaire de la protéine OmpA et des polymères amphiphiles, les amphipols. Application à l’étude structurale des protéines membranaires par RMN à haute résolution. Thèse de Doctorat, Université Paris-6, Paris
Zoonens M, Popot J-L (2014) Amphipols for each season J Membr Biol, in the press
Zoonens M, Catoire LJ, Giusti F, Popot J-L (2005) NMR study of a membrane protein in detergent-free aqueous solution. Proc Natl Acad Sci U S A 102:8893–8898
Zoonens M, Giusti F, Zito F, Popot J-L (2007) Dynamics of membrane protein/amphipol association studied by Förster resonance energy transfer. Implications for in vitro studies of amphipol-stabilized membrane proteins. Biochemistry 46:10392–10404
Zubay G (1973) In vitro synthesis of protein in microbial systems. Annu Rev Genet 7:267–287
Acknowledgments
We would like to thank L. J. Catoire for reading the protocol of APol-assisted MP folding. The development of amphipols has been mainly supported by the Centre National de la Recherche Scientifique, the Human Frontier Science Program Organization (RG00223/2000-M), and the European Community (BIO4-CT98-0269 and STREP LSHG-CT-2005-513770 Innovative Tools for Membrane Protein Structural Proteomics).
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2014 Springer Science+Business Media New York
About this chapter
Cite this chapter
Zoonens, M., Zito, F., Martinez, K., Popot, JL. (2014). Amphipols: A General Introduction and Some Protocols. In: Mus-Veteau, I. (eds) Membrane Proteins Production for Structural Analysis. Springer, New York, NY. https://doi.org/10.1007/978-1-4939-0662-8_7
Download citation
DOI: https://doi.org/10.1007/978-1-4939-0662-8_7
Published:
Publisher Name: Springer, New York, NY
Print ISBN: 978-1-4939-0661-1
Online ISBN: 978-1-4939-0662-8
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)