
Abstract
In this chapter, we will focus on physiological regulators of activated immune cells in cancerous tissue microenvironments. This consideration started when we were contemplating the molecular mechanism that would be responsible for the so-called Hellstrom Paradox. Indeed, it was not explained why cancer patients often have tumor-recognizing effector T cells without having tumor rejection. The latest great advances in identification of various immunological negative regulators of immune response still left room for tumor defense by physiological inhibitors of antitumor T and natural killer (NK) cells. We started by assuming that cancerous tissues could be misguidedly protected by the same mechanism, which saves lives by protecting vital tissues from collateral damage by overactive immune cells during the antipathogen immune response. In our search for a mechanism that protects tissues from collateral damage, we first focused on intracellular cyclic adenosine monophosphate (cAMP) which was long known to be immunosuppressive. It was important to identify which of the many Gs protein-coupled receptors is actually physiologically responsible for inhibition of immune response in tumor microenvironment. Levels of extracellular adenosine are high in inflamed and cancerous tissues corresponding to local hypoxia. A2A and A2B subtypes of adenosine receptor, which are coupled to cAMP-elevating Gs protein, are predominantly expressed in immune cells. Indeed, extracellular adenosine endogenously generated by degradation of adenosine triphosphate (ATP) could suppress immune response and immunoregulation by adenosine was notable in tumor microenvironment. Blockade of the hypoxia-adenosinergic immunosuppression may be a promising approach to eradicate cancer, especially when it is combined with adoptive immunotherapy or cancer vaccine.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others

Keywords
- Tumor microenvironment
- Hypoxia
- Adenosine
- A2A adenosine receptor
- A2B adenosine receptor
- T cell
- Regulatory T cell
- Myeloid-derived suppressor cells
- Adoptive immunotherapy
- Cancer vaccine
1 Hypoxia
1.1 Immunosuppression in Hypoxic Tissue Microenvironment
Tissue oxygen levels are not uniformly distributed in the same organ. Levels of oxygenation depend on diffusion of oxygen from blood vessels, distal area being more hypoxic [1], [2]. Pathological conditions in inflamed tissue and tumor are related to the formation of even less oxygenated microenvironments [2], [3]. Tissue inflammation inflicting damage to blood vessels reduces oxygen supply [3]. Combined with increased oxygen demand by accumulated inflammatory cells, inflamed tissue becomes deeply hypoxic [4].
Tumors are also often hypoxic for reasons different from those causing hypoxia in inflamed tissues. Oxygen demand is high in tumors because of aggressive proliferation of tumor cells. In addition, disorganized blood vessel formation in tumors is making blood flow sluggish, and therefore oxygen supply is low [5], [6]. Interface between tumor cells is tightly packed, preventing oxygen diffusion to the inside of tumor tissue [6], [7]. Hypoxia in tumors correlates with poor prognosis because hypoxic tumors are refractory to radiotherapy and chemotherapy [5]–[7]. Moreover, hypoxia is conductive to the establishment of tumor microenvironment , which is potentially suppressive to antitumor immune activities [8]–[10]. Hypoxia has been shown to suppress immune functions of T cells , natural killer (NK) cells, and antigen-presenting cells (APCs).
1.2 Suppression of T-cell Immunity Under Hypoxia
In vitro T-cell activation under hypoxia impairs proliferation of activated T cells and their effector functions such as cytotoxicity and cytokine production [1], [11]–[13]. Hypoxia blocks Ca2+ increase after stimulation of T-cell receptor (TCR) [14]. Whole body exposure of mice to hypoxic atmosphere inhibited T-cell activation in vivo [15]. In that study, the extent of T-cell activation correlated with the levels of oxygenation in the spleen. Indeed, degrees of T-cell activation were attenuated in poorly oxygenated environment as detected by covalent binding of nitroimidazole compound, Hypoxyprobe-1 [15].
Exposure to hypoxia induces cellular stress response to adapt energy deprivation. One of the most important events is stabilization of hypoxia-inducible factor-1α (HIF-1α), which upregulates glycolytic enzymes, angiogenesis, and erythropoiesis [16], [17]. In T cells , however, HIF-1α was reported to diminish TCR signaling [14]. Higher interferon gamma (IFN-γ) production and stronger cytotoxicity in T cells lacking HIF-1α suggest a negative regulatory role of HIF-1α [18], [19] .
2 Adenosine
2.1 Formation of Extracellular Adenosine in Tumor
Tumors have been found to contain high levels of extracellular adenosine [20], [21], one of the potential immunosuppressive molecules. Enzymatic degradation of extracellular adenosine triphosphate (ATP) leads to an increase of extracellular adenosine. 5’-Ecto-nucleotidases are responsible for this metabolism: CD39 converting ATP to adenosine diphosphate (ADP) and to adenosine monophosphate (AMP) and CD73 catalyzing adenosine formation from AMP. CD73-deficient mice maintain extracellular adenosine concentration at low levels physiologically and even after the induction of inflammation, suggesting that conversion of adenine nucleotides accounts for a large part of extracellular adenosine formation [22]–[24]. Extracellular adenosine may be removed by further metabolism to inosine by adenosine deaminase and by cellular uptake through nucleoside transporters. Adenosine kinase in the intracellular compartment metabolizes adenosine to AMP, making room for further adenosine uptake. Inhibitors of adenosine deaminase, nucleoside transporters, and adenosine kinase increase extracellular adenosine levels, indicating significance of these mechanisms in the regulation of extracellular adenosine [25]–[28].
Increase of extracellular adenosine levels has been observed during inflammation [29]–[32]. By causing tissue injury, inflammation is able to increase extracellular content of adenine nucleotides and facilitate metabolism to produce adenosine. Cellular damage is considered to cause leakage of adenine nucleotides to extracellular space [4]. Increased release of adenine nucleotides was reported in activated neutrophils and irritant-treated keratinocytes [33], [34].
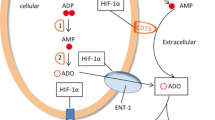
Subsequent to extracellular increase of adenine nucleotides, tissue hypoxia facilitates conversion to adenosine through upregulation of CD39 and CD73 levels [35], [36]. In parallel, hypoxia inhibits adenosine kinase [37]–[39]. Thus, tissue hypoxia is conductive to extracellular accumulation of adenosine [40] by increasing adenosine formation and by suppressing its removal. Intratumoral hypoxia caused by poor oxygen supply in spite of increasing demand of oxygen favors adenosine accumulation in the tumor. Various tumor cells expressing CD73 also contribute to the production of extracellular adenosine [41]–[44]. These findings correspond to the increase of adenosine levels in tumor tissue (Fig. 14.1).

The enhancement of extracellular adenosine generation in hypoxic tumor microenvironment. Adenine nucleosides (ATP, ADP, and AMP) in the extracellular compartment are catabolized to adenosine by the activities of CD39 and CD73 ecto-enzymes. In normoxic microenvironments (oxygen tension > ~ 3 %), the concentration of extracellular adenosine is kept low by, e.g., adenosine kinase and adenosine deaminase (not shown in this figure) and cellular uptake is regulated through nucleoside transporter (NT). However, hypoxia in tumor microenvironment can change the balance of extracellular adenosine formation and removal in favor of the accumulation of extracellular adenosine. Upregulation of CD39 and CD73 under hypoxia accelerates extracellular formation of adenosine. In addition, hypoxia down-regulates adenosine kinase (AK) and impairs removal of adenosine
2.2 Extracellular Adenosine as an Immunoregulatory Molecule
Adenosine is abundant in cells for its use in energy and nucleic acid metabolism. But its presence in the extracellular compartment results in distinctive effects on the cardiovascular system, neuronal cells, kidney, fat tissue, platelets, and leukocytes. In the mid-70s, incubation with adenosine was known to induce cyclic adenosine monophosphate (cAMP) in various cell types including T cells. The increase of cAMP in T cells led to speculation of receptor-mediated signaling, but at that time, the function of adenosine signaling was discussed in regard to energy production [45]–[47]. Effects of adenosine on T-cell function were reported in the 1980s, showing the inhibition of T-cell proliferation, interleukin-2 (IL-2) production, and B-cell helper function [48]–[50]. In parallel, the increase of cAMP was demonstrated to suppress IL-2 production, B-cell helper function, and cytotoxicity of T cells [51], [52]. These early studies implied that extracellular adenosine is inhibitory to T cells through the induction of cAMP.
Meanwhile, the presence of multiple adenosine receptor subtypes was speculated based on the different selectivity of synthetic adenosine derivatives [53], [54]. Since the first cloning of adenosine receptor in 1989, four different adenosine receptors have been identified to date. Among these, A2A and A2B adenosine receptors are cAMP-inducing receptors coupled to Gs protein, while A1 and A3 adenosine receptors are coupled with Gi protein to reduce cAMP levels [55], [56]. Indeed, adenosine A2A receptor (A2AR) is the predominant subtype in T cells [57, 58].
Similar to the inhibitory effects on polymorphonuclear cells and macrophages, suppression of T-cell activation by A2AR agonist was shown in two papers published in 1997 [57], [59]. In these papers, treatment with A2AR agonist resulted in decreased T-cell proliferation, downregulation of activation marker CD25, and decreased cytotoxicity with a reduced level of Fas ligand expression.
2.3 Mechanism of A2AR-mediated T-cell Inhibition
Subsequent studies revealed more details of T-cell suppression due to A2AR signaling. A2AR stimulation at the time of T-cell activation significantly reduced proliferation of T cells and their effector functions including cytotoxicity and production of cytokines such as IL-2, IFN-γ, and TNF-α [60]–[62]. Both CD4+ and CD8+ T cells are susceptible to this mechanism [62]. A2AR is also expressed in human T cells , and A2AR agonist was shown to be suppressive to effector functions of human T cells such as cytokine production and cytotoxicity [63], [64]. Inhibition of T-cell activation correlates well with the interruption of TCR signaling by A2AR stimulation [61], [65], [66]. A2AR agonist diminished phosphorylation of ZAP70 after TCR stimulation together with downstream phosphorylation of ERK. Inhibition of Akt phosphorylation by A2AR agonist also suggests interruption of the phosphatidylinositol-3-kinase pathway. Since A2AR stimulation induces cAMP, protein kinase A-dependent phosphorylation of COOH-terminal Src kinase may inhibit Lck activation in the early stage of TCR signaling [67].
The helper function of CD4+ T cells is important in activating both cellular immunity and humoral immunity depending on functional differentiation of CD4+ T cells into T helper 1 (Th1) and Th2 cells. Although A2AR agonist can inhibit development of both Th1 and Th2 cells [68], large declines in IFN-γ and IL-2 production by exposure to A2AR agonist indicated a strong suppression of Th1-type cellular immune responses [60]–[62]. Inhibition of Th1 cell development is consistent with changes in cytokine production from APCs in which A2AR agonist diminishes IL-12, but augments IL-10 [69], [70].
A2AR stimulation not only blocks activation of T cells immediately, but also elicits sustained inhibition of T-cell activities by inducing activated T cells with impaired effector functions. As mentioned above, T-cell activation in the presence of A2AR agonist reduced IFN-γ production from activated cells. However, when these cells were restimulated after the removal of A2AR agonist, IFN-γ-producing activity was still less than normal activated T cells [61], [62]. The induction of such anergic T cells suggests that the T-cell inhibitory effect of adenosine may be persistent even after clearance of adenosine (Fig. 14.2a). This property of A2AR signaling may be relevant to the memory of exposure to extracellular adenosine, where persistent elevation of cAMP was observed after transient exposure to adenosine [57].

Early stages of priming and activation of resting T cells are highly susceptible to the A2AR-mediated immunosuppression. a When resting T cells are stimulated in the presence of A2AR agonist, the activated T cells produce very low levels of IFN-γ. The impairment of IFN-γ-producing activity persists even after removal of A2AR agonist. b A2AR agonist can inhibit IFN-γ production from already activated T cells, but only in the very presence of A2AR agonist. After the removal of A2AR agonist, IFN-γ production from T cells returned to normal levels
Comparison between resting and activated T cells showed that already activated T cells are relatively resistant to A2AR-mediated inhibition (Fig. 14.2b). When activated T cells were restimulated, A2AR agonist still inhibited T-cell proliferation and IFN-γ production. After removal of the A2AR agonist, however, the effector function of these T cells came back to the same levels as in activated T cells that were never cultured with A2AR agonist [71], [72]. Therefore, although A2AR agonists can inhibit activities of the already activated T cells, the inhibitory effect did not persist after its removal. This result indicates that A2AR stimulation does not switch fully functional effector T cells to the anergic phenotype. The inhibition of activated effector cells by extracellular adenosine is highly territorial: only in extracellular adenosine-rich tissue microenvironment, but not in the neighboring adenosine-low microenvironment.
2.4 Adenosine Promotes Immunosuppressive Activity of Regulatory T Cells
Regulatory T cells (Treg) were initially identified as CD4+ T cells constitutively expressing CD25 at high levels . Activation of Treg follows the normal scheme of T-cell activation, but activated Treg spontaneously inhibit activation of other effector T cells. Since the lack of immunoregulation by Treg causes severe autoimmune diseases, Treg are indispensable for the control of immune activation against self-antigens in peripheral tissues [73], [74].
It was suggested that Treg development and effector functions are under control of the hypoxia-adenosinergic pathway, and the model was proposed to potentially unify the diverse functions of Treg [75]. A large body of published data are consistent with the model where Treg development and their immunoregulatory activity are mediated by the interplay of the cAMP-elevating adenosine receptors, HIF-1α, and subsequent cAMP response element (CRE)- and hypoxia response element (HRE)-mediated transcription in Treg and effector cells. Accordingly, HRE- and CRE-driven activities of Treg may be required to achieve a maximal level of immune suppression .
As a subset of T cells, Treg express functional A2AR as well [76]. In contrast to negative effects on activities of most T cells, A2AR stimulation rather promotes immunoregulatory activity of Treg [76]. In isolated spleen cells, containing both effector T cells and Treg at physiological ratio, T-cell stimulation in the presence of A2AR agonist inhibited activation of effector T cells but increased Treg population. A2AR stimulation not only increased the number of Treg but also augmented the T-cell inhibitory activity of Treg. Corresponding to the enhanced immunoregulatory activity, A2AR agonist upregulated cytotoxic T-lymphocyte antigen 4 (CTLA-4) expression in these Treg. The importance of CTLA-4 in the immunosuppressive activity of Treg was demonstrated by systemic lymphoproliferation and autoimmune disease in mice with Treg-specific deletion of CTLA-4 [77].
Adoptive transfer of Treg reduced ischemia-reperfusion injury in vivo, but pretreatment of Treg with A2AR agonist before transfer augmented the efficacy of this treatment [78]. Moreover, A2AR-deficient Treg were less effective compared to wild-type Treg, suggesting the in vivo significance of A2AR signaling in regulating the immunosuppressive activity of Treg.
Treg may develop either during T-cell maturation in the thymus (natural Treg) or in the peripherals by functional differentiation of mature T cells (inducible Treg). Analysis of A2AR-dependent Treg expansion showed the involvement of natural Treg proliferation and induction of new Treg [76]. The promotion of inducible Treg has been speculated from an upregulation of FoxP3 mRNA in T-cell culture treated with A2AR agonist [61]. FoxP3 is a transcription factor involved in the regulation of immunosuppressive activity. It was further confirmed that A2AR agonist expanded transforming growth factor beta (TGF-β)-inducible Treg both in vitro and in vivo [79]. Besides A2AR, adenosine 2B receptor (A2BR) may be also involved in the increase of inducible Treg [80] .
Upstream adenosine receptor signaling, hypoxia may be also involved in the regulation of Treg. Indeed, FoxP3 is inducible by hypoxia in T cells, and HIF-1α mediates hypoxic induction of FoxP3 [81], [82]. However, subsequent studies provided evidence for complicated role of HIF-1α in the regulation of Treg. Initially, studies using mice with HIF-1α-deficient T cells demonstrated HIF-1α-mediated downregulation of Treg and reciprocal increase of Th17 [83], [84]. In contrast, more recent papers showed that hypoxia induces FoxP3 and increases Treg abundance [82]. In this setting, HIF-1α is necessary for optimal immunosuppressive activity of Treg. Nonetheless, local oxygen levels may be an important regulator of Treg. Hypoxia in tumors may be relevant to increase of Treg population in tumor microenvironment .
While adenosine can control the immunoregulatory activity of Treg, Treg may utilize adenosine in their mechanism of immunosuppression. Treg express CD39 and CD73, extracellular nucleotidases that catalyze degradation of ATP to adenosine and increase extracellular adenosine concentration [85]–[87]. The produced adenosine, in turn, interacts with A2AR and blocks activation of T cells . This mechanism may explain why A2AR-deficient effector T cells were resistant to immunoregulatory cells [88] and why CD73-deficient Treg were less effective in inhibiting ischemia-reperfusion injury [78]. Furthermore, adenosine produced from Treg may autonomously target Treg to enhance their activity.
Thus, A2AR-mediated signaling promotes immunoregulation by Treg both quantitatively and qualitatively. The outcome of this effect is consistent with the direct inhibition of effector T-cell activation by A2AR-mediated signaling. In addition to the direct inhibition of T-cell activation, A2AR agonist also provides longer lasting T-cell inhibition by at least two different mechanisms. When present at the time of T-cell priming, A2AR agonist induces longer lasting inhibition of antigen-specific T-cell response by developing anergic effector T cells. In addition, when enforced in the presence of A2AR agonist, Treg may provide long-lasting suppression of antigen-specific T-cell response ([76], [78]; Fig. 14.3) .

Regulation of T cells’ effector functions by extracellular adenosine. Signals from A2AR on T cell’s surface directly inhibit the TCR-mediated activation. As a result, A2AR stimulation diminishes various T-cell functions including proliferation, cytokine production, and cytotoxicity. The impairment of effector functions in activated T cells can persist even after removal of agonist, suggesting the development of anergic T cells. Adenosine also indirectly influences T-cell activation by inducing alternative activation of antigen (Ag)-presenting cells. Macrophages and dendritic cells (DCs) activated in the presence of adenosine produce less IL-12 and more IL-10, changing cytokine milieu for functional differentiation of T cells. Moreover, A2AR stimulation promotes Treg expansion and their immunosuppressive function. Thus, adenosine signaling suppresses T-cell activation both directly and indirectly. Therefore, T-cell inhibitory effect of A2AR/A2BR-mediated immunosuppressive signaling is both immediate (i.e., by directly inhibiting the T-cell activation signal) and long-lasting (anergic T cells and Treg)
2.5 Myeloid-derived Suppressor Cells
Together with Treg, myeloid-derived suppressor cells (MDSCs) represent major immunoregulatory cells contributing to immunosuppressive environment in tumors [89] . Adenosine promotes expansion of MDSCs in A2BR-dependent manner. Indeed, the number of tumor-infiltrated MDSCs is low in A2BR-deficient mice [90]. Hypoxia also promotes differentiation and function of MDSCs [91], suggesting significance of the hypoxia-adenosine pathway in regulating MDSCs in tumors.
2.6 Antigen-presenting Cells
Adenosine receptor stimulation of APCs inhibits T-cell stimulating activity. A2AR agonists inhibit IL-12 production but induce IL-10 from dendritic cells [69], [70]. This change in cytokine milieu is suppressive to the induction of Th1 cells and therefore inhibitory to cellular immune responses. While A2AR stimulation suppresses activation of APCs to proinflammatory phenotype, adenosine induces alternative activation of APCs via A2BR [69], [70], [92], [93]. Alternative activation induces arginase, indoleamine-2,3-dioxygenase (IDO), TGF-β, and COX-2 in APCs, and such APCs inhibit optimal activation of T cells [94]. A2AR agonist also induces VEGF from macrophages [95], [96], suggesting adenosine switches APCs to tolerogenic and angiogenic phenotype.
Dendritic cells exposed to hypoxia express lesser levels of major histocompatibility complex (MHC) and co-stimulatory molecules [97], [98]. Hypoxia inhibits phagocytosis by dendritic cells, decreasing capture of antigen [99]. Therefore, those dendritic cells under hypoxia have impaired T-cell stimulatory capacity as APCs.
2.7 NK Cells
Adenosine suppresses NK cell activities. A2AR agonists are suppressive to IFN-γ production and cytotoxicity of lymphokine-activated killer (LAK) cells and NK cells from mice and humans [100]–[102].
Hypoxia also suppresses activity of NK cells [103]. Closely relevant to the inhibition of NK cell-dependent cytotoxicity, hypoxia downregulates NKG2D ligands including MHC class I chain-related molecules on tumor cells [104], [105]. Since NKG2D is an activating receptor of NK cells, hypoxic tumor cells are induced to be resistant to NK cell-dependent cytotoxicity. These observations suggest biological significance of oxygen tension in the regulation of antitumor immune responses.
3 Endogenous Adenosine as a Physiological Regulator of Immune Response
3.1 A2AR
A2AR stimulation suppresses immune responses through Gs protein-mediated cAMP increase. However, there are many other cAMP-elevating receptors on the surface of immune cells that can transduce immunosuppressive signals when activated pharmacologically. A brief inventory of such molecules includes prostaglandin E2, adrenaline, histamine, and small peptides such as vasoactive intestinal peptide (VIP) and pituitary adenylate cyclase activating peptide (PACAP). Adenosine has been regarded as just one of such anti-inflammatory small molecules, but in recent years, recognition of adenosine became prominent because of its nonredundancy as an endogenously produced immunoregulator [106]–[108].
One of the most important features of A2AR is its critical role in physiological regulation of immune responses. Acute hepatitis induction in A2AR-deficient mice resulted in remarkable exaggeration of liver damage and proinflammatory cytokine levels [109], [110]. The result demonstrated that (1) endogenously produced adenosine can control the intensity of immune response through A2AR and (2) A2AR signaling is critical to stop inflammation because other immunoregulatory mechanisms could not compensate for the lack of A2AR-mediated immunosuppression.
Adenosine-dependent immunoregulation may represent the tissue’s negative feedback response to overwhelming inflammation [106], [110]. Tissue damage inflicted by proinflammatory activities triggers an accumulation of extracellular adenosine . Indeed, an increase of adenosine levels was observed during inflammation [29]–[32]. Tissue hypoxia and nucleotidase activities of CD39 and CD73 are responsible, at least in part, for the increase in extracellular adenosine [22]–[24]. The increased adenosine transmits a signal to immune cells through A2AR to stop proinflammatory activities and prevent further tissue damage. Interruption of this sequence, e.g., A2AR-deficiency and A2AR antagonism, means loss of a brake on inflammation. Exaggerated inflammation in A2AR-deficient mice was demonstrated in various tissues and in various causes of inflammation [32], [111]–[114], suggesting that the adenosine–A2AR system is a universal mechanism in the vital body to prevent excessive tissue damage .
This discovery offered a solution to a clinically important issue that, as opposed to recruitment of anti-inflammatory effects by targeting A2AR with agonists, it is possible to enhance inflammation by blocking the action of endogenous adenosine by A2AR antagonist. Intake of A2AR antagonists may be detrimental to inflammatory disorders; however, we may take advantage of this mechanism in the treatment of cancer.
3.2 A2BR
Another Gs protein-coupled adenosine receptor is adenosine A2B receptor (A2BR) . Affinity of adenosine to A2BR is lower than A2AR; however, local adenosine levels in hypoxic tissue can be high enough to stimulate A2BR [115], [116]. A2BR is expressed on macrophages, dendritic cells, endothelial cells, epithelial cells, mast cells, and fibroblasts, and it has distinctive effects on inflammatory responses [115], [116]. A2BR agonist was shown to block inflammatory tissue injury in experimental models. Exacerbation of colitis, lung inflammation, and ischemia-reperfusion injury in A2BR-deficient mice suggests pathophysiological significance of endogenous adenosine signaling through A2BR [117]–[121]. Since A2BR stimulation changes the functions of macrophages and dendritic cells as APCs, T cells may receive indirect immunoregulatory effects from A2BR [92], [93], [115], [116]. Thus, increase of extracellular adenosine triggers anti-inflammatory negative feedback responses via A2AR and A2BR. The adenosine–A2AR/A2BR pathway may be vital as an immunoregulatory mechanism in tumor .
4 Cancer
Tremendous efforts by tumor immunologists have significantly advanced the understanding of tumor-associated antigens and improved induction of effector T cells recognizing tumor cells as foreign [122], [123]. It also became clear that the immunosuppressive environment in tumors is a potential problem in tumor eradication by immune cells. Tumors often have infiltration of T cells that can be reactive against the tumor cells, but the tumor-infiltrated T cells are inactive in attacking the tumor in vivo. In mice manipulated to express the same antigen in both normal and tumor tissues, the same effector T cells were disabled only in tumor [124]–[126]. Such studies provide a direct evidence for the existence of potentially immunosuppressive tumor microenvironment . Tumors may employ various mechanisms to evade immune response, e.g., Treg, MDSCs, anti-inflammatory cytokines, and IDO [9], [127]. Advances in T-cell technology developed methods of inducing antitumor effector T cells. However, efficacy of these antitumor T cells may be limited if they are sensitive to immunosuppression in tumor microenvironment. Disengagement of antitumor effectors from immunosuppressive mechanism in tumor will significantly improve the outcome of tumor immunotherapy.
Hypoxia , which is frequently observed in tumors, may play a role in the establishment of immunosuppressive environment. Hypoxia is conductive to the increase of extracellular adenosine levels, and indeed high levels of extracellular adenosine were observed in tumors [20], [21]. Various effects of hypoxia in vivo and in vitro are mediated by the interaction of extracellular adenosine with A2AR [32], [40, 128]–[132]. There is a similarity between tumor-infiltrated T cells and T cells activated in the presence of adenosine in terms of preferential suppression of effector functions [62]. Thus, adenosine may represent one of the potentially immunosuppressive mechanisms in tumors. This concept was established in a tumor inoculation study in which A2AR-deficient mice, but not wild-type mice, demonstrated regression in growing tumors [21]. Improvement of T cell-mediated tumor eradication upon inactivation of A2AR suggests nonredundance of the adenosine–A2AR pathway in the immunosuppressive tumor microenvironment . Tumors protect themselves utilizing the body’s common rule: You shall not take vengeance when you see adenosine .
The enhanced tumor regression in A2AR-deficient mice suggested that A2AR-antagonists might be useful to break immunosuppression in tumors and improve tumor immunotherapy. Indeed, A2AR antagonists such as caffeine, ZM241385, and SCH58261 blocked tumor growth by promoting antitumor immune responses [21], [42], [133]. Significant reduction of intratumoral blood vessels in A2AR antagonist-treated mice suggests that the treatment not only enhances antitumor immune response but also blocks adenosine-induced angiogenesis in tumors [21]. The countermeasure to immunosuppression in tumors in conjunction with successful induction of antitumor effector T cells may significantly improve the outcome of tumor immunotherapy.
In addition to A2AR, A2BR also participates in the protective mechanism of tumor against immune response. Retardation of tumor growth was observed in A2BR-deficient mice [134]. Treatment with A2BR antagonist is also inhibitory to tumor growth in wild-type mice, but not in T cell-deficient mice [135]. Enhanced T-cell infiltration into the tumor by A2BR antagonist suggests that extracellular adenosine in tumor discourages antitumor immune response through both A2AR and A2BR.
The critical role of adenosine-dependent immunosuppression in tumors was also demonstrated by the promotion of antitumor immunity in the absence of CD73 [136], [137]. The lack of CD73, ecto-nucleotidase, sharply decreases extracellular adenosine formation and promotes proinflammatory responses [22]–[24]. Neutralization of CD73 by the injection of antibody inhibited tumor growth and promoted antitumor immune response [42], [138]. CD73 expression on tumor cells plays an important role in immunosuppression and tumor metastasis. Indeed, tumor cells lacking CD73 are susceptible to antitumor immunity [42], [43]. Not only CD73 expression on tumor cells but also CD73 expression on normal cells plays a significant role. Inoculated tumors grow slower in CD73-deficient mice because of stronger antitumor T-cell response [139]. Moreover, CD73 deficiency resulted in the inhibition of carcinogenesis thanks to T cell- and NK cell-dependent immune response [140]. These studies suggest that, besides blockade of adenosine signaling by adenosine receptor antagonists, prevention of extracellular adenosine formation by targeting CD73 may be a promising countermeasure to immunosuppressive tumor microenvironment .
5 Natural A2AR Antagonists: Caffeine and Theophylline
Caffeine and theophylline are representative nature-derived methylxanthines, and they are the most widely consumed A2AR antagonists in the form of beverage, food, and medication. It is known that the psychostimulatory effect of caffeine is attributable to antagonism of the adenosine–A2AR interaction [141], [142]. Indeed, caffeine exacerbated inflammatory tissue damage in experimental acute hepatitis by blocking A2AR [133], [143]. While caffeine and theophylline block A2AR-mediated cAMP increase, a high concentration of these compounds actually increase cAMP levels by inhibiting cAMP phosphodiesterase. Therefore, while low doses of caffeine exacerbate inflammatory tissue damage, caffeine can be anti-inflammatory at high doses [133], [143]. Normal caffeine consumption in humans raises caffeine concentration enough to antagonize A2AR [141], [144], [145]. Since anti-inflammatory high dose may not be reproduced by normal caffeine consumption in humans, the immune-enhancing effect will be clinically more relevant.
In tumor immunotherapy, proinflammatory action of natural adenosine receptor antagonists may be beneficial in promoting antitumor immune response. Co-treatment with caffeine significantly improved tumor eradication by endogenously developed and adoptively transferred antitumor T cells [21]. The enhancement of antitumor activity by caffeine may be relevant to some epidemiological studies that have suggested inverse association between cancer incidence and coffee consumption. The statistics suggest that coffee consumption dose-dependently decreased incidence of breast, liver, colon, lung, skin, and endometrial cancer [146]–[154].
6 Conclusion
Hypoxia in tumors may be implicated to the establishment of immunosuppressive environment. Hypoxia inhibits diverse antitumor immune responses at least in part by upregulation of extracellular adenosine. Adenosine stops antitumor immune response through A2AR and A2BR on immune effector cells. This direct action of adenosine can immediately suppress immune responses in tumor microenvironment. Adenosine evokes longer lasting immunoregulation, which persists in immune cells even after the disappearance of adenosine. Cell activation in the presence of adenosine induces anergic T cells and alternative activation of APCs.
Furthermore, adenosine promotes cellular immunosuppressive activities. Adenosine promotes expansion of Treg and their immunoregulatory activity. MDSCs were also shown to increase in response to adenosine and hypoxia. The increase of professional immunoregulatory cells may be an important component of tumor microenvironment, which is harsh to immune effectors. Thus, the hypoxia-adenosine pathway involves direct inhibition of antitumor effector cells and long-term effect by developing tumor microenvironment favoring immunosuppression.
Treatment with adenosine receptor antagonist and CD73 inhibitor may be a promising approach to improve antitumor immunity. Since this treatment is compensatory to the current approach that focuses on the numerical increase of antitumor T cells, it will be more efficacious when combined with cancer vaccines and adoptive immunotherapy [21], [123], [155]. In cancer adoptive immunotherapy, downregulation of A2AR on the antitumor T cells is expected to promote their efficacy in vivo. In addition to A2AR antagonist treatment after cell transfer, transfer of cells that were created to be insensitive to adenosine may also be worth exploring [21], [72].
References
Caldwell CC, Kojima H, Lukashev D, Armstrong J, Farber M, Apasov SG, Sitkovsky MV (2001) Differential effects of physiologically relevant hypoxic conditions on T lymphocyte development and effector functions. J Immunol 167(11):6140–6149
Braun RD, Lanzen JL, Snyder SA, Dewhirst MW (2001) Comparison of tumor and normal tissue oxygen tension measurements using OxyLite or microelectrodes in rodents. Am J Physiol Heart Circ Physiol 280(6):H2533–2544
Karhausen J, Haase VH, Colgan SP (2005) Inflammatory hypoxia: role of hypoxia-inducible factor. Cell Cycle 4(2):256–258
Kominsky DJ, Campbell EL, Colgan SP (2010) Metabolic shifts in immunity and inflammation. J Immunol 184(8):4062–4068. doi:10.4049/jimmunol.0903002
Vaupel P, Mayer A (2007) Hypoxia in cancer: significance and impact on clinical outcome. Cancer Metastasis Rev 26(2):225–239. doi:10.1007/s10555-007-9055-1
Dewhirst MW, Cao Y, Moeller B (2008) Cycling hypoxia and free radicals regulate angiogenesis and radiotherapy response. Nat Rev Cancer 8(6):425–437. doi:10.1038/nrc2397
Minchinton AI, Tannock IF (2006) Drug penetration in solid tumours. Nat Rev Cancer 6(8):583–592. doi:10.1038/nrc1893
Finn OJ (2008) Cancer immunology. N Eng J Med 358(25):2704–2715. doi:10.1056/NEJMra072739
Mellor AL, Munn DH (2008) Creating immune privilege: active local suppression that benefits friends, but protects foes. Nature reviews. Immunology 8(1):74–80. doi:10.1038/nri2233
Sitkovsky MV, Kjaergaard J, Lukashev D, Ohta A (2008) Hypoxia-adenosinergic immunosuppression: tumor protection by T regulatory cells and cancerous tissue hypoxia. Clin Cancer Res 14(19):5947–5952. doi:10.1158/1078-0432.CCR-08-0229
Loeffler DA, Juneau PL, Masserant S (1992) Influence of tumour physico-chemical conditions on interleukin-2-stimulated lymphocyte proliferation. Br J Cancer 66(4):619–622
Naldini A, Carraro F, Silvestri S, Bocci V (1997) Hypoxia affects cytokine production and proliferative responses by human peripheral mononuclear cells. J Cell Physiol 173(3):335–342. doi:10.1002/(SICI)1097-4652(199712)173:3<335::AID-JCP5>3.0.CO;2-O
Atkuri KR, Herzenberg LA, Herzenberg LA (2005) Culturing at atmospheric oxygen levels impacts lymphocyte function. Proc Natl Acad Sci U S A 102(10):3756–3759. doi:10.1073/pnas.0409910102
Neumann AK, Yang J, Biju MP, Joseph SK, Johnson RS, Haase VH, Freedman BD, Turka LA (2005) Hypoxia inducible factor 1 alpha regulates T cell receptor signal transduction. Proc Natl Acad Sci U S A 102(47):17071–17076. doi:10.1073/pnas.0506070102
Ohta A, Diwanji R, Kini R, Subramanian M, Ohta A, Sitkovsky M (2011) In vivo T cell activation in lymphoid tissues is inhibited in the oxygen-poor microenvironment. Front Immunol 2:27. doi:10.3389/fimmu.2011.00027
Semenza GL (2007) Oxygen-dependent regulation of mitochondrial respiration by hypoxia-inducible factor 1. Biochem J 405(1):1–9. doi:10.1042/BJ20070389
Majmundar AJ, Wong WJ, Simon MC (2010) Hypoxia-inducible factors and the response to hypoxic stress. Mol Cell 40(2):294–309. doi:10.1016/j.molcel.2010.09.022
Lukashev D, Klebanov B, Kojima H, Grinberg A, Ohta A, Berenfeld L, Wenger RH, Ohta A, Sitkovsky M (2006) Cutting edge: hypoxia-inducible factor 1alpha and its activation-inducible short isoform I.1 negatively regulate functions of CD4+ and CD8+ T lymphocytes. J Immunol 177(8):4962–4965
Guo J, Lu W, Shimoda LA, Semenza GL, Georas SN (2009) Enhanced interferon-gamma gene expression in T Cells and reduced ovalbumin-dependent lung eosinophilia in hypoxia-inducible factor-1-alpha-deficient mice. Int Arch Allergy Immunol 149(2):98–102. doi:10.1159/000189191
Blay J, White TD, Hoskin DW (1997) The extracellular fluid of solid carcinomas contains immunosuppressive concentrations of adenosine. Cancer Res 57(13):2602–2605
Ohta A, Gorelik E, Prasad SJ, Ronchese F, Lukashev D, Wong MK, Huang X, Caldwell S, Liu K, Smith P, Chen JF, Jackson EK, Apasov S, Abrams S, Sitkovsky M (2006) A2A adenosine receptor protects tumors from antitumor T cells. Proc Natl Acad Sci U S A 103(35):13132–13137. doi:10.1073/pnas.0605251103
Colgan SP, Eltzschig HK, Eckle T, Thompson LF (2006) Physiological roles for ecto-5’-nucleotidase (CD73). Purinergic Signal 2(2):351–360. doi:10.1007/s11302-005-5302-5
Volmer JB, Thompson LF, Blackburn MR (2006) Ecto-5’-nucleotidase (CD73)-mediated adenosine production is tissue protective in a model of bleomycin-induced lung injury. J Immunol 176(7):4449–4458
Grenz A, Zhang H, Weingart J, von Wietersheim S, Eckle T, Schnermann J, Kohle C, Kloor D, Gleiter CH, Vallon V, Eltzschig HK, Osswald H (2007) Lack of effect of extracellular adenosine generation and signaling on renal erythropoietin secretion during hypoxia. Am J Physiol Renal Physio 293(5):F1501–1511. doi:10.1152/ajprenal.00243.2007
Resta R, Hooker SW, Laurent AB, Jamshedur Rahman SM, Franklin M, Knudsen TB, Nadon NL, Thompson LF (1997) Insights into thymic purine metabolism and adenosine deaminase deficiency revealed by transgenic mice overexpressing ecto-5’-nucleotidase (CD73). J Clin Invest 99(4):676–683. doi:10.1172/JCI119211
Tofovic SP, Zacharia L, Carcillo JA, Jackson EK (2001) Inhibition of adenosine deaminase attenuates endotoxin-induced release of cytokines in vivo in rats. Shock 16(3):196–202
Jarvis MF, Yu H, McGaraughty S, Wismer CT, Mikusa J, Zhu C, Chu K, Kohlhaas K, Cowart M, Lee CH, Stewart AO, Cox BF, Polakowski J, Kowaluk EA (2002) Analgesic and anti-inflammatory effects of A-286501, a novel orally active adenosine kinase inhibitor. Pain 96(1–2):107–118
Laghi-Pasini F, Guideri F, Petersen C, Lazzerini PE, Sicari R, Capecchi PL, Picano E (2003) Blunted increase in plasma adenosine levels following dipyridamole stress in dilated cardiomyopathy patients. J Internal Med 254(6):591–596
Driver AG, Kukoly CA, Ali S, Mustafa SJ (1993) Adenosine in bronchoalveolar lavage fluid in asthma. Am Rev Respir Dis 148(1):91–97. doi:10.1164/ajrccm/148.1.91
Nishiyama A, Miura K, Miyatake A, Fujisawa Y, Yue W, Fukui T, Kimura S, Abe Y (1999) Renal interstitial concentration of adenosine during endotoxin shock. Eur J Pharmacol 385(2–3):209–216
Martin C, Leone M, Viviand X, Ayem ML, Guieu R (2000) High adenosine plasma concentration as a prognostic index for outcome in patients with septic shock. Crit Care Med 28(9):3198–3202
Thiel M, Chouker A, Ohta A, Jackson E, Caldwell C, Smith P, Lukashev D, Bittmann I, Sitkovsky MV (2005) Oxygenation inhibits the physiological tissue-protecting mechanism and thereby exacerbates acute inflammatory lung injury. PLoS Biol 3(6):e174. doi:10.1371/journal.pbio.0030174
Lennon PF, Taylor CT, Stahl GL, Colgan SP (1998) Neutrophil-derived 5’-adenosine monophosphate promotes endothelial barrier function via CD73-mediated conversion to adenosine and endothelial A2B receptor activation. J Exp Med 188(8):1433–1443
Mizumoto N, Kumamoto T, Robson SC, Sevigny J, Matsue H, Enjyoji K, Takashima A (2002) CD39 is the dominant Langerhans cell-associated ecto-NTPDase: modulatory roles in inflammation and immune responsiveness. Nat Med 8(4):358–365. doi:10.1038/nm0402-358
Synnestvedt K, Furuta GT, Comerford KM, Louis N, Karhausen J, Eltzschig HK, Hansen KR, Thompson LF, Colgan SP (2002) Ecto-5’-nucleotidase (CD73) regulation by hypoxia-inducible factor-1 mediates permeability changes in intestinal epithelia. J Clin Invest 110(7):993–1002. doi:10.1172/JCI15337
Deaglio S, Robson SC (2011) Ectonucleotidases as regulators of purinergic signaling in thrombosis, inflammation, and immunity. Adv Pharmacol 61:301–332. doi:10.1016/B978-0-12-385526-8.00010-2
Decking UK, Schlieper G, Kroll K, Schrader J (1997) Hypoxia-induced inhibition of adenosine kinase potentiates cardiac adenosine release. Circ Res 81(2):154–164
Kobayashi S, Zimmermann H, Millhorn DE (2000) Chronic hypoxia enhances adenosine release in rat PC12 cells by altering adenosine metabolism and membrane transport. J Neurochem 74(2):621–632
Morote-Garcia JC, Rosenberger P, Kuhlicke J, Eltzschig HK (2008) HIF-1-dependent repression of adenosine kinase attenuates hypoxia-induced vascular leak. Blood 111(12):5571–5580. doi:10.1182/blood-2007-11-126763
Chouker A, Thiel M, Lukashev D, Ward JM, Kaufmann I, Apasov S, Sitkovsky MV, Ohta A (2008) Critical role of hypoxia and A2A adenosine receptors in liver tissue-protecting physiological anti-inflammatory pathway. Mol Med 14(3–4):116–123. doi:10.2119/2007-00075.Chouker
Ujhazy P, Berleth ES, Pietkiewicz JM, Kitano H, Skaar JR, Ehrke MJ, Mihich E (1996) Evidence for the involvement of ecto-5’-nucleotidase (CD73) in drug resistance. Int J Cancer (Journal international du cancer) 68(4):493–500. doi:10.1002/(SICI)1097-0215(19961115)68:4<493::AID-IJC15>3.0.CO;2–6
Jin D, Fan J, Wang L, Thompson LF, Liu A, Daniel BJ, Shin T, Curiel TJ, Zhang B (2010) CD73 on tumor cells impairs antitumor T-cell responses: a novel mechanism of tumor-induced immune suppression. Cancer Res 70(6):2245–2255. doi:10.1158/0008-5472.CAN-09-3109
Hausler SF, Montalban del Barrio I, Strohschein J, Anoop Chandran P, Engel JB, Honig A, Ossadnik M, Horn E, Fischer B, Krockenberger M, Heuer S, Seida AA, Junker M, Kneitz H, Kloor D, Klotz KN, Dietl J, Wischhusen J (2011) Ectonucleotidases CD39 and CD73 on OvCA cells are potent adenosine-generating enzymes responsible for adenosine receptor 2A-dependent suppression of T cell function and NK cell cytotoxicity. Cancer Immunol Immunother 60(10):1405–1418. doi:10.1007/s00262-011-1040-4
Serra S, Horenstein AL, Vaisitti T, Brusa D, Rossi D, Laurenti L, D’Arena G, Coscia M, Tripodo C, Inghirami G, Robson SC, Gaidano G, Malavasi F, Deaglio S (2011) CD73-generated extracellular adenosine in chronic lymphocytic leukemia creates local conditions counteracting drug-induced cell death. Blood 118(23):6141–6152. doi:10.1182/blood-2011-08-374728
Wolberg G, Zimmerman TP, Hiemstra K, Winston M, Chu LC (1975) Adenosine inhibition of lymphocyte-mediated cytolysis: possible role of cyclic adenosine monophosphate. Science 187(4180):957–959
Zenser TV (1975) Formation of adenosine 3’,5’-monophosphate from adenosine in mouse thymocytes. Biochimica et biophysica acta 404(2):202–213
Nordeen SK, Young DA (1976) Glucocorticoid action on rat thymic lymphocytes. Experiments utilizing adenosine to support cellular metabolism lead to a reassessment of catabolic hormone actions. J Biol Chem 251(23):7295–7303
Birch RE, Polmar SH (1982) Pharmacological modification of immunoregulatory T lymphocytes. I. Effect of adenosine, H1 and H2 histamine agonists upon T lymphocyte regulation of B lymphocyte differentiation in vitro. Clin Exp Immunol 48(1):218–230
Mandler R, Birch RE, Polmar SH, Kammer GM, Rudolph SA (1982) Abnormal adenosine-induced immunosuppression and cAMP metabolism in T lymphocytes of patients with systemic lupus erythematosus. Proc Natl Acad Sci U S A 79(23):7542–7546
DosReis GA, Nobrega AF, de Carvalho RP (1986) Purinergic modulation of T-lymphocyte activation: differential susceptibility of distinct activation steps and correlation with intracellular 3’,5’-cyclic adenosine monophosphate accumulation. Cell Immunol 101(1):213–231
Gilbert KM, Hoffmann MK (1985) cAMP is an essential signal in the induction of antibody production by B cells but inhibits helper function of T cells. J Immunol 135(3):2084–2089
Takayama H, Trenn G, Sitkovsky MV (1988) Locus of inhibitory action of cAMP-dependent protein kinase in the antigen receptor-triggered cytotoxic T lymphocyte activation pathway. J Biol Chem 263(5):2330–2336
van Calker D, Muller M, Hamprecht B (1979) Adenosine regulates via two different types of receptors, the accumulation of cyclic AMP in cultured brain cells. J Neurochem 33(5):999–1005
Olsson RA, Pearson JD (1990) Cardiovascular purinoceptors. Physiol Rev 70(3):761–845
Fredholm BB, AP IJ, Jacobson KA, Klotz KN, Linden J (2001) International Union of Pharmacology. XXV. Nomenclature and classification of adenosine receptors. Pharmacol Rev 53(4):527–552
Fredholm BB, AP IJ, Jacobson KA, Linden J, Muller CE (2011) International Union of Basic and Clinical Pharmacology. LXXXI. Nomenclature and classification of adenosine receptors–an update. Pharmacol Rev 63(1):1–34. doi:10.1124/pr.110.003285
Koshiba M, Kojima H, Huang S, Apasov S, Sitkovsky MV (1997) Memory of extracellular adenosine A2A purinergic receptor-mediated signaling in murine T cells. J Biol Chem 272(41):25881–25889
Lukashev DE, Smith PT, Caldwell CC, Ohta A, Apasov SG, Sitkovsky MV (2003) Analysis of A2a receptor-deficient mice reveals no significant compensatory increases in the expression of A2b, A1, and A3 adenosine receptors in lymphoid organs. Biochem Pharmacol 65(12):2081–2090
Huang S, Apasov S, Koshiba M, Sitkovsky M (1997) Role of A2a extracellular adenosine receptor-mediated signaling in adenosine-mediated inhibition of T-cell activation and expansion. Blood 90(4):1600–1610
Lappas CM, Rieger JM, Linden J (2005) A2A adenosine receptor induction inhibits IFN-gamma production in murine CD4+ T cells. J Immunol 174(2):1073–1080
Zarek PE, Huang CT, Lutz ER, Kowalski J, Horton MR, Linden J, Drake CG, Powell JD (2008) A2A receptor signaling promotes peripheral tolerance by inducing T-cell anergy and the generation of adaptive regulatory T cells. Blood 111(1):251–259. doi:10.1182/blood-2007-03-081646
Ohta A, Ohta A, Madasu M, Kini R, Subramanian M, Goel N, Sitkovsky M (2009) A2A adenosine receptor may allow expansion of T cells lacking effector functions in extracellular adenosine-rich microenvironments. J Immunol 183(9):5487–5493. doi:10.4049/jimmunol.0901247
Koshiba M, Rosin DL, Hayashi N, Linden J, Sitkovsky MV (1999) Patterns of A2A extracellular adenosine receptor expression in different functional subsets of human peripheral T cells. Flow cytometry studies with anti-A2A receptor monoclonal antibodies. Mol Pharmacol 55(3):614–624
Raskovalova T, Lokshin A, Huang X, Su Y, Mandic M, Zarour HM, Jackson EK, Gorelik E (2007) Inhibition of cytokine production and cytotoxic activity of human antimelanoma specific CD8+ and CD4+ T lymphocytes by adenosine-protein kinase A type I signaling. Cancer Res 67(12):5949–5956. doi:10.1158/0008-5472.CAN-06-4249
Sevigny CP, Li L, Awad AS, Huang L, McDuffie M, Linden J, Lobo PI, Okusa MD (2007) Activation of adenosine 2A receptors attenuates allograft rejection and alloantigen recognition. J Immunol 178(7):4240–4249
Linnemann C, Schildberg FA, Schurich A, Diehl L, Hegenbarth SI, Endl E, Lacher S, Muller CE, Frey J, Simeoni L, Schraven B, Stabenow D, Knolle PA (2009) Adenosine regulates CD8 T-cell priming by inhibition of membrane-proximal T-cell receptor signalling. Immunology 128(1 Suppl):e728–737. doi:10.1111/j.1365-2567.2009.03075.x
Vang T, Torgersen KM, Sundvold V, Saxena M, Levy FO, Skalhegg BS, Hansson V, Mustelin T, Tasken K (2001) Activation of the COOH-terminal Src kinase (Csk) by cAMP-dependent protein kinase inhibits signaling through the T cell receptor. J Exp Med 193(4):497–507
Csoka B, Himer L, Selmeczy Z, Vizi ES, Pacher P, Ledent C, Deitch EA, Spolarics Z, Nemeth ZH, Hasko G (2008) Adenosine A2A receptor activation inhibits T helper 1 and T helper 2 cell development and effector function. FASEB J 22(10):3491–3499. doi:10.1096/fj.08-107458
Hasko G, Kuhel DG, Chen JF, Schwarzschild MA, Deitch EA, Mabley JG, Marton A, Szabo C (2000) Adenosine inhibits IL-12 and TNF-[alpha] production via adenosine A2a receptor-dependent and independent mechanisms. FASEB J 14(13):2065–2074. doi:10.1096/fj.99-0508com
Panther E, Corinti S, Idzko M, Herouy Y, Napp M, la Sala A, Girolomoni G, Norgauer J (2003) Adenosine affects expression of membrane molecules, cytokine and chemokine release, and the T-cell stimulatory capacity of human dendritic cells. Blood 101(10):3985–3990. doi:10.1182/blood-2002-07-2113
Heijink IH, Vellenga E, Borger P, Postma DS, Monchy JG, Kauffman HF (2003) Polarized Th1 and Th2 cells are less responsive to negative feedback by receptors coupled to the AC/cAMP system compared to freshly isolated T cells. Brit J Pharmacol 138(8):1441–1450. doi:10.1038/sj.bjp.0705193
Ohta A, Kjaergaard J, Sharma S, Mohsin M, Goel N, Madasu M, Fradkov E, Ohta A, Sitkovsky M (2009) In vitro induction of T cells that are resistant to A2 adenosine receptor-mediated immunosuppression. Brit J Pharmacol 156(2):297–306. doi:10.1111/j.1476-5381.2008.00019.x
Rudensky AY (2011) Regulatory T cells and Foxp3. Immunol Rev 241(1):260–268. doi:10.1111/j.1600-065X.2011.01018.x
Sakaguchi S (2011) Regulatory T cells: history and perspective. Method Mol Biol 707:3–17. doi:10.1007/978-1-61737-979-6_1
Sitkovsky MV (2009) T regulatory cells: hypoxia-adenosinergic suppression and re-direction of the immune response. Trends Immunol 30(3):102–108. doi:10.1016/j.it.2008.12.002
Ohta A, Kini R, Ohta A, Subramanian M, Madasu M, Sitkovsky M (2012) The development and immunosuppressive functions of CD4(+) CD25(+) FoxP3(+) regulatory T cells are under influence of the adenosine-A2A adenosine receptor pathway. Front Immunol 3:190. doi:10.3389/fimmu.2012.00190
Wing K, Onishi Y, Prieto-Martin P, Yamaguchi T, Miyara M, Fehervari Z, Nomura T, Sakaguchi S (2008) CTLA-4 control over Foxp3+ regulatory T cell function. Science 322(5899):271–275. doi:10.1126/science.1160062
Kinsey GR, Huang L, Jaworska K, Khutsishvili K, Becker DA, Ye H, Lobo PI, Okusa MD (2012) Autocrine adenosine signaling promotes regulatory T cell-mediated renal protection. J Am Soc Nephrol 23(9):1528–1537. doi:10.1681/ASN.2012010070
Han KL, Thomas SV, Koontz SM, Changpriroa CM, Ha SK, Malech HL, Kang EM (2013) Adenosine A(2)A receptor agonist-mediated increase in donor-derived regulatory T cells suppresses development of graft-versus-host disease. J Immunol 190(1):458–468. doi:10.4049/jimmunol.1201325
Ehrentraut H, Westrich JA, Eltzschig HK, Clambey ET (2012) Adora2b adenosine receptor engagement enhances regulatory T cell abundance during endotoxin-induced pulmonary inflammation. PloS One 7(2):e32416. doi:10.1371/journal.pone.0032416
Ben-Shoshan J, Maysel-Auslender S, Mor A, Keren G, George J (2008) Hypoxia controls CD4+ CD25+ regulatory T-cell homeostasis via hypoxia-inducible factor-1alpha. Eur J Immunol 38(9):2412–2418. doi:10.1002/eji.200838318
Clambey ET, McNamee EN, Westrich JA, Glover LE, Campbell EL, Jedlicka P, de Zoeten EF, Cambier JC, Stenmark KR, Colgan SP, Eltzschig HK (2012) Hypoxia-inducible factor-1 alpha-dependent induction of FoxP3 drives regulatory T-cell abundance and function during inflammatory hypoxia of the mucosa. Proc Natl Acad Sci U S A 109(41):E2784–2793. doi:10.1073/pnas.1202366109
Dang EV, Barbi J, Yang HY, Jinasena D, Yu H, Zheng Y, Bordman Z, Fu J, Kim Y, Yen HR, Luo W, Zeller K, Shimoda L, Topalian SL, Semenza GL, Dang CV, Pardoll DM, Pan F (2011) Control of T(H)17/T(reg) balance by hypoxia-inducible factor 1. Cell 146(5):772–784. doi:10.1016/j.cell.2011.07.033
Shi LZ, Wang R, Huang G, Vogel P, Neale G, Green DR, Chi H (2011) HIF1alpha-dependent glycolytic pathway orchestrates a metabolic checkpoint for the differentiation of TH17 and Treg cells. J Exp Med 208(7):1367–1376. doi:10.1084/jem.20110278
Kobie JJ, Shah PR, Yang L, Rebhahn JA, Fowell DJ, Mosmann TR (2006) T regulatory and primed uncommitted CD4 T cells express CD73, which suppresses effector CD4 T cells by converting 5’-adenosine monophosphate to adenosine. J Immunol 177(10):6780–6786
Deaglio S, Dwyer KM, Gao W, Friedman D, Usheva A, Erat A, Chen JF, Enjyoji K, Linden J, Oukka M, Kuchroo VK, Strom TB, Robson SC (2007) Adenosine generation catalyzed by CD39 and CD73 expressed on regulatory T cells mediates immune suppression. J Exp Med 204(6):1257–1265. doi:10.1084/jem.20062512
Mandapathil M, Szczepanski MJ, Szajnik M, Ren J, Jackson EK, Johnson JT, Gorelik E, Lang S, Whiteside TL (2010) Adenosine and prostaglandin E2 cooperate in the suppression of immune responses mediated by adaptive regulatory T cells. J Biol Chem 285(36):27571–27580. doi:10.1074/jbc.M110.127100
Naganuma M, Wiznerowicz EB, Lappas CM, Linden J, Worthington MT, Ernst PB (2006) Cutting edge: critical role for A2A adenosine receptors in the T cell-mediated regulation of colitis. J Immunol 177(5):2765–2769
Nagaraj S, Gabrilovich DI (2008) Tumor escape mechanism governed by myeloid-derived suppressor cells. Cancer Res 68(8):2561–2563. doi:10.1158/0008-5472.CAN-07-6229
Ryzhov S, Novitskiy SV, Goldstein AE, Biktasova A, Blackburn MR, Biaggioni I, Dikov MM, Feoktistov I (2011) Adenosinergic regulation of the expansion and immunosuppressive activity of CD11b + Gr1 + cells. J Immunol 187(11):6120–6129. doi:10.4049/jimmunol.1101225
Corzo CA, Condamine T, Lu L, Cotter MJ, Youn JI, Cheng P, Cho HI, Celis E, Quiceno DG, Padhya T, McCaffrey TV, McCaffrey JC, Gabrilovich DI (2010) HIF-1alpha regulates function and differentiation of myeloid-derived suppressor cells in the tumor microenvironment. J Exp Med 207(11):2439–2453. doi:10.1084/jem.20100587
Ben Addi A, Lefort A, Hua X, Libert F, Communi D, Ledent C, Macours P, Tilley SL, Boeynaems JM, Robaye B (2008) Modulation of murine dendritic cell function by adenine nucleotides and adenosine: involvement of the A(2B) receptor. Eur J Immunol 38(6):1610–1620. doi:10.1002/eji.200737781
Novitskiy SV, Ryzhov S, Zaynagetdinov R, Goldstein AE, Huang Y, Tikhomirov OY, Blackburn MR, Biaggioni I, Carbone DP, Feoktistov I, Dikov MM (2008) Adenosine receptors in regulation of dendritic cell differentiation and function. Blood 112(5):1822–1831. doi:10.1182/blood-2008-02-136325
Gordon S, Martinez FO (2010) Alternative activation of macrophages: mechanism and functions. Immunity 32(5):593–604. doi:10.1016/j.immuni.2010.05.007
Leibovich SJ, Chen JF, Pinhal-Enfield G, Belem PC, Elson G, Rosania A, Ramanathan M, Montesinos C, Jacobson M, Schwarzschild MA, Fink JS, Cronstein B (2002) Synergistic up-regulation of vascular endothelial growth factor expression in murine macrophages by adenosine A(2A) receptor agonists and endotoxin. Am J Pathol 160 (6):2231–2244. doi:10.1016/S0002-9440(10)61170-4
Ramanathan M, Pinhal-Enfield G, Hao I, Leibovich SJ (2007) Synergistic up-regulation of vascular endothelial growth factor (VEGF) expression in macrophages by adenosine A2A receptor agonists and endotoxin involves transcriptional regulation via the hypoxia response element in the VEGF promoter. Mol Biol Cell 18(1):14–23. doi:10.1091/mbc.E06-07-0596
Mancino A, Schioppa T, Larghi P, Pasqualini F, Nebuloni M, Chen IH, Sozzani S, Austyn JM, Mantovani A, Sica A (2008) Divergent effects of hypoxia on dendritic cell functions. Blood 112(9):3723–3734. doi:10.1182/blood-2008-02-142091
Wang Q, Liu C, Zhu F, Liu F, Zhang P, Guo C, Wang X, Li H, Ma C, Sun W, Zhang Y, Chen W, Zhang L (2010) Reoxygenation of hypoxia-differentiated dentritic cells induces Th1 and Th17 cell differentiation. Mol Immunol 47(4):922–931. doi:10.1016/j.molimm.2009.09.038
Yang M, Ma C, Liu S, Sun J, Shao Q, Gao W, Zhang Y, Li Z, Xie Q, Dong Z, Qu X (2009) Hypoxia skews dendritic cells to a T helper type 2-stimulating phenotype and promotes tumour cell migration by dendritic cell-derived osteopontin. Immunology 128(1 Suppl):e237–249. doi:10.1111/j.1365-2567.2008.02954.x
Priebe T, Platsoucas CD, Nelson JA (1990) Adenosine receptors and modulation of natural killer cell activity by purine nucleosides. Cancer Res 50(14):4328–4331
Raskovalova T, Huang X, Sitkovsky M, Zacharia LC, Jackson EK, Gorelik E (2005) Gs protein-coupled adenosine receptor signaling and lytic function of activated NK cells. J Immunol 175(7):4383–4391
Lokshin A, Raskovalova T, Huang X, Zacharia LC, Jackson EK, Gorelik E (2006) Adenosine-mediated inhibition of the cytotoxic activity and cytokine production by activated natural killer cells. Cancer Res 66(15):7758–7765. doi:10.1158/0008-5472.CAN-06-0478
Fink T, Ebbesen P, Koppelhus U, Zachar V (2003) Natural killer cell-mediated basal and interferon-enhanced cytotoxicity against liver cancer cells is significantly impaired under in vivo oxygen conditions. Scand J Immunol 58(6):607–612
Siemens DR, Hu N, Sheikhi AK, Chung E, Frederiksen LJ, Pross H, Graham CH (2008) Hypoxia increases tumor cell shedding of MHC class I chain-related molecule: role of nitric oxide. Cancer Res 68(12):4746–4753. doi:10.1158/0008-5472.CAN-08-0054
Yamada N, Yamanegi K, Ohyama H, Hata M, Nakasho K, Futani H, Okamura H, Terada N (2012) Hypoxia downregulates the expression of cell surface MICA without increasing soluble MICA in osteosarcoma cells in a HIF-1alpha-dependent manner. Int J Oncol 41(6):2005–2012. doi:10.3892/ijo.2012.1630
Sitkovsky MV, Lukashev D, Apasov S, Kojima H, Koshiba M, Caldwell C, Ohta A, Thiel M (2004) Physiological control of immune response and inflammatory tissue damage by hypoxia-inducible factors and adenosine A2A receptors. Annu Rev Immunol 22:657–682. doi:10.1146/annurev.immunol.22.012703.104731
Eltzschig HK, Sitkovsky MV, Robson SC (2012) Purinergic signaling during inflammation. New Engl J Med 367(24):2322–2333. doi:10.1056/NEJMra1205750
Chen JF, Eltzschig HK, Fredholm BB (2013) Adenosine receptors as drug targets–what are the challenges? Nature Rev Drug Discov 12(4):265–286. doi:10.1038/nrd3955
Ohta A, Sitkovsky M (2001) Role of G-protein-coupled adenosine receptors in downregulation of inflammation and protection from tissue damage. Nature 414(6866):916–920. doi:10.1038/414916a
Sitkovsky MV, Ohta A (2005) The ‘danger’ sensors that STOP the immune response: the A2 adenosine receptors? Trends Immunol 26(6):299–304. doi:10.1016/j.it.2005.04.004
Lukashev D, Ohta A, Apasov S, Chen JF, Sitkovsky M (2004) Cutting edge: physiologic attenuation of proinflammatory transcription by the Gs protein-coupled A2A adenosine receptor in vivo. J Immunol 173(1):21–24
Mohsenin A, Mi T, Xia Y, Kellems RE, Chen JF, Blackburn MR (2007) Genetic removal of the A2A adenosine receptor enhances pulmonary inflammation, mucin production, and angiogenesis in adenosine deaminase-deficient mice. Am J Physiol Lung Cell Mol Physiol 293(3):L753–761. doi:10.1152/ajplung.00187.2007
Nadeem A, Fan M, Ansari HR, Ledent C, Jamal Mustafa S (2007) Enhanced airway reactivity and inflammation in A2A adenosine receptor-deficient allergic mice. Am J Physiol Lung Cell Mol Physiol 292(6):L1335–1344. doi:10.1152/ajplung.00416.2006
Alam MS, Kurtz CC, Wilson JM, Burnette BR, Wiznerowicz EB, Ross WG, Rieger JM, Figler RA, Linden J, Crowe SE, Ernst PB (2009) A2A adenosine receptor (AR) activation inhibits pro-inflammatory cytokine production by human CD4 + helper T cells and regulates Helicobacter-induced gastritis and bacterial persistence. Mucosal Immunol 2(3):232–242. doi:10.1038/mi.2009.4
Hasko G, Csoka B, Nemeth ZH, Vizi ES, Pacher P (2009) A(2B) adenosine receptors in immunity and inflammation. Trends Immunol 30(6):263–270. doi:10.1016/j.it.2009.04.001
Koeppen M, Eckle T, Eltzschig HK (2011) Interplay of hypoxia and A2B adenosine receptors in tissue protection. Adv Pharmacol 61:145–186. doi:10.1016/B978-0-12-385526-8.00006-0
Eckle T, Faigle M, Grenz A, Laucher S, Thompson LF, Eltzschig HK (2008) A2B adenosine receptor dampens hypoxia-induced vascular leak. Blood 111(4):2024–2035. doi:10.1182/blood-2007-10-117044
Frick JS, MacManus CF, Scully M, Glover LE, Eltzschig HK, Colgan SP (2009) Contribution of adenosine A2B receptors to inflammatory parameters of experimental colitis. J Immunol 182(8):4957–4964. doi:10.4049/jimmunol.0801324
Zhou Y, Mohsenin A, Morschl E, Young HW, Molina JG, Ma W, Sun CX, Martinez-Valdez H, Blackburn MR (2009) Enhanced airway inflammation and remodeling in adenosine deaminase-deficient mice lacking the A2B adenosine receptor. J Immunol 182(12):8037–8046. doi:10.4049/jimmunol.0900515
Schingnitz U, Hartmann K, Macmanus CF, Eckle T, Zug S, Colgan SP, Eltzschig HK (2010) Signaling through the A2B adenosine receptor dampens endotoxin-induced acute lung injury. J Immunol 184(9):5271–5279. doi:10.4049/jimmunol.0903035
Zhou Y, Schneider DJ, Morschl E, Song L, Pedroza M, Karmouty-Quintana H, Le T, Sun CX, Blackburn MR (2011) Distinct roles for the A2B adenosine receptor in acute and chronic stages of bleomycin-induced lung injury. J Immunol 186(2):1097–1106. doi:10.4049/jimmunol.1002907
Restifo NP, Dudley ME, Rosenberg SA (2012) Adoptive immunotherapy for cancer: harnessing the T cell response. Nature reviews. Immunology 12(4):269–281. doi:10.1038/nri3191
Vanneman M, Dranoff G (2012) Combining immunotherapy and targeted therapies in cancer treatment. Nature reviews. Cancer 12(4):237–251. doi:10.1038/nrc3237
Drake CG, Doody AD, Mihalyo MA, Huang CT, Kelleher E, Ravi S, Hipkiss EL, Flies DB, Kennedy EP, Long M, McGary PW, Coryell L, Nelson WG, Pardoll DM, Adler AJ (2005) Androgen ablation mitigates tolerance to a prostate/prostate cancer-restricted antigen. Cancer cell 7(3):239–249. doi:10.1016/j.ccr.2005.01.027
Bai A, Higham E, Eisen HN, Wittrup KD, Chen J (2008) Rapid tolerization of virus-activated tumor-specific CD8 + T cells in prostate tumors of TRAMP mice. Proc Natl Acad Sci U S A 105(35):13003–13008. doi:10.1073/pnas.0805599105
Kerkar SP, Restifo NP (2012) Cellular constituents of immune escape within the tumor microenvironment. Cancer Res 72(13):3125–3130. doi:10.1158/0008–5472.CAN-11-4094
Rabinovich GA, Gabrilovich D, Sotomayor EM (2007) Immunosuppressive strategies that are mediated by tumor cells. Annu Rev Immunol 25:267–296. doi:10.1146/annurev.immunol.25.022106.141609
Takagi H, King GL, Robinson GS, Ferrara N, Aiello LP (1996) Adenosine mediates hypoxic induction of vascular endothelial growth factor in retinal pericytes and endothelial cells. Invest Ophth Vis Sci 37(11):2165–2176
Coney AM, Marshall JM (1998) Role of adenosine and its receptors in the vasodilatation induced in the cerebral cortex of the rat by systemic hypoxia. J Physiol 509(Pt 2):507–518
Kobayashi S, Conforti L, Pun RY, Millhorn DE (1998) Adenosine modulates hypoxia-induced responses in rat PC12 cells via the A2A receptor. J Physiol 508(Pt 1):95–107
Fisher JW, Brookins J (2001) Adenosine A(2A) and A(2B) receptor activation of erythropoietin production. Am J Physiol Renal Physiol 281(5):F826–832
Koos BJ, Maeda T (2001) Adenosine A(2A) receptors mediate cardiovascular responses to hypoxia in fetal sheep. Am J Physiol Heart Circ Physiol 280(1):H83–89
Ohta A, Sitkovsky M (2011) Methylxanthines, inflammation, and cancer: fundamental mechanisms. Handb Exp Pharmacol (200):469–481. doi:10.1007/978-3-642-13443-2_19
Ryzhov S, Novitskiy SV, Zaynagetdinov R, Goldstein AE, Carbone DP, Biaggioni I, Dikov MM, Feoktistov I (2008) Host A(2B) adenosine receptors promote carcinoma growth. Neoplasia 10(9):987–995
Cekic C, Sag D, Li Y, Theodorescu D, Strieter RM, Linden J (2012) Adenosine A2B receptor blockade slows growth of bladder and breast tumors. J Immunol 188(1):198–205. doi:10.4049/jimmunol.1101845
Zhang B (2010) CD73: a novel target for cancer immunotherapy. Cancer Res 70(16):6407–6411. doi:10.1158/0008-5472.CAN-10-1544
Beavis PA, Stagg J, Darcy PK, Smyth MJ (2012) CD73: a potent suppressor of antitumor immune responses. Trends Immunol 33(5):231–237. doi:10.1016/j.it.2012.02.009
Stagg J, Divisekera U, McLaughlin N, Sharkey J, Pommey S, Denoyer D, Dwyer KM, Smyth MJ (2010) Anti-CD73 antibody therapy inhibits breast tumor growth and metastasis. Proc Natl Acad Sci U S A 107(4):1547–1552. doi:10.1073/pnas.0908801107
Stagg J, Divisekera U, Duret H, Sparwasser T, Teng MW, Darcy PK, Smyth MJ (2011) CD73-deficient mice have increased antitumor immunity and are resistant to experimental metastasis. Cancer Res 71(8):2892–2900. doi:10.1158/0008-5472.CAN-10-4246
Stagg J, Beavis PA, Divisekera U, Liu MC, Moller A, Darcy PK, Smyth MJ (2012) CD73-deficient mice are resistant to carcinogenesis. Cancer Res 72(9):2190–2196. doi:10.1158/0008-5472.CAN-12-0420
Fredholm BB, Battig K, Holmen J, Nehlig A, Zvartau EE (1999) Actions of caffeine in the brain with special reference to factors that contribute to its widespread use. Pharmacol Rev 51(1):83–133
Fredholm BB (2011) Notes on the history of caffeine use. Handb Exp Pharmacol (200):1–9. doi:10.1007/978-3-642-13443-2_1
Ohta A, Lukashev D, Jackson EK, Fredholm BB, Sitkovsky M (2007) 1,3,7-trimethylxanthine (caffeine) may exacerbate acute inflammatory liver injury by weakening the physiological immunosuppressive mechanism. J Immunol 179(11):7431–7438
Lelo A, Miners JO, Robson R, Birkett DJ (1986) Assessment of caffeine exposure: caffeine content of beverages, caffeine intake, and plasma concentrations of methylxanthines. Clin Pharmacol Ther 39(1):54–59
Cook DG, Peacock JL, Feyerabend C, Carey IM, Jarvis MJ, Anderson HR, Bland JM (1996) Relation of caffeine intake and blood caffeine concentrations during pregnancy to fetal growth: prospective population based study. Bmj 313(7069):1358–1362
Stensvold I, Jacobsen BK (1994) Coffee and cancer: a prospective study of 43,000 Norwegian men and women. Cancer Causes Control: CCC 5(5):401–408
Veierod MB, Thelle DS, Laake P (1997) Diet and risk of cutaneous malignant melanoma: a prospective study of 50,757 Norwegian men and women. Int J Cancer (Journal international du cancer) 71(4):600–604
Shimazu T, Tsubono Y, Kuriyama S, Ohmori K, Koizumi Y, Nishino Y, Shibuya D, Tsuji I (2005) Coffee consumption and the risk of primary liver cancer: pooled analysis of two prospective studies in Japan. Int J Cancer (Journal international du cancer) 116(1):150–154. doi:10.1002/ijc.20989
Nkondjock A, Ghadirian P, Kotsopoulos J, Lubinski J, Lynch H, Kim-Sing C, Horsman D, Rosen B, Isaacs C, Weber B, Foulkes W, Ainsworth P, Tung N, Eisen A, Friedman E, Eng C, Sun P, Narod SA (2006) Coffee consumption and breast cancer risk among BRCA1 and BRCA2 mutation carriers. Int J Cancer (Journal international du cancer) 118(1):103–107. doi:10.1002/ijc.21296
Hirose K, Niwa Y, Wakai K, Matsuo K, Nakanishi T, Tajima K (2007) Coffee consumption and the risk of endometrial cancer: evidence from a case-control study of female hormone-related cancers in Japan. Cancer Sci 98(3):411–415. doi:10.1111/j.1349-7006.2007.00391.x
Lee KJ, Inoue M, Otani T, Iwasaki M, Sasazuki S, Tsugane S, Group JS (2007) Coffee consumption and risk of colorectal cancer in a population-based prospective cohort of Japanese men and women. Int J Cancer (Journal international du cancer) 121(6):1312–1318. doi:10.1002/ijc.22778
Montella M, Polesel J, La Vecchia C, Dal Maso L, Crispo A, Crovatto M, Casarin P, Izzo F, Tommasi LG, Talamini R, Franceschi S (2007) Coffee and tea consumption and risk of hepatocellular carcinoma in Italy. Int J Cancer (Journal international du cancer) 120(7):1555–1559. doi:10.1002/ijc.22509
Ganmaa D, Willett WC, Li TY, Feskanich D, van Dam RM, Lopez-Garcia E, Hunter DJ, Holmes MD (2008) Coffee, tea, caffeine and risk of breast cancer: a 22-year follow-up. Int J Cancer (Journal international du cancer) 122(9):2071–2076. doi:10.1002/ijc.23336
Shimazu T, Inoue M, Sasazuki S, Iwasaki M, Kurahashi N, Yamaji T, Tsugane S, Study JSGMotJPHC-bP (2008) Coffee consumption and risk of endometrial cancer: a prospective study in Japan. Int J Cancer (Journal international du cancer) 123(10):2406–2410. doi:10.1002/ijc.23760
Waickman AT, Alme A, Senaldi L, Zarek PE, Horton M, Powell JD (2012) Enhancement of tumor immunotherapy by deletion of the A2A adenosine receptor. Cancer Immunol Immunother 61(6):917–926. doi:10.1007/s00262-011-1155-7
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2014 Springer Science+Business Media, LLC
About this chapter
Cite this chapter
Ohta, A., Sitkovsky, M. (2014). The Hypoxia-adenosinergic Immunosuppression and Redirection of Immune Response in Tumor Microenvironment. In: Gabrilovich, D., Hurwitz, A. (eds) Tumor-Induced Immune Suppression. Springer, New York, NY. https://doi.org/10.1007/978-1-4899-8056-4_14
Download citation
DOI: https://doi.org/10.1007/978-1-4899-8056-4_14
Published:
Publisher Name: Springer, New York, NY
Print ISBN: 978-1-4899-8055-7
Online ISBN: 978-1-4899-8056-4
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)