
Abstract
Over the past several years, it has become clear that alterations in the expression of microRNA (miRNA) genes contribute to the pathogenesis of most human malignancies. These alterations can be caused by various mechanisms, including deletions, amplifications, or mutations involving miRNA loci, epigenetic silencing, or the dysregulation of transcription factors that target specific miRNAs. Further, every cellular process is likely to be regulated by miRNAs and an aberrant miRNA expression signature is a hallmark of several diseases, including breast cancer. miRNA expression profiling has provided evidence of the association of these molecules with tumor development and progression. An increasing number of evidences have demonstrated that miRNAs can function as either potential oncogenes or tumor suppressor genes, depending on the cellular context and on the target genes they regulate. Here, we review our current knowledge about the involvement of miRNAs in breast cancer and their potential as diagnostic, prognostic, and therapeutic tools.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others


Keywords
1 Introduction
Cancer is a complex genetic disease involving structural and expression abnormalities of both coding and noncoding genes. For the past several decades, it has been thought that cancer is caused by genetic and/or epigenetic alterations to protein-coding oncogenes and tumor suppressor genes . These alterations result mainly from somatic genetic events that occur over long periods of time, particularly in solid malignancies like lung, breast, prostate, and gastrointestinal cancers [1]. These findings have enlightened the development of novel therapies, which are based on the specific genetic alterations that are involved in cancer pathogenesis. Although it is clear that tumors are initiated by somatic genetic alterations, it is also evident that many tumors show alterations in the expression of tumor suppressor genes because of epigenetic alterations, such as the methylation of CpG islands in their promoters, which lead to loss of function [2]. Therefore, therapies directed toward the reversal of the epigenetic changes that occur in various malignancies have also been developed. Even though significant progress has been made in identifying the genetic and epigenetic causes of cancer and identifying targets for therapy, several challenges still remain. For example, targeted therapies that are based on the identification of oncogenic mutations with causal roles in cancer have been developed, but the treatment of malignancies that are initiated through the loss of function of tumor suppressor genes is more difficult, because the lost gene function must be replaced in all the cancer cells and this should be a very challenging task to achieve. Thus, the identification of additional alterations that cause or contribute to malignancy is a high priority.
In 1993, Victor Ambros and colleagues discovered a gene, lin-4, that affected development in Caenorhabditis elegans and found that its product was a small noncoding RNA [3]. After these seminal findings, the cloning and characterization of small, 20–22-nucleotide-long members of the noncoding RNA family, called microRNAs (miRNAs), have led to the identification of ~ 1,000 miRNAs . After this initial discovery, the field of miRNAs has undergone a long period of silence and it took several more years to realize that these small RNA molecules are actually expressed in several organisms, including Homo sapiens. miRNAs are highly conserved across different species and are highly specific for tissue and developmental stages. In the past few years, miRNAs have taken their place in the complex circuitry of cell biology, revealing a key role as regulators of gene expression. miRNA genes represent approximately 1 % of the genome of different species, and each of them has hundreds of different conserved or non-conserved targets and it has been estimated that approximately 30 % of the genes are regulated by, at least, one miRNA [4]. Interestingly, more than 30 % of miRNAs have roles in the regulation of fundamental processes such as development, differentiation, cell proliferation, apoptosis, and stress responses in a range of organisms, including C. elegans, plants, Drosophila melanogaster, and mammals, including humans [5, 6].
miRNAs are transcribed for the most part by RNA polymerase II as long primary transcripts characterized by hairpin structures (pri-miRNAs) and processed into the nucleus by RNAse III Drosha into 70–100-nt-long pre-miRNAs. These precursor molecules are exported by an exportin-5 mediated mechanism to the cytoplasm, where an additional step mediated by the RNAse III Dicer generates a double-stranded RNA (dsRNA) of approximately 22 nts, named matured miRNA. The mature single-stranded miRNA product is then incorporated in the complex known as miRNA-containing ribonucleoprotein complex (miRNP) or miRNA-containing RNA-induced silencing complex (miRISC), whereas the other strand is likely subjected to degradation. In this complex, the mature miRNA is able to regulate gene expression at the posttranscriptional level, binding through partial complementarity for the most part to the 3′ untranslated region UTR of target messenger RNAs (mRNAs), and leading at the same time to some degree of mRNA degradation and translation inhibition (Fig. 7.1) [4, 7].

Processing of pri-microRNA and maturation of microRNAs (miRs). miRs are transcribed mainly by RNA polymerase II as long primary transcripts characterized by hairpin structures (pri-miRs) and processed in the nucleus by RNAse III Drosha in a 70-nucleotide-long pre-miR. This precursor molecule is exported by the exportin 5 to the cytoplasm, where RNAse III Dicer generates a dsRNA of approximately 22 nucleotides, named miR:miR*. The mature miRNA product is then incorporated in the complex known as miRNA-induced silencing complex (miRISC), whereas the other strand is likely subjected to degradation. As part of this complex, the mature miRNA is able to regulate gene expression binding through partial homology of the 3′UTR of target mRNAs and leading to mRNA degradation or translation inhibition
2 MicroRNA and Cancer
miRNA was initially identified in B cell chronic lymphocytic leukemia (CLL) and changes in the expression level of miRNAs have subsequently been detected in many types of human tumors, including breast cancer [8–14] . miRNAs have been proposed to contribute to oncogenesis because they can function as either tumor suppressors , like miR-15a and miR-16-1, or oncogenes, like miR-21 and miR-155, depending on the cellular context. In general, miRNAs are overexpressed in several human cancers and thus are considered as oncogenes [15–22] . In contrast, other miRNAs such as Let-7 are frequently downregulated in human malignancies including breast cancer and, in these contexts, miRNAs are functioning as a tumor suppressor gene [23–26] . Further, the genomic abnormalities found to influence the activity of miRNAs are the same as those described for protein-coding genes, such as chromosomal rearrangements, genomic amplifications, or deletions and mutations. In a specific set of tumors, abnormalities in both protein-coding genes and miRNAs can be identified [10]. Homozygous mutations or the combination of deletion plus mutation in miRNA genes is a rare event, and the functional consequences of heterozygous sequence variations of miRNAs in human cancers have not been identified [27, 28]. Furthermore, the role of polymorphisms in the complementary sites of target mRNAs in cancer patients or individuals with a predisposition to other hereditary diseases has also started to be understood [29, 30]. In addition, every type of tumor analyzed by miRNA profiling has shown significantly different miRNA profiles (mature and/or precursor miRNAs) compared with normal cells from the same tissue. The expression studies targeted to recognize the function of miRNAs revealed only a handful of these miRNAs in breast cancer . As in other cancers, some miRNAs function as tumor suppressors and other miRNAs as oncogenes . Therefore, tumor formation may occur by reduction or deletion of a tumor suppressor miRNA and/or by increased or overexpression of an oncogenic miRNA (Table 7.1). Two large profiling studies using various tumors and two distinct technologies to investigate genome-wide miRNA expression in 540 samples, including 363 from six of the most frequent human solid tumor types, and 177 normal controls found that cancer cells showed distinct miRNA profiles compared with normal cells that have been demonstrated [31, 32].
Further, studies have shown that miR-155 is upregulated in Burkitt’s lymphoma, diffuse large B cell lymphoma, primary mediastinal B cell lymphoma, and Hodgkin’s lymphoma [63, 64]. Mice overexpressing miR-155 in B-lymphocytes develop polyclonal preleukemic pre-B cell proliferation followed by a full-blown B cell malignancy [37]. More recently, two knockout mice models have demonstrated a critical role of miR-155 in immunity by showing that BIC/miR-155−/− have defective dendritic cell functions, impaired cytokine secretion, and TH cells intrinsically biased toward TH2 differentiation [65, 66]. Moreover, miR-155 could represent the connection between inflammation, immunity, and cancer, because its expression can be induced by mediators of inflammation and this is involved in response to endotoxic shock [67]. In contrast, members of the miR-29 family have been shown to be downregulated in aggressive CLL, invasive breast cancer, lung cancer, and cholangiocarcinoma [49, 51, 68, 69]. The transfection of miR-29b induces apoptosis in cholangiocarcinoma cell lines and reduces the tumorigenicity of lung cancer cells in nude mice. Further, it has been shown that rhabdomyosarcoma loses miR-29 expression because of an elevation of nuclear factor κB (NFκB) and YY1 levels, and introduction of miR-29s into the tumor delays rhabdomyosarcoma progression in mice [70]. miR-29s was also found to directly target myeloid cell leukemia 1 (Mcl-1) [49], an oncogene overexpressed in acute myeloid leukemia (AML), and the de novo DNA methyltransferases (DNMT)-3A and DNMT-3B and the maintenance of DNMT1 [51, 71] . Thus, the loss of miR-29 family member results in the constitutive overexpression of Mcl-1 and of DNMT, causing epigenetic changes characteristic of AML. These recent results suggest that the loss of miR-29s may be important, perhaps critical, for the pathogenesis of a major group of myelodysplastic syndromes and AMLs.
Breast Cancer
One of the first solid tumors to be profiled for miRNA expression was breast cancer . Iorio et al. [69] described the first miRNA signature for breast carcinoma and identified 13 miRNAs that discriminate breast tumors from normal tissues with 100 % accuracy. One of the most significant miRNAs differentially expressed between normal and breast tumor is miR-21, which is overexpressed in breast carcinoma. This miR-21 has been demonstrated to play a crucial role in regulation of cell survival and proliferation by directly targeting the tumor suppressor genes phosphatase and tensin homolog (PTEN), programmed cell death 4 (PDCD4), and tropomyosin 1 (TPM1). Further, this miR-21 has been associated with advanced clinical stage, lymph node metastasis, and poor patient prognosis [72, 73]. miR-21, is one of the first cancer miRNAs described, has been found overexpressed in a variety of other malignancies like glioblastoma, ovarian cancer, lung cancer, etc. [74–78]. Further, miR-21 overexpression is also associated with poor survival and poor therapeutic outcome in colorectal, pancreatic, endocrine, and exocrine tumors [79–81]. Conversely, downregulated miRNAs such as miR-125b and miR-205 regulate oncogenes like tyrosine kinase receptors human epidermal growth factor receptor (HER)-2 and HER-3 [82, 83] . Ectopic expression of miR-205 in a breast cancer cell line decreases proliferation and improves the responsiveness to tyrosine kinase inhibitors like gefitinib and lapatinib [84]. miRNA expression is also related to some histopathologic features of breast carcinoma, such as estrogen receptor (ER) and progesterone receptor (PR) expression, grade and stage, and presence of invasion [69]. Further studies have shown the correlation between miRNA expression and the classification in different subtypes of breast cancer [85]. In addition, studies have shown the correlation between miRNAs and ER status in human breast cancer. For example, miR-206 directly targets ER-α, and miR-221 and miR-222 confer tamoxifen resistance by regulating p27 and ER-α [86, 87]. Further, studies have also shown that there is a regulatory loop between ER-α and miR-221 and miR-222: The two miRNAs are able to directly target ER-α receptor, which in turn negatively regulates their expression, binding estrogen-responsive elements on their promoter region [88] .
3 MicroRNAs are Regulated by Transcription Factors in Breast Cancer
Breast cancer is the second leading cause of death among women in the Western world and its molecular pathogenesis is still not fully understood. Several lines of evidence indicate that ERα-negative breast tumors, which are highly aggressive and unresponsive to hormonal therapy, arise from ERα-positive precursors through different molecular pathways. High levels of miR-221 and miR-222 were found in ERα-negative cells and in primary breast tumor samples. Overexpression of miR-221 and miR-222 in ERα-positive cells suppressed ERα protein and luciferase assays confirmed that ERα is a target of miR-221 and miR-222. ERα was also found to negatively regulate the expression of miR-221 and miR-222 by promoter binding [88]. Therefore, silencing ERα, either by methylation or by dysregulating miR-221 and miR-222, results in the constitutive activation of miR-221 and miR-222 and the subsequent inhibition of the tumor suppressors like p27, p57, PTEN, and tissue inhibitor of metalloproteinases-3 (TIMP3), which in turn contribute to the development of the invasive phenotype [45] . This regulatory feedback loop seems to be involved in the development of ERα-negative breast cancers [89]. Further, studies have shown that the family of miR-34, which consists of miR-34a, miR-34b, and miR-34c, is induced directly by the p53 tumor suppressor and suggested that some p53 effects could be mediated by these miRNAs [46–48] . By using various cellular models, these authors compared miRNA expression in cells with high or low levels of p53 expression and found that the expression levels of p53 correlated with the levels of expression of miR-34 family members [46]. In addition, chromatin immunoprecipitation experiments showed that p53 binds to the miR-34 promoters [47, 48]. The introduction of miR-34 family members into cells that had lost miR-34 expression resulted in cell cycle arrest.
Therefore, miRNAs could be dysregulated by transcription factors and, therefore, genetic or epigenetic alterations that result in the dysregulation of transcription factors can cause miRNA dysregulation, which contributes to malignant transformation. Further, Volinia and coworkers have defined miRNA expression signatures, which distinguish cancerous tissues from normal tissues [32]. Interestingly, they observed that some miRNA genes were dysregulated not just in one tumor type but also in many tumors, suggesting that these miRNAs may be downstream targets of pathways that are commonly dysregulated in cancer. Thus, if miRNAs are downstream targets of pathways that are commonly dysregulated in human cancer they become excellent targets for therapeutic intervention. It is known that proliferative signals lead to the activation of the c-Myc transcription factor (that integrates the cell cycle machinery), which positively and negatively regulates the expression of many miRNA genes, including miR-17-92 cluster (c-Myc regulates positively) and miR-15a and miR-16-1 (c-Myc regulates negatively). Therefore, it seems that the mechanism of action of the activated oncogene is, at least partly, miRNA dysregulation and thereby it causes the downregulation of tumor suppressors and upregulation of oncogenes . Therefore, genetic or epigenetic alterations in protein-coding cancer genes or in miRNA genes may have similar consequences. Until now, it has not been possible to target the overexpression of c-Myc in tumors with drugs, but it is possible to target the miRNAs that are dysregulated c-Myc by treating the cancer cells with anti-miR-17-92 or with miR-15a and miR-16-1 . Recently, Bonci et al. have shown that the miR-15a-miR-16-1 cluster can control prostate cancer by targeting multiple oncogenic activities [90]. In general, in tumors with alterations in protein-coding cancer genes, it should be possible to induce tumor regression using miRNAs and/or anti-miRNAs. In fact, recent studies have shown that c-Myc-induced hepatocellular carcinomas in mice are regressed by targeting miRNA that is induced by c-Myc [91].
4 Role of Cancer Epigenetics in Regulation of miRNAs
The most-studied epigenetic changes in cancer cells are the methylation of cytosines in the dinucleotide CpG in DNA [92]. Such “methylable” sites, known as CpG islands, are preferentially located in the 5′ region (which consists of the promoter, 5′ UTR, and exon 1) of many genes and are non-methylated in normal cells but are transcribed in the presence of the appropriate transcription factors. Methylation of the CpG islands of tumor suppressors results in their silencing and contributes to malignant transformation [92] . miRNAs are affected by genetic changes, such as deletion, gene amplification, and mutation, and by transcription factors. In addition, the expression of miRNAs is affected by epigenetic changes, such as methylation of the CpG islands of their promoters. Saito et al. [93] reported that miR-127 is silenced by promoter methylation in bladder tumors and that its expression could be restored by demethylating agents such as 5-azacitidine. miR-127 targets B cell lymphoma 6 (Bcl6), an oncogene that is involved in the regulation of apoptosis signaling and in the development of B cell lymphoma . Therefore, silencing of miR-127 may lead to the overexpression of Bcl6, which leads to malignant transformation. Further, miRNAs can also regulate enzymes that are involved in the methylation of the CpG islands of tumor suppressor genes . For example, the overexpression of DNMTs is a poor prognostic indicator in several human malignancies, and the miR-29 family targets de novo DNMT3A and DNMT3B [94]. However, recent studies have also shown that miR-29 not only targets DNMT3A and DNMT3B but also targets indirectly DNMT1 [71]. Interestingly, the introduction of miR-29 into cancer cell lines caused demethylation of the CpG islands in the promoter regions of tumor suppressor genes, which allowed their reactivation and resulted in the loss of tumorigenicity [71]. The introduction of miR-29 into cancer cells resulted in the loss of expression of the oncogene Mcl1 and the three DNMTs with reactivation of the p16 tumor suppressor [71]. Further, the loss of miR-29 cluster in Dicer-deficient mouse embryonic stem cells leads to downregulation of DNMT1, DNMT3a, and DNMT3b through the modulation of their repressor, RBL-2, a proven target of miR-290 [95, 96] . Therefore, treatment of the malignancies with miR-29 family members caused the reversion of epigenetic changes that contribute to malignant transformation in human cancers. Further, miRNAs also target histone deacetylases (HDACs) and other proteins that are involved in chromatin structure and function [97] . Therefore, all these observations indicate that alterations in the expression of miRNAs could be responsible for some of the epigenetic changes that are observed in cancer cells, and that such miRNAs could provide novel targets for cancer therapy.
5 Role of MicroRNAs in Regulation of Cancer-Initiating Stem Cells in Breast Cancer
Cancer stem cells (CSC) are a small subpopulation of cells capable of self-renewal, differentiation, and tumor initiation . The prevailing view is that CSCs are the root of cancer origin and recurrence . Although not all cancers have been found to contain CSC populations, to date, the data on the roles of miRNAs in CSCs are consistent with the existence of CSCs in breast, prostate, lung, pancreatic, and liver cancers . It has been well established that miRNAs regulate tumor development, prognosis, and metastasis either as oncogenes or as tumor suppressors [98, 99] . In addition, emerging evidence suggests that miRNAs also play essential roles in stem cell self-renewal and differentiation by negatively regulating the expression of key stem cell-regulating genes [100]. Evidence for a role of miRNAs in stem cell maintenance and differentiation is accumulating from analysis of mutations in key RNA interference (RNAi) components. For example, Dicer-mutant mice die early in development with a loss of Oct4-positive multipotent stem cells [101]. Further, abnormal miRNA expression may result in dysregulation of self-renewal in CSCs during cancer progression [102, 103]. Silber et al. reported that miR-124 and miR-137 induce differentiation of neural and glioblastoma stem cells and induce cell cycle arrest [104]. These results suggest that the targeted delivery of miR-124 and miR-137 to glioblastoma cells may be therapeutically efficacious for the glioblastoma treatment. Further, studies have shown that prostate cancer stem and/or progenitor cell populations have lower levels of miR-34a and Let-7b compared to bulk tumor cells [105]. In addition, studies have also shown that miR34a targets CD44, resulting in impaired tumor growth and decreased metastases in mouse models of prostate cancer. The increased survival of mice treated with systemically delivered miR34a suggests a novel strategy to target prostate CSCs, thereby inhibiting tumor growth and metastasis [105]. Further, a number of studies describing the role of miRNA in the regulation of normal and malignant breast stem cells have been conducted. Recent studies have shown that both miR-205 and miR-22 are highly expressed in mouse mammary stem/progenitor cells, whereas miR-93 and Let-7 are depleted in this population [106]. Further, studies have also reported that miR-205 overexpression in mouse mammary cells led to an expansion of the progenitor cell population, decreased cell size, and increased cellular proliferation [107]. More recent studies have shown that overexpression of miR-200c reduced the clonogenic and tumor-initiation activities of breast cancer stem cells (BCSCs) and suppressed mammary duct formation by normal mammary stem cells. This occurred through the downregulation of the polycomb gene Bmi-1, a target of miR-200c. This work demonstrated a molecular link between normal breast stem cells and BCSCs [23]. Yu et al. showed that Let-7 is decreased in BCSCs and that overexpression of Let-7 inhibits the cell proliferation, mammosphere formation, BCSC self-renewal and differentiation, and tumor formation and metastasis in nonobese diabetic/severe combined immunodeficiency (NOD/SCID) mice [108]. These effects were shown to be mediated through downregulation of the Let-7 targets H-Ras and HMGA2 [108]. This group also demonstrated that expression of miR-30 markedly reduced BCSCs by targeting ubiquitin-conjugating enzyme 9 (UBC9) and integrin b3 (ITGB3). A more complete inhibition of self-renewal and mammosphere formation of BCSCs was observed when both Let-7 and miR-30 were simultaneously introduced compared to each miRNA individually [108]. The ability of these miRNAs to target BCSCs suggests that they may have significant therapeutic potential. All these studies show that miRNA plays a crucial role in the regulation of stem cells and progenitor and cancer-initiating stem cells . Thus, the most effective cancer therapy must be directed against both the small, quiescent pool of CSCs as well as the more actively proliferating bulk tumor mass . This may be possible if specific CSC signals are inhibited using molecularly targeted therapy, while simultaneously attacking proliferating cells by conventional therapies like chemotherapy and radiotherapy. To attain this goal, developments in miRNA regulation present exciting new prospects.
6 MicroRNAs in Breast Cancer Invasion, Angiogenesis, and Metastasis
The leading cause of death in breast cancer (BC) patients is not the primary tumor in the breast per se, but metastasis to distant organs. Metastasis accounts for more than 90 % of the deaths in BC patients. Millions of cells are released from the primary tumor into the blood circulation, but only a small portion of these cells survive and colonize on distant organs. The development of cancer metastases depends on multiple factors, including miRNAs [109–112], epithelial–mesenchymal transition (EMT) [113–116], CSCs [117, 118], etc . miRNAs have been implicated not only in the development of primary tumors, but also in affecting progression and in the metastatic phase of the disease. Indeed, several lines of evidence show how miRNAs are involved in the regulation of biologic processes leading to the acquisition of metastatic potential, as adhesion, migration and invasion, and angiogenesis. Further, the connection between miRNAs and EMT has also been well established. Numerous studies have revealed that miRNAs are dysregulated in cancer versus normal tissue, and in noninvasive versus invasive forms [119, 120]. Discordant miRNA expression between normal and breast tumor tissues and between ERα-positive and ERα-negative tumors is also well established [69]. In general, the levels of the most mature (processed) miRNAs are lower in cancer versus normal tissue, and in ERα-negative versus ERα-positive breast cancer, and both Drosha and Dicer levels are subnormal in several cancers [121, 122]. Interestingly, estrogen (E2) induces Dicer expression in MCF-7 cells, suggesting that the loss of ERα and estrogen signaling in breast cancers may contribute to the decreased expression of Dicer and consequently lower levels of miRNA expression [123] . Some evidence supports a role of miRNAs in dampening Dicer expression. Martello et al. [124] showed that miR-103/107 targets Dicer mRNA and inhibits Dicer expression, thereby promoting EMT in breast cancer cell lines and the metastatic spread in mice . These investigators also provided evidence that the miR-103/107-induced decrease in Dicer leads to compromised processing of pre-miRNAs belonging to the EMT-inhibitory miR-200 family. Cochrane et al. [121] demonstrated that miR-221, miR-222, and miR-29a, which are elevated in ERα-negative breast cancer cell lines, target Dicer. Upregulation of miR-200c increased Dicer in two ERα-negative cell lines. Although the majority of miRNAs are decreased after neoplastic transformation, some miRNAs clearly show an increase [119].
To perform miRNA effector functions, miRNAs must be incorporated into Argonaute (AGO)-containing complexes [125] . The expression of the miRNA effector AGO proteins is also altered in cancer. The AGO1 gene is frequently deleted in several cancers, including breast cancer [126]. However, studies have also shown that both AGO1 and AGO2 were elevated in ERα-negative versus ERα-positive breast cancer [127). ]. Further, forced expression of AGO2 reduced E-cadherin expression and enhanced motility in epithelial breast cancer cell lines [128]. Additional work is needed to examine whether increased AGO2 expression induces a complete EMT in breast cancer. Specific miRNAs have been demonstrated to promote EMT and metastasis. One of the seminal studies by the Weinberg Group [129] showed that Twist (but not Snail) induced expression of miR-10b. They observed that miR-10b was downmodulated in all the breast carcinomas from metastasis-free patients, but surprisingly, 50 % of metastasis-positive patients had elevated miR-10b levels in their primary tumors. miR-10b targets and represses the homeobox D10 (HOXD10) expression, thereby relieving transcriptional inhibition of the prometastatic Ras homolog gene family member C (RHOC) and thus leading to tumor cell invasion and metastasis. Further, as a functional screen that aimed to discover miRNAs that promote cell migration in vitro, Huang et al. [38] identified miR-373 and validated its metastatic potential in tumor transplantation experiments using breast cancer cells. More recently, several “metastamiRs” have been characterized [130]. Conversely, several miRNAs have been characterized as suppressors of metastasis [130]. In particular, members of the miR-200 family of miRNAs and miR-205 have been shown to reduce cell migration and invasiveness by targeting zinc-finger E-box binding (ZEB) transcription factors, which are known inducers of EMT [131, 132]. Further, the oncogenic miR-21 stimulates invasion, extravasation, and metastasis in different tumor types, including breast cancer , whereas oncosuppressor miR-205 has opposite effects, reducing invasion in vitro and suppressing lung metastasis in vivo [133, 84]. With the same aim of searching for regulators of breast cancer metastasis, Tavazoie et al. [134] identified miR-126 and miR-335 as metastasis suppressors: Reduced levels of the two miRNAs are associated with poor metastasis-free survival of patients with breast cancer, whereas their re-expression inhibits metastasis in a cell transplantation model. Interestingly, it has been recently observed that primary tumors and metastasis from the same tissue show a similar pattern of miRNAs expression [135]. Being a more accurate classifier than mRNA expression studies, miRNA profiling has thus revealed the potential to solve one of the most demanding issues in cancer diagnostics: the origin of metastasis of unknown primary tumors.
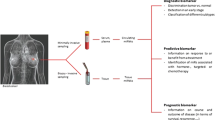
7 MicroRNAs Are a New Clinical Tool
As active players in important oncogenic signaling pathways, miRNAs should affect cancer diagnosis and prognosis .
miRNA Profiling as a Diagnostic Tool
Metastatic cancer of unknown primary site (CUP) is one of the ten most frequent cancer diagnoses worldwide, and constitutes 3–5 % of all human malignancies [136]. Patients with CUP present with metastases (late-stage disease) without an established primary tumor (a site at which the tumor has initially developed and from which it has metastasized). The study by Lu et al. [31] produces an important advance in the diagnosis of this peculiar type of cancer. Analyzing 17 poorly differentiated tumors with nondiagnostic histological appearance, they showed that the miRNA-based classifier was much better at establishing the correct diagnosis of the samples than the mRNA classifier [31]. This result is exciting because profiling a few hundred miRNAs has a much better predictive power for CUP diagnosis than profiling several tens of thousands of mRNAs. As miRNA expression changes with differentiation, the poorly differentiated tumors have lower global expression levels of miRNAs compared with well-differentiated tumors from control groups [31]. The reduced expression levels of miRNAs in poorly differentiated tumors reveal why miRNA profiling is effective in the diagnosis of CUP.
miRNA Profiling as a Prognostic Tool
Breast cancer is the leading cause of death from cancer in women worldwide . Therefore, the identification of new prognostic markers (markers that correlate with disease evolution) could be a significant advance for the identification of patients that would benefit from more aggressive therapy. In univariate analyses, the expression of both miR-155 (high levels) and Let-7a-2 (low levels) has been shown to correlate with poor survival in 104 patients; in multivariate analyses, the expression of miR-155 also correlated with a poor prognosis when all clinical variables were considered together.
8 MicroRNAs and Anti-MicroRNA in Cancer Treatment
The evidence collected to date demonstrate how microRNAs could represent valid diagnostic, prognostic, and predictive markers in cancer. Indeed, the aberrant miRNA expression is correlated with specific biopathologic features, disease outcome, and response to specific therapies in different tumor types. Considering the importance of miRNAs in development, progression, and treatment of cancer, the potential usefulness of an miRNA-based therapy in cancer is now being exploited, with the attempt to modulate their expression, reintroducing miRNAs lost in cancer, or inhibiting oncogenic miRNAs by using anti-miRNA oligonucleotides. For example, the transfection of miR-15a/16–1 induces apoptosis in leukemic MEG01 cells and inhibits tumor growth in vivo in a xenograft model [137], whereas the inhibition of miR-21 with antisense oligonucleotides generates a proapoptotic and antiproliferative response in vitro in different cellular models and reduces tumor development and metastatic potential in vivo [138].
Moreover, miRNAs involved in specific networks, such as the apoptotic, proliferation, or receptor-driven pathways, could likely influence the response to targeted therapies or to chemotherapy: Inhibition of miR-21 and miR-200b enhances sensitivity to gemcitabine, probably by modulation of CLOCK, PTEN, and PTPN12, whereas reintroduction of miR-205 in breast cancer cells can improve the responsiveness to tyrosine kinase inhibitors through HER-3 silencing [139, 83] . Besides targeted therapies and chemotherapy, miRNAs could also alter the sensitivity to radiotherapy; the Let-7 family of miRNAs can suppress the resistance to anticancer radiation therapy, probably through RAS regulation [140]. Evidence described to date represents the experimental bases for the use of miRNAs as both targets and tools in anticancer therapy, but there are at least two primary issues to address to translate these fundamental research advances into medical practice: the development of engineered animal models to study cancer-associated miRNAs and the improvement of the efficiency of miRNAs/anti-miRNAs delivery in vivo . To this aim, modified miRNA molecules with longer half-lives and efficiency have been developed, such as anti-miRNA oligonucleotides, locked nucleic acid-modified oligonucleotides, and cholesterol-conjugated antagomirs [141–143]. Interestingly, Ebert et al. [144] have recently described a new approach to inhibit miRNAs function: Synthetic mRNAs containing multiple binding sites for a specific miRNA, called miRNA sponges, are able to bind up the miRNA, preventing its association with endogenous targets. To improve the in vivo delivery of either miRNAs or anti-miRNAs, the methods that have been tested in preclinical studies over the past decades for short-interfering RNAs (siRNA) or short heteroduplex RNA (shRNA) could be applied also to miRNAs. Moreover, the advantage of miRNAs over siRNA/shRNA is their ability to affect multiple targets with a single hit, thus regulating a whole network of interacting molecules.
9 Future Perspective
It has been unequivocally proven that miRNA dysregulation occurs in many human malignancies including the most common human malignancies like lung, breast, prostate, and gastrointestinal cancers. Such dysregulation, like the dysregulation of oncogenes and tumor suppressor genes , can be caused by multiple mechanisms, such as deletion, amplification, mutation, transcriptional dysregulation, and epigenetic changes. As miRNAs have multiple targets, their function in tumorigenesis could be due to their regulation of a few specific targets, possibly even one, or many targets. A future challenge would be to identify all of the targets of the miRNAs involved in cancer and establish their contribution to malignant transformation. An additional challenge would be the identification of all of the miRNAs that are dysregulated by pathways consistently dysregulated in various types of human cancers. If these miRNA targets are crucial for the expression of the malignant phenotype and the cancer cells depend on their dysregulation for proliferation and survival, we can expect that the use of miRNAs or anti-miRNAs will result in tumor regression. Over the past several years, we have observed a shift from conventional chemotherapy to targeted therapies, and miRNAs and anti-miRNAs could contribute extensively in the forthcoming years.
References
Croce CM. Oncogenes and cancer. N Engl J Med. 2008;358:502–11.
Jones PA, Baylin SB. The epigenomics of cancer. Cell. 2007;128:683–92.
Lee RC, Feinbaum RL, Ambros V. The C. elegans heterochronic gene lin-4 encodes small RNAs with anti-sense complementarity to lin-14. Cell. 1993;75:843–54.
Bartel DP. MicroRNAs: genomics, biogenesis, mechanism, and function. Cell. 2004;116:281–97.
Lagos-Quintana M, Rauhut R, Lendeckel W, Tuschl T. Identification of novel genes coding for small expressed RNAs. Science. 2001;294:853–8.
Calin GA, Croce CM. MicroRNA signatures in human cancers. Nature Rev Cancer. 2006;6:857–66.
Gregory RI, Shiekhattar R. MicroRNA biogenesis and cancer. Cancer Res. 2005;65:3509–12.
Calin GA, Dumitry CD, Shimizu M, Bichi R, et al. Frequent deletions and downregulation of micro-RNA genes miR15 and miR16 at 13q14 in chronic lymphocytic leukemia. Proc Natl Acad Sci U S A. 2002;99:15524–9.
Caldas C, Brenton JD. Sizing up miRNAs as cancer genes. Nature Med. 2005;11:712–4.
Calin GA, Croce CM. MicroRNA-cancer connection: the beginning of a new tale. Cancer Res. 2006;66:7390–4.
Chen CZ. MicroRNAs as oncogenes and tumor suppressors. N Engl J Med. 2005;353:1768–71.
Hwang HW, Mendell JT. MicroRNAs in cell proliferation, cell death, and tumorigenesis. Br J Cancer. 2006;94:776–80.
Hammond SM. MicroRNAs as oncogenes. Curr Opin Genet Dev. 2006;16:4–9.
Esquela-Kerscher A, Slack FJ. Oncomirs-microRNAs with a role in cancer. Nature Rev Cancer. 2006;6:259–69.
Petrocca F, Vecchione A, Croce CM. Emerging role of miR-106b-25/miR-17-92 clusters in the control of transforming growth factor beta signaling. Cancer Res. 2008;68:8191–4.
Hayashita Y, Osada H, Tatematsu Y, Yamada H, et al. A polycistronic microRNA cluster, miR-17-92, is overexpressed in human lung cancers and enhances cell proliferation. Cancer Res. 2005;65:9628–32.
Mendell JT. miRiad roles for the miR-17-92 cluster in development and disease. Cell. 2008;133:217–22.
Bandres E, Agirre X, Ramirez N, Zarate R, Garcia-Foncillas J. MicroRNAs as cancer players: potential clinical and biological effects. DNA Cell Biol. 2007;26:273–82.
Hernando E. microRNAs and cancer: role in tumorigenesis, patient classification and therapy. Clin Transl Oncol. 2007;9:155–60.
Negrini M, Ferracin M, Sabbioni S, Croce CM. MicroRNAs in human cancer: from research to therapy. J Cell Sci. 2007;120:1833–40.
Wiemer EA. The role of microRNAs in cancer: no small matter. Eur J Cancer. 2007;43:1529–44.
Wijnhoven BP, Michael MZ, Watson DI. MicroRNAs and cancer. Br J Surg. 2007;94:23–30.
Yu F, Yao H, Zhu P, Zhang X, et al. let-7 regulates self-renewal and tumorigenicity of breast cancer cells. Cell. 2007;131:1109–23.
Shimono Y, Zabala M, Cho RW, Lobo N, et al. Downregulation of miRNA-200c links breast cancer stem cells with normal stem cells. Cell. 2009;138:592–603.
Esquela-Kerscher A, Trang P, Wiggins JF, Patrawala L, et al. The let-7 microRNA reduces tumor growth in mouse models of lung cancer. Cell Cycle. 2008;7:759–64.
Cimmino A, Calin GA, Fabbri M, Iorio MV, et al. miR-15 and miR-16 induce apoptosis by targeting BCL2. Proc Natl Acad Sci U S A. 2006;102:13944–9.
Calin GA, Farracin M, Cimmino A, Di Lava G, et al. A unique microRNA signature associated with prognostic factors and disease progression in B cell chronic lymphocytic leukemia. N Engl J Med. 2005;352:1667–76.
Diederichs S, Haber DA. Sequence variations of microRNAs in human cancer: alterations in predicted secondary structure do not affect processing. Cancer Res. 2006;66:6097–104.
He H, Jazdzewki K, Li W, Liyanarachchi S, et al. The role of microRNA genes in papillary thyroid carcinoma. Proc Natl Acad Sci U S A. 2005;102:19075–80.
Abelson JF, Kwan KY, O’Roak BJ, Baek DY, et al. Sequence variants in SLITRK1 are associated with Tourette’s syndrome. Science. 2006;310:317–20.
Lu J, Getz G, Miska EA, Alvarez-Saavedra E, et al. MicroRNA expression profiles classify human cancers. Nature. 2005;435:834–8.
Volinia S, Calin GA, Liu CG, Ambs S, et al. A microRNA expression signature of human solid tumors define cancer gene targets. Proc Natl Acad Sci U S A. 2006;103:2257–61.
Meng F, Henson R, Wehbe-Janek H, Ghoshal K et al. MicroRNA-21 regulates expression of the PTEN tumor suppressor gene in human hepatocellular cancer. Gastroenterology. 2007;133:647–58.
Frankel LB, Chrisoffersen NR, Jacobsen A, Lindow M et al. Programmed cell death 4 (PDCD4) is an important functional target of the microRNA miR-21 in breast cancer cells. J Biol Chem. 2008;283:1026–33.
Zhu S, Si M-L, Win H, Mo Y-Y. MicroRNA-21 targets the tumor suppressor gene tropomyosin 1 (TPM1). J Biol Chem. 2007;282:14328–36.
Eis PS, Tam W, Sun L, Chadburn AZ et al. Accumulation of miR-155 and BIC RNA in human B cell lymphomas. Proc Natl Acad Sci U S A. 2005;102:3627–32.
Costinean S, Zanesi N, Pekarsh Y, Tili E et al. Pre-B cell proliferation and lymphoblastic leukemia/high grade lymphoma in Eμ-miR155 transgenic mice. Proc Natl Acad Sci U S A. 2006;103:7024–9.
Huang Q, Gumireddy K, Schrier M, le Sage C et al. The microRNAs miR-373 and miR-520c promote tumor invasion and metastasis. Nat Cell Biol. 2008;10:202–10.
Yan GR, Xu SH, Tan ZL, Liu L, He QY. Global identification of miR-373-regulated genes in breast cancer by quantitative proteomics. Proteomics. 2011;11:912–20.
Voorhoeve PM, le Sage C, Schrier M, Gillis AJ et al. A genetic screen implicates miRNA-372 and miRNA-373 as oncogenes in testicular germ cell tumors. Cell. 2006;124:1169–81.
Ventura AG, Young AG, Winslow MM, Meissner A, et al. Targeted deletion reveals essential and overlapping functions of the miR-17 through 92 family of miRNA clusters. Cell. 2008;132:875–86.
Xiao C, Srinivasan L, Calado DP, Patterson HC, et al. Lymphoproliferative disease and autoimmunity in mice with increased miR-17-92 expression in lymphocytes. Nature Immunol. 2008;9:405–14.
Ota A, Tagawa H, Karnan S, Tsuzuki S, et al. Identification and characterization of a novel gene, C13orf25, as a target for 13q31-q32 amplification in malignant lymphoma. Cancer Res. 2004;64:3087–95.
Ma L, Teruya-Feldstein J, Weinberg RA. Tumour invasion and metastasis initiated by microRNA-10b in breast cancer. Nature. 2007;449:682–8.
Garofalo M, Di Leva G, Romano G, Suh SS, et al. MiR-221 and 222 regulate TRAIL resistance and enhance tumorigenicity through PTEN and TIMP3 downregulation. Cancer Cell. 2009;16:498–509.
He L, He X, Lim LP, de Stanchina E, et al. A microRNA component of the p53 tumour suppressor network. Nature. 2007;447:1130–4.
Raver-Shapira N, Marciano E, Meiri E, Spectpr Y, et al. Transcriptional activation of miR-34a contributes to p53-mediated apoptosis. Mol Cell. 2007;26:731–43.
Chang TC, Wentzel EA, Kent OA, Ramachandran K, et al. Transactivation of miR-34a by p53 broadly influences gene expression and promotes apoptosis. Mol Cell. 2007;26:745–52.
Mott JL, Kobayashi S, Bronk SF, Gores GJ. miR-29 regulates Mcl-1 protein expression and apoptosis. Oncogene. 2007;26:6133–40.
Pekarsky Y, Santanam U, Cimmino A, Palamarchuk A, et al. Tcl1 expression in chronic lymphocytic leukemia is regulated by miR-29 and miR-181. Cancer Res. 2006;66:11590–3.
Fabbri M, Garzon R, Cimmino A, Liu Z, et al. MicroRNA family reverts aberrant methylation in lung cancer by targeting DNA methyltransferases 3A and 3B. Proc Natl Acad Sci U S A. 2007;104:15805–10. (2007.
Johnson SM, Grosshans H, Shingara J, Byrom M, et al. RAS is regulated by the let-7 microRNA family. Cell. 2005;120:635–47.
Lee YA, Dutta A. The tumor suppressor microRNA let-7 represses the HMGA2 oncogene. Genes Dev. 2007;21:1025–30.
Iorio MV, Ferracin M, Liu CG, Veronese A, Spizzo R, Sabbioni S, Magri E, Pedriali M, Fabbri M, Campiglio M, Menard S, Palazzo JP, Rosenberg A, Musiani P, Volinia S, Nenci I, Calin GA, Querzoli P, Negrini M, Croce CM. MicroRNA gene expression deregulation in human breast cancer. Cancer Res. 2005;65:7065–70.
Adams BD, Furneaux H, White BA. The micro-ribonucleic acid (miRNA) miR-206 targets the human estrogen receptor-alpha (ERalpha) and represses ERalpha messenger RNA and protein expression in breast cancer cell lines. Mol Endocrinol. 2007;21:1132–47.
Hossain A, Kuo MT, Saunders GF. Mir-17-5p regulates breast cancer cell proliferation by inhibiting translation of AIB1 mRNA. Mol Cell Biol. 2006;26:8191–201.
Yu Z, Wang C, Wang M, Li Z, Casimiro MC, Liu M, Wu K, Whittle J, Ju X, Hyslop T, McCue P, Pestell RG. A cyclin D1/microRNA 17/20 regulatory feedback loop in control of breast cancer cell proliferation. J Cell Biol. 2008;182:509–17.
Mattie MD, Benz CC, Bowers J, Sensinger K, Wong L, Scott GK, Fedele V, Ginzinger D, Getts R, Haqq C. Optimized high-throughput microRNA expression profi ling provides novel biomarker assessment of clinical prostate and breast cancer biopsies. Mol Cancer. 2006;5:24.
Scott GK, Goga A, Bhaumik D, Berger CE, Sullivan CS, Benz CC. Coordinate suppression of ERBB2 and ERBB3 by enforced expression of micro-RNA miR-125a or miR-125b. J Biol Chem. 2007;282:1479–86.
Gregory PA, Bert AG, Paterson EL, Barry SC, Tsykin A, Farshid G, Vadas MA, Khew-Goodall Y, Goodall GJ. The miR-200 family and miR-205 regulate epithelial to mesenchymal transition by targeting ZEB1 and SIP1. Nat Cell Biol. 2008;10:593–601.
Dykxhoorn DM, Wu Y, Xie H, Yu F, Lal A, Petrocca F, Martinvalet D, Song E, Lim B, Lieberman J. miR-200 enhances mouse breast cancer cell colonization to form distant metastases. PLoS One. 2009;4:e7181.
Valastyan S, Reinhardt F, Benaich N, Calogrias D, Szasz AM, Wang ZC, Brock JE, Richardson AL, Weinberg RA. A pleiotropically acting microRNA, miR-31, inhibits breast cancer metastasis. Cell. 2009;137:1032–46.
Metzler M, Wilda M, Busch K, et al. High expression of precursor microRNA-155/BIC RNA in children with Burkitt lymphoma. Genes Chromosomes Cancer. 2004;39:167–9.
Kluiver J, Poppema S, de JD, et al. BIC and miR-155 are highly expressed in Hodgkin, primary mediastinal and diffuse large B cell lymphomas. J Pathol. 2005;207:243–9.
Thai TH, Calado DP, Casola S, et al. Regulation of the germinal center response by microRNA-155. Science. 2007;316:604–8.
Rodriguez A, Vigorito E, Clare S, et al. Requirement of bic/microRNA-155 for normal immune function. Science. 2007;316:608–11.
Tili E, Michaille JJ, Cimino A, et al. Modulation of miR-155 and miR-125b levels following lipopolysaccharide/TNF-alpha stimulation and their possible roles in regulating the response to endotoxin shock. J Immunol. 2007;179:5082–9.
Calin GA, Ferracin M, Cimmino A, et al. A MicroRNA signature associated with prognosis and progression in chronic lymphocytic leukemia. N Engl J Med. 2005;353:1793–801.
Iorio MV, Ferracin M, Liu CG, et al. MicroRNA gene expression deregulation in human breast cancer. Cancer Res. 2005;65:7065–70.
Wang H, Garzon R, Sun H, et al. NF-kappaB YY1-miR-29 regulatory circuitry in skeletal myogenesis and rhabdomyosarcoma. Cancer Cell. 2008;14:369–81.
Garzon R, Liu S, Fabbri M, et al. MicroRNA-29b induces global DNA hypomethylation and tumor suppressor gene re-expression in acute myeloid leukemia by targeting directly DNMT3A and 3B and indirectly DNMT1. Blood. 2009;113:6411–8.
Yan LX, Huang XF, Shao Q, et al. MicroRNA miR-21 overexpression in human breast cancer is associated with advanced clinical stage, lymph node metastasis and patient poor prognosis. RNA. 2008;14:2348–60.
Qian B, Katsaros D, Lu L, et al. High miR-21 expression in breast cancer associated with poor disease-free survival in early stage disease and high TGF-beta1. Breast Cancer Res Treat. 2009;117:131–40.
Chan JA, Krichevsky AM, Kosik KS. MicroRNA-21 is an antiapoptotic factor in human glioblastoma cells. Cancer Res. 2005;65:6029–33.
Ciafre SA, Galardi S, Mangiola A, et al. Extensive modulation of a set of microRNAs in primary glioblastoma. Biochem Biophys Res Commun. 2005;334:1351–8.
Iorio MV, Visone R, Di LG, et al. MicroRNA signatures in human ovarian cancer. Cancer Res. 2007;67:8699–707.
Yanaihara N, Caplen N, Bowman E, Seike M, et al. Unique microRNA molecular profiles in lung cancer diagnosis and prognosis. Cancer Cell. 2006;9:189–98.
Markou A, Tsaroucha EG, Kaklamanis L, Fotinou M, et al. Prognostic value of mature microRNA-21 and microRNA-205 overexpression in non-small cell lung cancer by quantitative real-time RT-PCR. Clin Chem. 2008;54:1696–704.
Schetter AJ, Leung SY, Sohn JJ, Zanetti KA, et al. MicroRNA expression profiles associated with prognosis and therapeutic outcome in colon adenocarcinoma. JAMA. 2008;299:425–36.
Roldo C, Missiaglia E, Hagan JP, Falconi M, et al. MicroRNA expression abnormalities in pancreatic endocrine and acinar tumors are associated with distinctive pathologic features and clinical behavior. J Clin Oncol. 2006;24:4677–84.
Bloomston M, Frankel WL, Petrocca F, Volinia S, et al. MicroRNA expression patterns to differentiate pancreatic adenocarcinoma from normal pancreas and chronic pancreatitis. JAMA. 2007;297:1901–8.
Scott GK, Goga A, Bhaumik D, Berger CE, et al. Coordinate suppression of ERBB2 and ERBB3 by enforced expression of micro-RNA miR-125a or miR-125b. J Biol Chem. 2007;282:1479–86.
Iorio MV, Casalini P, Piovan C, Di Leva G, et al. MicroRNA-205 regulates HER3 in human breast cancer. Cancer Res. 2009;69:2195–200.
Wu H, Zhu S, Mo YY. Suppression of cell growth and invasion by miR-205 in breast cancer. Cell Res. 2009;19:439–48.
Blenkiron C, Goldstein LD, Thorne NP, Spiteri I, et al. MicroRNA expression profiling of human breast cancer identifies new markers of tumor subtype. Genome Biol. 2007;8:R214.
Miller TE, Ghoshal K, Ramaswamy B, Roy S, et al. MicroRNA-221/222 confers tamoxifen resistance in breast cancer by targeting p27Kip1. J Biol Chem. 2008;283:29897–903.
Zhao JJ, Lin J, Yang H, Kong W, et al. MicroRNA-221/222 negatively regulates estrogen receptor alpha and is associated with tamoxifen resistance in breast cancer. J Biol Chem. 2008;283:31079–86.
Di Leva G, Gasparini P, Piovan C, Ngankeu A, et al. MicroRNA cluster 221-222 and estrogen receptor alpha interactions in breast cancer. J Natl Cancer Inst. 2010;102:706–21.
Derynck R, Akhurst RJ, Balmain A. TGF-β signaling in tumor suppression and cancer progression. Nature Genet. 2001;29:117–29.
Bonci D, Coppola V, Musumeci M, Adddario A, et al. The miR-15a-miR-16-1 cluster controls prostate cancer by targeting multiple oncogenic activities. Nature Med. 2008;14:1271–7.
Kota J, Chivukula RR, O’Donnell KA, Wentzel EA, et al. Therapeutic microRNA delivery suppresses tumorigenesis in a murine liver cancer model. Cell. 2009;137:1005–17.
Esteller M. Cancer epigenomics: DNA methylomes and histone-modification maps. Nature Rev Genet. 2007;8:286–98.
Saito Y, Liang G, Egger G, Friedman JM, et al. Specific activation of microRNA-127 with downregulation of the proto-oncogene BCL6 by chromatin-modifying drugs in human cancer cells. Cancer Cell. 2006;9:435–43.
Fabbri M, Garzon R, Cimmino A, Liu Z, et al. MicroRNA family reverts aberrant methylation in lung cancer by targeting DNA methyltransferases 3A and 3B. Proc Natl Acad Sci U S A. 2007;104:15805–10.
Benetti R, Gonzalo S, Jaco I, Munoz P, et al. A mammalian microRNA cluster controls DNA methylation and telomere recombination via Rbl2-dependent regulation of DNA methyltransferases. Nat Struct Mol Biol. 2008;15:268–79.
Sinkkonen L, Hugenschmidt T, Berninger P, Gaidatzis D, et al. MicroRNAs control de novo DNA methylation through regulation of transcriptional repressors in mouse embryonic stem cells. Nat Struct Mol Biol. 2008;15:259–67.
Noonan EJ, Place RF, Pookot D, Basak S, et al. miR-449a targets HDAC-1 and induces growth arrest in prostate cancer. Oncogene. 2009;28:1714–24.
Lowery AJ, Miller N, McNeill RE, Kerin MJ. MicroRNAs as prognostic indicators and therapeutic targets: potential effect on breast cancer management. Clin Cancer Res. 2008;14:360–5.
Wiemer EA. The role of microRNAs in cancer: no small matter. Eur J Cancer. 2007;43:1529–44.
Hatfield S, Ruohola-Baker H. microRNA and stem cell function. Cell Tissue Res. 2008;331:57–66.
Bernstein E, Kim SY, Carmell MA, Murchison EP, et al. Dicer is essential for mouse development. Nat Genet. 2003;35:215–7.
Croce CM, Calin GA. miRNAs, cancer, and stem cell division. Cell. 2005;122:6–7.
Yu F, Yao H, Zhu P, Zhang X, et al. let-7 regulates self renewal and tumorigenicity of breast cancer cells. Cell. 2007;131:1109–23.
Silber J, Lim DA, Petritsch C, Persson AI, et al. miR-124 and miR-137 inhibit proliferation of glioblastoma multiforme cells and induce differentiation of brain tumor stem cells. BMC Med. 2008;6:14–31.
Liu C, Kelnar K, Liu B, Chen X, et al. The microRNA miR-34a inhibits prostate cancer stem cells and metastasis by directly repressing CD44. Nat Med. 2011;17:211–5.
Ibarra I, Erlich Y, Muthuswamy SK, Sachidanandam R, et al. A role for microRNAs in maintenance of mouse mammary epithelial progenitor cells. Genes Dev. 2007;21:3238–43.
Greene SB, Gunaratne PH, Hammond SM, Rosen JM. A putative role for microRNA-205 in mammary epithelial cell progenitors. J Cell Sci. 2010;123:606–18.
Yu F, Deng H, Yao H, Liu Q, et al. Mir-30 reduction maintains self-renewal and inhibits apoptosis in breast tumor initiating cells. Oncogene. 2010;29:4194–204.
Olson P, Lu J, Zhang H, Shai A, et al. MicroRNA dynamics in the stages of tumorigenesis correlate with hallmark capabilities of cancer. Genes Dev. 2009;23:2152–65.
Farazi TA, Horlings HM, Ten Hoeve JJ, Mihailovic A, et al. MicroRNA sequence and expression analysis in breast tumors by deep sequencing. Cancer Res. 2011;71:4443–53.
Baranwal S, Alahari SK. miRNA control of tumor cell invasion and metastasis. Int J Cancer. 2010;126:1283–90.
Ceppi P, Mudduluru G, Kumarswamy R, Rapa I, et al. Loss of miR-200c expression induces an aggressive, invasive, and chemoresistant phenotype in non-small cell lung cancer. Mol Cancer Res. 2010;8:1207–16.
Thiery JP, Sleeman JP. Complex networks orchestrate epithelial-mesenchymal transitions. Natl Rev Mol Cell Biol. 2006;7:131–42.
Thiery JP. Epithelial-mesenchymal transitions in tumor progression. Nat Rev. Cancer. 2002;2:442–54.
Brabletz T, Jung A, Spaderna S, Hlubek F, et al. Opinion: migrating cancer stem cells-an integrated concept of malignant tumor progression. Nat Rev Cancer. 2005;5:744–9.
Lee JM, Dedhar S, Kalluri R, Thompson EW. The epithelial- mesenchymal transition: new insights in signaling, development, and disease. J Cell Biol. 2006;172:973–81.
Hurt EM, Farrar WL. Cancer stem cells: the seeds of metastasis? Mol Interv. 2008;8:140–2.
Visvader JE, Lindeman GJ. Cancer stem cells in solid tumors: accumulating evidence and unresolved questions. Nat Rev Cancer. 2008;8:755–68.
Garofalo M, Croce CM. MicroRNAs: master regulators as potential therapeutics in cancer. Annu Rev Pharmacol Toxicol. 2011;51:25–43.
Adams BD, Guttilla IK, White BA. Involvement of microRNAs in breast cancer. Semin Reprod Med. 2008;26:522–36.
Cochrane DR, Cittelly DM, Howe EN, Spoelstra NS, et al. MicroRNAs link estrogen receptor alpha status and Dicer levels in breast cancer. Horm Cancer. 2010;1:306–19.
Krol J et al. The widespread regulation of microRNAs biogenesis, function and decay. Nat Rev Genet. 2010;11:597–610.
Bhat-Nakshatri P, Wang G, Collins NR, Thomson MJ, et al. Estradiol-regulated microRNAs control estradiol response in breast cancer cells. Nucleic Acids Res. 2009;37:4850–61.
Martello G, Rosato A, Ferrari F, Manfrin A, et al. A MicroRNA targeting dicer for metastasis control. Cell. 2010;141:1195–207.
Ender C, Meister G. Argonaute proteins at a glance. J Cell Sci. 2010;123:1819–23.
Davis-Dusenbery BN, Hata A. Mechanisms of control of microRNA biogenesis. J Biochem. 2010;148:381–92.
Cheng C, Fu X, Alves P, Gerstein M. mRNA expression profiles show differential regulatory effects of microRNAs between estrogen receptor-positive and estrogen receptor-negative breast cancer. Genome Biol. 2009;10:R90.
Adams BD, Claffery KP, White BA. Argonaute-2 expression is regulated by epidermal growth factor receptor and mitogen-activated protein kinase signaling and correlates with a transformed phenotype in breast cancer cells. Endocrinology. 2009;150:14–23.
Ma L, Teruya-Feldstein J, Weinberg RA. Tumour invasion and metastasis initiated by microRNA-10b in breast cancer. Nature. 2007;449:682–8.
Wang L, Wang J. MicroRNA-mediated breast cancer metastasis: from primary site to distant organs. Oncogene. 2011;31:2499–511.
Gregory PA, Bracken CP, Bert AG, Goodall GJ. MicroRNAs as regulators of epithelial-mesenchymal transition. Cell Cycle. 2008;7:3112–8.
Park SM, Gaur AB, Lengyel E, Peter ME. The miR-200 family determines the epithelial phenotype of cancer cells by targeting the E-cadherin repressors ZEB1 and ZEB2. Genes Dev. 2008;22:894–907.
Zhu S, Wu H, Wu F, Nie D, et al. MicroRNA-21 targets tumor suppressor genes in invasion and metastasis. Cell Res. 2008;18:350–9.
Tavazoie SF, Alarcon C, Oskarsson T, Padua D, et al. Endogenous human microRNAs that suppress breast cancer metastasis. Nature. 2008;451:147–52.
Rosenfeld N, Aharonov R, Meiri E, Rosenwald S, et al. MicroRNAs accurately identify cancer tissue origin. Nat Biotechnol. 2008;26:462–9.
Pavlidisa N, Fizazib K. Cancer of unknown primary (CUP). Crit Rev Oncol Hematol. 2008;54:243–50.
Calin GA, Cimmino A, Fabbri M, Ferracin M, et al. MiR-15a and miR-16-1 cluster functions in human leukemia. Proc Natl Acad Sci U S A. 2008;105:5166–71.
Si ML, Zhu S, Wu H, Lu Z, et al. MiR-21-mediated tumor growth. Oncogene. 2007;26:2799–803.
Meng F, Henson R, Lang M, Wehbe H, et al. Involvement of human micro-RNA in growth and response to chemotherapy in human cholangiocarcinoma cell lines. Gastroenterology. 2006;130:2113–29.
Weidhaas JB, Babar I, Nallur SM, Trang P, et al. MicroRNAs as potential agents to alter resistance to cytotoxic anticancer therapy. Cancer Res. 2007;67:11111–6.
Weiler J, Hunziker J, Hall J. Anti-miRNA oligonucleotides (AMOs): Ammunition to target miRNAs implicated in human disease? Gene Ther. 2006;13:496–502.
Orom UA, Kauppinen S, Lund AH. LNA modified oligonucleotides mediate specific inhibition of microRNA function. Gene. 2006;372:137–41.
Krutzfeldt J, Rajewsky N, Braich R, Rajeev KG, et al. Silencing of microRNAs in vivo with ‘antagomirs’. Nature 2005;438:685–689.
Ebert MS, Neilson JR, Sharp PA. MicroRNA sponges: competitive inhibitors of small RNAs in mammalian cells. Nat Methods. 2007;4:721–6.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2014 Springer Science+Business Media New York
About this chapter
Cite this chapter
Thangraju, M., Jain, A. (2014). MicroRNAs in Development and Progression of Breast Cancer. In: Singh, S., Rameshwar, P. (eds) MicroRNA in Development and in the Progression of Cancer. Springer, New York, NY. https://doi.org/10.1007/978-1-4899-8065-6_7
Download citation
DOI: https://doi.org/10.1007/978-1-4899-8065-6_7
Published:
Publisher Name: Springer, New York, NY
Print ISBN: 978-1-4899-8064-9
Online ISBN: 978-1-4899-8065-6
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)