
Abstract
Diabetic cardiomyopathy, which is defined as cardiac disease independent of vascular complications, is considered one of the consequences of the altered metabolic milieu during diabetes. Constant requirement for energy in the form of ATP is fulfilled mainly by utilizing carbohydrates (glucose and lactate) and fatty acids in the heart. Only minor differences exist between species, and the healthy adult heart relies on the oxidation of fatty acids for ATP production. Utilization of energetic substrates depends on many factors and hormones play a major role in the process. Insulin deficiency, for example, affects the levels of circulating glucose as well as fatty acids and, most certainly, these alterations contribute to the utilization of these substrates. In the past few decades, adipose tissue-originated hormones, such as leptin and adiponectin with major effects on metabolism, have been identified. Not only the amounts of hormones or substrate supply but also subcellular modifications seem to determine the heart’s preference for certain substrates during physiological and pathological transitions. Among them, in diabetes, the preference of the heart changes, or perhaps the heart becomes obligated to adapt to dramatic shifts in hormones, substrate supply, and subcellular alterations. This chapter summarizes the contribution of energetic substrate metabolism to the development of diabetic cardiomyopathy.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others


Keywords
1 Introduction
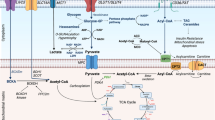
The heart has a very high energy demand because of the continuous work performed by the cardiac muscle. Under physiological conditions, the main substrates for energy production are fatty acids (60–90 % of ATP production), glucose, and lactate. Under nonischemic conditions, more than 95 % of the energy requirement is supplied by the oxidation of fatty acids and carbohydrates, and myocardial energy expenditure can be estimated from oxygen consumption. The contribution of these substrates to overall energy production is a dynamic process, and physiological adaptations such as fetal-to-newborn transition [1–3], and alterations related to disease states have been well established [4–8]. The heart exerts a metabolic flexibility, and myocardial substrate utilization depends on substrate availability, nutritional status, and exercise level. With glucose as the more energetically efficient substrate, the healthy heart is able to switch to glucose under stress conditions, such as ischemia, pressure overload, or in heart failure. Interestingly, interventions such as increasing fatty acid uptake [9, 10], or fatty acid oxidation [11, 12], result in alterations that resemble diabetic cardiomyopathy, and normalization of substrate metabolism in diabetic models reverses these alterations [13]. These studies indicate an important contributing role for substrate metabolism in the development of diabetic cardiomyopathy.
Although excessively available during diabetes, utilization of carbohydrates is compromised. Thus, the myocardium is forced to use alternative sources for ATP production such as fatty acids, which are high in diabetes as well [14–18]. The consequences of this pathological switch occur at multiple levels, as is discussed here.
From the mechanical efficiency point of view, an increase in fatty acid usage at the expense of carbohydrates results in ATP hydrolysis for noncontractile purposes and is attributed to a lower phosphate/oxygen (P/O) ratio for fatty acid metabolism, increased uncoupling of mitochondrial oxidative phosphorylation, and futile cycling (reviewed in [4]).
High levels of fatty acids further lower glucose usage as first defined by Randle et al. [19]. We now know that intracellular changes occur during diabetes and contribute to the aforementioned switch as well as development of diabetic cardiomyopathy.
2 Alterations in Cardiac Carbohydrate Utilization
Two glucose transporters, GLUT1 and GLUT4, are involved in basal and insulin-mediated glucose uptake. GLUT1 shows sarcolemmal localization and represents basal cardiac uptake. GLUT4, on the other hand, is located in the intracellular pool, and insulin facilitates the localization of this transporter to the sarcolemma [20]. More recently, AMP-dependent protein kinase (AMPK)-mediated and insulin-independent uptake of glucose by this transporter has been documented [21]. A decrease in the number and translocation of GLUT4 to the sarcolemma has been suggested to play a major role in the reduction of glucose metabolism in diabetes. A reduction in both glycolysis and glucose oxidation was reported in db/db mice in which cardiac dysfunction has been already defined [13]. Because both metabolic parameters and cardiac function were normalized in transgenic mice that overexpressed GLUT4, it has been concluded that there was a causative relationship between impaired substrate metabolism and diabetic cardiomyopathy [13]. A key enzyme in the regulation of glycolysis is phosphofructokinase (PFK)-I, the enzyme that catalyzes the phosphorylation of fructose-6-phosphate to produce fructose-1,6-bisphosphate. PFK-I activity is inhibited by citrate and acetyl CoA and activated by a low ATP/ADP ratio [22]. An increase in citrate levels as a result of increased fatty acid oxidation in the diabetic heart probably contributes to the inhibition of PFK-I and therefore glycolysis. At the transcriptional level of glucose uptake and metabolism, Finck et al. reported that both GLUT4 and PFK expression were lower and that PDK4 expression was higher in transgenic mice that overexpress peroxisome proliferator-activated receptor (PPAR)-α [12]. The inhibition of GLUT4 and PFK was probably not a direct result of PPAR-α overexpression, but associated with alterations in substrate metabolism that were mediated by PPAR-α. The increase in PDK4, on the other hand, was likely linked to PPAR-α overexpression because PPAR-α ligands were previously shown to activate this enzyme [23]. Another member of the PPAR family of transcription factors is PPAR-δ. PPAR-δ is the predominant form in the heart and regulates cardiac substrate metabolism [24]. A decrease in PPAR-δ expression was reported in diabetic heart [25]. However, another similar study reported that mice overexpressing PPAR-β/δ did not accumulate myocardial lipid and had normal cardiac function [26]. Conversely, cardiac glucose transport and glycolytic enzymes were activated in PPAR-β/δ transgenics.
Another limiting step in myocardial glucose metabolism is pyruvate dehydrogenase complex (PDH), which catalyzes the irreversible conversion of pyruvate to acetyl CoA. The amount of active dephosphorylated PDH is reduced when phosphorylated by PDH kinase (PDK) and induced also by PDH phosphatase. The rate of pyruvate oxidation depends not only on the phosphorylation state but also on the concentrations of its substrates (pyruvate, NAD+, and CoA) and products (NADH and acetyl CoA). Thus, an increase in mitochondrial acetyl CoA, through an increase in fatty acid oxidation, for example, inhibits pyruvate oxidation. Indeed, the active dephosphorylated form of the PDH is reduced in diabetes models [7]. Moreover, PDK-4 is one of the targets of PPAR-α, and upregulation of PDK-4 in mice overexpressing PPARα is associated with reduced glucose oxidation [27].
Inhibition of pyruvate conversion into acetyl CoA results in accumulation and diversion of glycolytic intermediates into diacylglycerol biosynthesis, which contributes to the activation of diacylglycerol-sensitive protein kinase C (PKC) isoforms. Recently, inhibition of one of the PKC isoforms, PKC-beta, was shown to preserve cardiac function in a transgenic mice model of diabetic diastolic failure [28].
The reports on carbohydrate utilization in the human diabetic heart are controversial. Studies in type 1 diabetic patients reported lower [29, 30] or unchanged [31] uptake of carbohydrates by the myocardium. In type 2 diabetes, GLUT4 protein levels in diabetics were about 30 % lower compared to control patients [32]. However, other studies reported that cardiac glucose uptake was not compromised in type 2 diabetes [33–35] and reduced only in type 2 diabetics with hypertriglyceridemia [36] and increased plasma fatty acids. Because glucose can still enter the cell through mass action, as evidenced by a high glucose pool in the type 1 diabetic heart [22], glucose metabolism is unlikely to be regulated at the level of uptake in diabetes despite a deficiency in or resistance to insulin action.
Lactate is another potential substrate for myocardial ATP production in vivo [37], but the information on diabetes-related alterations in lactate oxidation is relatively scant. A greater decrease in lactate oxidation relative to glucose oxidation in hearts from diabetic rats was observed when lactate and glucose were the only substrates in the perfusate for ATP production. Under these conditions, a specific inhibition of lactate oxidation independent of pyruvate dehydrogenase was suggested [38]. Hearts from ZDF rats also showed lower lactate oxidation [39, 40]. The contribution of lactate in diabetic cardiomyopathy clearly needs further study.
3 Alterations in Cardiac Fatty Acid Utilization
An increase of fatty acids that are supplied as free acids bound to albumin and as esters in chylomicrons and very low density lipoproteins have been reported in diabetes [7]. The effects of elevated levels of lipoproteins on myocardial fatty acid metabolism are not clear, and neither is the relative contribution of cardiac lipoprotein lipase (LPL) activity to the delivery of free fatty acids to the diabetic heart. Unchanged, increased, and decreased levels of LPL protein and activity were reported in the diabetic heart, and this discrepancy was suggested to be related to the diversity in rat strains, the dosage of diabetogenic agent, and the duration of diabetes [41].
Free fatty acids enter the cardiac myocyte either by passive diffusion or via a protein carrier-mediated pathway. These protein carriers include fatty acid translocase (FAT)/CD36, the plasma membrane isoform of fatty acid-binding protein (FABPpm), and fatty acid transport protein (FATP) 1/6. FAT/CD36 plays a major role in the translocation of fatty acid across the sarcolemmal membrane of cardiac myocytes as this protein was shown to mediate 50–60 % of fatty acid and transport of the heart. Also, FAT/CD36 is able to translocate between intracellular endosomes and the sarcolemmal membrane and thereby regulate fatty acid uptake [4].
Fatty acid uptake is increased in diabetes and leads to increased fatty acid oxidation and triacylglycerol (TAG) storage. In the streptozotocin (STZ)-induced model of type 1 diabetes, this increase is facilitated by fatty acid translocase (FAT/CD36) [42]. In type 2 diabetic models, an increase in FAT/CD36 and fatty acid-binding protein (FATP1) [43] and a permanent relocation of FAT/CD36 to the cardiomyocyte membrane was shown to increase fatty acid uptake [44, 45]. Interestingly, insulin was suggested to upregulate FAT/CD36 and translocate it to the sarcolemma [46].
The majority (70–90 %) of the fatty acids that enter cardiomyocyte is oxidized for energy production; the rest is converted to TAGs [47]. Excessive accumulation of lipids, or lipotoxicity, within nonadipose tissue provides substrates for nonoxidative processes such as ceramide and diacylglycerol synthesis, which can lead to apoptosis [48, 49]. Accumulation of TAG within the myocardium of insulin-resistant rats was associated with contractile dysfunction [50]. We also have shown that insulin-resistant rats have increased TAG accumulation, which reduced insulin-stimulated glucose metabolism [51]. Although the exact mechanism of lipotoxicity-induced cardiac dysfunction is not known, it seems to be related to a combination of apoptotic cell death and impaired substrate metabolism.
The most important step in the regulation of fatty acid oxidation is the transport of fatty acids into the mitochondria for further metabolism. The activation of short- and medium-chain fatty acids occurs in the matrix and does not require carnitine. However, long-chain fatty acids are shuttled into the mitochondria by three carnitine-dependant enzymes. Carnitine palmitoyltransferase (CPT)-I catalyzes the conversion of long-chain acyl CoA to long-chain acylcarnitine. Carnitine:acylcarnitine translocase (CAT) transports long-chain acylcarnitine across the inner mitochondrial membrane, and CPT-II regenerates long-chain acyl-CoA in the mitochondrial matrix [52]. Of these, CPT-I is the master regulator of mitochondrial uptake of fatty acids and is allosterically inhibited by malonyl CoA [53]. The turnover of malonyl CoA in the heart is fast. Therefore, myocardial malonyl CoA concentrations are dependent on the balance between its synthesis from acetyl CoA via acetyl CoA carboxylase (ACC) and its degradation via malonyl CoA decarboxylase (MCD) [4]. A good correlation has been established between levels of malonyl CoA and rates of fatty acid oxidation, and a reduction in malonyl CoA levels is almost consistent in situations in which fatty acid oxidation is increased [18]. The decrease in malonyl CoA levels seems to be related to increased degradation of malonyl CoA by MCD [18]. MCD is transcriptionally regulated by PPAR-α [54, 55]. In addition to diabetes, activity and expression of cardiac MCD was increased in fasting, high-fat-fed, and newborn hearts [56–59]. Moreover, PPAR-α null mice had increased rates of glucose oxidation and decreased expression and activity of MCD [56].
Increase in circulating fatty acids directly modifies the enzymes in substrate metabolism because fatty acids and their derivatives are ligands for the PPAR family of nuclear receptors, of which PPAR-α and its coactivator peroxisome proliferator-activated receptor-gamma coactivator (PGC)-1 are particularly important in the heart [60–62]. PPAR-α signaling was increased in 15-week-old ob/ob and db/db mice [60]. Other studies reported an increase in the expressions of PPAR-α, PGC-1, and their targets in models of insulin resistance and type 2 diabetes [12, 63–65].
Once in the mitochondrial matrix, long-chain fatty acyl CoAs pass through the β-oxidation enzyme system to produce one acetyl CoA at each cycle, one NADH, and one FADH. The key enzyme in the β-oxidation pathway is β-hydroxyacyl-CoA dehydrogenase. The activity of this enzyme was shown to be normal [66] or high [67] in diabetic rat mitochondria. Higher expression of another enzyme, 3-ketoacyl-CoA thiolase, has also been shown in streptozotocin-diabetic rat hearts [8]. Thus, a combination of high circulating levels of fatty acids, decreased inhibition of fatty acid uptake by the mitochondria, and a normal or accelerated β-oxidation pathway results in a large proportion of acetyl CoA for the tricarboxylic acid (TCA) cycle being supplied from fatty acid oxidation.
4 Alterations in Diabetic Mitochondria
Acetyl CoA derived from both β-oxidation of fatty acids and PDH enters the TCA cycle. The myocardial TCA cycle does not appear to be altered in diabetes because measurements of TCA cycle enzyme activities are similar in diabetic and control hearts [7].
Mitochondrial proteins are produced in the mitochondria and are regulated under the mitochondrial transcriptional factor A (TFAM) by mitochondrial DNA. A recent study reported that overexpression of TFAM improved ATP content and SERCA2a content, both of which were deteriorated by exposing neonatal rat myocytes to hyperglycemic conditions [68].
5 Ketone Body Metabolism
Plasma ketone bodies (β-hydroxybutyrate and acetoacetate) are formed from fatty acids in the liver, and their plasma concentration is normally very low. Thus, ketone bodies are normally a minor substrate for the myocardium. When plasma levels increase during extreme conditions, such as poorly controlled diabetes and starvation, the heart extracts and oxidizes ketone bodies [29]. Ketone bodies inhibit the uptake and metabolism of other energetic substrates [69–74]. The biochemical mechanisms responsible for the inhibition of glucose, lactate, or fatty acid oxidation is not clearly understood.
6 Potential Drugs with Metabolic Effects in the Treatment of Diabetes
Our knowledge of the adipose tissue-originated hormones—adipokines—and their biological effects has increased tremendously since the discovery of the OB gene product, leptin, in 1994 [75]. Two of these, leptin and adiponectin, have direct effects on substrate metabolism as well as insulin secretion. Despite the presence of some conflicting reports, leptin seems to regulate pancreatic cell function and thus insulin secretion [76–79]. Acute exposure of myocytes [80] and isolated working hearts [51] to leptin stimulates fatty acid oxidation. In contrast, type 2 diabetic models, ob/ob and db/db mice, also display increased myocardial fatty acid β-oxidation [60, 81, 82], which is probably related to other alterations in these genetic models. Moreover, leptin administration to subjects with lipodystrophy, abnormal distribution of adipose tissue in which leptin levels are usually decreased [83, 84], has been shown to improve metabolic abnormalities such as hypertriglyceridemia and impaired glucose control, which often are resistant to maximum doses of insulin sensitizers or very high doses of insulin [85–87]. Taken together, these studies suggest that leptin has the potential to become an effective antidiabetic agent with effects on substrate metabolism.
Described by Scherer et al. in 1995 [88], adiponectin is another antidiabetic adipokine. Adiponectin possibly affects β-cell function as adiponectin receptors are expressed in β cells [89–91]. More recently, adiponectin was shown to protect against caspase-8-mediated apoptosis in β cells [92]. As for the effects on substrate metabolism, globular head domain of adiponectin (gAd) stimulates fatty acid oxidation in isolated working 1-day-old rabbit heart [93]. Similar effects on fatty acid oxidation with both the high molecular weight hexameric form of adiponectin and gAd have been reported for cardiac myocytes [94].
Finally, it is worth mentioning other drugs that have beneficial effects on substrate metabolism. Beta-adrenergic receptor antagonists (β-blockers) are used in the treatment of many disease states such as hypertension, ischemic heart disease, arrhythmia, heart failure, glaucoma, and migraine, as well as to reduce the symptoms related to anxiety and hyperthyroidism. Depending on their receptor subtype specificity, β-blockers have different effects on substrate usage. As clearly shown by clinical trials, long-term use of nonselective β-blockers such as proporanolol increases the incidence of diabetes [95, 96], and cardioselective β-blockers such as atenolol and metoprolol reduce insulin sensitivity [97, 98], whereas carvedilol improves serum lipid profile, reduces insulin resistance, and decreases the risk for diabetes [99, 100]. Based on these reports, nonselective and cardioselective β-blockers are accepted as “pro-insulin resistant” whereas carvedilol, the drug that also blocks α1-adrenergic receptors, is known to have opposite effects (reviewed in [101, 102]). The beneficial properties of carvedilol seem to be related to its direct effects on substrate metabolism because carvedilol was shown to reduce the myocardial uptake of long-chain fatty acids and improved left ventricular ejection fraction in heart failure patients [103]. More recently, carvedilol treatment reduced fatty acid oxidation and increased glycolysis in C2C12 cells [104]. The possible effects of carvedilol on diabetic cardiomyopathy need future investigations. However, the fact that carvedilol has beneficial effects on insulin resistance warrants an improvement in substrate utilization and hence diabetic cardiomyopathy.
7 Conclusion
Diabetes is a complex disorder. Its contributors exist at multiple levels. Hormones predominantly determine the levels of energetic substrates, and their major impact on energetic substrate utilization has been well documented. However, modulation of subcellular steps that are involved in ATP-producing processes seems to be just as important in the regulation of substrate metabolism and is expected to have beneficial effects on the mechanical function of the heart. Future studies will reveal such interventions that improve the hormonal milieu or deteriorated substrate metabolism, and leptin and adiponectin agonists are good candidates.
References
Onay-Besikci A (2006) Regulation of cardiac energy metabolism in newborn. Mol Cell Biochem 287:1–11
Lopaschuk GD, Spafford MA (1990) Energy substrate utilization by isolated working hearts from newborn rabbits. Am J Physiol 258:H1274–H1280
Onay-Besikci A, Campbell FM, Hopkins TA et al (2003) Relative importance of malonyl CoA and carnitine in maturation of fatty acid oxidation in newborn rabbit heart. Am J Physiol Heart Circ Physiol 284:H283–H289
Lopaschuk GD, Ussher JR, Folmes CD et al (2010) Myocardial fatty acid metabolism in health and disease. Physiol Rev 90:207–258
Paolisso G, Gambardella A, Galzerano D et al (1994) Total-body and myocardial substrate oxidation in congestive heart failure. Metabolism 43:174–179
Taylor M, Wallhaus TR, Degrado TR et al (2001) An evaluation of myocardial fatty acid and glucose uptake using PET with 18F-fluoro-6-thia-heptadecanoic acid and 18F-FDG in patients with congestive heart failure. J Nucl Med 42:55–62
Stanley WC, Lopaschuk GD, McCormack JG (1997) Regulation of energy substrate metabolism in the diabetic heart. Cardiovasc Res 34:25–33
Onay-Besikci A, Guner S, Arioglu E et al (2007) The effects of chronic trimetazidine treatment on mechanical function and fatty acid oxidation in diabetic rat hearts. Can J Physiol Pharmacol 85:527–535
Yagyu H, Chen G, Yokoyama M et al (2003) Lipoprotein lipase (LpL) on the surface of cardiomyocytes increases lipid uptake and produces a cardiomyopathy. J Clin Invest 111: 419–426
Chiu HC, Kovacs A, Blanton RM et al (2005) Transgenic expression of fatty acid transport protein 1 in the heart causes lipotoxic cardiomyopathy. Circ Res 96:225–233
Chiu HC, Kovacs A, Ford DA et al (2001) A novel mouse model of lipotoxic cardiomyopathy. J Clin Invest 107:813–822
Finck BN, Lehman JJ, Leone TC et al (2002) The cardiac phenotype induced by PPAR-alpha overexpression mimics that caused by diabetes mellitus. J Clin Invest 109:121–130
Belke DD, Larsen TS, Gibbs EM et al (2000) Altered metabolism causes cardiac dysfunction in perfused hearts from diabetic (db/db) mice. Am J Physiol Endocrinol Metab 279: E1104–E1113
Randle PJ, Garland PB, Hales CN et al (1963) The glucose fatty-acid cycle. Its role in insulin sensitivity and the metabolic disturbances of diabetes mellitus. Lancet 1:785–789
Goodale WT, Olson RE, Hackel DB (1959) The effects of fasting and diabetes mellitus on myocardial metabolism in man. Am J Med 27:212–220
Wall SR, Lopaschuk GD (1989) Glucose oxidation rates in fatty acid-perfused isolated working hearts from diabetic rats. Biochim Biophys Acta 1006:97–103
Reaven GM, Hollenbeck C, Jeng CY et al (1988) Measurement of plasma glucose, free fatty acid, lactate, and insulin for 24 h in patients with NIDDM. Diabetes 37:1020–1024
Sakamoto J, Barr RL, Kavanagh KM et al (2000) Contribution of malonyl-CoA decarboxylase to the high fatty acid oxidation rates seen in the diabetic heart. Am J Physiol Heart Circ Physiol 278:H1196–H1204
Randle PJ, Garland PB, Hales CN et al (1966) Interactions of metabolism and the physiological role of insulin. Recent Prog Horm Res 22:1–48
Luiken JJ, Coort SL, Koonen DP et al (2004) Regulation of cardiac long-chain fatty acid and glucose uptake by translocation of substrate transporters. Pflugers Arch 448:1–15
Li J, Hu X, Selvakumar P et al (2004) Role of the nitric oxide pathway in AMPK-mediated glucose uptake and GLUT4 translocation in heart muscle. Am J Physiol Endocrinol Metab 287:E834–E841
Isfort M, Stevens SC, Schaffer S et al (2013) Metabolic dysfunction in diabetic cardiomyopathy. Heart Fail Rev
Wu P, Inskeep K, Bowker-Kinley MM et al (1999) Mechanism responsible for inactivation of skeletal muscle pyruvate dehydrogenase complex in starvation and diabetes. Diabetes 48: 1593–1599
Cheng L, Ding G, Qin Q et al (2004) Cardiomyocyte-restricted peroxisome proliferator-activated receptor-delta deletion perturbs myocardial fatty acid oxidation and leads to cardiomyopathy. Nat Med 10:1245–1250
Yu BC, Chang CK, Ou HY et al (2008) Decrease of peroxisome proliferator-activated receptor delta expression in cardiomyopathy of streptozotocin-induced diabetic rats. Cardiovasc Res 80:78–87
Burkart EM, Sambandam N, Han X et al (2007) Nuclear receptors PPAR-beta/delta and PPAR-alpha direct distinct metabolic regulatory programs in the mouse heart. J Clin Invest 117:3930–3939
Hopkins TA, Sugden MC, Holness MJ et al (2003) Control of cardiac pyruvate dehydrogenase activity in peroxisome proliferator-activated receptor-alpha transgenic mice. Am J Physiol Heart Circ Physiol 285:H270–H276
Connelly KA, Kelly DJ, Zhang Y et al (2009) Inhibition of protein kinase C-beta by ruboxistaurin preserves cardiac function and reduces extracellular matrix production in diabetic cardiomyopathy. Circ Heart Fail 2:129–137
Avogaro A, Nosadini R, Doria A et al (1990) Myocardial metabolism in insulin-deficient diabetic humans without coronary artery disease. Am J Physiol 258:E606–E618
Doria A, Nosadini R, Avogaro A et al (1991) Myocardial metabolism in type 1 diabetic patients without coronary artery disease. Diabet Med 8(spec no):S104–S107
Nuutila P, Knuuti J, Ruotsalainen U et al (1993) Insulin resistance is localized to skeletal but not heart muscle in type 1 diabetes. Am J Physiol 264:E756–E762
Armoni M, Harel C, Bar-Yoseph F et al (2005) Free fatty acids repress the GLUT4 gene expression in cardiac muscle via novel response elements. J Biol Chem 280:34786–34795
Jagasia D, Whiting JM, Concato J et al (2001) Effect of non-insulin-dependent diabetes mellitus on myocardial insulin responsiveness in patients with ischemic heart disease. Circulation 103:1734–1739
Maki M, Nuutila P, Laine H et al (1997) Myocardial glucose uptake in patients with NIDDM and stable coronary artery disease. Diabetes 46:1491–1496
Utriainen T, Takala T, Luotolahti M et al (1998) Insulin resistance characterizes glucose uptake in skeletal muscle but not in the heart in NIDDM. Diabetologia 41:555–559
Monti LD, Landoni C, Setola E et al (2004) Myocardial insulin resistance associated with chronic hypertriglyceridemia and increased FFA levels in type 2 diabetic patients. Am J Physiol Heart Circ Physiol 287:H1225–H1231
Chatham JC (2002) Lactate – the forgotten fuel! J Physiol 542:333
Chatham JC, Gao ZP, Bonen A et al (1999) Preferential inhibition of lactate oxidation relative to glucose oxidation in the rat heart following diabetes. Cardiovasc Res 43:96–106
Chatham JC, Seymour AM (2002) Cardiac carbohydrate metabolism in Zucker diabetic fatty rats. Cardiovasc Res 55:104–112
Wang P, Lloyd SG, Zeng H et al (2005) Impact of altered substrate utilization on cardiac function in isolated hearts from Zucker diabetic fatty rats. Am J Physiol Heart Circ Physiol 288:H2102–H2110
An D, Rodrigues B (2006) Role of changes in cardiac metabolism in development of diabetic cardiomyopathy. Am J Physiol Heart Circ Physiol 291:H1489–H1506
Luiken JJ, Arumugam Y, Bell RC et al (2002) Changes in fatty acid transport and transporters are related to the severity of insulin deficiency. Am J Physiol Endocrinol Metab 283: E612–E621
Luiken JJ, Arumugam Y, Dyck DJ et al (2001) Increased rates of fatty acid uptake and plasmalemmal fatty acid transporters in obese Zucker rats. J Biol Chem 276:40567–40573
Coort SL, Hasselbaink DM, Koonen DP et al (2004) Enhanced sarcolemmal FAT/CD36 content and triacylglycerol storage in cardiac myocytes from obese Zucker rats. Diabetes 53:1655–1663
Coort SL, Luiken JJ, van der Vusse GJ et al (2004) Increased FAT (fatty acid translocase)/CD36-mediated long-chain fatty acid uptake in cardiac myocytes from obese Zucker rats. Biochem Soc Trans 32:83–85
Chabowski A, Coort SL, Calles-Escandon J et al (2004) Insulin stimulates fatty acid transport by regulating expression of FAT/CD36 but not FABPpm. Am J Physiol Endocrinol Metab 287:E781–E789
Stanley WC, Recchia FA, Lopaschuk GD (2005) Myocardial substrate metabolism in the normal and failing heart. Physiol Rev 85:1093–1129
Unger RH, Orci L (2000) Lipotoxic diseases of nonadipose tissues in obesity. Int J Obes Relat Metab Disord 24(suppl 4):S28–S32
Zhou YT, Grayburn P, Karim A et al (2000) Lipotoxic heart disease in obese rats: implications for human obesity. Proc Natl Acad Sci USA 97:1784–1789
Finck BN, Han X, Courtois M et al (2003) A critical role for PPARalpha-mediated lipotoxicity in the pathogenesis of diabetic cardiomyopathy: modulation by dietary fat content. Proc Natl Acad Sci USA 100:1226–1231
Atkinson LL, Kozak R, Kelly SE et al (2003) Potential mechanisms and consequences of cardiac triacylglycerol accumulation in insulin-resistant rats. Am J Physiol Endocrinol Metab 284:E923–E930
McGarry JD, Brown NF (1997) The mitochondrial carnitine palmitoyltransferase system. From concept to molecular analysis. Eur J Biochem/FEBS 244:1–14
McGarry JD, Mills SE, Long CS et al (1983) Observations on the affinity for carnitine, and malonyl-CoA sensitivity, of carnitine palmitoyltransferase I in animal and human tissues. Demonstration of the presence of malonyl-CoA in non-hepatic tissues of the rat. Biochem J 214:21–28
Kim SK, Zhao ZS, Lee YJ et al (2003) Left-ventricular diastolic dysfunction may be prevented by chronic treatment with PPAR-alpha or -gamma agonists in a type 2 diabetic animal model. Diabetes Metab Res Rev 19:487–493
Lee GY, Kim NH, Zhao ZS et al (2004) Peroxisomal-proliferator-activated receptor alpha activates transcription of the rat hepatic malonyl-CoA decarboxylase gene: a key regulation of malonyl-CoA level. Biochem J 378:983–990
Campbell FM, Kozak R, Wagner A et al (2002) A role for peroxisome proliferator-activated receptor alpha (PPAR-alpha) in the control of cardiac malonyl-CoA levels: reduced fatty acid oxidation rates and increased glucose oxidation rates in the hearts of mice lacking PPAR-alpha are associated with higher concentrations of malonyl-CoA and reduced expression of malonyl-CoA decarboxylase. J Biol Chem 277:4098–4103
Dyck JR, Berthiaume LG, Thomas PD et al (2000) Characterization of rat liver malonyl-CoA decarboxylase and the study of its role in regulating fatty acid metabolism. Biochem J 350 (Pt 2):599–608
Goodwin GW, Taegtmeyer H (1999) Regulation of fatty acid oxidation of the heart by MCD and ACC during contractile stimulation. Am J Physiol 277:E772–E777
Young ME, Goodwin GW, Ying J et al (2001) Regulation of cardiac and skeletal muscle malonyl-CoA decarboxylase by fatty acids. Am J Physiol Endocrinol Metab 280:E471–E479
Buchanan J, Mazumder PK, Hu P et al (2005) Reduced cardiac efficiency and altered substrate metabolism precedes the onset of hyperglycemia and contractile dysfunction in two mouse models of insulin resistance and obesity. Endocrinology 146:5341–5349
Gilde AJ, van der Lee KA, Willemsen PH et al (2003) Peroxisome proliferator-activated receptor (PPAR) alpha and PPAR-beta/delta, but not PPAR-gamma, modulate the expression of genes involved in cardiac lipid metabolism. Circ Res 92:518–524
Murray AJ, Panagia M, Hauton D et al (2005) Plasma free fatty acids and peroxisome proliferator-activated receptor alpha in the control of myocardial uncoupling protein levels. Diabetes 54:3496–3502
Duncan JG, Fong JL, Medeiros DM et al (2007) Insulin-resistant heart exhibits a mitochondrial biogenic response driven by the peroxisome proliferator-activated receptor-alpha/PGC-1alpha gene regulatory pathway. Circulation 115:909–917
Bernal-Mizrachi C, Weng S, Feng C et al (2003) Dexamethasone induction of hypertension and diabetes is PPAR-alpha dependent in LDL receptor-null mice. Nat Med 9:1069–1075
Sharma S, Adrogue JV, Golfman L et al (2004) Intramyocardial lipid accumulation in the failing human heart resembles the lipotoxic rat heart. FASEB J 18:1692–1700
Glatz JF, van Breda E, Keizer HA et al (1994) Rat heart fatty acid-binding protein content is increased in experimental diabetes. Biochem Biophys Res Commun 199:639–646
Chen V, Ianuzzo CD, Fong BC et al (1984) The effects of acute and chronic diabetes on myocardial metabolism in rats. Diabetes 33:1078–1084
Suarez J, Hu Y, Makino A et al (2008) Alterations in mitochondrial function and cytosolic calcium induced by hyperglycemia are restored by mitochondrial transcription factor A in cardiomyocytes. Am J Physiol Cell Physiol 295:C1561–C1568
Bing RJ, Siegel A, Ungar I et al (1954) Metabolism of the human heart. II. Studies on fat, ketone and amino acid metabolism. Am J Med 16:504–515
Stanley WC, Meadows SR, Kivilo KM et al (2003) Beta-hydroxybutyrate inhibits myocardial fatty acid oxidation in vivo independent of changes in malonyl-CoA content. Am J Physiol Heart Circ Physiol 285:H1626–H1631
Sultan AM (1992) Effects of diabetes and insulin on ketone bodies metabolism in heart. Mol Cell Biochem 110:17–23
Chen V, Wagner G, Spitzer JJ (1984) Regulation of substrate oxidation in isolated myocardial cells by beta-hydroxybutyrate. Horm Metab Res 16:243–247
Forsey RG, Reid K, Brosnan JT (1987) Competition between fatty acids and carbohydrate or ketone bodies as metabolic fuels for the isolated perfused heart. Can J Physiol Pharmacol 65:401–406
Hasselbaink DM, Glatz JF, Luiken JJ et al (2003) Ketone bodies disturb fatty acid handling in isolated cardiomyocytes derived from control and diabetic rats. Biochem J 371:753–760
Zhang Y, Proenca R, Maffei M et al (1994) Positional cloning of the mouse obese gene and its human homologue. Nature 372:425–432
Ahren B, Larsson H (1997) Leptin–a regulator of islet function? Its plasma levels correlate with glucagon and insulin secretion in healthy women. Metabolism 46:1477–1481
Kulkarni RN, Wang ZL, Wang RM et al (1997) Leptin rapidly suppresses insulin release from insulinoma cells, rat and human islets and, in vivo, in mice. J Clin Invest 100:2729–2736
Brown JE, Thomas S, Digby JE et al (2002) Glucose induces and leptin decreases expression of uncoupling protein-2 mRNA in human islets. FEBS Lett 513:189–192
Tanizawa Y, Okuya S, Ishihara H et al (1997) Direct stimulation of basal insulin secretion by physiological concentrations of leptin in pancreatic beta cells. Endocrinology 138: 4513–4516
Palanivel R, Eguchi M, Shuralyova I et al (2006) Distinct effects of short- and long-term leptin treatment on glucose and fatty acid uptake and metabolism in HL-1 cardiomyocytes. Metabolism 55:1067–1075
Mazumder PK, O’Neill BT, Roberts MW et al (2004) Impaired cardiac efficiency and increased fatty acid oxidation in insulin-resistant ob/ob mouse hearts. Diabetes 53:2366–2374
Carley AN, Semeniuk LM, Shimoni Y et al (2004) Treatment of type 2 diabetic db/db mice with a novel PPAR-gamma agonist improves cardiac metabolism but not contractile function. Am J Physiol Endocrinol Metab 286:E449–E455
Pardini VC, Victoria IM, Rocha SM et al (1998) Leptin levels, beta-cell function, and insulin sensitivity in families with congenital and acquired generalized lipoatropic diabetes. J Clin Endocrinol Metab 83:503–508
Nagy GS, Tsiodras S, Martin LD et al (2003) Human immunodeficiency virus type 1-related lipoatrophy and lipohypertrophy are associated with serum concentrations of leptin. Clin Infect Dis 36:795–802
Musso C, Cochran E, Javor E et al (2005) The long-term effect of recombinant methionyl human leptin therapy on hyperandrogenism and menstrual function in female and pituitary function in male and female hypoleptinemic lipodystrophic patients. Metabolism 54: 255–263
Oral EA, Simha V, Ruiz E et al (2002) Leptin-replacement therapy for lipodystrophy. N Engl J Med 346:570–578
Chong AY, Lupsa BC, Cochran EK et al (2010) Efficacy of leptin therapy in the different forms of human lipodystrophy. Diabetologia 53:27–35
Scherer PE, Williams S, Fogliano M et al (1995) A novel serum protein similar to C1q, produced exclusively in adipocytes. J Biol Chem 270:26746–26749
Kharroubi I, Rasschaert J, Eizirik DL et al (2003) Expression of adiponectin receptors in pancreatic beta cells. Biochem Biophys Res Commun 312:1118–1122
Brown JE, Conner AC, Digby JE et al (2010) Regulation of beta-cell viability and gene expression by distinct agonist fragments of adiponectin. Peptides 31:944–949
Wijesekara N, Krishnamurthy M, Bhattacharjee A et al (2010) Adiponectin-induced ERK and Akt phosphorylation protects against pancreatic beta cell apoptosis and increases insulin gene expression and secretion. J Biol Chem 285:33623–33631
Holland WL, Miller RA, Wang ZV et al (2011) Receptor-mediated activation of ceramidase activity initiates the pleiotropic actions of adiponectin. Nat Med 17:55–63
Onay-Besikci A, Altarejos JY, Lopaschuk GD (2004) gAd-globular head domain of adiponectin increases fatty acid oxidation in newborn rabbit hearts. J Biol Chem 279:44320–44326
Palanivel R, Fang X, Park M et al (2007) Globular and full-length forms of adiponectin mediate specific changes in glucose and fatty acid uptake and metabolism in cardiomyocytes. Cardiovasc Res 75:148–157
Samuelsson O, Hedner T, Berglund G et al (1994) Diabetes mellitus in treated hypertension: incidence, predictive factors and the impact of non-selective beta-blockers and thiazide diuretics during 15 years treatment of middle-aged hypertensive men in the Primary Prevention Trial Goteborg, Sweden. J Hum Hypertens 8:257–263
Gress TW, Nieto FJ, Shahar E et al (2000) Hypertension and antihypertensive therapy as risk factors for type 2 diabetes mellitus. N Engl J Med 342:905–912
Pollare T, Lithell H, Morlin C et al (1989) Metabolic effects of diltiazem and atenolol: results from a randomized, double-blind-study with parallel groups. J Hypertens 7:551–559
Pollare T, Lithell H, Selinus I et al (1989) Sensitivity to insulin during treatment with atenolol and metoprolol: a randomized, double-blind study of effects on carbohydrate and lipoprotein metabolism in hypertensive patients. Br Med J 298:1152–1157
Jacob S, Rett K, Wicklmayr M et al (1996) Differential effect of chronic treatment with two beta-blocking agents on insulin sensitivity: the carvedilol-metoprolol study. J Hypertens 14:489–494
Torp-Pederson C, Cleland J, Di Lenarda A et al (2005) Carvedilol reduces the risk for new onset of diabetes related adverse events in heart failure compared to metoprolol: results of the COMET study. J Am Coll Cardiol 45:187a
Ashrafian H, Frenneaux MP, Opie LH (2007) Metabolic mechanisms in heart failure. Circulation 116:434–448
Kostis JB, Sanders M (2005) The association of heart failure with insulin resistance and the development of type 2 diabetes. Am J Hypertens 18:731–737
Wallhaus TR, Taylor M, DeGrado TR et al (2001) Myocardial free fatty acid and glucose use after carvedilol treatment in patients with congestive heart failure. Circulation 103: 2441–2446
Onay-Besikci A, Suzmecelik E, Ozcelikay AT (2012) Carvedilol suppresses fatty acid oxidation and stimulates glycolysis in C2C12 cells. Can J Physiol Pharmacol 90:1087–1093
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2014 Springer Science+Business Media New York
About this chapter
Cite this chapter
Onay-Besikci, A. (2014). Substrate Metabolism in the Diabetic Heart. In: Turan, B., Dhalla, N. (eds) Diabetic Cardiomyopathy. Advances in Biochemistry in Health and Disease, vol 9. Springer, New York, NY. https://doi.org/10.1007/978-1-4614-9317-4_4
Download citation
DOI: https://doi.org/10.1007/978-1-4614-9317-4_4
Published:
Publisher Name: Springer, New York, NY
Print ISBN: 978-1-4614-9316-7
Online ISBN: 978-1-4614-9317-4
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)