
Abstract
Human growth hormone (GH) is a peptide hormone physiologically secreted in the anterior pituitary gland. GH is well known to affect linear height growth. GH treatment for its effect of growth in patients with many types of disorders of short stature has been expanding. In childhood, longitudinal bones become thick at the same time as long axis elongation, and bone quantity and bone mineral density (BMD) are elevated continuously until the adolescent stage. Because patients with GH deficiency have a short stature and relatively low BMD, GH likely plays a critical role in bone mineral metabolism. To improve linear growth and bone mineral metabolism, some recent studies have challenged GH treatment in patients with metabolic bone diseases, in addition to approved diseases. In this chapter, we describe the physiology of GH action, efficacy, and trials of GH treatment. The especially on bone mineral metabolism.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others


Keywords
These keywords were added by machine and not by the authors. This process is experimental and the keywords may be updated as the learning algorithm improves.
Physiology of Growth Hormone
Regulatory Mechanism of GH Secretion
Daily rhythmical growth hormone (GH) secretion controls its serum concentration. A total of 70 % of daily GH secretion occurs with the first episode of slow-wave sleep [1]. GH secretion is also affected by serum glucose density, amino acids, free fatty acids, drugs, and GH itself. These various conditions stimulate the hypothalamus and regulate secretion of three hypothalamic hormones, growth hormone releasing hormone (GHRH), ghrelin, and somatostatin. These signals eventually control GH secretion from GH-producing cells, expressing the specific receptors for them, in the pituitary gland [2] (Fig. 8.1).

Stimulation and action of intrinsic GH. GH is secreted in the pituitary gland and stimulated by hypothalamic hormones, such as GHRH and ghrelin, and is suppressed by somatostatin. GH acts on many peripheral tissues and plays a role in linear growth, bone metabolism, adipose metabolism, protein metabolism, and saccarometabolism
GHRH selectively induces GH secretion from the pituitary gland through the GHRH receptor [3, 4]. Somatostatin suppresses GH pulse amplitude and frequency, and inhibits central GHRH release via direct synaptic connections with hypothalamic neurons, but does not affect GH biosynthesis [2]. Hypothalamic GHRH and somatostatin are secreted in independent waves and interact to generate pulsatile GH release together with additional GH secretagogues (GHS). Ghrelin binds to the GHS receptor to induce hypothalamic GHRH and pituitary GH [5, 6]. The greatest amount of ghrelin is secreted from gastric cells rather than from the hypothalamus. Plasma ghrelin concentrations increase when fasting, and decrease after food intake [7].
Mechanism of GH Action
GH stimulates linear body growth through differentiation and proliferation of the cells in the growth plate in children. GH also acts on many peripheral tissues other than the growth plate, and plays important roles in homeostasis, such as glycemic effects, hydration, protein anabolism, and lipid degradation [8] (Fig. 8.1).
At least part of the growth effect by GH is through endocrine, autocrine, and paracrine mechanisms of insulin-like growth factor I (IGF-I). GH action in body growth may be explained through three pathways involving IGF-I. In one pathway, GH acts through GH receptor (GHR) expression in hepatocytes and generation of IGF-I [9]. Consequently, serum IGF-I levels increase and IGF-I acts on peripheral tissues as a hormone by an endocrine mechanism. In a second pathway, GH acts on peripheral tissues, not the liver, promoting IGF-I generation, and this IGF-I affects local tissues by an autocrine/paracrine system [10]. Expression of GHR, IGF-I, and IGF-I receptor has been detected in chondrocytes, osteoblasts, osteoclasts, myocytes, and adipocytes. In a third pathway, GH affects peripheral tissues directly. For Laron syndrome in which there is deficiency of GHR, extrinsic IGF-I administration does not have a sufficient effect on growth in spite of its biological activities, such as improvement of hyperglycemia [11]. This phenomenon implies a direct action of GH.
GH Treatment
History
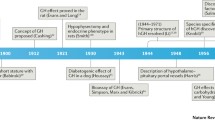
In the 1950s, human GH was first used to stimulate linear growth in a child with hypopituitarism [12]. At that time, GH was extracted and purified from the pituitary grand. Because the supply of the extracted GH was limited, GH treatment was restricted to children with the most severe and unequivocal GH deficiency (GHD). Delays in diagnosis and treatment, interruptions in treatments, and dosage restrictions were common during this time. Consequently, while GH accelerated growth of these individuals, adult height was usually less than average [13–15].
In 1985, Creutzfeldt–Jakob disease (CJD) was recognized in patients who had received GH. Distribution of pituitary-derived GH was stopped. Subsequently, in the United States, CJD was diagnosed in seven recipients of GH [16, 17]. Fortunately, 192- and 191-amino-acid biosynthetic GHs were approved in 1985 and 1987, respectively. The production of GH by biological systems transplanted with the GH gene yields a virtually unlimited supply of GH.
Biosynthetic GH treatment eliminated the risk of CJD and offered children with severe GHD an opportunity for optimal treatment. Children with milder forms of inadequate GH secretion, previously excluded from receiving GH, could become treated. In addition, metabolic effects of GH, apart from linear growth promotion, are now being studied extensively, leading to new indications for GH treatment [18].
Approved Disorders and the Efficacy of GH Treatment
Approved disorders for GH treatment have been expanding in the world in spite of its high cost, with expectations of promoting linear growth. Currently, these growth disorders are GHD, short children with small for gestational age (SGA), Turner syndrome (TS), chronic renal insufficiency (CRI), Prader–Willi syndrome (PWS), short stature homeobox (SHOX) haploinsufficiency, achondroplasia (ACH), hypochondroplasia (HCH), Noonan syndrome (NS), and idiopathic short stature (ISS) (Table 8.1). The type of approved disorders, criteria of diagnosis, and treatment dose vary and depend on the country.
GH treatment was started primarily for classical GHD patients to promote linear growth. Untreated patients with GHD have profound short stature, averaging nearly −5 standard deviation (SD) [19–21]. In many countries, pediatric endocrinologists have developed guidelines for diagnosis, criteria for starting treatment, treatment regimens, criteria for continuing treatment, and criteria for finishing treatment. GH treatment in GHD patients gradually improved their adult height SD score by approximately −1.3 SD, although most patients failed to reach their genetic target heights [22, 23].
SGA is a term used to describe a neonate’s birth size based upon appropriate auxological standards for healthy infants. Approximately 86 % of SGA children achieve a length within the normal range by 12 months [24, 25]. Catch-up growth in the normal range is virtually always complete by 2 years of age [26]. Overall, 8–14 % of SGA infants become short in stature with an adult height of approximately 1 SD [27, 28]. SGA children achieve a final height within the normal height range after 7.8 years of GH treatment [29]. The effects of GH extend beyond linear growth and potentially include important effects on body composition, muscle mass and function, bone mass, metabolism, behavior, and cognitive function, and even quality of life, IQ, and bone mineral content [30, 31].
TS is characterized by short stature, cubitus valgus, webbing of the neck, and sexual infantilism [32]. Over 95 % of TS patients eventually fall below the −2 SD, and their adult height is typically approximately 20 cm below the mean for females of their respective ethnic group. GH treatment in TS patients improves their final height to 8.5 cm above the mean projected adult height and there is a mean height gain due to GH of +7.2 cm [33, 34].
Growth failure is still a major obstacle to successful rehabilitation of children with CRI. The mean height SD score at the start of renal replacement therapy is approximately −2, indicating that half of the patients have a short stature [35, 36]. Similarly, the mean final height SD score of CRI patients is reported to be significantly reduced and varies between −1.4 in girls and −2.2 in boys in various reports [37, 38]. GH treatment for short stature in CRI became available approximately 20 years ago [39]. The final height of CRI patients after extended GH treatment appears to be an average of 1.0–1.5 SD [40, 41].
PWS is a neurogenetic disorder characterized by mental and physical abnormalities. The mean adult height achieved by men and women with PWS is 155–162 and 148–150 cm, respectively [42, 43]. The GH-deficient state commonly associated with PWS, as evidenced by reduced GH secretion, low serum IGF-I levels, and clinical features typical of GHD, has provided a rationale for trials assessing the efficacy of GH treatment. However, currently, the duration of treatment is limited. Longitudinal growth has been shown to increase by GH treatment in PWS [44, 45]. Some reports have shown that growth continues to improve by GH treatment in PWS, with the result that the target height SD score can be reached [46].
NS is a genetic syndrome with many features similar to TS, and is characterized by pulmonary valvular stenosis, visual problems, clotting disorders, and short stature [47]. Although approximately half of NS patients will reach an adult height within 2 SD of the population mean, the mean adult height of NS is approximately 162.5 cm and 153 cm for males and females, respectively [48]. GH treatment in NS improves the final height SD score to 1.7 [49]. Additionally, pretreatment baseline cortical bone mineral density (BMD) is reported to be in the low–normal range and it increases over 2 years of GH therapy [50]. In the majority of reports, GH treatment induced catch-up growth in most of the NS patients. First data on long-term outcome demonstrate an effect comparable with or even better than that in TS.
ISS is a purely descriptive term that refers to a child, adolescent, or adult with a height below the age reference for population and sex, in whom, with current diagnostic tools, no etiological diagnosis is made [51]. The mean final height is similar to the mean predicted height in ISS. There is a large interindividual variation that is primarily correlated with the initial height SD score and bone age delay at start of GH treatment [52]. GH for ISS in a supraphysiological dosage increases the final height by approximately 7 cm, but for the individual child, the height gain is difficult to predict.
Side Effects
Recombinant biosynthetic GH preparations are highly purified and free of contaminants. The possibility of viral transmission through GH has been virtually eliminated. Antigenicity of GH preparations is also low, although GH antibodies can be detected in 10–30 % of treated children [53]. With rare exceptions (less than 0.1 %), these antibodies do not impede effects of GH.
Laboratory indications of hypothyroidism can be found in as many as 25 % of GHD children treated with GH [54]. GHD patients, who display subnormal nocturnal thyroid-stimulating hormone surges, signifying preexisting central hypothyroidism, are more likely to display subnormal T4 and free T4 levels during GH therapy [55]. However, most studies have indicated that children with normal thyroid function before treatment do not develop significant perturbations in thyroid hormone metabolism during GH therapy.
Administration of unphysiological high concentrations of GH may lead to defects in glucose metabolism [56]. When intrinsic GH secretion is increased, as in sleep, oral or intravenous glucose tolerance tests show a defect in glucose metabolism [57]. This defect of glucose metabolism lasts even after finishing GH treatment and normalization of serum GH concentrations.
Edema and sodium retention rarely occur early in the course of GH therapy, which is attributable to an anti-natriuretic effect on the renal tubules of GH and/or IGF-I. Minor elevations in plasma renin activity and aldosterone observed in the first 3 days of treatment resolve within 1 or 2 weeks [58]. Occasionally, fluid shifts within the central nervous system are sufficient to cause benign intracranial hypertension, with symptoms of headache, visual loss, vomiting, and papilledema. Direct fluid-retaining properties of GH and/or action of locally produced IGF-I on cerebrospinal fluid production are speculated to be causative. Cessation of GH therapy reverses symptoms in spite of continued GH treatment [59]. Resumption of GH treatment has been successfully accomplished with re-initiation at a lower dosage and a gradual return to the initial dosage. Performing a fundoscopic examination is recommended in all patients before initiation of GH therapy and periodically thereafter [60].
Growth Hormone and Bone
Effect of GH on Bone and Cartilage Metabolism
GH acts directly on the perichondrial layer in the growth plate of growing bones, and promotes proliferation and differentiation of pre-chondrocytes, as well as promotes IGF-I synthesis (Fig. 8.2). Pre-chondrocytes proliferate and differentiate to chondrocytes in the proliferative zone of growth plates by acquiring the ability for reaction to IGF-I and for generation of IGF-I [61].

Schematic representation of bone metabolism by GH. GH stimulates differentiation and proliferation of chondrocytes directly and through IGF-I synthesis. This endochondral ossification leads to linear growth. GH also stimulates differentiation and proliferation in osteoblasts and osteoclasts. Consequently, GH affects bone metabolism and, consequently, linear growth and bone mineral density
Osteoblasts express GHR and IGF-I receptor, and have the ability of generating IGF-I [62]. Therefore, GH promotes synthesis of IGF-I in osteoblasts, and IGF-I acts on these cells through the autocrine/paracrine system. In osteoblastic culture, IGF-I stimulates differentiation of osteoblasts to osteocytes by promoting proliferation of osteoblasts, expression of type I collagen, and activation of alkaline phosphatase, and by suppressing expression of matrix metalloproteinase 1 [63]. However, it is still unclear whether GH action on osteoblasts occurs through IGF-I or there is a direct pathway.
GH action is also detected in osteoclasts. Precursors of osteoclasts express GHR, and GH promotes their differentiation to osteoclasts. Factors promoting osteoclast differentiation are generated by GH-stimulated osteoblasts and bone marrow cells [64]. These findings show that GH promotes bone resorption through its direct effect on bone marrow cells or through osteoblasts. In fact, when GH is administered in pediatric patients, bone resorption markers are elevated before growth is detected and bone formation markers are elevated.
GH promotes bone turnover, including bone generation and bone resorption; GH consequently promotes longitudinal bone growth while maintaining BMD suitable for increasing quantities of bone (Fig. 8.2). Since GH also increases mass and strength of the skeletal muscles, mechanical stress may be another factor for GH effect on increasing BMD [65]. Although the effect of GH on BMD is still controversial in certain conditions such as burn injury, BMD is indeed correlated with nocturnal GH secretion in young healthy men and acromegaly [66, 67]. Moreover lumbar BMD is reduced in pediatric GHD patients, and GH treatment increases BMD in GHD and other diseases [68–71].
For the considerable variability in response to GH treatment, several prediction models that attempt to estimate the growth response to GH treatment have been developed [72–75]. As a result, in growing children, markers of bone metabolism reflect skeletal growth and development. For example, urinary deoxypyridinoline and serum pyridinoline, bone resorption markers, are strongly related to height velocity. These results imply that bone metabolism and linear growth are closely related to each other.
Approved GH Treatment in Skeletal Dysplasia
Skeletal dysplasia is a heterogeneous group of diseases affecting the skeleton. The estimated incidence is 30–45 in every 100,000 newborns. The final height differs substantially between the various disorders, but is often in the range of 110–130 cm [76]. Currently, although a remarkably short stature has been detected in various skeletal dysplasias, only three skeletal dysplasias have been approved for GH treatment: ACH, HCH and SHOX haploinsufficiency [77] (Table 8.1).
ACH is the most common type of rhizomelic short-limb dwarfism caused by activating point mutations in the fibroblast growth factor receptor 3 (FGFR3) gene [78, 79]. The incidence of ACH is estimated as 1 in 25,000 live births. The average adult height of ACH is approximately 132 cm (−6.8 SD) for males and 124 cm (−6.4 SD) for females [80]. FGFR3 is expressed in the growth plate, and its activation suppresses IGF-I expression and cell proliferation, and promotes apoptosis of chondrocytes. GH administration increases IGF-I expression in chondrocytic cell lines expressing mutated FGFR3 and prevents these cells from apoptosis [81]. This could explain one of the mechanisms by which GH therapy improves disturbed bone growth in ACH.
GH treatment in ACH has been approved only in Japan, since 1997. As a short-term effect, GH administration increases height velocity from (mean ± SD) 3.8 ± 0.9 to 6.6 ± 1.6 cm/year in patients with ACH for at least 6 months [81]. In longer-term studies, GH treatment in ACH patients promotes their height velocity in the first treatment year and promotes their linear growth, with a gain of 1–1.5 SD over 3–6 years, although height velocity is low after the second year of treatment (Fig. 8.3) [82–84]. More than 15 years have passed since approval, but reports on the long-term effect of GH on ACH regarding the prognosis of height and bone mineral metabolism have still not been published.

Short-term effect of GH treatment in achondroplasia (ACH). The graph shows the percentage increase in height in ACH patients. Growth velocity is increased when GH treatment is started and is maintained at a higher level than that before GH treatment during 3 years [83]
HCH is also mainly caused by mutations of the FGFR3 gene and is characterized by short stature and abnormal body proportions, although not as severe as in ACH. The final height in HCH is compromised and in the range of 132–147 cm [85, 86]. GH treatment for HCH has been approved only in Japan at the same time as ACH in 1997. Several reports have shown that the median height SD score is approximately −3.2 SD at the start of GH therapy for HCH and it improves plus 1 SD after 2–5 years of GH treatment [87, 88]. Because some HCH patients have no mutation in the FGFR3 gene, but characteristic facial features, bone deformities, and disproportionate short stature are observed, there are still some doubts as to the certainty of the diagnosis in some of the patients diagnosed with HCH. Therefore, clinical studies of GH treatment, including genetic background data, are required.
Dyschondrosteosis, or Leri–Weill syndrome, is a mesomelic skeletal disorder caused by a deletion or mutation in the SHOX gene [89]. In dyschondrosteosis, there are abnormal proportions due to short legs, and the adult height in these individuals is variable, but in most patients it is reduced. However, a reduction in height appears to be sex-specific, with a greater loss of height in females compared with males [90]. Isolated SHOX haploinsufficiency is observed in 56–100 % of patients with Leri–Weill dyschondrosteosis and in 1–14 % of ISS [91]. Short stature observed in patients with TS is partially explained by haploinsufficiency of the SHOX gene [92]. Because GH treatment in TS improves the final height SD score, GH treatment in patients with SHOX haploinsufficiency has been approved in some countries. Prepubertal children with isolated SHOX defects treated with GH during 2 years present with a similar growth response to that of TS patients [93] and reach their final height with a height SD score gain of 1.1 ± 0.7 after 4.7 years [94]. The gain in the height SD score during the first year of GH therapy for patients with SHOX haploinsufficiency shows an increase of 0.7 SD [95]. The sitting height ratio SD score does not change during 1 year of GH treatment in patients with SHOX haploinsufficiency. Adult height in GH treatment for dyschondrosteosis has not been published yet.
Challenging Trials of GH Treatment
GH treatment has been attempted in many diseases with short stature, such as Down syndrome, Cornelia de Lange syndrome, Kabuki syndrome, Fanconi anemia, Rubinstein–Taybi syndrome, Klippel–Feil syndrome, Diamond–Blackfan anemia, and skeletal dysplasia. We discuss below regarding GH treatment in skeletal dysplasia, focusing on GH and bone, such as osteogenesis imperfecta (OI) and X-linked hypophosphatemic (XLH) rickets. Because the final height of each disorder has not been determined yet, further evidence of GH treatment in all challenging disorders needs to be gathered.
Osteogenesis Imperfecta
OI is an autosomal dominant disorder caused by dysfunction of type I collagen proteins. OI is characterized by congenital-decreased BMD, bone fragility, short stature, blue sclerae, progressive bone deformities, and dentinogenesis imperfecta [96, 97]. Clinical severity varies widely from lethal to mild with non-deformity. Recently, OI patients were classified into eight types according to their severity [98, 99].
The most popular internal treatment of OI is bisphosphonates suppressing bone resorption. Bisphosphonates in OI children increase BMD and result in dramatically decreased bone fractures [97]. Because growth deficiency is constantly present in severe OI and common in mild to moderate forms of OI, GH could be used in OI for stimulating bone metabolism or for increasing linear growth [100, 101].
Although there are few reports of GH treatment in patients with OI, GH action positively affects bone growth and bone turnover by stimulating osteoblasts, collagen synthesis, and longitudinal bone growth [102, 103]. A recent study also suggested that combined bisphosphonate and GH treatment in OI patients for 1 year positively increases BMD and growth velocity, and does not affect the peripheral fracture rate [104]. Although GH treatment in OI has not been approved yet, GH may be expected to improve symptoms of OI patients.
X-Linked Hypophosphatemic Rickets
XLH rickets is the most common form of hereditary rickets and is characterized by short stature, rickets, osteomalacia, and hypophosphatemia [105]. XLH rickets is due to mutations in the phosphate-regulating gene with homologies to endopeptidases on the X chromosome (PHEX), which encodes a membrane-bound endopeptidase expressed in mineralizing tissues (i.e., bone and teeth) [106]. Although the precise function of PHEX protein still remains to be determined, inactivation of PHEX reduces serum phosphate levels by suppressing proximal tubular phosphate reabsorption and intestinal phosphate absorption though synthesis of fibroblast growth factor 23 [107]. This has been shown to be causative for renal phosphate wasting and diminished 1α-hydroxylation of 25(OH) vitamin D [108, 109].
Combined treatment with oral phosphates and activated vitamin D (calcitriol) has been shown to improve growth and skeletal abnormalities in XLH rickets [110–112]. Even with optimal medical treatment, many XLH rickets patients do not demonstrate catch-up growth to achieve normal stature [113–115]. Mean adult height in cohorts of treated XLH rickets patients ranges from −2.8 to −1.7 SD [116].
IGF-I increases Phex expression in bone and sodium-dependent phosphate cotransporter mRNA expression in the kidney, and increases circulating phosphate concentration through these two mechanisms [117]. Previous studies have shown that administration of GH increases renal tubular phosphate reabsorption and serum concentrations of 1,25(OH)2D, suggesting that GH is involved in phosphate homeostasis and in renal 1α-hydroxylation of vitamin D through IGF-I [118, 119]. Although some studies have shown that GH treatment increases the growth rate of XLH rickets patients, some studies have suggested that GH might make deformities of XLH rickets worse. The effect of GH in XLH rickets is still controversial [118–122]. Further investigation may be able to add GH treatment to the standard choice in the future.
Other Skeletal Dysplasias
Metaphyseal chondrodysplasia (MCD) Schmid type is caused by inactive mutations in the type X collagen gene and treatment-free adult height is approximately 130–160 cm [123]. One-year GH treatment in patients with MCD Schmid type was reported to improve height from −3.2 SD to −2.7 SD [124]. No final heights from GH treatment in patients with MCD Schmid type are available.
MCD McKusick type, also known as cartilage-hair hypoplasia (CHH), is characterized by short-limbed short stature, hypoplastic hair, defective immunity, and diminished erythrocyte generation [125]. CHH is caused by several mutations in the RNA component of mitochondrial RNA processing endoribonuclease gene [126]. Adult height of CHH patients is reported as approximately 131.1 cm in males and 122.5 cm in females [127]. GH treatment increased height of a CHH patient from −4.2 to −2.1 SD together with limb lengthening [128], but another report showed no benefit of the treatment [129].
Spondyloepiphyseal dysplasia is caused by the mutation or deletion of the type II collagen (COL2A1) gene and is characterized by severe short stature and a markedly short trunk, with a final height of 100–125 cm [130]. GH treatment in 17 patients with spondyloepiphyseal dysplasia did not result in a significant increase in the height SD score during the first year of treatment, although some patients did appear to benefit [123]. However, the sitting height SD score was improved from 2.7 to 1.8. No adult heights from GH treatment in patients with spondyloepiphyseal dysplasia are available.
Pseudoachondroplasia is caused by mutation in the cartilage oligomeric matrix protein (COMP) gene and is characterized by severe short stature with a waddling gait, deformity of the legs, short fingers, loose joints, and ligamentous laxity [131]. The final height is approximately 80–130 cm. Only one report described that GH treatment was given to four patients with pseudoachondroplasia, and that GH did not increase annual height gain in the first year of the treatment [124]. The increase in height of seven patients with pseudoachondroplasia was not significant, with a median gain of 0.2 SD [123]. No adult heights in patients with pseudoachondroplasia who had GH treatment have been reported.
Conclusion
Physiological GH secreted by the pituitary gland and extrinsic recombinant GH have the ability to promote bone elongation and linear growth of children, as well as regulate bone mineral metabolism with accelerating bone turnover. Currently, GH treatment has been established for many disorders. However, there are many types of diseases with short stature, and treatment of these patients is still limited. Although more evidence is required, GH has the possibility to be useful for other skeletal dysplasias, including metabolic bone diseases, because of its potential capacity for regulating bone mineral metabolism.
References
Van Cauter E, Plat L, Copinschi G. Interrelations between sleep and the somatotropic axis. Sleep. 1998;21:553–66.
Melmed S, Kleinberg D, Ho K. Pituitary physiology and diagnostic evaluation. In: Kronenberg HM, Melmed S, Polonsky KS, Larsen PR, editors. Williams textbook of endocrinology. 12th ed. Philadelphia, PA: Saunders Elsevier; 2011. p. 186–97. Chapter 8.
Barinaga M, Yamonoto G, Rivier C, Vale W, Evans R, Rosenfeld MG. Transcriptional regulation of growth hormone gene expression by growth hormone-releasing factor. Nature. 1983;306:84–5.
Gaylinn BD. Molecular and cell biology of the growth hormone-releasing hormone receptor. Growth Horm IGF Res. 1999;9:37–44.
Kojima M, Hosoda H, Date Y, Nakazato M, Matsumoto H, Kangawa K. Ghrelin is a growth-hormone-releasing acylated peptide from stomach. Nature. 1999;402:656–60.
Howard AD, Feighner SD, Cully DF, Arena JP, Liberator PA, Rosenblum CI, Hamelin M, Hreniuk DL, Palyha OC, Anderson J, Paress PS, Diaz C, Chou M, Liu KK, McKee KK, Pong SS, Chaung LY, Elbrecht A, Dashkevicz M, Heavens R, Rigby M, Sirinathsinghji DJ, Dean DC, Melillo DG, Patchett AA, Nargund R, Griffin PR, DeMartino JA, Gupta SK, Schaeffer JM, Smith RG, Van der Ploeg LH. A receptor in pituitary and hypothalamus that functions in growth hormone release. Science. 1996;273:974–7.
Cummings DE, Purnell JQ, Frayo RS, Schmidova K, Wisse BE, Weigle DS. A preprandial rise in plasma ghrelin levels suggests a role in meal initiation in humans. Diabetes. 2011;50:1714–9.
Feldt-Rasmussen U. Metabolic effects of growth hormone. In: Ranke MB, Price DA, Reiter EO, editors. Growth hormone therapy in pediatrics-20 years of KIGS. Basel: Karger; 2007. p. 477–84.
Le Roith D. Seminars in medicine of the Beth Israel Deaconess Medical Center. Insulin-like growth factors. N Engl J Med. 1997;336:633–40.
Yakar S, Liu JL, Stannard B, Butler A, Accili D, Sauer B, LeRoith D. Normal growth and development in the absence of hepatic insulin-like growth factor I. Proc Natl Acad Sci U S A. 1999;96:7324–9.
Ohlsson C, Nilsson A, Isaksson O, Lindahl A. Growth hormone induces multiplication of the slowly cycling germinal cells of the rat tibial growth plate. Proc Natl Acad Sci U S A. 1992;89:9826–30.
Raben MS. Treatment of a pituitary dwarf with human growth hormone [Letter]. J Clin Endocrinol. 1958;18:901.
Burns EC, Tnner JM, Preece MA, Cameron N. Final height and pubertal development in 55 children with idiopathic growth hormone deficiency, treated for between 2 and 15 years with human growth hormone. Eur J Pediatr. 1981;137:155–64.
Bundak R, Hindmarsh PC, Smith PJ, Brook CGD. Long term auxologic effects of human growth hormone. J Pediatr. 1988;112:875–9.
Joss E, Juppinger K, Schwarz HP, Roten G. Final height of patients with pituitary growth failure and changes in growth variables after long-term hormonal therapy. Pediatr Res. 1983;17:676–9.
Fradkin JE, Schonberger LB, Mills JL, Gunn WJ, Piper JM, Wysowski DK, Thomson R, Durako S, Brown P. Creutzfeldt-Jakob disease in pituitary growth hormone recipients in the United States. JAMA. 1991;265:880–4.
Lawson Wilkins Pediatric Endocrine Society Committee, Underwood LE, Fisher DA, Frasier SD, Gertner JM, Kaplan SL, Kirkland RT, Lippe BM, Salvatore R. Degenerative neurologic disease in patients formerly treated with human growth hormone-Report of the Committee on Growth Hormone Use of the Lawson Wilkins Pediatric Endocrine Society, May 1985. J Pediatr. 1985;107:10.
Allen DB. Growth hormone treatment. In: Lifshitz F, editor. Pediatric endocrinology. 4th ed. CRC: Boca Raton; 2006. p. 87.
Timoin DL, Merimee TJ, Rabinowitz D, McKusick VA. Genetic aspects of clinical endocrinology. Recent Prog Horm Res. 1968;24:365–437.
Ranke MB. A note on adults with growth hormone deficiency. Acta Paediatr Scand Suppl. 1987;331:80–2.
Van der Wefften Bosch JJ, Bot A. Growth of males with idiopathic hypopituitarism without growth hormone treatment. Clin Endocrinol. 1990;32:707–17.
August GP, Julius JR, Blethwn SL. Adult height in children with growth hormone deficiency who are treated with biosynthetic growth hormone: the National Cooperative Growth Study experience. Pediatrics. 1998;102:512–6.
Cutfield WS, Lindberg A, Chatelain P, Price DA, Albertsson-Wikland K, Wilton P, Ranke MB. Final height following growth hormone treatment of idiopathic growth hormone deficiency in KIGS. In: Ranke BM, Wilton P, editors. Growth hormone therapy in KIGS—10 years’ experience. Heidelberg-Leipzig: Johann Ambrosius Barth; 1999. p. 93–110.
Karlberg J, Albertsson-Wikland K. Growth in full-term small-for-gestational-age infants: from birth to final height. Pediatr Res. 1995;38:733–9.
Tenovuo A, Kero P, Piekkala P, Korvenranta H, Sillanpaa M, Erkkola R. Growth of 519 small for gestational age infants during the first two years of life. Acta Paediatr Scand. 1987;76:636–46.
Hokken-Koelega AC, De Ridder MA, Lemmen RJ, Den Hartog H, De Muinck Keizer-Schrama SM, Drop SL. Children born small for gestational age: do they catch up? Pediatr Res. 1995;38:267–71.
Albertsson-Wikland K, Karlberg J. Natural growth in children born small for gestational age with and without catchup growth. Acta Paediatr. 1994;399:64–70.
Leger J, Levy-Marchal C, Bloch J, Pinet A, Chevenne K, Porquet D, Collin D, Czernichow P. Reduced final height and indications for insulin resistance in 20 year olds born small for gestational age; regional cohort study. BMJ. 1997;315:341–7.
VanPareren Y, Mulder P, Houdijk M, Jansen M, Reeser M, Hokken-Koelega A. Adult height after long-term, continuous growth hormone (GH) treatment in short children born small for gestational age; results of a randomized, double-blind dose–response GH trial. J Clin Endocrinol Metab. 2003;88:3584–90.
Bannink EMN, van Pareren Y, Teunissen NCM, Raat H, Mulder PGM, HokkenKoelega ACS. Quality of life in adolescents born small for gestational age: dose growth hormone make a difference. Horm Res. 2005;64:166–74.
Hokken-Koelega ACS, Vam Pareren Y, Sas T, Arends N. Final height data, body composition and glucose metabolism in growth hormone-treated short children born small for gestational age. Horm Res. 2003;60:113–4.
Turner H. A syndrome of infantilism, congenital webbed neck, and cubitus valgus. Endocrinology. 1938;28:566–74.
Soriano-Guillen L, Coste J, Ecosse E, Leger J, Tauber M, Cabrol S, Nicolio M, Brauner R, Chaussain JL, Carel JC. Adult height and pubertal growth in Turner syndrome after treatment with recombinant growth hormone. J Clin Endocrinol Metab. 2005;90:5197–204.
Canadian Growth Hormone Advisory Committee. Impact of growth hormone supplementation on adult height in Turner syndrome: results of the Canadian randomized controlled trial. J Clin Endocrinol Metab. 2005;90:3360–6.
Gorman G, Fivush B, Frankenfield D, Warady B, Watkins S, Brem A, Neu A. Short stature and growth hormone use in pediatric hemodialysis patients. Pediatr Nephrol. 2005;20:1794–800.
Shroff R, Rees L, Trompeter R, Hutchinson C, Ledermann S. Long-term outcome of chronic dialysis children. Pediatr Nephrol. 2006;21:257–64.
Hokken-Koelega AC, Van Zaal MA, van Bergen W, de Ridder MA, Stijnen T, Wolff ED, de Jong RC, Donckerwolcke RA, de Muinck Keizer-Schrama SM, Drop SL. Final height and its predictive factors after renal transplantation in childhood. Pediatr Res. 1994;36:323–8.
Andre JL, Bourquard R, Guillemin F, Krier MJ, Briancon S. Final height in children with chronic renal failure who have not received growth hormone. Pediatr Nephrol. 2003;18:685–9.
Mehls O, Lindberg A, Nissel R, Wuhl E, Schaefer F, Tonshoff B, Haffner D. Growth hormone treatment in short children with chronic kidney disease. In: Ranke MB, Price DA, Reiter EO, editors. Growth hormone therapy in pediatrics—20 years of KIGS. Basel: Karger; 2007. p. 407–21.
Fine RN, Stablein D. Long-term use of recombinant human growth hormone in pediatric allograft recipients: a report of the NAPRTCS Transplant Registry. Pediatr Nephrol. 2005;20:404–8.
Crompton CH, Australian and New Zealand Paediatric Nephrology Association. A long-term recombinant human growth hormone use in Australian children with renal disease. Nephrology (Carlton). 2004;9:325–30.
Wollmann HA, Schltz U, Grauer ML, Ranke MB. Reference values for height and weight in Prader-Willi syndrome based on 315 patients. Eur J Pediatr. 1998;157:634–42.
Hauffa BP, Schlippe G, Roos M, Gillessen-Kaesbach G, Gasser T. Spontaneous growth in German children and adolescents with genetically confirmed Prader-Willi syndrome. Acta Paediatr. 2000;89:1302–11.
Lindgren AC, Hagenas L, Muller J, Blichfeldt S, Rosenborn M, Brismar T, Ritzen EM. Growth hormone treatment of children with Prader-Willi syndrome affects linear growth and body composition favourably. Acta Paediatr. 1998;87:28–31.
Davies PSW, Evens S, Broomhead S, Clough H, Day JME, Laidlaw A, Barnes ND. Effect of growth hormone on height, weight, and body composition in Prader-Willi syndrome. Arch Dis Child. 1998;78:474–6.
Eiholzer U, L’Allemand D. Growth hormone normalizes height, prediction of final height and length in children with Prader-Willi syndrome after 4 years of therapy. Horm Res. 2000;53:185–92.
Noonan JA. Hypertelorism with Turner phenotype. A new syndrome with associated congenital heart disease. Am J Dis Child. 1968;116:373–80.
Ranke M, Heidemann P, Knupfer C, Enders H, Schmaltz AA, Bierich JR. Noonan syndrome: growth and clinical manifestations in 144 cases. Eur J Pediatr. 1988;148:220–7.
Oiso D, Dahlgren J, Wikland KA, Westphal O. Improved final height with long-term growth hormone treatment in Noonan syndrome. Acta Paediatr. 2005;94:1232–7.
Limal JM, Parfait B, Cabrol S, Bonnet D, Leheup B, Lyonnet S, Vidaud M, Le Bouc Y. Noonan syndrome; relationships between genotype, growth and growth factors. J Clin Endocrinol Metab. 2006;91:300–6.
Wit JM. Idiopathic short stature: definition, spontaneous growth and response to treatment. In: Ranke MB, Price DA, Reiter EO, editors. Growth hormone therapy in pediatrics-20 years of KIGS. Basel: Karger; 2007. p. 309–18.
Wit JM, Rekers-Mombarg LT. Final height gain by GH therapy in children with idiopathic short stature is dose dependent. J Clin Endocrinol Metab. 2002;87:604–11.
Buzi F, Buchanan C, Morrell DJ, Preece MA. Antigenicity and efficacy of authentic sequence recombinant human growth hormone (somatropin): first year experience in the United Kingdom. Clin Endocrinol. 1989;30:531–8.
Sato T, Suzuki Y, Taketani T. Enhanced peripheral conversion of thyroxine to triiodothyronine during GH therapy in GH deficient children. J Clin Endocrinol Metab. 1977;45:324–9.
Municchi G, Malozowski S, Nisula BC, Cristiano A, Rose SR. Nocturnal thyrotropin surge in growth hormone-deficient children. J Pediatr. 1992;121:214–20.
Sherwin RS, Schulman GA, Hendler R, et al. Effect of growth hormone on oral glucose tolerance and circulating metabolic fuels in man. Diabetologia. 1983;24:155.
Schnure JJ, Raskin P, Lipman RL. Growth hormone secretion during sleep: impairment in glucose tolerance and nonsuppressibility by hyper glycemia. J Clin Endocrinol Metab. 1971;33:234.
Lampit M, Nave T, Hochberg X. Water and sodium retention during short-term administration of growth hormone to short normal children. Horm Res. 1998;50:83–8.
Otten BJ, Rotteveel JJ, Cruysberg JRM. Pseudotumor cerebri following treatment with growth hormone. Horm Res. 1992;37 Suppl 4:16.
Maneatis T, Baptista J, Connelly K, Blethen S. Growth hormone safety update from the national cooperative growth study. J Pediatr Endocrinol. 2000;13:1035–44.
Skottner A, Clark RG, Robinson IC, Fryklund L. Recombinant human insulin-like growth factor: testing the somatomedin hypothesis in hypophysectomized rats. J Endocrinol. 1987;112:123–32.
Leung K, Rajkovic IA, Reters E, Markus I, Van Wyk JJ, Ho KK. Insulin-like growth factor I and insulin down-regulate growth hormone (GH) receptors in rat osteoblasts: evidence for a peripheral feedback loop regulating GH action. Endocrinology. 1996;137:2694–702.
Hock JM, Centrella M, Canalis E. Insulin-like growth factor I has independent effects on bone matrix formation and cell replication. Endocrinology. 1988;122:254–60.
Nishiyama K, Sugimoto T, Kaji H, Kanatani M, Kobayashi T, Chihara K. Stimulatory effect of growth hormone on bone resorption and osteoclast differentiation. Endocrinology. 1996;137:35–41.
Klefter O, Feldt-Rasmussen U. Is increase in bone mineral content caused by increase in skeletal muscle mass/strength in adult patients with GH-treated GH deficiency? A systematic literature analysis. Eur J Endocrinol. 2009;161:213–21.
Ho PJ, Fig LM, Barkan AL, Shapiro B. Bone mineral density of the axial skeleton in acromegaly. J Nucl Med. 1992;33:1608–12.
Russell-Aulet M, Shapiro B, Jaffe CA, Gross MD, Barkan AL. Peak bone mass in young healthy men is correlated with the magnitude of endogenous growth hormone secretion. J Clin Endocrinol Metab. 1998;83:3463–8.
Baroncelli GI, Bertelloni S, Ceccarelli C, Saggese G. Measurement of volumetric bone mineral density accurately determines degree of lumbar undermineralization in children with growth hormone deficiency. J Clin Endocrinol Metab. 1998;83:3150–4.
Saggese G, Baroncelli GI, Bertelloni S, Barsanti S. The effect of long-term growth hormone (GH) treatment on bone mineral density in children with GH deficiency. Role of GH in the attainment of peak bone mass. J Clin Endocrinol Metab. 1996;81:3077–83.
Sass TC, De Muinck Keizer-Schrama SM, Stijnen T, Asarfi A, Van Leeuwen WJ, Van Teunenbroek A, Van Rijn RR, Drop SL. A longitudinal study on bone mineral density until adulthood in girls with Turner’s syndrome participating in a growth hormone injection frequency-response trial. Clin Endocrinol (Oxf). 2000;52:531–6.
Carrel AL, Myers SE, Whitman BY, Allen DB. Sustained benefits of growth hormone on body composition, fat utilization, physical strength and agility, and growth in Prader-Willi syndrome are dose-dependent. J Pediatr Endocrinol Metab. 2001;14:1097–105.
Ranke MB, Guilbaud O, Lindberg A, Cole T. Prediction of the growth response in children with various growth disorders treated with growth hormone: analyses of data from the Kabi Pharmacia International growth study. International Board of the Kabi Pharmacia International Growth Study. Acta Paediatr. 1993;82 Suppl 392:82–8.
Schönau E, Westermann F, Rauch F, Stabrey A, Wassmer G, Keller E, Brämswig J, Blum WF, German Lilly Growth Response Study Group. A new and accurate prediction model for growth response to growth hormone treatment in children with growth hormone deficiency. Eur J Endocrinol. 2001;144:13–20.
Fujimoto S, Kubo T, Tanaka H, Miura M, Seino Y. Urinary pyridinoline and deoxypyridinoline in healthy children and in children with growth hormone deficiency. J Clin Endocrinol Metab. 1995;80:1922–8.
Seino Y, Yamashita S, Morisaki Y, Tanaka H, Chihara K, Tanaka T. Japanese growth prediction model for prepubertal children with growth hormone deficiency. J Pediatr Endocrinol Metab. 2012;25:909–15.
Scott CI Jr. Dwarfism. Clin Symp 1988;1–32.
Hertel T, Seino Y. Skeletal dysplasia. In: Novo Nordisk A/S, editor. Growth hormone therapy in children and adults. Bagsvaerd: Novo Nordisk A/S; 2004. p. 61–74.
Shiang RTL, Zhu YZ, Church DM, Fielder TJ, Bocia M, Winodur ST, Wasmuth MM. Mutations in the transmembrane domain of FGFR3 cause the most common genetic form of dwarfism, achondroplasia. Cell. 1994;78:335–42.
Rousseau FBJ, Legeai ML, Pelet A, Rozet JM, Marotearx P, Le MM, Munnich A. Mutations in the gene encoding fibroblast growth factor receptor-3 in achondroplasia. Nature. 1994;371:252–4.
Horton WA, Rotter JI, Rimoin DL, Scott Jr CI, Hall JG. Standard growth curves for achondroplasia. J Pediatr. 1978;93:435–8.
Koike M, Yamanaka Y, Inoue M, Tanaka H, Nishimura R, Seino Y. Insulin-like growth factor-1 rescues the mutated FGF receptor 3 (G380R) expressing ATDC5 cells from apoptosis through phosphatidylinositol 3-kinase and MAPK. J Bone Miner Res. 2003;18:2043–51.
Seino Y, Yamate T, Kanzaki S, Kubo T, Tanaka H. Achondroplasia: effect of growth hormone in 40 patients. Clin Pediatr Endocrinol. 1994;3:41–5.
Tanaka H, Kubo T, Yamate T, Ono T, Kanzaki S, Seino Y. Effect of growth hormone therapy in children with achondroplasia: growth pattern, hypothalamic-pituitary function, and genotype. Eur J Endocrinol. 1998;138:276–80.
Ramaswami U, Rumsby G, Spoudeas HA, Hindmarsh PC, Brook CGD. Treatment of achondroplasia with growth hormone: six year of experience. Pediatr Res. 1999;46:435–9.
Hagenas L, Aagenaes O, Eklof O, Hertel T, Kaitila I, Mohnike K, Perhentuupa J, Ritzen M, Sipila I, Muller J. Growth hormone treatment in achondroplasia: 2 year resuols of a dose–response study. Clin Pediatr Endocrinol. 1997;6:93–8.
Riepe FG, Krone N, Sippell WG. Disproportionate stature but normal height in hypochondroplasia. Eur J Pediatr. 2005;164:397–9.
Appan S, Laurent S, Chapman M, Hindmarsh PC, Brook CGD. Growth and growth hormone therapy in hypochondroplasia. Acta Paediatr Scand. 1990;79:796–803.
Bridges NA, Hindmarsh P, Brook CGD. Growth of children with hypochondroplasia treated with growth hormone for up to three years. Horm Res. 1991;36:56–60.
Belin V, Cusin VM, Viot G, Girlich D, Toutain A, Monla A, Vekemans M, Le Marrer M, Munnich A, Cormier-Daire V. SHOX mutations in dyschondrosteosis (Leri-Weill syndrome). Nat Genet. 1998;19:67–9.
Ross JL, Sott Jr C, Marttila P, Kowal K, Nass A, Papenhausen P, Abboudi J, Osterman L, Kushner H, Carter P, Ezaki M, Elder F, Wei F, Chen H, Zinn AR. Phenotypes associated with SHOX deficiency. J Clin Endocrinol Metab. 2001;86:5674–80.
Jorge AA, Souza SC, Nishi MY, Billerbeck AE, Liborio DC, Kim CA, Arnhold IJ, Mendonca BB. SHOX mutations in idiopathic short stature and Leri-Weill dyschondrosteosis: frequency and phenotypic variability. Clin Endocrinol (Oxf). 2007;66:130–5.
Kosho T, Muroya K, Nagai T, Fujimoto M, Yokoya S, Sakamoto H, Hirano T, Terasaki H, Ohashi H, Nishimura G, Sato S, Matsuo N, Ogata T. Skeletal features and growth pattern sin 14 patients with haploinsufficiency of SHOX: implications for the development of Turner syndrome. J Clin Endocrinol Metab. 1999;84:4613–21.
Blum WF, Crowe BJ, Quigley CA, Jung H, Cao D, Ross JL, Braun L, Rappold G. Growth hormone is effective in treatment of short stature associated with short stature homeobox-containing gene deficiency: two-year results of a randomized, controlled, multicenter trial. J Clin Endocrinol Metab. 2007;92:219–28.
Blum WF, Cao D, Hesse V, Fricke-Otto S, Ross JL, Jones C, Quigley CA, Binder G. Height gains in response to GH treatment to final height are similar in patients with SHOX deficiency and Turner syndrome. Horm Res. 2009;71:167–72.
Hertel T. Growth hormone treatment in skeletal dysplasias: the KIGS experience. In: Ranke MB, Price DA, Reiter EO, editors. Growth hormone therapy in pediatrics-20 years of KIGS. Basel: Karger; 2007. p. 356–68.
Rauch F, Glorieux FH. Osteogenesis imperfect. Lancet. 2004;363:1377–85.
Cheung MS, Glorieux FH. Osteogenesis imperfect: update on presentation and management. Rev Endocr Metab Disord. 2008;9:153–60.
Sillence DO, Senn A, Dannks DM. Genetic heterogeneity in osteogenesis imperfecta. J Med Genet. 1979;16:101–16.
Van Dijk FS, Pals G, van Rijn RR, Nikkels PG, Cobbben JM. Classification of osteogenesis imperfect revisited. Eur J Med Genet. 2010;53(1):1–5.
Lund AM, Muller J, Skovby F. Anthropometry of patients with osteogenesis imperfect. Arch Dis Child. 1999;80:524–8.
Monti E, Mottes M, Franschini P, Brunelli PC, Forlino A, Venturi G, Doro F, Perlini S, Cavarzere P, Antoniazzi F. Current and emerging treatments for the management of osteogenesis imperfect. Therapeut Clin Risk Manage. 2010;6:376–81.
Antoniaxxi F, Bertoldo F, Mottes M, Sirpresi S, Zamboni G, Valentini R, Tató L. Growth hormone treatment in osteogenesis imperfect with quantitative defect of type I collagen synthesis. J Pediatr. 1996;129:432–9.
Marini JC, Hopkins E, Glorieux FH, Chrousos GP, Reynorlds JC, Gundberg CM, Reing CM. Positive linear growth and bone responses to growth hormone treatment in children with types III and IV osteogenesis imperfect: high predictive value of the carboxyterminal propeptide of type I procollagen. J Bone Min Metab. 2003;18:237–43.
Antoniazzi F, Monti E, Venturi F, Franceschi R, Doro F, Gatti D, Zamboni G, Tato L. GH in combination with bisphosphonate treatment in osteogenesis imperfect. Eur J Endocrinol. 2010;163:479–87.
Alon US. Hypophosphatemic vitamin-D resistant rickets. In: Favus MJ, editor. Primer on the metabolic bone diseases and disorders of mineral metabolism. Washington, DC: American Society of Bone and Mineral Research; 2006. p. 342–5.
HYP Consortium. A gene (PEX) with homologies to endopeptidases is mutated in patients with X-linked hypophosphatemic rickets. The HYP Consortium. Nat Genet. 1995;11:130–6.
Fukumoto S. Physiological regulation and disorders of phosphate metabolism—pivotal role of fibroblast growth factor 23. Inter Med. 2008;47:337–43.
Aono Y, Yamazaki Y, Yasutake J, Kawata T, Hasegawa H, Urakawa I, Fujita T, Wada M, Yamashita T, Fukumoto S, Shimada T. Therapeutic effects of anti-FGF23 antibodies in hypophosphatemic rickets/osteomalacia. J Bone Miner Res. 2008;24:1879–88.
de Beur SM J, Levine MA. Molecular pathogenesis of hypophosphatemic rickets. J Clin Endocrinol Metab. 2002;87:2467–73.
Glorieux FH, Scriver CR, Reade TM, Goldman H, Roseborough A. Use of phosphate and vitamin D to prevent dwarfism and rickets in X-linked hypophosphatemia. N Engl J Med. 1972;287:481–7.
Harrell RM, Lyles KW, Harrelson JM, Friedman NE, Drezner MK. Healing of bone disease in X-linked hypophosphatemic rickets/osteomalacia. Induction and maintenance with phosphorus and calcitriol. J Clin Invest. 1985;75:1858–68.
Verge CF, Lam A, Simpson JM, Cowell CT, Howard NJ, Silind M. Effects of therapy in X-linked hypophosphatemic rickets. N Engl J Med. 1991;325:1843–8.
Chesney RW, Mazess RB, Rose P, Hamstra AJ, DeLuca HF, Breed AL. Long-term influence of calcitriol (1,25-dihydroxyvatamin D) and supplemental phosphorus in X-linked hypophosphatemic rickets. Pediatrics. 1983;83:81–7.
Friedman NE, Lobaugh B, Drezner MK. Effects of calcitriol and phosphorus therapy on the growth of patients with X-linked hypophosphatemia. J Clin Endocrinol Metab. 1993;76:839–44.
Makitie O, Doria A, Kooh SW, Cole WG, Daneman A, Sochett E. Early treatment improves growth and biochemical and radiographic outcome in X-linked hypophosphatemic rickets. J Clin Endocrinol Metab. 2003;88:3591–7.
Baroncelli GI, Bertelloni S, Ceccarelli C, Saggese G. Effect of growth hormone treatment on final height, phosphate metabolism, and bone mineral density in children with X-linked hypophosphatemic rickets. J Pediatr. 2001;138:236–43.
Zoidis E, Zapf J, Schmid C. Phex cDNA cloning from rat bone and studies on phex mRNA expression: tissue specificity, age dependency, and regulation by insulin-like growth factor I in vivo. Mol Cell Endocrinol. 2000;168:41–51.
Bianda T, Glatz Y, Bouillon R, Froesch ER, Schmid C. Effects of short-term insulin-like growth factor-I or Gh treatment on bone metabolism and on production of 1,25-dihydroxycholeaclciferol in GH-deficient adults. J Clin Endocrinol Metab. 1998;83:81–7.
Caverzasio J, Montessuit C, Bonjour JP. Stimulatory effect of insulin-like growth factor-I on renal Pi transport and plasma 1,25-dihydroxyvitamin D3. Endocrinology. 1990;127:453–9.
Saggesse G, Baroncelli GI, Bertelloni S, Perri G. Long-term growth hormone treatment in children with renal hypophosphatemic rickets: effects on growth, mineral metabolism, and bone density. J Pediatr. 1995;127:395–402.
Haffner D, Wuhl E, Blum WF, Schaefer F, Mehls O. Disproportionate growth following long-term growth hormone treatment in short children with X-linked hypophosphatemia. Eur J Pediatr. 1995;154:610–3.
Seikaly MG, Brown R, Baum M. The effect of recombinant human growth hormone in children with X-linked hypophosphatemia. Pediatrics. 1997;100:879–84.
Hertel T. Growth hormone treatment in skeletal dysplasias: the KIGS experience. In: Ranke MB, Price DA, Reiter EO, editors. Growth hormone therapy in pediatrics-20 years of KIGS. Basel: Karger; 2007. p. 477–84.
Kanazawa H, Tanaka H, Inoue M, Yamanaka Y, Namba N, Seino Y. Efficacy of growth hormone therapy for patients with skeletal dysplasia. J Bone Miner Metab. 2003;21:307–10.
McKusick VA, Eldridge R, Hostetler JA, Egeland JA, Ruangwit U. Dwarfism in the Amish. 2. Cartilage-hair hypoplasia. Bull Johns Hopkins Hosp. 1965;116:285–326.
Ridanpaa M, van Eenennaam H, Pelin K, Chadwick R, Johnson C, Yuan B, van Venrooij W, Pruijn G, Salmela R, Rockas R, Makitie O, Kaitila I, de la Chapella A. Mutations in the RNA component of RNase MRP cause a pleotropic human disease, cartilage-hair dysplasia. Cell. 2001;104:195–203.
Makitie O, Perheentupa J, Kaitila I. Growth in cartilage-hair hypoplasia. Pediatr Res. 1992;31:176–80.
Harada D, Yamanaka Y, Ueda K, Shimizu J, Inoue M, Seino Y, Tanaka H. An effective case of growth hormone treatment on cartilage-hair hypoplasia. Bone. 2005;36:317–22.
Bocca G, Weenaes CM, van der Burgt I, Otten BJ. Growth hormone treatment in cartilage-hair hypoplasia: effect on growth and the immune system. J Pediatr Endocrinol Metab. 2004;17:47–54.
Nishimura G, Haga N, Kitoh H, Tanaka Y, Sonoda T, Kitamura M, Shirahama S, Itoh T, Nakashima E, Ohashi H, Ikegawa S. The phenotypic spectrum of COL2A1 mutations. Hum Mutat. 2005;26:36–43.
Briggs MD, Hoffmann SMG, King LM, Olsen AS, Mohrenweser H, Leroy JG, Mortier GR, Rimoin DL, Lachman RS, Gaines ES, Cekleniak JA, Knowlton RG, Cohn DH. Pseudoachondroplasia and multiple epiphyseal dysplasia due to mutations in the cartilage oligomeric matrix protein gene. Nat Genet. 1995;10:330–6.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2014 Springer Science+Business Media New York
About this chapter
Cite this chapter
Harada, D., Seino, Y. (2014). Growth Hormone and Bone. In: Klein, G. (eds) Bone Drugs in Pediatrics. Springer, Boston, MA. https://doi.org/10.1007/978-1-4899-7436-5_8
Download citation
DOI: https://doi.org/10.1007/978-1-4899-7436-5_8
Published:
Publisher Name: Springer, Boston, MA
Print ISBN: 978-1-4899-7435-8
Online ISBN: 978-1-4899-7436-5
eBook Packages: MedicineMedicine (R0)