
Abstract
The aging population and healthcare costs for heart failure (HF) therapy in the elderly (age ≥65 years) are increasing worldwide. Cardiovascular (CV) diseases, including myocardial infarction (MI), hypertension (HTN), and HF, are all more prevalent in the elderly. While the renin–angiotensin system (RAS) has critical functions in CV physiology, an upregulated RAS plays a critical role in CV pathophysiology, including post-MI dilative remodeling and HF associated with low ejection fraction, and in hypertrophic remodeling and fibrosis associated with HTN and HF with preserved ejection fraction. Accordingly, components of the RAS are important targets for CV disease and HF pharmacotherapy. Angiotensin II (AngII) is the primary effector molecule of the RAS, and RAS/AngII inhibitors form the basis of therapy for both elderly and non-elderly HF patients. However, clinical studies indicate that elderly post-MI patients are at higher risk for adverse events despite therapy with RAS/AngII inhibitors. Remodeling of the RAS with aging may account for the poor outcome in the elderly. Aging is associated with increased AngII and other components of the RAS. Enhanced upregulation and/or dysregulation of the RAS and renin–angiotensin–aldosterone system (RAAS) pathways with aging may play a critical role in the accelerated march to HF and the increasing HF burden despite conventional therapy in the elderly. Increased AngII may also explain increased cytosolic and mitochondrial oxidant production, mitochondrial dysfunction, and increased extracellular matrix deposition associated with aging. Disruption of the AngII type 1 receptor confers protection from CV morbidity and mortality and promotes longevity in animal models. More research into the biology of aging-related remodeling of the RAS/RAAS and related pathways (such as kinins, ACE2/Ang (1–7) , and mineralocorticoids) may lead to discovery and development of improved therapies for post-MI and post-HTN cardiac remodeling and HF in the elderly.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others

Keywords
- Mineralocorticoid Receptor
- Leave Ventricular Remodel
- Secretory Leukocyte Protease Inhibitor
- Adverse Leave Ventricular Remodel
- Mineralocorticoid Receptor Activation
These keywords were added by machine and not by the authors. This process is experimental and the keywords may be updated as the learning algorithm improves.
Introduction: Statement of the Problem
Human aging is a natural biological process that is progressive and is associated with cardiovascular (CV) and other biological changes that impact disease expression and response to injury and therapy [1–10]. As expected, progress, prosperity, and improved health care have been accompanied by prolonged longevity with an increase in people aged ≥65 years in industrialized developed countries and most developing countries are following this pattern [11, 12]. As reviewed in the first two chapters of this book, the definition of elderly by the chronological age ≥65 years was driven by pension legislations in the Europe of the 1870s, presumably to reduce the socioeconomic burden and political pressure when very few people were expected to survive to that age. This nearly 150-year-old definition has persisted into the twenty-first century despite the changing demographics, increased longevity, progress in medical therapies, and prosperity [13–16] and certainly mandates upward adjustment in keeping with modern trends. Progress and prosperity over these years have been accompanied by expansion of an elderly population burdened with cardiovascular disease (CVD) including hypertension (HTN) and coronary heart disease (CHD), comorbidities that impact the CV system, and heart failure (HF), the known ultimate result of CVD [1, 14, 17]. While medical progress and clinical trials since the mid-1970s have improved therapy of CVDs in the non-elderly (aged <65 years), there is an alarming knowledge gap about the pathobiology and pathophysiology of CVDs and their therapy in the elderly people aged ≥65 years. The negative impact of that knowledge gap is already clearly apparent. The burden of CVD and HF is known to be greatest in the elderly, with escalating morbidity/mortality and related healthcare costs [18–24].
One reason for this global problem is that therapies for the non-elderly may not be optimal for the elderly because aging may result in remodeling of major pathways leading to CVD [6, 10, 17, 25, 26]. This chapter focuses on the aging-related remodeling of the renin–angiotensin system (RAS) [27] and related pathways including the renin–angiotensin–aldosterone system (RAAS) which play central roles in the pathophysiology and pharmacotherapy of CVDs and HF.
Aging and the RAS
Population studies have shown that myocardial infarction (MI), HTN, and HF are more prevalent in the elderly [6, 11, 24, 28]. The healthcare costs for post-MI HF in the elderly are high and increasing [6, 11, 17–19, 28]. While the RAS has critical functions in CV physiology, an upregulated RAS leads to CV pathology in both non-elderly and elderly populations [6]. Accordingly, components of the RAS have become important targets for CVD and HF pharmacotherapy since the mid-1980s [6, 28, 29]. Angiotensin II (AngII) is the primary effector molecule of the RAS. Since the 1990s, AngII inhibitors have formed the basis of therapy for both elderly and non-elderly HF patients [6, 29]. However, several clinical studies indicate that elderly post-MI patients are at higher risk for adverse events despite recommended therapy with RAS/AngII inhibitors compared to younger patients [6, 28]. Aging is associated with increased AngII and other components of the RAS [6, 30–33]. Emerging evidence suggests that the RAS may also become further upregulated and/or dysregulated during aging [6, 9, 29]. This adverse remodeling of the RAS may account for poor outcome and increased HF burden in the elderly despite conventional therapies [27].
Aging and Remodeling of the RAS and Left Ventricle Post-MI and Post-HTN
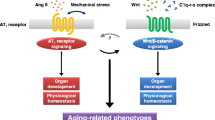
Aging of the CV system is associated with fundamental physiological, biological, and structural changes that lead to increased extracellular matrix (ECM) and fibrosis, increased ventricular-arterial stiffening, left ventricular (LV) diastolic dysfunction, and HF associated with preserved ejection fraction (HF/PEF) [6, 7, 17] (Fig. 18.1). The aging process is progressive and marches in parallel with the CVD continuum towards HF [7, 17]. In this aging-HF continuum [14], CVD risk factors and comorbidities as well as the upregulated RAS [27] and other related pathways can fuel progression towards HF in the elderly (Fig. 18.1). It is important to recognize that the biological changes during aging lead to profound structural remodeling of the CV system that impact cardiac reserve. Even in the absence of CVD, the progressive changes during aging can themselves lead to HF. However, exposure to CV risk factors and interaction with comorbidities during aging together with the effect of increased RAS and reactive oxygen species (ROS) tend to accelerate progression to both categories of HF, namely, HF associated with low ejection fraction (HF/low EF) and HF/PEF in the elderly (Fig. 18.2).

Cardiovascular aging and role of the RAS in the march to heart failure. While age is considered to be a nonmodifiable risk factor, the age-related changes in cardiovascular vascular structure and function and associated physiological, biochemical, cellular/subcellular, and pathophysiological changes provide the substrate for heart failure with preserved ejection fraction (HF/PEF). Dysregulation of the RAS and related pathways and other unrelated pathways influence the aging-related changes and can be modified by appropriate interventions. RAS renin–angiotensin system; AngII angiotensin II

Role of risk factors, comorbidities, ROS, and RAS in heart failure. Interactions between cardiovascular (CV) risk factors, comorbidities, and increased ROS and RAS spur the march to heart failure during CV aging. RAS renin–angiotensin system; ROS reactive oxygen species
Upregulation of the RAS is especially pertinent to the two major categories of HF (i.e., HF/low EF and HF/PEF) that contribute nearly equally to the HF burden in the elderly [7, 17] (Fig. 18.3). Some studies suggest that the prevalence of HF/PEF in the elderly is higher than 50 %, especially the very old [34–36]. Based on evidence suggesting that aging is associated with increased AngII and other components of the RAS [30–33], it is reasonable to speculate that remodeling of the RAS with aging may play a critical role in increased fibrosis in elderly hearts [37, 38] as well as hypertrophic remodeling and enhanced fibrosis in HF/PEF associated with hypertensive disease and enhanced dilative remodeling and HF/low-EF post-MI (Fig. 18.3).

The two major categories of heart failure in the elderly. Role of risk factors, comorbidities, ROS, and RAS. Aging-related increase in RAS and ROS play major roles in dilative and concentric LV remodeling, leading to the two main categories of heart failure. CAD coronary artery disease; CV cardiovascular; EF ejection fraction; HF heart failure; LV left ventricular; LVH LV hypertrophy; MI myocardial infarction; PEF preserved EF; RAS renin–angiotensin system; ROS reactive oxygen species
Additionally, increased AngII with aging can also lead to amplified pro-inflammatory and pro-remodeling effects after injury. Aging has been shown to result in impaired repair mechanisms after reperfused MI [6–9]. Increased RAS/AngII and oxidative stress/ROS with aging [6] aggravate myocardial and matrix damage post-MI [9] and exacerbate adverse LV remodeling and dysfunction post-MI and progression to HF/low EF [7–10, 17]. Taken together, the aging-related increase in AngII, other components of the RAS, ROS, and oxidative stress may explain both the increased fibrosis in elderly hearts and HF/PEF and the enhanced postinfarction adverse LV remodeling and associated HF/low EF [6, 10].
It follows that there is need for a paradigm shift in HF management, going beyond targeting the RAS with respect to HF therapy for the elderly. There is need for identifying new therapeutic targets and strategies through translational research for optimizing HF therapy in the elderly. Because responses to major causes of HF such as HTN and MI may be amplified in the elderly, different therapeutic strategies may be needed in the elderly compared to the young. Because biological changes with aging may alter responses to therapies that were tested in mostly young and non-elderly patients with HF, caution should be exercised and appropriate adjustments of medications (i.e., type, dosage, and timing) should be done when applying those in the elderly.
Aging and AngII, AT1/AT2 Pathways
Traditional concepts of RAS inhibition have been based on research studies that were done in predominantly young animals and humans. While the fundamental concepts and pathways are similar, there are some important differences with evidence of aging-related RAS dysregulation that may profoundly impact therapy of HF in the elderly.
Pharmacology of the RAS/RAAS in the Young and Non-elderly
The pertinent aspects of RAS/RAAS pharmacology have been reviewed elsewhere [39]. Briefly, the RAS/RAAS regulates blood volume and systemic vascular resistance, thereby modulating cardiac output and blood pressure (BP) through a cascade of events (Fig. 18.4). Renin, released primarily from the kidneys, cleaves circulating angiotensinogen to the decapeptide angiotensin I (AngI), which is further cleaved by the angiotensin-converting enzyme (ACE), found primarily in pulmonary vascular endothelium, to the octapeptide molecule AngII. The latter is considered to be the major effector polypeptide molecule of the RAS/RAAS. It is also a pleiotropic cytokine that exerts several important physiological actions, including vasoconstriction, release of neurohumoral agonists (such as aldosterone, vasopressin, and norepinephrine), drinking, secretion of prolactin and adrenocorticotropic hormone, and glycogenolysis. AngII also plays a critical role in the pathophysiology of CVD. Thus, chronically increased AngII induces increased vasoconstriction (leading to high BP and HTN), stimulates growth (leading to cardiac and vascular hypertrophy), contributes to LV dysfunction and progression of HF, mediates adverse structural cardiac and vascular remodeling [40], and causes deleterious activation of other neurohumoral agonists including endothelin. Sodium and water homeostasis becomes dysregulated, especially in the elderly. Since high AngII levels and low cardiac output stimulate thirst, the elderly patient with high AngII, HF, and low cardiac output is prone to develop hyponatremia despite reduced water intake.

The major pathways in the RAS and RAAS cascades. ACE angiotensin-converting enzyme; ACE-I ACE inhibitor; ARB angiotensin receptor blocker; cGMP cyclic guanosine 3′ 5′ monophosphate; EDHF endothelin-derived hyperpolarizing factor; eNOS endothelial nitric oxide synthase; MRA mineralocorticoid receptor antagonist; PAI-1 plasminogen activator inhibitor-1; PGI 2 prostacyclin; PKCε protein kinase Cε; RAS renin–angiotensin system; RAAS renin–angiotensin–aldosterone system; t-PA tissue plasminogen activator
While the RAS was initially regarded as an endocrine system, a steady flow of new evidence since the 1990s has expanded the paradigm into that of a multifunctional endocrine, paracrine, and intracrine system with circulating and local/tissue components [40–42]. While AngII is produced in the circulation and tissues and acts on the AngII type 1 (AT1) and AngII type 2 (AT2) receptors [39, 41], most of the effects of AngII are normally mediated through the AT1 receptor. However, in the presence of CV pathology—such as cardiac hypertrophy, vascular injury, MI, HF, and wound healing—the AT2 receptor is upregulated and may mediate some CV effects of AngII through AT2. Since there is a decrease in AT1 and an increase in AT2 receptors in HF, it was proposed that the antiproliferative and vasodilatory effects of AT2 counterbalance the growth-stimulating and vasoconstricting effects of AT1. In that construct, it follows that an AT1 receptor blocker (ARB) would completely block effects of AngII via AT1 and result in unopposed AT2 receptor stimulation that might augment its beneficial effects [43]. Although, there is controversy about the role of AT2 in humans [44], it may be important in the elderly. However, the role of AT3 and AT4 receptors with aging remains unclear and needs study.
Cardioprotective Effect of RAS Inhibition: Role of Kallikrein–Kinin System
Cumulative evidence supports the view that the cardioprotective effects of ACE inhibitors are due not only to the inhibition of AngII formation via ACE but also to inhibition of breakdown of bradykinin and other tachykinins related to ACE’s kininase II activity. In this construct, ACE inhibition decreases the amount of AngII that is presented to both AT1 and AT2 receptors, at least initially, so that decreased but balanced AT1 and AT2 effects is expected. However, increased bradykinin during ACE inhibition leads to stimulation of nitric oxide (NO), prostaglandins such as prostacyclin (PGI2,), endothelial-derived hyperpolarizing factor (EDHF) and tissue-thromboplastin activator (t-PA), thereby contributing to vasodilation, CV protection, and other favorable CV effects of ACE inhibitors [45]. The increased bradykinin also contributes to the hypotensive effect of ACE inhibitors which may be pertinent for the elderly.
In contrast to ACE inhibitors, the cardioprotective effect of ARBs is mediated through at least three pathways: (1) the primary pathway involves selective AT1 receptor blockade, (2) a secondary pathway involves AT2 receptor activation, and (3) a third pathway involves the release of kinins and stimulation of kinin B1 or B2 receptors [46–49] and/or direct AT2-mediated signaling via protein kinase C (PKCε), NO, and cyclic guanosine monophosphate (cGMP) [50–53]. Coupling between the AT2 receptor and kallikrein during AT1 blockade has been demonstrated in young mice with myocardial ischemia–reperfusion injury [49].
However, while kinins contribute to the protective effects of ACE inhibitors and ARBs acting through the AT2 receptor and NO contribute to myocardial and vascular protection, the kinin system is also involved in inflammation and the pathogenesis of inflammatory diseases such as arthritis ([54], for review). The kinins are known to act as mediators of inflammation by promoting maturation of dendritic cells which activate the body’s adaptive immune system and thereby promote inflammation. This may also be pertinent in elderly HF patients.
Role of Chymase and Other Non-ACE Pathways in Angiotensin II Generation
The discovery, in the 1990s, of non-ACE pathways that can generate AngII during ACE inhibition [55–60] has important implications for optimal therapy of HF in both non-elderly and elderly patients. Data in mostly non-elderly patients indicated that ACE inhibitors do not block all AngII formation. This includes AngII from AngI via the serine protease chymase and other non-ACE enzymes and/or that from angiotensinogen via non-renin pathways. Several studies showed that AngII levels persist during long-term ACE-inhibitor therapy, suggesting incomplete RAS blockade or the so-called ACE-inhibitor escape due to a reactive rise in active plasma renin [57–60]. This finding not only supported the use of ARBs for blocking the effects of AngII at the AT1 receptor but also fueled the push for using combined ACE inhibition and AT1 receptor blockade for more complete blockade of the deleterious effects of AngII and therefore anticipated greater benefits. However, support for the concept of dual ACE and AT1 receptor blockade came from experimental [61] and clinical [62] studies that were done in mostly the young with HF. For example, in young rats with post-MI HF, the ACE inhibitor fosinopril combined with the ARB valsartan resulted in suppression of histopathologic evidence of remodeling and normalized collagen I, macrophages, and myofibroblasts [63].
Two additional findings deserve emphasis. First, the AngII-forming capacity of chymase from AngI was found to be 20-fold higher than that for ACE [55, 56]. Second, chymase was also shown to activate the Kallikrein–kinin pathway [64, 65]. These findings imply that the combination of a chymase inhibitor with either an ACE inhibitor or an ARB may potentially double the beneficial effects through decrease of AngII and increased kinins. Dual chymase and ACE inhibition was proposed for HTN and HF to boost the efficacy of ACE inhibition and prevent ACE-inhibitor escape.
In fact, chymase inhibitors have been tested in young experimental animal models. They were shown to improve diastolic LV function and prevent fibrosis in a canine model of tachycardia-induced HF [66], reduce arrhythmias and LV dysfunction after MI [67, 68], attenuate LV remodeling in a mouse model of intermittent hypoxia [69], and decrease infarct size after ischemia–reperfusion through attenuation of matrix metalloproteinase (MMP)-9 and pro-inflammatory cytokines in a porcine model of reperfused MI [70]. In the mouse model of intermittent hypoxia, decrease in LV hypertrophy and fibrosis was associated with decrease in LV chymase, AngII, oxidative stress, interleukin (IL)-6, tumor necrosis factor (TNF)-α, and transforming growth factor (TGF)-β [69]. In a hamster model of post-MI HF, combined ACE and chymase inhibition decreased infarct size and LV remodeling and dysfunction compared to ACE inhibition alone [71]. In the canine model of LV volume overload due to mitral regurgitation, the anti-remodeling effect of chymase inhibition appears to be in part due to inhibition of MMP and kallikrein activation and fibronectin degradation [72], thereby attenuating loss of ECM and cell-ECM connections, and cell death. The multiple actions of chymase on tissue remodeling support its role in adverse LV remodeling and HF post-MI [73–75] atherosclerosis [76–79].
Whether the AngII generation by non-ACE pathways is increased with aging and in elderly HF patients needs investigation. Whether chymase inhibitors might be beneficial in older animals and patients with HF needs study. Recent evidence indicates that chymase is upregulated in coronary and renal arteries of diabetic patients [80] and implicates chymase in the intracellular formation of AngII in cardiomyocytes, fibroblasts, and renal mesangial and vascular smooth muscle cells under hyperglycemic conditions [81]. This finding may be especially pertinent in the context of aging as diabetes is a major comorbidity in elderly patients with HF.
Interactions Between the RAS and the Endothelin System
Extending the concept of dual inhibition one step further, the ARB valsartan combined with the endothelin (ET) blocker bosentan was shown to produce added benefits on neurohumoral activity and LV loading and performance end-points in pigs with HF induced by rapid atrial pacing [82]. Both the RAS acting through AngII and the endothelin system acting through ET-1 produce powerful vasoconstrictor and vasopressor effects that promote adverse LV remodeling and progression of HF ([83–85] for review). As AngII, both circulating and myocardial tissue ET levels are increased in animals and humans with HF, interactions between the RAS and ET system have been described [83]; AngII stimulates ET production through transcriptional regulation while ET inhibits renin synthesis and stimulates aldosterone secretion. Both AngII and ET stimulate matrix synthesis. In animal models of HF, both nonselective ET-A and ET-B antagonists and selective ET-A antagonists exert beneficial CV effects. However, ET-A antagonist therapy in experimental HF results in augmentation of the RAS and sustained sodium retention [86]. Furthermore, in patients with HF, short-term ET antagonist therapy improved hemodynamics but long-term therapy did not improve combined morbidity/mortality end-points [87]. In the EARTH trial, the ET antagonist darusentan as an add-on to an ACE inhibitor, β-adrenergic blocker, or aldosterone antagonist in patients with chronic HF failed to improve LV remodeling, clinical symptoms, or outcomes [87]. Long-term trials of the ET antagonist bosentan in HF were prematurely terminated due to increased adverse events [88]. Despite anecdotal evidence of success with bosentan in the subgroup of patients with pulmonary artery hypertension secondary to HF, a recent trial showed no improvement [89]. Despite further anecdotal reports of short-term benefit in patients with severe pulmonary hypertension awaiting heart transplantation, the routine use of ET antagonists is not recommended by the World Health Organization (WHO) Pulmonary Hypertension group 2 [90]. Whether ET antagonists may have an application in elderly HF patients has not been studied.
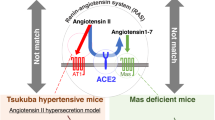
Role of the Counter-Regulatory ACE2 and Ang-(1–7) Arm of the RAS
While the RAS became recognized as a central regulator of CV and renal function with a major role in pathophysiology of CVD and HF, the discovery of angiotensin-converting enzyme 2 (ACE2) in 2000 [91, 92] further modified the traditional concepts about the RAS. Subsequent studies viewed ACE2 as an essential regulator of cardiac function [93] and several studies underscored the importance of AngII degradation by the carboxypeptidase ACE2 to Ang-(1–7), a vasodilator, antitrophic and antifibrotic heptapeptide that functions as an endogenous inhibitor of AngII [94, 95]. Both ACE2 and Ang-(1–7) were demonstrated in rat and human cardiomyocytes. Later, ACE2/Ang-(1–7) was also demonstrated in other tissues, including blood vessels, kidneys, lungs, and brain, and was implicated in CV homeostasis.
Experimentally in rats, ACE inhibition decreased AngII formation and increased Ang-(1–7), while AT1 blockade increased AngII and Ang-(1–7) [96]. The increase in Ang-(1–7) with ACE inhibition was attributed to increased AngI and inhibition of Ang-(1–7) metabolism, while the increase with AT1 blockade was attributed to formation from increased AngI [96]. After MI in rats, AT1 blockade was shown to upregulate ACE2 [97], which may contribute to its cardioprotective effect via Ang-(1–7) formation as verified by Ang-(1–7) infusion [98]. In the late phase of LV dysfunction after MI in rats, the ACE inhibitor enalapril was shown to attenuate downregulation of ACE2 [99]. In hypertensive rats, the ARB telmisartan attenuated aortic hypertrophy through modulation of ACE2 [100].
Preliminary data from our laboratory showed that the ARB candesartan and the vasopeptidase inhibitor omapatrilat attenuated LV remodeling and dysfunction during healing after reperfused MI in rats through modulation of ACE2 as well as MMP-9, inflammatory cytokine IL-6, TNF-α, TGF-β, N-acetyl-seryl-aspartyl-lysyl-proline (Ac-SDKP), collagens, and fibrosis [101]. In that study, increased ACE2 and Ang-(1–7) levels, associated with enhanced AT2 receptor signaling and suppression of TGF-β1 and smad-2 signaling, inflammatory cytokines, and AngII signaling via AT1 receptors, effectively limited fibrosis, adverse remodeling, and dysfunction during healing after reperfused MI [101]. Recent evidence suggests that the ACE2-Ang-(1–7)-Mas receptor can counter-regulate remodeling effects of AngII and inhibit hypertrophy and fibrosis [102].
It appears that whereas the RAS through ACE-AngII-AT1 receptor axis promotes adverse remodeling, the ACE2/Ang-(1–7) system through ACE2-Ang-(1–7)-Mas receptor axis is counter-regulatory ([96, 102, 103] for review) and prevents adverse remodeling. The increase in ACE2/Ang-(1–7) mediated by ACE inhibitors and ARBs may contribute to the anti-remodeling and other beneficial effects in HF ([102, 103] for review). However, ACE2/Ang-(1–7) is upregulated in human HF [104] and overexpression of ACE2 attenuates LV remodeling post-MI in young rats [105].
In human HF, plasma ACE2 activity is elevated and correlates with poor outcome [106, 107], suggesting a compensatory response to LV dysfunction. ACE2 expression in rat lung decreases with aging [108]. Responses to Ang-(1–7) differ in young, aged, and diabetic rabbit corpus cavernosum [109]. Whether a loss of ACE2 function with aging might lead to amplified activation of RAS and more adverse remodeling and explain the worse clinical outcome in the elderly with HF [6, 8, 14] needs study. Preliminary data from our laboratory showed that more severe age-related LV remodeling and dysfunction after acute reperfused ST-segment elevation MI was associated with downregulation of myocardial AT2 receptors, ACE2, Ang-(1–7), Ac-SDKP, and smad-2 in the dog model [110].
Since Ang-(1–7) is a substrate for inactivation by ACE, it competes with AngI and bradykinin for degradation, thereby inhibiting AngII formation and augmenting bradykinin activity and its vasodilatory effects [111]. Increased Ang-(1–7) with ACE inhibition may further augment bradykinin activity. Later, AT1 blockade was shown to increase bradykinin levels in hypertensive humans, probably due to decreased metabolism by ACE and neutral endopeptidase [112]. The authors warned that the increased bradykinin with ARBs may augment their therapeutic actions but may also lead to angioedema. Collectively, these findings indicated that ACE inhibitors and ARBs increase both Ang-(1–7) and bradykinin. Whether these effects are amplified in the elderly needs study. An alternative strategy needing study for slowing progression of HF with aging would be to increase levels of Ang-(1–7) as opposed to reducing levels of AngII with ACE inhibitors and ARBs, thereby avoiding side effects related to increased bradykinin such as coughing, dizziness, and angioedema.
Role of the Chymase/Ang-(1–12) Axis in Renin-Independent Generation of AngII
Recent evidence since 2006 suggests that Ang-(1–12), a propeptide cleaved from angiotensinogen, may represent an alternate substrate for the formation of angiotensins including AngII [113, 114]. Ang-(1–12) was shown to be increased in cardiomyocytes of adult spontaneously hypertensive rats [115]. Cardiac Ang-(1–12) as well as AngI and AngII was increased in myocardium of rats with bilateral nephrectomy and absence of circulating renin while plasma levels decreased [116]. In a rat model of ischemia–reperfusion injury, pro-angiotensin 12 (PA12) was suggested to act as a circulating substrate for a chymase-mediated AngII production [117]. Recently, this renin-independent mechanism of AngII generation was demonstrated in human left atrial tissue from patients undergoing the MAZE surgical procedure for chronic atrial fibrillation [118]. This pathway has also been demonstrated in the normal and diseased human LV tissue [119, 120].
Whether this system is augmented in the elderly needs study.
RAS Inhibition for Heart Failure: ACE Inhibitors and ARBs
The introduction of RAS inhibition with ACE inhibitors and ARBs for the treatment of chronic HF represents one of the most significant advances in CV medicine during the latter half of the twentieth century. When the role of the RAS in CV disease was first recognized in the 1950s, the focus was on HTN and the neurohumoral paradigm. Several major large-scale, multicenter randomized clinical trials (RCTs) of ACE inhibitors since the mid-1980s helped to establish its use for improving the survival of patients with HF and acute MI [121–126] (Table 18.1). The rationale for using ACE inhibitors was to inhibit ACE and thereby decrease the formation of AngII and its adverse effects. Subsequently, RCTs since the mid-1990s investigated the benefits of using ARBs in patients with HF and MI [127–136] (Table 18.2). The main rationale for ARBs was to achieve specific and selective blockade of the effects of AngII via the AT1 receptor [137].
Three points about those and subsequent RCTs need emphasis. First, most of the early RCTs recruited patients aged ≤65 or 18–65 years as reflected in the mean ages in Tables 18.1 and 18.2. Elderly patients were excluded. Second, since the benefits of ACE inhibitors in hypertension, HF, and MI were already established when ARBs were introduced, it became necessary to demonstrate that ARBs were superior to them or equally effective in patients intolerant to them and receiving other background therapies in RCTs rather than relative to a true placebo group. Third, ACE inhibitors and ARBs were used on top of background contemporary therapy that often included β-blockers in patients with LV systolic dysfunction and HF, and β-blockers were known to reduce renin [138] and AngII [139] and produce effects additive to that of ACE inhibitors [140].
Additionally, three reasons were proposed as justification for using ARBs as an add-on or alternative to already established ACE inhibitors. First, compared to ACE inhibitors, ARBs were expected to provide more complete inhibition of AngII derived from all sources, including non-ACE and non-renin pathways, especially as the latter is increased during ACE inhibition [43, 55]; however, ARBs were subsequently found to increase renin, AngI, and AngII as well as Ang-(1–7) levels [94, 96]. Second, since ARBs do not inhibit kininase II, or via this mechanism increase systemic peptides of the inflammatory response such as bradykinin, substance P, and other tachykinins known to produce cough and angioedema side effects associated with ACE inhibitors [141, 142], these side effects would be avoided; however, as discussed before, ARBs can also increase release of kinins and stimulation of kinin B1 or B2 receptors [46–49]. Third, ARBs might produce unopposed AT2 receptor stimulation resulting in added benefits, including long-term CV structural changes over that seen with ACE inhibitors [43]. These arguments led to RCTs on the effects of valsartan in post-MI LV systolic dysfunction and/or HF [130] and chronic HF [144], respectively. However, in that study (VALIANT), there was no upper age limit and valsartan was shown to be not superior to the ACE inhibitor captopril and the combination increased the risk of adverse events in elderly patients [135]. Importantly with increasing age (<65, 65–74, 75–84, and ≥85 years), the 3-year mortality increased fourfold (13 %, 26 %,36 %, and 52 %, respectively), the composite end-point events increased more than twofold (25 %, 41 %, 52 %, and 67 %, respectively), and HF admissions increased threefold (12 %, 23 %, 31 %, and 35 %, respectively) in VALIANT [135]. These findings further underscore the fact that the elderly represent a high-risk group for acute MI and HF/low EF with disproportionately high mortality and morbidity in need of improved therapies.
RAS Inhibitors for Hypertension: ACE Inhibitors and ARBs
The central role of RAS in the regulation of BP, fluid and electrolyte balance, and pathophysiology of CV disease is well recognized (Fig. 18.1) [24, 144–147]. AngII not only increases BP but also promotes vascular inflammation leading to endothelial dysfunction and atherosclerosis, stimulates vascular smooth muscle hypertrophy and vascular remodeling, and stimulates myocardial fibrosis and hypertrophy leading to cardiac remodeling [144–147]. Importantly, it also increases aldosterone which stimulates fibrosis and CV remodeling (Fig. 18.1). The fact that most of the effects of AngII are mediated via AT1 receptors provides the rationale for ACE inhibition and AT1 receptor blockade (Fig. 18.1). Since aging is associated with increased AngII and other RAS components which in turn contribute to increased CV remodeling and CV risk in the elderly [6], RAS inhibition with ACE inhibitors and ARBs is recommended in that group [24].
ACE inhibitors have also been studied for reducing CV risk. Indeed, they effectively control BP in patients with hypertension and have additional benefits on CHD, stroke, MI, HF, diabetes, or chronic kidney disease. Several RCTs have established that ACE inhibitors reduce rates of death, MI, and stroke in patients with HF [123], LV dysfunction [148], vascular disease [149–152], or high-risk diabetes [153]. The HOPE trial with the ACE inhibitor ramipril showed improved prognosis; decreased rate of death, MI, and stroke in high-risk patients for CV events and without low EF or HF; and decreased new-onset diabetes and complications of diabetes [154]. The EUROPA trial, which included lower risk patients than HOPE, showed improvement in the composite end-point of CV mortality, MI, and resuscitation [149]. However, QUIET which included low-risk patients showed no significant benefit [150]. The PEACE trial, which also included low-risk patients and used the ACE inhibitor trandolapril, showed no benefit [151].
Although the dose of ACE inhibitors in QUIET and PEACE may not have been optimal, a meta-analysis of the trials, with pooled data in 31,600 patients, showed that ACE inhibitors are effective in preventing CV events, with 26 % reduction in the risk of HF or stroke and 13–18 % reduction in total and CV mortality, and MI compared to placebo [152]. Other studies suggested that ACE inhibitors not only control BP and reduce stroke but also prevent renal complications of diabetes [155]. In the HOPE/The Ongoing Outcomes (TOO) study, development of diabetes in the follow-up phase decreased, suggesting an added benefit of long-term ramipril [156]. In the MICRO-HOPE substudy [153], ramipril was beneficial for CV events and overt nephropathy in patients with diabetes. In the ADVANCE trial, the ACE inhibitor perindopril together with the diuretic indapamide reduced the risks of major vascular events and death in type 2 diabetes [157].
As discussed before, the ability of ARBs to selectively block AngII at the AT1 receptor resulting in more complete inhibition was considered advantageous (Fig. 18.4). ARBs also do not increase bradykinin by suppressing its degradation as ACE inhibitors do, thereby enhancing vasodilation but also increasing cough and angioneurotic edema that troubles ~20 % of patients, especially women and Asians [29, 153, 154]. ARBs may result in unopposed AT2 receptor activation and enhance vasodilation via downstream AT2-mediated signaling (Fig. 18.4). Apart from blocking deleterious effects of AngII and controlling BP, ARBs might have similar protective effects as ACE inhibitors. RCTs showed that ARBs effectively control BP in HTN and are well tolerated [158]. However, despite well-known arguments for using ARBs [29], whether ARBs are as effective as ACE inhibitors in reducing events such as stroke and MI has been questioned [159]. Moreover, ARBs can also release kinins and increase bradykinin levels in hypertensive patients [112, 158] and thereby mediate CV protection. Such ARB-induced increase in bradykinin can augment therapeutic actions but also lead to cough and angioedema [112, 158]. As discussed before, both ACE inhibitors and ARBs can increase Ang-(1–7) in the counter-regulatory arm of the RAS.
A complicating factor with the chronic use of ACE inhibitors in HF patients is that AngII levels increase and symptoms worsen [128]. However, studies in HTN have shown that ARBs such as losartan and valsartan are as effective as ACE-Is in lowering BP [160, 161]. In hypertensive patients with ACE-I-induced cough, this complication is less frequent with ARBs [162]. In patients with HF/low EF, an ARB was shown to reduce the rate of death or hospitalization relative to placebo in those patients who could not tolerate an ACE inhibitor [133] or were already receiving it [130, 132]. In the LIFE study, compared to β-blockers, ARBs reduced vascular events in high-risk patients with hypertension and LV hypertrophy (LVH) [163]. Taken together, these studies suggest that an ARB is an effective and well-tolerated alternative to an ACE inhibitor for CV protection.
Since ACE inhibitors preceded ARBs for treating HTN and HF, it has become necessary in clinical trials to demonstrate non-inferiority or superiority of an ARB over an ACE inhibitor as comparator. In patients with MI, two studies comparing an ARB with an ACE inhibitor produced different results. OPTIMAAL [164] and VALIANT [143] compared the ARBs losartan and valsartan, respectively, to the captopril in patients with signs of HF within 10 days of MI. In OPTIMAAL, the ARB was not superior and the non-inferiority criteria were not met; in fact, there was an increase in CV mortality after a 2.7-year mean follow-up [164]. In VALIANT, the ARB was non-superior and non-inferior for mortality and the composite end-point of fatal and nonfatal events; the study established that valsartan was as effective as an ACE inhibitor in reducing mortality in high-risk survivors of MI [143]. A meta-analysis of 54,254 patients from 11 trials showed a potential 18 % increase in MI with ARBs compared to placebo and a possible increase compared with other active therapy [165]. In a separate meta-analysis of 55,050 patients from 11 trials that compared ARBs with either placebo or an active comparator, ARBs were found to reduce event rates for stroke, not to reduce event rates for global death, and to increase rates of MI by 8 % [159]. The cloud of doubt cast by these reports has been partly dispelled by studies with the ARB telmisartan [166–169].
Aging, RAS Dysregulation, and AngII Inhibition with ACE Inhibitors and ARBs
The contribution of the RAS to CV changes during aging has been confirmed in experimental studies. As mentioned previously, aging alters all RAS components and results in increased angiotensinogen, AngII, AT1 and AT2 receptors, and ACE in rat hearts [30, 31]. Increased AngII and other RAS components with aging may explain increased cytosolic and mitochondrial oxidant production, mitochondrial dysfunction, and increased ECM deposition associated with aging [32]. Short-term AngII was shown to downregulate AT1 receptor mRNA in fibroblasts from aged rat myocardium [170]. A study with long-term AngII inhibition with the ACE inhibitor enalapril or the ARB losartan protected against CV effects of aging and prolonged life in rats [171], implying a harmful effect of increased RAS and AngII effects during aging. Furthermore, disruption of the AT1 receptor in aging mice was shown to protect from CV morbidity and mortality and promote longevity [33].
As discussed above, clinical studies of RAS inhibition with using ACE inhibitors and ARBs in predominantly non-elderly patients with HF/low EF produce undeniable benefits. However, evidence from some of these clinical studies has established that elderly patients with post-MI HF are at higher risk despite therapy and a dominant mechanism is persistent adverse LV remodeling [6, 8, 10]. While reperfusion is widely used in acute MI, data on healing and remodeling post-reperfused MI in the elderly is lacking [6]. Post-MI survivors who develop HF on therapy have a 10-fold greater risk of dying [172], and the risk is even greater in the elderly [28, 173, 174]. The survivors with persistent post-ischemic LV dysfunction after reperfused MI remain at risk for remodeling and its consequences including HF despite receiving optimal therapy [135, 175–182]. Evidence suggests that aging-related impaired or defective healing/repair may be a major culprit resulting in adverse remodeling and poor outcome [6, 181].
Recent evidence from our laboratory suggests that the RAS may become dysregulated with aging [6, 9]. Physiological, cellular, and molecular changes that occur with CV aging appear to negatively impact the healing/repair response to injury including reperfused MI [6]. Timed release of several factors after injury modulate healing/repair [6, 26, 38, 176, 177]; the factors include AngII, ROS, chemokines, inflammatory cytokines, growth factors, MMPs, and other matrix proteins such as healing-specific matrix and matricellular proteins (HSMPs) including secretory leukocyte protease inhibitor (SLPI), secreted protein acidic and rich in cysteine (SPARC), and osteopontin (OPN) [6]. Together, they orchestrate inflammation, ECM remodeling and fibrosis, and adverse LV remodeling [6, 175–177]. In 2008, Bujak et al. first reported defective infarct healing and increased adverse remodeling after reperfused MI in old versus young mice [7, 8]. Since therapy for optimizing healing is lacking [6, 8], our laboratory considered the hypothesis that in aging hearts, increased myocardial AngII, through its pro-inflammatory, pro-oxidant, and pro-remodeling effects, may amplify increases in pro-inflammatory cytokines, MMPs, and oxidative markers and contribute to impaired healing/repair and adverse LV remodeling [6]. We postulated that aging results in a dysregulation of matrix, inflammation, and fibrosis pathways, leading to impaired healing and adverse LV remodeling post-reperfused MI (Fig. 18.3). In this construct, aging-related impaired or defective healing post-MI may be the major culprit leading to defective infarct fibrosis that in turn might result in amplified adverse maladaptive LV remodeling, increased progressive LV enlargement, and increased disability and/or death in older patients. Evidence from our laboratory supports the idea that aging-related adverse remodeling may be due in part to impaired healing/repair mechanisms after reperfused MI [6, 9, 182], but further research is needed.
Our preliminary data in dog, rat, and mouse models of post-reperfused MI suggest that concurrent upregulation of the three HSMPs (SLPI, SPARC, and OPN) may interact with concurrently upregulated ECM-proteolytic, inflammation, and fibrosis pathways and contribute to remodeling in young animals [9, 182] and upregulation of the HSMPs and proteolytic, inflammation, and fibrosis pathways is amplified in old animals that develop more severe LV remodeling and dysfunction [9, 182, 183]. Importantly, the ARB candesartan attenuated these changes across the old groups albeit with a trend towards lesser benefit in the oldest [9], implicating AngII and dysregulation of inflammatory and ECM-proteolytic pathways in the augmented remodeling.
Taken together, our data suggest that the HSMPs SLPI, SPARC, and OPN that are increased post-reperfused MI may interact with inflammation and fibrosis pathways and improve healing and LV remodeling in the young, but the pathways become dysregulated in the old [9]. Thus, aging amplifies responses in the critical cellular signaling and ECM-proteolytic pathways, and interaction with the AngII/AT1 receptor pathway after reperfused MI in the clinically relevant dog model [182]. Our preliminary data during healing after reperfused MI in rats [183] confirm that aging amplifies the increased expression of HSMPs, MMPs, and inflammatory and fibrogenic cytokines in infarct zones during healing after reperfused MI. In that study [183], SLPI, SPARC, and OPN were colocalized in macrophages and monocytes. Importantly, candesartan during healing suppressed these changes in the HSMPs and remodeling in young and old rats, implying regulation by AngII with aging. Candesartan also attenuated increases in MMPs, inflammatory and fibrogenic cytokines, and iNOS in the young and old rats, implying regulation by AngII during healing. The data also showed age-related increase in tissue myeloperoxidase (MPO, oxidant activity marker), and MPO-positive granulocytes and CD68 and MAC387-positive macrophages at day 25, implying persistent inflammation and granulation tissue with impaired healing in the late phase after reperfused MI.
In summary, our data suggest that aging upregulates two critical pathways: (1) increased AngII/ROS → increased iNOS-NO → peroxynitrite → MMP and HSMP activation → adverse remodeling and dysfunction and (2) increased AngII/ROS → increased inflammatory cytokines → MMP and HSMP activation → adverse remodeling and dysfunction.
Dysregulation of AT2 Pathway with Aging and Effect of ACE Inhibitors and ARBs
Evidence also suggests that other components of the RAS are altered with aging. Normally in adults, most actions of AngII are mediated through AT1 and AT2 receptor expression is low. However, AT2 is reexpressed in CV disease, and during AT1 blockade, AngII induces AT2-mediated vasodilation through the bradykinin/NO/cGMP pathway [184]. Paradoxically with aging, AT2 activation leads to vasoconstriction via ROS rather than vasodilation in old rats [185], which explains the aging-induced AT2R paradox in resistance arteries. This may also explain the so-called ARB-MI paradox suggesting that increased AT2 receptor signaling during ARB therapy may prove harmful in elderly patients [159]. In the study by Pinaud et al. [185], the resistance arteries of the old rats had impaired flow- and NO-mediated vasodilation and reduced expression of endothelial NO synthase compared to young rats. Importantly, aging increased AT2 expression in vascular smooth muscle rather than endothelial cells, and AT2 receptor blockade improved flow-mediated dilation, implying AT2-receptor-mediated vasoconstriction [185]. In addition, treatment with the vasodilator hydralazine attenuated AT2 receptor induction of ROS and direct vasoconstriction, thereby enhancing flow-mediated vasodilation [185]. In a study of microvascular AT2 receptor expression in hypertensive patients with type 2 diabetes receiving therapy with an ARB, AT2 appears to mediate vasodilation [186]. Whether aging in humans leads to (1) a similar molecular switch from AT2-receptor-mediated vasodilation to vasoconstriction, (2) impaired responsiveness of the bradykinin/NO/cGMP vasodilator cascade to AT2 receptor activation, and a cell signaling switch that converts AT2 receptor inhibition of phosphorylation of mitogen-activated protein kinase (extracellular signal-related kinase 1/2) into stimulation needs verification [184]. Whether AT2 receptor blockers and hydralazine might be beneficial in the elderly HF patient needs study.
Aging and Remodeling of ACE-2/Ang-(1–7) and Other Pathways
Interestingly, besides AngII receptor remodeling with aging [184], in the counter-regulatory arm of the RAS, decrease of ACE-2 in null mice is associated with decreased cardiac function [93] and ACE-2 also decreases with aging [108–110]. Whether increasing the ACE2/Ang-(1–7) pathway might be beneficial in the elderly HF patient needs study. Our studies with ACE inhibition and AT1 blockade in aging hearts with HF post-reperfused MI suggest that beneficial effect of RAS inhibition may be blunted in elderly [6, 8–10]. Remodeling of other RAS-related pathways with aging has also been described, including the inflammation and Kallikrein–kinin pathways [187], the β-adrenergic pathway [188], and the AngII-aldosterone pathway [189]. It should be noted that precautions in using both ACE inhibitors and ARBs are needed in the elderly with MI and hypertension [24, 28, 159, 190–193].
Taken together, remodeling of RAS with aging may account for the reported poor outcome post-MI despite optimal use of evidence-based therapy in the elderly [6, 27, 28]. As discussed before, clinical studies indicate that elderly post-MI patients are at higher risk for adverse events despite contemporary therapy including RAS inhibitors. Since AngII is the primary effector molecule of the RAS and AngII inhibitors form the basis of therapy for both elderly and non-elderly HF patients, this aging-induced remodeling of the RAS may have important implications for therapy based on AngII inhibitors for the elderly with post-MI HF.
RAS, Aldosterone, and the Mineralocorticoid Receptor Antagonists
The RAS was expanded to RAAS in order to emphasize the importance of the AngII-/AT1-receptor-mediated activation of aldosterone which promotes sodium retention, loss of magnesium and potassium, sympathetic activation, parasympathetic inhibition, myocardial and vascular fibrosis, baroreceptor dysfunction, vascular damage, and impaired arterial compliance. The rationale for using mineralocorticoid receptor (MR) antagonists (MRAs) is that AngII stimulates the release of aldosterone from the adrenal cortex, thereby activating MRs whose activation persists despite treatment with ACE inhibitors, ARBs, and β-adrenergic blockers. Several trials with MRAs have shown that MRAs effectively reduce total mortality in patients with HF/low EF [194–199] (Table 18.3). Based on evidence from two of the first two RCTs [194, 196], the addition of an MRA is reasonable in patients with moderate to severe HF/low EF provided renal function (serum creatinine ≤2.5 mg/dL in men and ≤2.0 mg/dL in women) and serum potassium (≤5.0 mEq/L) are carefully monitored. In RALES [194], the MRA spironolactone as an add-on to background therapy with ACE inhibitor, β-adrenergic blocker, diuretic, and digoxin in patients with moderate to severe HF (EF < 35 %) was prematurely terminated due to an early finding of a 30 % reduction in all-cause mortality and reduced morbidity and hospitalization. However, gynecomastia occurred in 10 % and hyperkalemia in 2 % of treated patients. In EPHESUS [196], the MRA eplerenone that selectively blocks the MR but not glucocorticoid, progesterone, or androgen receptors was assessed as an add-on to optimal medical therapy in post-MI HF patients with EF ≤ 40%. EPHESUS [196] reported a 14 % reduction in all-cause mortality, 17 % in CV mortality, and 15 % in risk of hospitalization. However, serious hyperkalemia occurred in 5.5 % and increased to 10 % in patients with baseline renal dysfunction defined as creatinine clearance <50 mL/min [196]. In a recent trial in patients with systolic HF and mild symptoms, eplerenone reduced both the risk of CV death and HF hospitalization but the trial was terminated prematurely according to prespecified rules [199]. In that study [199], serum potassium was >5.5 mmol/L in 11.8 % of patients receiving eplerenone compared to 7.2 % receiving placebo (P < 0.001).
In a substudy of RALES [195], the MRA spironolactone increased levels of markers of collagen synthesis, suggesting that limitation of excessive extracellular matrix (ECM) turnover contributed to the benefits. In a substudy of EPHESUS [197], early initiation of eplerenone was reported to reduce the 30-day all-cause mortality after acute MI. In a study of HF/PEF patients, eplerenone prevented progressive increase in procollagen type-III aminopeptide but had no impact on other markers of collagen turnover or diastolic function [198]. A recent meta-analysis of RCTs with MRAs spironolactone, canrenoate, and eplerenone demonstrated that MRAs exert beneficial effects on reversal of LV remodeling and dysfunction [200].
Close monitoring of renal function and serum potassium should be done when using MRAs, especially in the elderly. Both RALES and EPHESUS excluded patients with serum creatinine >2.5 mg/dL. Since use of aldosterone antagonists in patients with renal dysfunction (creatinine clearance <50 mL/min) increases the risk of hyperkalemia and this risk is greater in elderly patients and those receiving an ACE inhibitor or ARB concurrently, spironolactone or eplerenone should be used at a low dose under those circumstances and avoided in those with creatinine clearance <30 mL/min. In patients already receiving a long-term diuretic and potassium supplements, the latter should be reduced or discontinued.
Aging, RAAS, and Mineralocorticoid Receptor Antagonists
Whereas AngII is increased AngII [30, 31] with aging and AngII stimulates aldosterone secretion, aging in healthy humans results in decreased plasma aldosterone levels [201]. While this finding implies a dysregulated response in the AngII–aldosterone axis with aging and would suggest a reduced need for MRAs in elderly patients with HF, several lines of evidence suggest that aging may in fact result in enhanced activation of the MR. First, in a study of steroid hormone metabolites in hypertensive patients aged 18–84 years, aging was associated with decreased 11β-hydroxysteroid dehydrogenase type 2 (11βHSD2) activity [202]. This might explain the rising prevalence of HTN in the elderly. Importantly, the enzyme 11βHSD2 has been shown to convert cortisol and corticosterone to cortisone and 11-dehydrocorticosterone which are MR-inactive 11-keto derivatives [203, 204]. More importantly, cortisol not only activates MR but has greater affinity for MRs than aldosterone [205]. Since 11βHSD2 in renal tubular epithelium converts most of the cortisol into cortisone, aldosterone is the main endogenous agonist and activator of MRs in renal tubules normally and in the young [205]. Normally, cortisol seems to occupy epithelial and nonepithelial cells in tonic inhibitory fashion but, in the presence of tissue damage, becomes an MR agonist; this may explain both vascular and myocardial MR activation in HTN and HF and efficacy of MRAs despite low aldosterone levels [206]. Moreover, in the elderly with HTN [202] and non-elderly patients with essential HTN aged 40–60 years [207] with low aldosterone levels, decreased renal 11βHSD2 activity and increased cortisol may explain enhanced MR activation and justify use of MRAs. While data from mostly non-elderly patients show that the gene encoding aldosterone synthase (CYP11B2) is associated with high aldosterone and HTN and the adjacent gene (CYP11B1) encoding 11β-hydroxylase is associated with altered adrenal 11-hydroxylation efficiency (deoxycortisol to cortisol), more research is needed to determine their relative roles in the elderly and very old with HTN.
Second, while MRs are also present in myocardium, most cardiac aldosterone seems to come from the adrenals via the circulation, and glucocorticoids and aldosterone may serve as endogenous cardiac MR agonists [208]. As discussed before, low myocardial 11βHSD2 and increased cortisol in the elderly may lead to enhanced myocardial MR activation. In addition, increased AngII, ROS, and oxidative stress in the elderly and HF may act in synergy and contribute to increased MR activation, inflammation, fibrosis, myocardial hypertrophy and apoptosis, and thence HF progression [6, 189].
Third, a study in rats showed that aging is also associated with increased MR activity in vascular smooth muscle cells which promotes inflammation via extracellular signal-regulated kinase 1/2 mitogen-activated protein kinase and epidermal growth factor receptor-dependent pathways [209]. Fourth, since enhanced MR activation in the elderly leads to increased expression of tissue ACE and upregulation of AT1 receptors [210], combined AngII and aldosterone blockade or inhibition has been proposed for increasing benefits [211]. Both these studies [210, 211] used telomere length in white blood cells (WBCs), an index of CV aging and the burden of oxidative stress. Fifth, the three early clinical trials of MRAs in HF/low EF included the elderly and showed efficacy in both young and old patients with [194–196]. The mean age was 65 years in RALES with spironolactone [194] and 64 and 69 years, respectively, in EPHESUS [196] and EMPHASIS-HF [199] with eplerenone. Importantly, the decrease in total mortality and HF hospitalizations was similar in the elderly and non-elderly groups.
Sixth, MRAs may be especially effective in elderly patients with HF/PEF, where the dominant pathology is HTN associated with LV hypertrophy and myocardial fibrosis. Therapy with ACE inhibitors, ARBs, and β-adrenergic blockers fail to reduce mortality in that group. Specific therapy for diastolic dysfunction is lacking in all age groups and myocardial fibrosis is a major cause of diastolic dysfunction in the elderly even in the absence of HTN. Since RAAS inhibitors are powerful antifibrotic agents, it is reasonable to hypothesize that elderly patients might benefit from them unless the pathways are dysregulated [6].
As discussed before, the emerging paradigm is that aldosterone-induced MR activation leads to myocardial and vascular fibrosis in the non-elderly, while amplified cortisol-induced MR activation leads to amplified stimulation of fibrosis in the elderly and patients with essential HTN. In a substudy of RALES in patients with HF/low EF, spironolactone limited fibrosis and reduced procollagen I and III levels [195] and the mortality benefit was mainly in patients with continuing collagen turnover [189]. In the 4E trial of older adult patients with essential HTN, LV hypertrophy, and mean ages between 59 and 60 years [212], combination therapy with the ACE inhibitor enalapril and the MRA eplerenone was more effective than monotherapy in decreasing LV mass and albuminuria despite normal plasma aldosterone levels, endorsing the hypothesis that the MRA may have blocked cortisol-induced MRA activation in the face of decreased 11β-HSD2. In the small study of elderly patients with HF/PEF (N = 44; mean age 80 ± 8 years), eplerenone prevented progressive increase in collagen turnover assessed by procollagen III aminoterminal peptide and despite background therapy with ACE inhibitors in 64 %, ARBs in 34 %, and β-adrenergic blockers in 68 % [198]. In a small study of older adult patients with HF/PEF and mean age 62 ± 6 years, spironolactone limited diastolic dysfunction [213]. In another small study of patients with HTN aged between 30 and 70 years, eplerenone reduced vascular stiffness more than the β-adrenergic blocker atenolol [214]. However, the mean ages in the three groups in that study (controls 51, eplerenone 54, atenolol 44 years, respectively) suggested that most patients were non-elderly [214]. In a small study of patients with HTN, regression of LV mass correlated with the decrease in plasma aldosterone, and the ARB losartan reduced LV mass more than the calcium channel blocker amlodipine by reducing plasma aldosterone in addition to BP [215]. In the Aldo-DHF multicenter, prospective, randomized, double-blind, and placebo-controlled 6-year (2007–2012) trial of spironolactone in HF/PEF patients aged ≥50 years (mean 67 ± 8 years), spironolactone improved diastolic function without improving exercise capacity or quality of life [216].
In the ongoing 6-year (2006–2013) phase 3 treatment of preserved cardiac function heart failure with an aldosterone antagonist (TOPCAT) trial of spironolactone on CV mortality and HF hospitalization in 3,445 older adult and elderly patients with HF/PEF (age ≥50 years; mean 69 ± 10 years), 91 % have HTN [217]. This trial is also multicenter, prospective, randomized, double-blind, and placebo controlled. Comorbidities typically include coronary artery disease (57 %), atrial fibrillation (35 %), chronic kidney disease (38 %), and diabetes mellitus (32 %) [217].
MRAs in Very Old Elderly Patients with HF/PEF
Elderly and especially very old (aged ≥80 years) patients with HF/PEF frequently have concomitant comorbidities including obesity, sleep apnea, coronary artery disease, atrial fibrillation, chronic renal disease, and diabetes that may benefit from MRAs [189, 217]. Several lines of evidence provide justification for blockade of effects of aldosterone and MR activation in epithelial and nonepithelial tissues of these patients. First, evidence from studies of telomere length in white blood cells (WBCs) suggests that aldosterone accelerates CV aging through mechanisms that generate ROS and telomere length may serve as an index of the burden of oxidative stress. In a study of normotensive and mild hypertensive males, plasma aldosterone was inversely related to telomere length, suggesting that aldosterone is not only pro-oxidant but increased levels might be linked to accelerated telomere shortening and increased biological aging [211].
Second, other studies suggest that aldosterone and MR activation exert CV and renal pleiotropic effects that extend well beyond the classical renal regulation of sodium balance. Mechanisms include oxidative stress, inflammation, impaired vascular reactivity, and endothelial-mediated vasorelaxation, downregulation of proteins in insulin metabolic pathways, and impaired renal podocyte and mesangial cell integrity. Elevated plasma aldosterone in patients with metabolic syndrome [218], kidney fibrosis [219], sleep apnea [220], and CHD [221, 222] has been implicated in the pathophysiology of these conditions. Interestingly in the latter study [222], plasma aldosterone was directly related to BMI (body mass index), HTN, and NYHA class besides mortality and ischemic events and inversely to age, creatinine clearance, and use of β-blockers.
Third, adipocytes which protect against urinary protein loss were shown to release Rac-1 and other proteins that stimulate adrenal release of aldosterone [223]. Interestingly, while obese patients with metabolic syndrome often have salt-sensitive HTN and only ~33 % have elevated plasma aldosterone, MRAs prevent salt-induced cardiorenal damage suggesting that high salt and aldosterone contribute to the damage [224]. This is explained by the discovery of an alternative pathway of MR activation through the small GTP-binding protein Rac-1 which is activated by both high salt and hyperglycemia [224]. Fourth, evidence from patients with primary aldosteronism also supports the hypothesis that aldosterone suppresses pancreatic beta-cell function leading to insulin resistance with hyperglycemia and diabetes [225]. Fifth, aldosterone was shown to impair vascular reactivity by reducing glucose-6-phosphate dehydrogenase (G6PD) expression thereby decreasing glutathione, antioxidant reserve, nitric oxide (NO) generation, and NO availability [226].
Sixth, aldosterone was shown to increase signaling via nuclear factor κB (NFκB) and transcription factor activator protein-1 (AP-1) pathways, inflammation, and cytokine expression in vascular smooth muscle cells, thereby contributing to endothelial dysfunction and atherosclerosis [227, 228]. These and other studies of the aldosterone → oxidative stress → inflammation → endothelial dysfunction axis and the aldosterone → oxidative stress → hypertrophic remodeling/fibrosis axis suggest that many of the effects of aldosterone are mediated by genomic and non-genomic pathways in MR-dependent or independent manner [227]. Seventh, macrophages in the vascular wall and atherosclerotic plaques have been shown to express MRs [229]; aldosterone also stimulates vascular NADPH oxidase and p38MAP kinase and release of MMPs that mediate progression of atherosclerosis and plaque rupture [189, 227, 228]. Eighth, other studies in monocyte-derived macrophages from patients with congestive HF suggested that the protective role of MRAs is partly due to increased generation of Ang-(1–7) and ACE2 and decreased AngII formation, and the effects were mediated by NADPH oxidase [230]. Interestingly, aldosterone and/or cortisol-induced of MR is associated with upregulation of the AT1 receptor as well as downregulation of ACE2 [231].
Ninth, in the EMPHASIS-HF study of elderly patients with systolic HF and mild symptoms, eplerenone not only reduces mortality and hospitalization [199] but also reduces the incidence of new atrial fibrillation/flutter likely via attenuation of atrial remodeling and fibrosis [232]. Tenth, MRAs also block the aldosterone-induced renal podocyte injury associated with decreased NADPH oxidase, increased oxidative stress, and enhanced aldosterone effector kinase Sgk1, thereby decreasing podocyte damage, albuminuria, and mesangial fibrosis [219, 233]. MRAs also inhibit MR-mediated kidney DNA damage in DOCA-salt hypertensive rats [234]. Eleventh, MRAs also attenuate LV hypertrophy and vascular stiffness in chronic kidney disease [223].
Twelfth, although many studies show that MRAs limit remodeling and fibrosis in various models of chronic HF, the mechanisms of benefit are not completely clear. In one study of MI in rats, angiotensin and aldosterone blockade inhibited osteopontin expression, LV remodeling, and fibrosis [235]. Enhanced MR signaling induced by transgenic expression of 11βHSD2 to drive cardiac hypertrophy and HF results in severe cardiomyopathy, fibrosis, and increased mortality that are partially improved by eplerenone treatment [236]. Transgenic overexpression of aldosterone synthase in cardiomyocytes causes coronary dysfunction but prevents harmful effects of diabetes by preserving capillary density [237]. Ablation of MRs in cardiomyocytes but not cardiac fibroblasts preserves cardiac function and limits cardiac dilation in chronic pressure overload [238]. Studies of selective ablation of MR expression in different cardiac cell types may help to clarify mechanisms of cardioprotection by MRAs [238]. More systematic studies of MRAs in the old and very old versus young groups are needed to uncover ancillary pathways.
MRAs and Hyperkalemia in the Elderly and Very Old Elderly Patients with HF
While MRAs are considered beneficial in elderly and very old patients, the risk of hyperkalemia is greater in very old elderly HF patients [189] and those with chronic kidney disease and/or diabetes [239]. It is therefore prudent to closely monitor serum potassium and reduce the dose of MRAs in these patients. In TOPCAT, spironolactone was initiated at a lower dose of 15 mg daily, with escalation to 45 mg daily provided serum potassium is <5 mEq/L [217]. Hyperkalemia can be treated by the new potassium binding polymer RLY5016 [240]. This was evaluated in patients with mean age of 68 years, chronic kidney disease, and a history of hyperkalemia resulting in discontinuation of RAAS therapy in the PEARL-HF trial [240]. The patients received 30 g/day of RLY5016 on top on spironolactone 25–50 mg/day; adverse drug reactions (ADRs) with RLY5016 were seen in 7 % (versus 6 % in placebo) while hypokalemia (K+ < 3.5 mEq/L) occurred in 6 % (versus 0 % in placebo) [240]. More cardioselective MRAs with better Na+/K+ ratio than spironolactone and eplerenone may be safer in elderly and very old patients [189].
Novel RAAS Therapies in Hypertension: Aldosterone Synthase Inhibition
A meta-analysis of RCTs of RAAS inhibitors in hypertensive patients suggested that in patients with HTN, ACE-inhibitor treatment, but not ARB treatment, resulted in further reduction of all-cause mortality [241]. However, the authors did not discuss impact of age. Control of BP and end-organ damage with therapy of HTN with conventional RAAS inhibitors can be challenging but a number of novel approaches hold promise for treatment of resistant HTN [242] and need evaluation in the elderly. Resistant or uncontrolled HTN despite three antihypertensive agents of different classes [243] is considered a trigger of cardiac decompensation in very old patients with HF/PEF and comorbidities such as diabetes and chronic kidney disease [189]. While there are proponents of triple therapy with combined RAAS inhibition using combined ACE inhibitor, ARB, and MRA for resistant HTN, such triple therapy is not recommended for all patients along the cardiorenovascular continuum; rather, in congestive HF patients with incomplete neuroendocrine blockade evidenced by repeated bouts of cardiac decompensation, dual therapy can be tried with close attention to patient safety [244].
The rationale for aldosterone synthase (CYP11B2) inhibitors is to inhibit aldosterone formation and thereby prevent increase in aldosterone levels and their MR-independent effects. Evidence suggested that the aldosterone synthase inhibitor LCI99 was modestly effective in patients with primary aldosteronism (mean age 50 years), decreased BP, corrected hypokalemia, and produced latent inhibition of cortisol synthesis [245]. Its development was stopped in favor of the search for more specific inhibitors [242]. The aldosterone synthase inhibitor FAD286 was shown to reduce mortality, cardiac hypertrophy, albuminuria, cell infiltration, and matrix deposition in the heart and kidney of double transgenic renin and angiotensinogen (dTGR) rats without profound effect on BP [246]; reduce cardiac and renal fibrosis induced by AngII and high salt in uninephrectomized rats [247]; and improve LV hemodynamics, remodeling, function, and redox status in rats with HF [248]. Data on the aldosterone synthase inhibitors in older groups is lacking.
Novel RAAS Therapies in Hypertension: Renin Inhibition
Renin as a target has been debated at least since the 1990s, since it catalyzes the key rate limiting step in the RAAS cascade. The selective renin inhibitor aliskiren has been shown to reduce AngI and AngII levels [249] and attenuate BP comparable to β-blockers [250], diuretics [251], ACE inhibitors [252, 253], and ARBs [254]. It attenuates plasma renin activity that is increased by ACE inhibitors and ARBs [255]. However, high renin levels due to escape during aliskiren therapy is a concern [256] as aliskiren does not prevent binding of renin to pro(renin) and activation of pro(renin) receptors [257]. Aliskiren, in a dose that did not reduce BP, improved LV dysfunction and remodeling after MI in mice and decreased apoptosis [258]. In patients with symptomatic HF, HYHA class II–IV, a history of HTN, elevated BNP, and mean age of 68 years, aliskiren on top of an ACE inhibitor or ARB and β-blocker had favorable neurohumoral effects [259]. The ASTRONAUT study will test whether aliskiren on top of standard therapy will reduce post-discharge mortality and rehospitalization in patients with worsening HF/low EF [260]. The ATMOSPHERE study will determine whether aliskiren added to or as an alternative to ACE inhibition in patients with chronic systolic HF improves outcomes [261]. In the ASPIRE study, adding aliskiren to standard therapy including a RAAS inhibitor in high-risk post-MI patients with LV systolic dysfunction did not result in further attenuation of LV remodeling and was associated with more adverse effects [262]. The ASPIRE HIGHER program should provide data on protection from target organ damage and CV morbidity/mortality in a range of cardiorenal conditions including HF, post-MI, and diabetic nephropathy [263]. The AVOID study showed that aliskiren may be renoprotective and reduced albuminuria in patients with type 2 diabetes, kidney disease, and HTN [264]. Several of those studies included the elderly. The ALTITUDE study which aimed to determine whether aliskiren on top of an ACE inhibitor or ARB therapy delays cardiorenal complications in patients with type 2 diabetes at high risk for cardiorenal events was stopped in 2012 because of no apparent benefit and an increase in adverse events including hyperkalemia (aliskiren 11 % versus placebo 7 %) and hypotension (12 % versus 8 %) [265]. Whether aliskiren might benefit the elderly with HTN and HF/PEF remains to be addressed.
Dual-Action Molecules in Elderly Patients with HF/PEF
Dual inhibition of ACE and neutral endopeptidase (NEP) pathways in a single molecule such as omapatrilat (OMA) was studied in patients with HTN, but despite superior antihypertensive efficacy over ACE inhibition and equal anti-remodeling efficacy in HF patients [266], the Federal Drug Administration (FDA) bureau did not approve OMA for patients with HTN because of troubling angioedema. The concept of dual-action molecules was recently revisited with LCZ696, which combines neprilysin (NEP) and the ARB valsartan in a phase 2 trial and was shown to be beneficial in patients with HF/PEF [267] and is being evaluated in patients with HF/low EF [267, 268]. Whether dual pathway inhibition may be more effective in elderly HF patients needs study.
Conclusion
The RAS/RAAS has critical functions in CV physiology and CV pathophysiology. Evidence over three decades since the 1980s indicates that RAS/RAAS upregulation plays a major role in CV pathophysiology. Thus, the RAS/RAAS play critical roles in both post-MI dilative remodeling associated with HF/low EF and hypertrophic remodeling and fibrosis associated with hypertensive disease and HF/PEF. Aging is associated with enhanced dysregulation of the RAS evidenced by increased AngII and several components of the RAS. Evidence suggests that dysregulation of the RAS may contribute to CVD and the RAS dysregulation may be further amplified with aging. This enhanced remodeling of the RAS may account for the poor outcome in elderly post-MI patients. Emerging evidence suggests aging-related dysregulation in the RAAS with reduced plasma aldosterone and enhanced MR activation related to cortisol and other pathways that may benefit from MRAs. Enhanced RAS/RAAS remodeling may have important implications for therapy based on AngII inhibitors and MRAs in elderly patients with post-MI HF. It is important to remember that these therapies were tested in mostly non-elderly patients. More research into the biology of aging-induced remodeling of the RAS/RAAS and related pathways may lead to discovery and development of improved therapies for post-MI HF and post-HTN HF in different age groups and tailored for the young, adult, and elderly patient.
References
Jugdutt BI. Prevention of heart failure in the elderly: when, where and how to begin. Heart Fail Rev. 2012;15:531–44.
Weisfeldt ML. Left ventricular function. In: Weisfeldt ML, editor. The aging heart: its function and response to stress. New York: Raven; 1980. p. 297–316.
Lakatta EG, Gerstenblith G, Weisfeldt ML. The aging heart: structure, function, and disease. In: Braunwald E, editor. Heart disease. Philadelphia, PA: Saunders; 1997. p. 1687–700.
Lakatta EG, Levy D. Arterial and cardiac aging: major shareholders in cardiovascular disease enterprises. Part I. Aging arteries: a “set up” for vascular disease. Circulation. 2003;107:139–46.
Lakatta EG. Arterial and cardiac aging: major shareholders in cardiovascular disease enterprises. Part II. Circulation. 2003;107:346–54.
Jugdutt BI. Aging and remodeling during healing of the wounded heart: current therapies and novel drug targets. Curr Drug Targets. 2008;9:325–44.
Bujak M, Kweon HJ, Chatila K, et al. Aging-related defects are associated with adverse cardiac remodeling in a mouse model of reperfused myocardial infarction. J Am Coll Cardiol. 2008;51:1384–92.
Jugdutt BI, Jelani A. Aging and defective healing, adverse remodeling and blunted postconditioning in the reperfused wounded heart. J Am Coll Cardiol. 2008;51:1399–403.
Jugdutt BI, Jelani A, Palaniyappan A, et al. Aging-related early changes in markers of ventricular and matrix remodeling after reperfused ST-segment elevation myocardial infarction in the canine model. Effect of early therapy with an angiotensin II type 1 receptor blocker. Circulation. 2010;122:341–51.
Jelani A, Jugdutt BI. STEMI and heart failure in the elderly: role of adverse remodeling. Heart Fail Rev. 2010;15:513–21.
Lloyd-Jones D, Adams RJ, Brown TM, et al. Heart disease and stroke statistics – 2010 update: a report from the American Heart Association Statistics Committee and Stroke Statistics Subcommittee. Circulation. 2010;121:e46–215.
Centers for Disease Control and Prevention. Public health and aging: trends in aging: United States and worldwide. MMRW Morb Mortal Wkly Rep. 2003;52:101–6. http://www.cdc.gov/mmwr/preview/mmwrhtml/mm5206a2.htm. Accessed 18 Aug 2011.
World Health Organization (WHO). Definition of an older or elderly person. http://www.who.int/healthinfo/survey/ageingdefnolder/en/print.html. Accessed 30 Dec 2009.
Jugdutt BI. Aging and heart failure: changing demographics and implications for therapy in the elderly. Heart Fail Rev. 2010;15:401–5.
Roebuck J. When does old age begin? The evolution of the English definition. J Soc Hist. 1979;12:416–28.
Holborn, H. A history of modern Germany – 1840–1945. Princeton University Press; 1969. p. 291–3.
Jugdutt BI. Heart failure in the elderly: advances and challenges. Expert Rev Cardiovasc Ther. 2010;8:695–715.
Hunt SA, Abraham WT, Chin MH, et al. ACC/AHA 2005 guideline update for the diagnosis and management of chronic heart failure in the adult: a report of the American College of Cardiology/American Heart Association Task Force on Practice Guidelines (writing committee to update the 2001 guidelines for the evaluation and management of heart failure. Circulation. 2005;112:e154–235.
Jessup M, Abraham WT, Casey DE, et al. 2009 focused update: ACCF/AHA guidelines for the diagnosis and management of heart failure in adults: a report of the American College of Cardiology Foundation/American Heart Association Task Force on Practice Guidelines: developed in collaboration with the International Society for Heart and Lung Transplantation. Circulation. 2009;119:1977–2016.
Johansen H, Strauss B, Arnold MO, Moe G, Liu P. On the rise: the current and projected future burden of congestive heart failure hospitalization in Canada. Can J Cardiol. 2003;19:430–5.
Arnold MO, Liu P, Demers C, et al. Canadian Cardiovascular Society consensus conference recommendations on heart failure 2006: diagnosis and treatment. Can J Cardiol. 2006;22:23–45.
Dickstein K, Cohen-Solal A, Filippatos G, et al. ESC guidelines for the diagnosis and treatment of acute and chronic heart failure 2008: the task force for the diagnosis and treatment of acute and chronic heart failure 2008 of the European Society of Cardiology. Eur J Heart Fail. 2008;10:933–89.
McMurray J, Adamopoulos S, Anker S, et al. ESC guidelines for the diagnosis and treatment of acute and chronic heart failure 2012 – the task force for the diagnosis and treatment of acute and chronic heart failure 2012 of the European Society of Cardiology. Developed in collaboration with the Heart Failure Association (HFA) of the ESC. Eur J Heart Fail. 2012;14:803–69.
Aronow WS, Fleg JL, Pepine CJ, et al. ACCF/AHA 2011 expert consensus document on hypertension in the elderly. J Am Coll Cardiol. 2011;57:2037–114.
Jugdutt BI. Optimal medical therapy for optimal healing. In: Lewis BS, Flugelman MY, Halon DA, editors. Proceedings 9th International Congress on Coronary Artery disease. Coronary artery diseases 2011 update– from Prevention to Intervention, Venice 2011. Bologna, Italy: Medimond; 2011. p. 243–7.
Jugdutt BI, Jelani A. Aging and markers of adverse remodeling after myocardial infarction. In: Jugdutt BI, Dhalla NS, editors. Cardiac remodeling. Molecular mechanisms. New York: Springer; 2013. p. 487–512.
Jugdutt BI. Aging and remodeling of the renin-angiotensin-system post infarction. In: Kimchi A, editor. Proceedings 15th World congress on Heart Disease, Vancouver 2010. Bologna, Italy: Medimond; 2010. p. 87–91.
Alexander KP, Newby LK, Armstrong PW, et al. American Heart Association Council on Clinical Cardiology; Society of Geriatric Cardiology. Acute coronary care in the elderly, Part II. ST-segment-elevation myocardial infarction. A scientific statement for healthcare professionals from the American Heart Association Council for Clinical Cardiology. Circulation. 2007;115:2570–89.
Jugdutt BI. Valsartan in the treatment of heart attack survivors. Vasc Health Risk Manag. 2006;2:125–38.
Heymes C, Silvestre JS, Llorens-Cortes C, et al. Cardiac senescence is associated with enhanced expression of angiotensin II receptor subtypes. Endocrinology. 1998;139:2579–87.
Cao XJ, Li YF. Alteration of messenger RNA and protein levels of cardiac alpha(1)-adrenergic receptor and angiotensin II receptor subtypes during aging in rats. Can J Cardiol. 2009;25:415–20.
De Cavanagh EM, Ferder M, Inserra F, Ferder L. Angiotensin II, mitochondria, cytoskeletal, and extracellular matrix connections: an integrating viewpoint. Am J Physiol Heart Circ Physiol. 2009;296:H550–8.
Benigni A, Corna D, Zoja C, et al. Disruption of the Ang II type 1 receptor promotes longevity in mice. J Clin Invest. 2009;119:524–30.
Fleg JL, Lakatta EG. Normal aging of the cardiovascular system. In: Aronow WS, Fleg JL, Rich MW, editors. Cardiovascular disease in the elderly. 4th ed. New York, NY: Informa; 2008. p. 1–43.
McCullough PA, Khandelwal AK, McKinnon JE, et al. Outcomes and prognostic factors of systolic as compared with diastolic heart failure in urban America. Congest Heart Fail. 2005;11:6–11.
McDonald K. Diastolic heart failure in the elderly: underlying mechanisms and clinical relevance. Int J Cardiol. 2008;125:197–202.
Jugdutt BI. Extracellular matrix and cardiac remodeling. In: Villarreal FJ, editor. Interstitial fibrosis in heart failure. New York, NY: Springer; 2004. p. 23–55.
Jugdutt BI. Regulation of fibrosis after myocardial infarction: implications for ventricular remodeling. In: Jugdutt BI, Dhalla NS, editors. Cardiac remodeling. Molecular mechanisms. New York, NY: Springer; 2013. p. 525–45.
Jugdutt BI. Angiotensin II, receptor blockers. In: Crawford MH, editor. Cardiology clinics annual of drug therapy, vol. 2. Philadelphia, PA: W.B. Saunders; 1998. p. 1–17.
Dzau VJ. Tissue renin-angiotensin system in myocardial hypertrophy and failure. Arch Intern Med. 1993;153:937–42.
Dzau VJ. Theodore Cooper lecture: tissue angiotensin and pathobiology of vascular disease: a unifying hypothesis. Hypertension. 2001;37:1047–52.
Kumar R, Thomas CM, Yong QC, Chen W, Baker KM. The intracrine renin-angiotensin system. Clin Sci (Lond). 2012;123:273–84.
de Gasparo M, Levens N. Does blockade of angiotensin II receptors offer clinical benefits over inhibition of angiotensin-converting enzyme? Pharmacol Toxicol. 1998;82:257–71.
Opie LH, Sack MN. Enhanced angiotensin II activity in heart failure: reevaluation of the counterregulatory hypothesis of receptor subtypes. Circ Res. 2001;88:654–8.
Drexler H. Endothelial dysfunction in heart failure and potential for reversal by ACE inhibition. Br Heart J. 1994;72(3 Suppl):S11–4.
Seyedi N, Xu X, Nasjletti A, et al. Coronary kinin generation mediates nitric oxide release after angiotensin receptor stimulation. Hypertension. 1995;26:164–70.
Liu YH, Yang XP, Sharov VG, et al. Effects of angiotensin-converting enzyme inhibitors and angiotensin II type 1 receptor antagonists in rats with heart failure. Role of kinins and angiotensin II type 2 receptors. J Clin Invest. 1997;99:1926–35.
Liu YH, Yang XP, Shesely EG, Sankey SS, Carretero OA. Role of angiotensin II type 2 receptors and kinins in the cardioprotective effect of angiotensin II type 1 receptor antagonists in rats with heart failure. J Am Coll Cardiol. 2004;43:1473–80.
Messadi-Laribi E, Griol-Charhbill V, Pizard A, et al. Tissue kallikrein is involved in the cardioprotective effect of AT1-receptor blockade in acute myocardial ischemia. J Pharmacol Exp Ther. 2007;323:210–6.
Xu Y, Menon V, Jugdutt BI. Cardioprotection after angiotensin II type 1 blockade involves angiotensin II type 2 receptor expression and activation of protein kinase C-epsilon in acutely reperfused myocardial infarction in the dog. Effect of UP269-6 and losartan on AT1 and AT2-receptor expression and IP3 receptor and PKCε proteins. J Renin Angiotensin Aldosterone Syst. 2000;1:184–95.
Jugdutt BI, Balghith M. Enhanced regional AT2-receptor and PKCε expression during cardioprotection induced by AT1-receptor blockade after reperfused myocardial infarction. J Renin Angiotensin Aldosterone Syst. 2001;2:134–40.
Jugdutt BI, Menon V. AT1 receptor blockade limits myocardial injury and upregulates AT2 receptors during reperfused myocardial infarction. Mol Cell Biochem. 2004;260:111–8.
Jugdutt BI, Menon V. Valsartan-induced cardioprotection involves angiotensin II type 2 receptor upregulation in dog and rat in vivo models of reperfused myocardial infarction. J Cardiac Fail. 2004;10:74–82.
Rhaleb N-E, Yang X-P, Carretero OA. The kallikrein‐kinin system as a regulator of cardiovascular and renal function. Compr Physiol. 2011;1:971–93.
Urata H, Kinoshita A, Misono KS, Bumpus FM, Husain A. Identification of a highly specific chymase as the major angiotensin II-forming enzyme in the human heart. J Biol Chem. 1990;265:22348–57.
Urata H, Healy B, Stewart RW, Bumpus FM, Husain A. Angiotensin II-forming pathways in normal and failing human hearts. Circ Res. 1990;66:883–90.
Kawamura M, Imanashi M, Matsushima Y, et al. Circulating angiotensin II levels under repeated administration of lisinopril in normal subjects. Clin Exp Pharmacol Physiol. 1992;19:547–53.
Jorde UP, Ennezat PV, Lisker J, et al. Maximally recommended doses of angiotensin-converting enzyme (ACE) inhibitors do not completely prevent ACE-mediated formation of angiotensin II in chronic heart failure. Circulation. 2000;101:844–6.
Wolny A, Clozel JP, Rein J, et al. Functional and biochemical analysis of angiotensin II-forming pathways in the human heart. Circ Res. 1997;80:219–27.
Azizi M, Menard J. Combined blockade of the renin-angiotensin system with angiotensin-converting enzyme inhibitors and angiotensin II type 1 receptor antagonists. Circulation. 2004;109:2492–9.
Spinale FG, de Gasparo M, Whitebread S, et al. Modulation of the renin-angiotensin pathway through enzyme inhibition and specific receptor blockade in pacing-induced heart failure: I. Effects on left ventricular performance and neurohormonal systems. Circulation. 1997;96:2385–96.
Hamroff G, Katz SD, Mancini D, et al. Addition of angiotensin II receptor blockade to maximal angiotensin-converting enzyme inhibition improves exercise capacity in patients with severe congestive heart failure. Circulation. 1999;99:990–2.
Yu CM, Tipoe GL, Wing-Hon Lai K, et al. Effects of combination of angiotensin-converting enzyme inhibitor and angiotensin receptor antagonist on inflammatory cellular infiltration and myocardial interstitial fibrosis after acute myocardial infarction. J Am Coll Cardiol. 2001;38:1207–15.
Forteza R, Lauredo I, Abraham WM, Conner GE. Bronchial tissue kallikrein activity is regulated by hyaluronic acid binding. Am J Respir Cell Mol Biol. 1999;21:666–74.
Hara M, Ono K, Hwang MW, et al. Evidence for a role of mast cells in the evolution to congestive heart failure. J Exp Med. 2002;195:375–81.
Matsumoto T, Wada A, Tsutamoto T, et al. Chymase inhibition prevents cardiac fibrosis and improves diastolic dysfunction in the progression of heart failure. Circulation. 2003;107:2555–8.
Jin D, Takai S, Sakaguchi M, Okamoto Y, Muramatsu M, Miyazaki M. An antiarrhythmic effect of a chymase inhibitor after myocardial infarction. J Pharmacol Exp Ther. 2004;309:490–7.
Jin D, Takai S, Yamada M, et al. Impact of chymase inhibitor on cardiac function and survival after myocardial infarction. Cardiovasc Res. 2003;60:413–20.
Matsumoto C, Hayashi T, Kitada K, et al. Chymase plays an important role in left ventricular remodeling influenced by intermittent hypoxia in mice. Hypertension. 2009;54:164–71.
Oyamada S, Bianchi C, Takai S, Chu LM, Selke FW. Chymase inhibition reduces infarction and matrix metalloproteinase-9 activation and attenuates inflammation and fibrosis after acute myocardial ischemia/reperfusion. J Pharmacol Exp Ther. 2011;339:143–51.
Wei CC, Hase N, Inoue Y, et al. Mast cell chymase limits the cardiac efficacy of Ang I-converting enzyme inhibitor therapy in rodents. J Clin Invest. 2010;120:1229–39.
Pat B, Chen Y, Killingsworth C, Gladden JD, et al. Chymase inhibition prevents fibronectin and myofibrillar loss and improves cardiomyocyte function and LV torsion angle in dogs with isolated mitral regurgitation. Circulation. 2011;122:1488–95.
Okumura K, Takai S, Muramatsu M, et al. Human chymase degrades human fibronectin. Clin Chim Acta. 2004;347:223–5.
Hoshino F, Urata H, Inoue Y, et al. Chymase inhibitor improves survival in hamsters with myocardial infarction. J Cardiovasc Pharmacol. 2003;41 Suppl 1:S11–8.
Ihara M, Urata H, Shirai K, et al. High cardiac angiotensin-II-forming activity in infarcted and non-infarcted human myocardium. Cardiology. 2000;94:247–53.
Ihara M, Urata H, Kinoshita A, et al. Increased chymase-dependent angiotensin II formation in human atherosclerotic aorta. Hypertension. 1999;33:1399–405.
Arakawa K, Urata H. Hypothesis regarding the pathophysiological role of alternative pathways of angiotensin II formation in atherosclerosis. Hypertension. 2000;36:638–41.
Uehara Y, Urata H, Sasaguri M, et al. Increased chymase activity in internal thoracic artery of patients with hypercholesterolemia. Hypertension. 2000;35:55–60.
Uehara Y, Urata H, Ideishi M, Arakawa K, Saku K. Chymase inhibition suppresses high-cholesterol diet-induced lipid accumulation in the hamster aorta. Cardiovasc Res. 2002;55:870–6.
Koka V, Wang W, Huang XR, et al. Advanced glycation end products activate a chymase-dependent angiotensin II-generating pathway in diabetic complications. Circulation. 2006;113:1353–60.
Singh VP, Baker KM, Kumar R. Activation of the intracellular renin angiotensin system in cardiac fibroblasts by high glucose: role in extracellular matrix production. Am J Physiol. 2008;294:H1675–84.
New RB, Sampson AC, King MK, et al. Effects of combined angiotensin II and endothelin receptor blockade with developing heart failure: effects on left ventricular performance. Circulation. 2000;102:1447–53.
Rossi GP, Sacchetto A, Cesari M, Pessina AC. Interactions between endothelin-1 and the renin-angiotensin-aldosterone system. Cardiovasc Res. 1999;43:300–7.
Luscher TF, Barton M. Endothelins and endothelin receptor antagonists: therapeutic considerations for a novel class of cardiovascular drugs. Circulation. 2000;102:2434–40.
Teerlink JR. Endothelins: pathophysiology and treatment implications in chronic heart failure. Curr Heart Fail Rep. 2005;2:191–7.
Schirger JA, Chen HH, Jougasaki M, et al. Endothelin A receptor antagonism in experimental congestive heart failure results in augmentation of the renin-angiotensin system and sustained sodium retention. Circulation. 2004;109:249–54.
Anand I, McMurray J, Cohn JN, et al. Long-term effects of darusentan on left-ventricular remodelling and clinical outcomes in the Endothelin A Receptor Antagonist Trial in Heart Failure (EARTH): randomised, double-blind, placebo-controlled trial. Lancet. 2004;364:347–54.
Moraes DL, Colucci WS, Givertz MM. Secondary pulmonary hypertension in chronic heart failure: the role of the endothelium in pathophysiology and management. Circulation. 2000;102:1718–23.
Jiang BH, Tardif J-C, Shi Y, Dupuis J. Bosentan does not improve pulmonary hypertension and lung remodeling in heart failure. Eur Respir J. 2011;37:578–86.
Fang JC, DeMarco T, Givertz MM, et al. World Health Organization Pulmonary Hypertension group 2: pulmonary hypertension due to left heart disease in the adult – a summary statement from the Pulmonary Hypertension Council of the International Society for heart and Lung Transplantation. J Heart Lung Transplant. 2012;31:913–33.
Tipnis SR, Hooper NM, Hyde R, et al. A human homolog of angiotensin-converting enzyme. Cloning and functional expression as a captopril-insensitive carboxypeptidase. J Biol Chem. 2000;275:33238–43.
Donoghue M, Hsieh F, Baronas E, et al. A novel angiotensin-converting enzyme-related carboxypeptidase (ACE2) converts angiotensin I to angiotensin 1-9. Circ Res. 2000;87:E1–9.
Crackower MA, Sarao R, Oudit GY, et al. Angiotensin-converting enzyme 2 as an essential regulator of heart function. Nature. 2002;417:822–8.
Ferrario CM, Trask AJ, Jessup JA. Advances in biochemical and functional roles of angiotensin-converting enzyme 2 and angiotensin-(1-7) in regulation of cardiovascular function. Am J Physiol. 2005;289:H2281–90.
Iwata M, Cowling RT, Gurantz D, et al. Angiotensin-(1-7) binds to specific receptors on cardiac fibroblasts to initiate antifibrotic and antitrophic effects. Am J Physiol. 2005;289:H2356–63.
Ferrario CM, Jessup J, Chappell MC, et al. Effect of angiotensin-converting enzyme inhibition and angiotensin II receptor blockers on cardiac angiotensin-converting enzyme 2. Circulation. 2005;111:2605–10.
Ishiyama Y, Gallagher PE, Averill DB, et al. Upregulation of angiotensin-converting enzyme 2 after myocardial infarction by blockade of angiotensin II receptors. Hypertension. 2004;43:970–6.
Loot AE, Roks AJ, Henning RH, et al. Angiotensin-(1-7) attenuates the development of heart failure after myocardial infarction in rats. Circulation. 2002;105:1548–50.
Ocaranza MP, Godoy I, Jalil JE, et al. Enalapril attenuates downregulation of angiotensin-converting enzyme 2 in the late phase of ventricular dysfunction in myocardial infarcted rat. Hypertension. 2006;48:572–8.
Zhong JC, Ye JY, Jin HY, et al. Telmisartan attenuates aortic hypertrophy in hypertensive rats by the modulation of ACE2 and profiling-1 expression. Regul Pept. 2011;166:90–7.
Jugdutt B, Palaniyappan A, Idikio H. Role of ACE2 and Ang (1-7) in limiting fibrosis and remodeling during healing after reperfused myocardial infarction. J Mol Cell Cardiol. 2009;57:S24 (Abstract).
Cowling RT, Goldberg BH. The ACE2/Ang-(1-7) pathway in cardiac fibroblasts as a potential target for cardiac remodeling. In: Jugdutt BI, Dhalla NS, editors. Cardiac remodeling. Molecular mechanisms. New York, NY: Springer; 2013. p. 547–57.
Wang W, Bodiga S, Das SK, et al. Role of ACE2 in diastolic and systolic heart failure. Heart Fail Rev. 2012;17:683–9.
Zisman LS, Keller RS, Weaver B, et al. Increased angiotensin-(1-7)-forming activity in failing human heart ventricles: evidence for upregulation of the angiotensin-converting enzyme Homologue ACE2. Circulation. 2003;108:1707–12.
Zhao YX, Yin HQ, Yu QT, et al. ACE2 overexpression ameliorates left ventricular remodeling and dysfunction in a rat model of myocardial infarction. Hum Gene Ther. 2010;21:1545–54.
Epelman S, Shrestha K, Troughton RW, et al. Soluble angiotensin-converting enzyme 2 in human heart failure: relation with myocardial function and clinical outcomes. J Card Fail. 2009;15:565–71.
Wang Y, Moreira Mda C, Heringer-Walther S, et al. Plasma ACE2 activity is an independent prognostic marker in Chagas’ disease and equally potent as BNP. J Card Fail. 2010;16:157–63.
Xie X, Chen J, Wang X, et al. Age- and gender-related difference of ACE2 expression in a rat lung. Life Sci. 2006;78:2166–71.
Yousif MH, Kehinde EO, Benter IF. Different responses to angiotensin-(1-7) in young, aged and diabetic rabbit corpus cavernosum. Pharmacol Res. 2007;56:209–16.
Palaniyappan A, Idikio H, Jugdutt BI. Effect of age on expression of AT1 and AT2 receptors and ACE-2 and Angiotensin (1-7), Ac-SDKP and Smad-2 proteins after acute reperfused ST-segment myocardial infarction. Circulation. 2008;118 Suppl 2:S547 (Abstract).
Tom B, de Vries R, Saxena PR, et al. Bradykinin potentiation by angiotensin-(1-7) and ACE inhibitors correlates with ACE C- and N-domain blockade. Hypertension. 2001;38:95–9.
Campbell DJ, Krum H, Esler MD. Losartan increases bradykinin levels in hypertensive humans. Circulation. 2005;111:315–20.
Nagata S, Kato J, Sasaki K, et al. Isolation and identification of proangiotensin-12, a possible component of the renin-angiotensin system. Biochem Biophys Res Commun. 2006;350:1026–31.
Jessup JA, Trask AJ, Chappell MC, et al. Localization of the novel angiotensin peptide, angiotensin-(1-12), in heart and kidney of hypertensive and normotensive rats. Am J Physiol. 2008;294:H2614–8.
Trask AJ, Jessup JA, Chappell MC, Ferrario CM. Angiotensin-(1-12) is an alternate substrate for angiotensin peptide production in the heart. Am J Physiol. 2008;294:H2242–7.
Ferrario C, Varagic J, Hanini J, et al. Differential regulation of angiotensin-(1-12) in plasma and cardiac tissue in response to bilateral nephrectomy. Am J Physiol. 2009;296:H1184–92.
Prosser HC, Forster ME, Richards AM, Pemberton CJ. Cardiac chymase converts rat proAngiotensin-12 (PA12) to angiotensin II: effects of PA12 upon cardiac haemodynamics. Cardiovasc Res. 2009;82:40–50.
Ahmad S, Simmons T, Varagic J, et al. Chymase-dependent generation of angiotensin II from angiotensin-(1-12) in human atrial tissue. PLoS One. 2011;6:e28501.
Ahmad S, Wei CC, Tallaj J, et al. Chymase mediates angiotensin-(1-12) metabolism in human hearts. J Am Soc Hypertens. 2013;7:128–36.
Moniwa N, Wei C-C, dell’Italia LJ, et al. Chymase-mediated angiotensin II generation from angiotensin-(1-12) in left ventricular tissue of normal and diseased human subjects. J Clin Hypertens (Greenwich). 2012;14 Suppl 1:149 (Abstract).
The CONSENSUS Trial Study Group. Effects of enalapril on mortality in severe congestive heart failure: results of the Cooperative North Scandinavian Enalapril Survival Study (CONSENSUS). N Engl J Med. 1987;316:1429–35.
Giles TD, Katz R, Sullivan JM, et al. Short- and long-acting angiotensin-converting enzyme inhibitors: a randomized trial of lisinopril versus captopril in the treatment of congestive heart failure. The Multicenter Lisinopril-Captopril Congestive Heart Failure Study Group. J Am Coll Cardiol. 1989;13:1240–7.
The SOLVD Investigators. Effect of enalapril on survival in patients with reduced left ventricular ejection fractions and congestive heart failure. N Engl J Med. 1991;325:293–302.
The SOLVD Investigators. Effect of enalapril on mortality and the development of heart failure in asymptomatic patients with reduced left ventricular ejection fractions. N Engl J Med. 1992;327:685–91.
Ryden L, Armstrong PW, Cleland JG, et al. Efficacy and safety of high-dose lisinopril in chronic heart failure patients at high cardiovascular risk, including those with diabetes mellitus. Results from the ATLAS trial. Eur Heart J. 2000;21:1967–78.
Cleland JG, Tendera M, Adamus J, The PEPCHF Investigators, et al. The perindopril in elderly people with chronic heart failure (PEP-CHF) study. Eur Heart J. 2006;27:2338–45.
Pitt B, Segal R, Martinez FA, et al. Randomised trial of losartan versus captopril in patients over 65 with heart failure (Evaluation of Losartan in the Elderly Study, ELITE). Lancet. 1997;349:747–52.
McKelvie RS, Yusuf S, Pericak D, et al. Comparison of candesartan, enalapril, and their combination in congestive heart failure: randomized evaluation of strategies for left ventricular dysfunction (RESOLVD) pilot study. The RESOLVD Pilot Study Investigators. Circulation. 1999;100:1056–64.
Pitt B, Poole-Wilson PA, Segal R, et al. Effect of losartan compared with captopril on mortality in patients with symptomatic heart failure: randomised trial−the Losartan Heart Failure Survival Study ELITE II. Lancet. 2000;355:1582–7.
Cohn JN, Tognoni G, Valsartan Heart Failure Trial Investigators. A randomized trial of angiotensin receptor blocker valsartan in chronic heart failure. N Engl J Med. 2001;345:1667–75.
Pfeffer MA, Swedberg K, Granger CB, et al. CHARM Investigators and Committees. Effects of candesartan on mortality and morbidity in patients with chronic heart failure: the CHARM-Overall programme. Lancet. 2003;362:759–66.
McMurray JJ, Ostergren J, Swedberg K, et al. CHARM Investigators and Committees. Effects of candesartan in patients with chronic heart failure and reduced left-ventricular systolic function taking angiotensin-converting-enzyme inhibitors: the CHARM-Added trial. Lancet. 2003;362:767–71.
Granger CB, McMurray JJ, Yusuf S, et al. CHARM Investigators and Committees. Effects of candesartan in patients with chronic heart failure and reduced left-ventricular systolic function intolerant to angiotensin-converting-enzyme inhibitors: the CHARM-Alternative trial. Lancet. 2003;362:772–6.
Yusuf S, Pfeffer MA, Swedberg K, et al. CHARM Investigators and Committees. Effects of candesartan in patients with chronic heart failure and preserved left-ventricular ejection fraction: the CHARM-preserved trial. Lancet. 2003;362:777–81.
White HD, Aylward PE, Huang Z, et al. Mortality and morbidity remain high despite captopril and/or valsartan therapy in elderly patients with left ventricular systolic dysfunction, heart failure, or both after acute myocardial infarction: results of the Valsartan in Acute Myocardial Infarction Trial (VALIANT). Circulation. 2005;112:3391–9.
Massie BM, Carson PE, McMurray JJ, et al. I-PRESERVE Investigators. Irbesartan in patients with heart failure and preserved ejection fraction. N Engl J Med. 2008;359:2456–67.
Timmermans PB, Carini DJ, Chiu AT, et al. The discovery of a new class of highly specific nonpeptide angiotensin II receptor antagonists. Am J Hypertens. 1991;4:275S–81.
Buhler FR, Laragh JH, Baer L, et al. Propranolol inhibition of renin secretion. A specific approach to diagnosis and treatment of renin-dependent hypertensive diseases. N Engl J Med. 1972;287:1209–14.
Campbell DJ, Aggarwal A, Esler M, et al. β-blockers, angiotensin II, and ACE inhibitors in patients with heart failure. Lancet. 2001;358:1609–10.
Sharpe N. Benefit of beta-blockers for heart failure: proven in 1999. Lancet. 1999;353:1988–9.
Benz J, Oshrain C, Henry D, et al. Valsartan, a new angiotensin II receptor antagonist: a double-blind study comparing the incidence of cough with lisinopril and hydrochlorothiazide. J Clin Pharmacol. 1997;37:101–7.
Howes LG, Tran D. Can angiotensin receptor antagonists be used safely in patients with previous ACE inhibitor-induced angioedema? Drug Saf. 2002;25:73–6.
Pfeffer MA, McMurray JJ, Velazquez EJ, et al. Valsartan in Acute Myocardial Infarction Trial Investigators. Valsartan, captopril, or both in myocardial infarction complicated by heart failure, left ventricular dysfunction, or both. N Engl J Med. 2003;349:1893–906.
Chobanian AV, Bakris GL, Black HR, et al. Joint National Committee on Prevention, Detection, Evaluation, and Treatment of High Blood Pressure. National Heart, Lung, and Blood Institute; National High Blood Pressure Education Program Coordinating Committee. Seventh report of the Joint National Committee on prevention, detection, evaluation, and treatment of high blood pressure. Hypertension. 2003;42:1206–52.
Chobanian AV, Bakris GL, Black HR, et al. National Heart, Lung, and Blood Institute Joint National Committee on Prevention, Detection, Evaluation, and Treatment of High Blood Pressure; National High Blood Pressure Education Program Coordinating Committee. The seventh report of the Joint National Committee on prevention, detection, evaluation, and treatment of high blood pressure: the JNC 7 report. JAMA. 2003;289:2560–72.
Rosendorff C, Black HR, Cannon CP, et al. American Heart Association Council for High Blood Pressure Research; American Heart Association Council on Clinical Cardiology; American Heart Association Council on Epidemiology and Prevention. Treatment of hypertension in the prevention and management of ischemic heart disease: a scientific statement from the American Heart Association Council for High Blood Pressure Research and the Councils on Clinical Cardiology and Epidemiology and Prevention. Circulation. 2007;115:2761–88.
Mancia G, De Backer G, Dominiczak A, et al. Guidelines for the management of arterial hypertension: the task force for the management of arterial hypertension of the European Society of Hypertension (ESH) and of the European Society of Cardiology (ESC). Eur Heart J. 2007;28:1462–536.
Pfeffer MA, Braunwald E, Moyé LA, et al. Effect of captopril on mortality and morbidity in patients with left ventricular dysfunction after myocardial infarction. Results of the survival and ventricular enlargement trial. The SAVE Investigators. N Engl J Med. 1992;327:669–77.
Fox KM, The EURopean trial On reduction of cardiac events with Perindopril in stable coronary Artery disease Investigators. Efficacy of perindopril in reduction of cardiovascular events among patients with stable coronary artery disease: randomised, double-blind, placebo-controlled, multicentre trial (the EUROPA study). Lancet. 2003;362:782–8.
Pitt B, O’Neill B, Feldman R, et al. QUIET Study Group. The QUinapril Ischemic Event Trial (QUIET): evaluation of chronic ACE inhibitor therapy in patients with ischemic heart disease and preserved left ventricular function. Am J Cardiol. 2001;87:1058–63.
Braunwald E, Domanski MJ, Fowler SE, et al. PEACE Trial Investigators. Angiotensin-converting-enzyme inhibition in stable coronary artery disease. N Engl J Med. 2003;362:782–8.
Dagenais GR, Pogue J, Fox K, Simoons ML, Yusuf S. Angiotensin-converting-enzyme inhibitors in stable vascular disease without left ventricular systolic dysfunction or heart failure: a combined analysis of three trials. Lancet. 2006;368:581–8.
Heart Outcomes Prevention Evaluation Study Investigators. Effects of ramipril on cardiovascular and microvascular outcomes in people with diabetes mellitus: results of the HOPE study and MICRO-HOPE substudy. Lancet. 2000;355:253–9.
Yusuf S, Sleight P, Pogue J, et al. HOPE/HOPE-TOO Study Investigators. Effects of an angiotensin-converting-enzyme inhibitor, ramipril, on cardiovascular events in high-risk patients. The Heart Outcomes Prevention Evaluation Study Investigators. N Engl J Med. 2000;342:145–53.
Lewis EJ, Hunsicker LG, Bain RP, Rohde RD. The effect of angiotensin-converting-enzyme inhibition on diabetic nephropathy. The Collaborative Study Group. N Engl J Med. 1993;329:1456–62.
Bosch J, Lonn E, Pogue J, et al. HOPE/HOPE_TOO Study Investigators. Long-term effects of ramipril on cardiovascular events and on diabetes: results of the HOPE study extension. Circulation. 2005;112:1339–46.
Patel A, MacMahon S, Chalmers J, et al. ADVANCE Collaborative Group. Effects of a fixed combination of perindopril and indapamide on macrovascular and microvascular outcomes in patients with type 2 diabetes mellitus (the ADVANCE trial): a randomised controlled trial. Lancet. 2007;370:829–40.
Kjeldsen SE, Lyle PA, Tershakovec AM, et al. Targeting the renin-angiotensin system for the reduction of cardiovascular outcomes in hypertension: angiotensin-converting enzyme inhibitors and angiotensin receptor blockers. Expert Opin Emerg Drugs. 2005;10:729–45.
Strauss MH, Hall AS. Angiotensin receptor blockers may increase risk of myocardial infarction: unraveling the ARB-MI paradox. Circulation. 2006;114:838–54.
Tikkanen I, Omvik P, Jensen HA. Comparison of the angiotensin II antagonist losartan with the angiotensin converting enzyme inhibitor enalapril in patients with essential hypertension. J Hypertens. 1995;13:1343–51.
Holwerda NJ, Fogari R, Angeli P, et al. Valsartan, a new angiotensin II antagonist for the treatment of essential hypertension: efficacy and safety compared with placebo and enalapril. J Hypertens. 1996;14:1147–51.
Chan P, Tomlinson B, Huang TY, et al. Double-blind comparison of losartan, lisinopril, and metolazone in elderly hypertensive patients with previous angiotensin-converting enzyme inhibitor-induced cough. J Clin Pharmacol. 1997;37:253–7.
Dahlof B, Devereux RB, Kjeldsen SE, et al. LIFE Study Group. Cardiovascular morbidity and mortality in the Losartan Intervention For Endpoint reduction in hypertension study (LIFE): a randomised trial against atenolol. Lancet. 2002;359:995–1003.
Dickstein K, Kjekshus J, OPTIMAAL Steering Committee of the OPTIMAAL Study Group. Effects of losartan and captopril on mortality and morbidity in high-risk patients after acute myocardial infarction: the OPTIMAAL randomised trial. Optimal Trial in Myocardial Infarction with Angiotensin II Antagonist Losartan. Lancet. 2002;360:752–60.
Volpe M, Mancia G, Trimarco B. Angiotensin II receptor blockers and myocardial infarction: deeds and misdeeds. J Hypertens. 2005;23:2113–8.
Yusuf S, Teo KK, Pogue J, et al. ONTARGET Investigators. Telmisartan, ramipril, or both in patients at high risk for vascular events. N Engl J Med. 2008;358:1547–59.
Yusuf S, Diener HC, Sacco RL, et al. PROFESS Study Group. Telmisartan to prevent recurrent stroke and cardiovascular events. N Engl J Med. 2008;359:1225–37.
Yusuf S, Teo K, Anderson C, et al. Telmisartan Randomised AssessmeNt Study in ACE iNtolerant subjects with cardiovascular Disease (TRANSCEND) Investigators. Effects of the angiotensin-receptor blocker telmisartan on cardiovascular events in high-risk patients intolerant to angiotensin-converting enzyme inhibitors: a randomised controlled trial. Lancet. 2008;372:1174–83.
Verdecchia P, Sleight P, Mancia G, et al. ONTARGET/TRANSCEND Investigators. Effects of telmisartan, ramipril, and their combination on left ventricular hypertrophy in individuals at high vascular risk in the Ongoing Telmisartan Alone and in Combination With Ramipril Global End point Trial and the Telmisartan Randomized Assessment Study in ACE Intolerant Subjects With Cardiovascular Disease. Circulation. 2009;120:1380–9.
Shivakumar K, Dostal DE, Boheler K, et al. Differential response of cardiac fibroblasts from young and senescent rats to ANG II. Am J Physiol Heart Circ Physiol. 2003;284:H1454–9.
Basso N, Cini R, Pietrelli A, et al. Protective effect of long-term angiotensin II inhibition. Am J Physiol Heart Circ Physiol. 2007;293:H1351–8.
Lewis EF, Moye LA, Rouleau JL, et al. Predictors of late development of heart failure in stable survivors of myocardial infarction: the CARE study. J Am Coll Cardiol. 2003;42:1446–53.
St John Sutton M, Pfeffer MA, Moye L, et al. Cardiovascular death and left ventricular remodeling two years after myocardial infarction: baseline predictors and impact of long-term use of captopril: information from the Survival and Ventricular Enlargement (SAVE) trial. Circulation. 1997;96:3294–9.
Maggioni AP, Maseri A, Fresco C, et al. Age-related increase in mortality among patients with first myocardial infarctions treated with thrombolysis. The investigators of the gruppo Italiano per lo Studio della supravvivenza nell’Infarcto Miocardico (GISSI-2). N Engl J Med. 1993;329:1442–8.
Jugdutt BI. Prevention of ventricular remodeling after myocardial infarction and in congestive heart failure. Heart Fail Rev. 1996;1:115–29.
Jugdutt BI. Ventricular remodeling post-infarction and the extracellular collagen matrix. When is enough enough? Circulation. 2003;108:1395–403.
Jugdutt BI. Remodeling of the myocardium and potential targets in the collagen degradation and synthesis pathways. Curr Drug Targets Cardiovasc Haematol Disord. 2003;3:1–30.
Kim CB, Braunwald E. Potential benefits of late reperfusion of infarcted myocardium. The open artery hypothesis. Circulation. 1993;88:2426–36.
Bolognese L, Neskovic AN, Parodi G, et al. Left ventricular remodeling after primary coronary angioplasty: patterns of left ventricular dilation and long-term prognostic implications. Circulation. 2002;106:2351–7.
Bolognese L, Carrabba N, Parodi G, et al. Impact of microvascular dysfunction on left ventricular remodeling and long-term clinical outcome after primary coronary angioplasty for acute myocardial infarction. Circulation. 2004;109:1121–6.
Ferrari R, for the PREAMI Investigators. Effects of angiotensin-converting enzyme inhibition with peridopril on left ventricular remodeling and clinical outcome. Results of the randomized Perindopril and Remodeling Elderly with Acute Myocardial Infarction (PREAMI) study. Arch Intern Med. 2006;166:659–66.
Jugdutt BI, Palaniyappan A, Uwiera RRE, Idikio H. Role of healing-specific-matricellular proteins and matrix metalloproteinases in age-related enhanced early remodeling after reperfused STEMI in dogs. Mol Cell Biochem. 2009;322:25–36.
Palaniyappan A, Idikio H, Jugdutt BI. Secretory leucocyte protease inhibitor and matricellular protein modulation of post reperfused myocardial infarction healing, fibrosis and remodeling in rat model. Effect of candesartan and omapatrilat. Circulation. 2009;120 Suppl 2:S837 (Abstract).
Carey RM. Angiotensin receptors and aging. Hypertension. 2007;50:33–4.
Pinaud F, Bocquet A, Dumont O, et al. Paradoxical role of angiotensin II type 2 receptors in resistance arteries of old rats. Hypertension. 2007;50:96–102.
Savoia C, Touyz RM, Volpe M, Schiffrin EL. Angiotensin type 2 receptor in resistance arteries of type 2 diabetic hypertensive patients. Hypertension. 2007;49:341–6.
Chen W, Frangogiannis NG. The role of inflammatory and fibrogenic pathways in heart failure associated with aging. Heart Fail Rev. 2010;15:415–22.
Ho D, Yan L, Iwatsubo K, Vatner DE, Varner SF. Modulation of β-adrenergic receptor signaling in heart failure and longevity: targeting adenyl cyclase type 5. Heart Fail Rev. 2010;15:495–512.
Pitt B. The role of mineralocorticoid receptor antagonists (MRAs) in very old patients with heart failure. Heart Fail Rev. 2012;17:573–9.
Cruickshank JM, Thorp JM, Zacharias FJ. Benefits and potential harm of lowering high blood pressure. Lancet. 1987;1(8533):581–4.
Jugdutt BI. Intravenous nitroglycerin unloading in acute myocardial infarction. Am J Cardiol. 1991;68(14):52D–63.
Verma S, Strauss M. Angiotensin receptor blockers and myocardial infarction. BMJ. 2004;329:1248–9.
Thomas GN, Chan P, Tomlinson B. The role of angiotensin II type 1 receptor antagonists in elderly patients with hypertension. Drugs Aging. 2006;23:131–55.
Pitt B, Zannad F, Remme WJ, et al. The effect of spironolactone on morbidity and mortality in patients with severe heart failure. Randomized Aldactone Evaluation Study Investigators. N Engl J Med. 1999;341:709–17.
Zannad F, Alla F, Dousset B, Perez A, Pitt B. Limitation of excessive extracellular matrix turnover may contribute to survival benefit of spironolactone therapy in patients with congestive heart failure: insights from the randomized aldactone evaluation study (RALES). Rales Investigators. Circulation. 2000;102:2700–6.
Pitt B, Remme W, Zannad F, et al. Eplerenone Post-Acute Myocardial Infarction Heart Failure Efficacy and Survival Study Investigators. Eplerenone, a selective aldosterone blocker, in patients with left ventricular dysfunction after myocardial infarction. N Engl J Med. 2003;348:1309–21.
Pitt B, White H, Nicolau J, et al. EPHESUS Investigators. Eplerenone reduces mortality 30 days after randomization following acute myocardial infarction in patients with left ventricular systolic dysfunction and heart failure. J Am Coll Cardiol. 2005;46:425–31.
Mak GJ, Ledwidge MT, Watson CJ, et al. Natural history of markers of collagen turnover in patients with early diastolic dysfunction and impact of eplerenone. J Am Coll Cardiol. 2009;54:1674–82.
Zannad F, McMurray JJ, Krum H, et al. Eplerenone in patients with systolic heart failure and mild symptoms. N Engl J Med. 2011;364:11–21.
Li X, Qi Y, Li Y, et al. Impact of mineralocorticoid receptor antagonists on changes in cardiac structure and function of left ventricular dysfunction. A meta-analysis of randomized controlled trials. Circ Heart Fail. 2013;6:156–65.
Weidmann P, de Myttenaere-Bursztein S, Maxwell MK, de Lima J. Effect of aging on plasma renin and aldosterone in normal man. Kidney Int. 2008;8:325–33.
Henschkowski J, Stuck AE, Frey BM, et al. Age-dependent decrease 11 beta-hydroxysteroid dehydrogenase type 2 (11 beta-HSD2) activity in hypertensive patients. Am J Hypertens. 2008;21:644–9.
Funder JW, Pearce P, Smith R, Smith AL. Mineralocorticoid action: target–tissue specificity is enzyme, not receptor mediated. Science. 1988;242:583–5.
Edwards CR, Stewart PM, Burt D, et al. Localization of 11 beta-hydroxysteroid dehydrogenase-tissue specific receptor of the mineralocorticoid receptor. Lancet. 1988;2:986–9.
Funder JW. Rales, ephesus and redox. J Steroid Biochem Mol Biol. 2005;93:121–5.
Funder WF. Reconsidering the roles of the mineralocorticoid receptor. Hypertension. 2008;53(Pt 2):286–90.
Bocchi B, Kenouch S, Lamarre-Cliche M, et al. Impaired 11-beta hydroxysteroid dehydrogenase type 2 activity in sweat gland ducts in human essential hypertension. Hypertension. 2004;43:803–8.
Chai W, Danser AHJ. Why are mineralocorticoid receptor antagonists cardioprotective? Naunyn Schmiedebergs Arch Pharmacol. 2006;374:153–62.
Krug AW, Allenhofer L, Monticone R, et al. Elevated mineralocorticoid receptor activity in aged rat vascular smooth muscle cells promotes a proinflammatory phenotype via extracellular signal-regulated kinase 1/2 mitogen-activated protein kinase and epidermal growth factor receptor-dependent pathways. Hypertension. 2010;55:1476–83.
Vasan RS, Demisse S, Kimura M, et al. Association of leucocyte telomere length with circulating biomarkers of the renin-angiotensin-aldosterone system: the Framingham Heart Study. Circulation. 2008;117:1138–44.
Benetos A, Gardner JP, Kimura M, et al. Aldosterone and telomere length in white blood cells. J Gerontol A Biol Sci Med Sci. 2005;60:1593–6.
Pitt B, Reichek N, Willenbrock R, et al. Effects of eplerenone, enalapril, and eplerenone/enalapril in patients with essential hypertension and left ventricular hypertrophy: the 4E-left ventricular hypertrophy study. Circulation. 2003;108:1831–8.
Mottram PM, Haluska B, Leano R, et al. Effect of aldosterone antagonism on myocardial dysfunction in hypertensive patients with diastolic heart failure. Circulation. 2004;110:558–65.
Savoia C, Touyz RM, Amiri F, Schiffrin EL. Selective mineralocorticoid receptor blocker eplerenone reduces resistance artery stiffness in hypertensive patients. Hypertension. 2008;51:432–9.
Yoshida C, Goda A, Naito Y, et al. Role of plasma aldosterone concentration in regression of left-ventricular mass following antihypertensive medication. J Hypertens. 2011;29:357–63.
Edelmann F, Wachter R, Schmidt AG, et al. Effect of spironolactone on diastolic function and exercise capacity in patients with heart failure with preserved ejection fraction: the Aldo-DHF randomized controlled trial. JAMA. 2013;309:781–91.
Shah SJ, Heitner JF, Sweitzer NK, et al. Baseline characteristics of patients in the treatment of preserved cardiac function with an aldosterone antagonist trial. Circ Heart Fail. 2013;6:184–92.
Bochud M, Nussberger J, Bovet P, et al. Plasma aldosterone is independently associated with the metabolic syndrome. Hypertension. 2006;48:239–45.
Remuzzi G, Cattaneo D, Perico N. The aggravating mechanisms of aldosterone on kidney fibrosis. J Am Soc Nephrol. 2008;19:1459–62.
Pratt-Ubanama MN, Nishizaka MK, Boedefeld RL, et al. Plasma aldosterone is related to severity of obstructive sleep apnea in subjects with resistant hypertension. Chest. 2007;131:453–9.
Tomaschitz A, Pilz S, Ritz E, et al. Plasma aldosterone levels are associated with increased cardiovascular mortality: the Ludwigshafen Risk and Cardiovascular (LURIC) health study. Eur Heart J. 2010;31:1237–47.
Ivanes F, Susen S, Mouquet F, et al. Aldosterone, mortality, and acute ischemic events in coronary artery disease patients outside the setting of acute myocardial infarction or heart failure. Eur Heart J. 2012;33:191–202.
Edwards NC, Steeds RP, Stewart PM, et al. Effect of spironolactone on left ventricular mass and aortic stiffness in early-stage chronic kidney disease: a randomized controlled trial. J Am Coll Cardiol. 2009;54:505–12.
Fujita T. Mineralocorticoid receptors, salt-sensitive hypertension, and metabolic syndrome. Hypertension. 2010;55:813–8.
Mosso LM, Carvajal CA, Maiz A, et al. A possible association between primary aldosteronism and a lower beta-cell function. Hypertension. 2007;25:2125–30.
Leopold JA, Dam A, Maron BA, et al. Aldosterone impairs vascular reactivity by decreasing glucose-6-phosphate dehydrogenase activity. Nat Med. 2007;13:189–97.
Rocha R, Funder JW. The pathophysiology of aldosterone in the cardiovascular system. Ann NY Acad Sci. 2002;970:89–100.
Callera GE, Touyz RM, Tostes RC, et al. Aldosterone activates vascular p38MAP kinase and NADPH oxidase via c-Src. Hypertension. 2005;45:773–9.
Usher MG, Duan SZ, Ivaschenko CY, et al. Myeloid mineralocorticoid receptor controls macrophage polarization and cardiovascular hypertrophy and remodeling in mice. J Clin Invest. 2010;120:3350–64.
Keidar S, Gamliel-Lazarovich A, Kaplan M, et al. Mineralocorticoid receptor blocker increases angiotensin-converting enzyme 2 activity in congestive heart failure patients. Circ Res. 2005;97:946–53.
Yamamuro M, Yoshimura M, Nakayama M, et al. Aldosterone, but not angiotensin II, reduces angiotensin converting enzyme 2 gene expression levels in cultured neonatal rats cardiomyocytes. Circ J. 2008;72:1346–50.
Swedberg K, Zannad F, McMurray JJ, et al. EMPHASIS-HF Study Investigators. Eplerenone and atrial fibrillation in mild systolic heart failure: results from the EMPHASIS-HF (eplerenone in mild patients hospitalization and survival study in heart failure) study. J Am Coll Cardiol. 2012;59:1598–603.
Shibata S, Nagase M, Yoshida S, Kawachi H, Fujita T. Podocyte as the target for aldosterone: roles of oxidative stress and Sgk1. Hypertension. 2007;49:355–64.
Schupp N, Kolkhof P, Queisser N, et al. Mineralocorticoid receptor-mediated DNA damage in kidneys of DOCA-salt hypertensive rats. FASEB J. 2011;25:968–78.
Zhang Y-L, Zhou S-X, Lei J, Yuan G-Y, Wang J-F. Blockades of angiotensin and aldosterone reduce osteopontin expression and interstitial fibrosis infiltration in rats with myocardial infarction. Chin Med J. 2008;121:2192–6.
Qin W, Rudolph AE, Bond BR, et al. Transgenic model of aldosterone–driven cardiac hypertrophy and heart failure. Circ Res. 2003;93:69–76.
Messaoudi S, Milliez P, Samuel JL, Delcayre C. Cardiac aldosterone overexpression prevents harmful effects of diabetes in the mouse heart by preserving capillary density. FASEB J. 2009;23:2176–85.
Lother A, Berger S, Gilsbach R, et al. Ablation of mineralocorticoid receptors in myocytes but not in fibroblasts preserves cardiac function. Hypertension. 2011;57:746–54.
Palmer BF. Managing hyperkalemia caused by inhibition of the renin-angiotensin-aldosterone system. N Engl J Med. 2004;351:585–92.
Pitt B, Anker SD, Bushinsky DA, Kitzman DW, Zannad F, Huang IZ, the PEARL-HF investigators. Evaluation of the efficacy and safety of RLY5016, a polymeric potassium binder, in a double-blind, placebo-controlled study in patients with chronic heart failure (the PEARL-HF) trial. Eur Heart J. 2011;32:820–8.
van Vark LC, Bertrand M, Akkerhuis KM, et al. Angiotensin-converting enzyme inhibitors reduce mortality in hypertension: a meta-analysis of randomized clinical trials of renin-angiotensin-aldosterone system inhibitors involving 158998 patients. Eur Heart J. 2012;33:2088–97.
Unger T, Paulis L, Sica DA. Therapeutic perspectives in hypertension: novel means for renin-angiotensin-aldosterone system modulation and emerging device-based approaches. Eur Heart J. 2011;32:2739–47.
Calhoun DA, Jones D, Textor S, et al. Resistant hypertension: diagnosis, evaluation, and treatment. A scientific statement from the American Heart Association Professional Education Committee of the Council for High Blood Pressure Research. Hypertension. 2008;51:1403–19.
Werner C, Poss J, Bohm M. Optimal antagonism of the renin-angiotensin-aldosterone system: do we need dual or triple therapy? Drugs. 2010;70:1215–30.
Amar L, Azizi M, Menard J, et al. Aldosterone synthase inhibition with LCI699. A proof-of-concept study in patients with primary aldosteronism. Hypertension. 2010;56:831–8.
Fiebeler A, Nussberger J, Shagdarsuren E, et al. Aldosterone synthase inhibitor ameliorates angiotensin-II induced organ damage. Circulation. 2005;111:3087–94.
Lea WB, Kwak ES, Luther JM, et al. Aldosterone antagonism or synthase inhibition reduces end-organ damage induced by treatment with angiotensin and high salt. Kidney Int. 2009;75:936–44.
Mulder P, Mellin V, Favre J, et al. Aldosterone synthase inhibition improves cardiovascular function and structure in rats with heart failure: a comparison with spironolactone. Eur Heart J. 2008;29:2171–9.
Nussberger J, Wuerzner G, Jensen C, Brunner HR. Angiotensin II suppression in humans by the orally active renin inhibitor Aliskiren (SPP100): comparison with enalapril. Hypertension. 2002;39:E1–8.
Dietz R, Dechend R, Yu CM, et al. Effects of the direct renin inhibitor aliskiren and atenolol alone or in combination in patients with hypertension. J Renin Angiotensin Aldosterone Syst. 2008;9:163–75.
Schmieder RE, Philipp T, Guerediaga J, et al. Long-term antihypertensive efficacy and safety of the oral direct renin inhibitor aliskiren: a 12-month randomized, double-blind comparator trial with hydrochlorothiazide. Circulation. 2009;119:417–25.
Andersen K, Weinberger MH, Egan B, et al. Comparative efficacy and safety of aliskiren, an oral direct renin inhibitor, and ramipril in hypertension: a 6-month, randomized, double-blind trial. J Hypertens. 2008;26:589–99.
Duprez DA, Munger MA, Botha J, Keefe DL, Charney AN. Aliskiren for geriatric lowering of systolic hypertension: a randomized controlled trial. J Hum Hypertens. 2010;24:600–8.
Stanton A, Jensen C, Nussberger J, O’Brien E. Blood pressure lowering in essential hypertension with an oral renin inhibitor, aliskiren. Hypertension. 2003;42:1137–43.
Menard J, Campbell DJ, Azizi M, Gonzales MF. Synergistic effects of ACE inhibition and AngII antagonism on blood pressure, cardiac weight, and renin in spontaneously hypertensive rats. Circulation. 1997;96:3072–8.
Sealey JE, Laragh JH. Aliskiren, the first renin inhibitor for treating hypertension: reactive renin secretion may limit its effectiveness. Am J Hypertens. 2007;20:587–97.
Schefe JH, Neumann C, Goebel M, et al. Prorenin engages the pro(renin) receptor like renin and both ligand activities are unopposed by aliskiren. J Hypertens. 2008;26:1787–94.
Westermann D, Riad A, Lettau O, et al. Renin inhibition improves cardiac function and remodeling after myocardial infarction independent of blood pressure. Hypertension. 2008;52:1068–75.
McMurray JJV, Pitt B, Latini R, et al. Effects of the oral renin inhibitor aliskiren in patients with symptomatic HF. Circ Heart Fail. 2008;1:17–24.
Gheorghiade M, Albaghdadi M, Zannad F, et al. On behalf of the ASTRONAUT investigators and study coordinators. Rationale and design of the multicentre, randomized, double-blind, placebo-controlled aliskiren trial on acute heart failure outcomes (ASTRONAUT). Eur J Heart Fail. 2011;13:100–6.
Krum H, Massie B, Abraham WT, et al. ATMOSPHERE investigators. Direct renin inhibition in addition to or as an alternative to angiotensin converting enzyme inhibition in patients with chronic systolic heart failure: rationale and design of the aliskiren trial to minimize outcomes in patients with heart failure (ATMOSPHERE) study. Eur J Heart Fail. 2011;13:107–14.
Solomon SD, Shin SH, Shah A, et al. Aliskiren Study in Post-MI Patients to Reduce Remodeling (ASPIRE) Investigators. Effect of the direct renin inhibitor aliskiren on left ventricular remodelling following myocardial infarction with systolic dysfunction. Eur Heart J. 2011;32:1227–34.
Rasilez® ASPIRE HIGHER Clinical Program Expands to 35,000 Patients in 14 Trials, The Largest Cardio-renal Outcomes Program Ever. Medical News Today, 20 June 2008. http://www.medicalnewstoday.com/releases/112086.php. Accessed 31 July 2013.
Parving HH, Persson F, Lewis JB, et al. AVOID study investigators. Aliskiren combined with losartan in type 2 diabetes and nephropathy. N Engl J Med. 2008;358:2433–46.
Parving HH, Brenner BM, McMurray JJ, et al. ALTITUDE Investigators. Cardiorenal end points in a trial of aliskiren for type 2 diabetes. N Engl J Med. 2012;367:2204–22.
Solomon SD, Skali H, Bourgoun M, et al. Effect of angiotensin-converting enzyme or vasopeptidase inhibition on ventricular size and function in patients with heart failure: the omapatrilat versus enalapril randomized trial of utility in reducing events (OVERTURE) echocardiographic study. Am Heart J. 2005;150:257–62.
Solomon SD, Zile M, Pieske B, et al. Prospective comparison of ARNI with ARB on Management Of heart failure with preserved ejectioN fracTion. PARAMOUNT investigators. The angiotensin receptor neprilysin inhibitor LCZ696 in heart failure with preserved ejection fraction: a phase 2 double-blind randomized clinical trial. Lancet. 2012;380(9851):1387–95.
McMurray JJ, Packer M, Desai AS, et al. On behalf of the PARADIGM-HF Committees and Investigators. Dual angiotensin receptor and neprilysin inhibition as an alternative to angiotensin-converting enzyme inhibition in patients with chronic systolic heart failure: rationale for and design of the Prospective comparison of ARNI with ACEI to determine impact on global mortality and morbidity in patients with heart failure trial (PARADIGM-HF). Eur J Heart Fail. 2013;15:1062–73.
Acknowledgement
This work was supported in part by grant # IAP99003 from the Canadian Institutes of Health Research, Ottawa, Ontario. I am indebted to Catherine Jugdutt for expert assistance.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2014 Springer Science+Business Media New York
About this chapter
Cite this chapter
Jugdutt, B.I. (2014). Aging and Remodeling of the RAS and RAAS and Related Pathways: Implications for Heart Failure Therapy. In: Jugdutt, B. (eds) Aging and Heart Failure. Springer, New York, NY. https://doi.org/10.1007/978-1-4939-0268-2_18
Download citation
DOI: https://doi.org/10.1007/978-1-4939-0268-2_18
Published:
Publisher Name: Springer, New York, NY
Print ISBN: 978-1-4939-0267-5
Online ISBN: 978-1-4939-0268-2
eBook Packages: MedicineMedicine (R0)