
Abstract
Cardiovascular diseases are the leading cause of death and disability among diabetic patients. Diabetes-induced cardiovascular complications are primarily caused by impaired NO bioavailability, eventually leading to endothelial dysfunction and a sequela of cardiovascular disorders. Phosphodiesterase-5 (PDE-5) inhibitors are class erectile dysfunction (ED) drugs that are shown to induce powerful cardiovascular benefits against ischemia–reperfusion injury, myocardial infarction, heart transplantation, cardiac hypertrophy, heart failure, and doxorubicin-induced cardiotoxicity. The use of PDE-5 inhibitors, including sildenafil (Viagra), vardenafil (Levitra), and tadalafil (Cialis), represent a potential therapeutic strategy to reduce the incidence of heart diseases in diabetic patients because these compounds prevent damage to the vascular endothelium by upregulating eNOS, iNOS, and increased NO production. This review provides new insights into the potential benefits of PDE-5 inhibitors for diabetic patients and discusses the multiple molecular mechanisms by which these drugs protect the diabetic hearts.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others

Keywords
1 Introduction
Diabetes is a major public health concern affecting nearly 170 million people worldwide [1]. Its incidence is dramatically on the rise, with a global estimate of more than 300 million diabetic patients by 2025 [1, 2]. Diabetes is associated with increased risk for a wide variety of long-term health problems including macrovascular/microvascular complications that induce damage to major organs such as brain, heart, kidney, eye, peripheral vascular system, and nervous system. Among the various diabetes-related complications, heart diseases and associated cardiovascular complications continue to be the leading cause of morbidity and mortality, accounting for nearly 80 % of the deaths in the diabetic population [3–6]. Both type 1 and type 2 diabetes have been closely linked to cardiovascular disease (CVD). Diabetic patients exhibit increased risk for multiple cardiovascular complications such as endothelial dysfunction, coronary atherosclerosis, microangiopathy, and hypertension [7, 8]. They also exhibit enhanced risk for cardiac dysfunction that occurs independently of other risk factors such as hypertension and coronary artery disease. Several factors exaggerate the risk of heart failure and stroke in diabetic patients, such as hypertension, insulin resistance, hyperinsulinemia, hyperamilinemia, dyslipidemia, and coagulation system disorder and hyperhomosisteinemia. It is estimated that two of three diabetes patients develop heart failure and eventually die of myocardial infarction or stroke [9, 10]. In addition, diabetics are known to experience worse outcomes following acute myocardial infarction, coronary angioplasty, and cardiac surgery. The CVD treatment accounts for a large part of the healthcare costs attributable to management of diabetic complications, averaging nearly 50 % of the total healthcare expenditure of diabetic patients [11]. These concerns all denote the overwhelming significance of CVD in diabetic patients [12].
2 Pathophysiology of Cardiovascular Disease in Diabetic Patients
Cardiovascular risk actually begins with insulin resistance, a condition that occurs well before diabetes mellitus. Reduced nitric oxide (NO) bioavailability and enhanced reactive oxygen species (ROS) formation within the vascular wall results in an imbalance between NO and ROS, contributing to impaired insulin utilization and thus leading to insulin resistance [13]. Insulin resistance causes a number of micro- and macrovascular insults leading to retinopathy, nephropathy, and painful neuropathy, and eventually to more adverse complications such as atherosclerosis, coronary artery disease, and cerebrovascular disease. The increased incidence of these complications has been attributed to higher levels of inflammatory cytokines, chronic hyperglycemia leading to formation of advanced glycation end products (AGEs), and elevated levels of oxidative stress that lead to endothelial dysfunction. Vascular NO is critical for normal vasodilatation and endothelial function, and impairment of NO bioavailability is known to cause endothelial dysfunction [14]. In a large meta-analysis, it was reported that endothelial dysfunction is a significant independent risk factor for cardiac death, myocardial infarction, stroke, and the need for coronary revascularization [15]. A number of clinical studies have shown that hyperglycemia and increased AGEs are key factors in potentiating vascular inflammation and increasing levels of ROS and oxidative stress [16, 17]. This vascular milieu of elevated inflammation, impaired NO bioavailability, and oxidative stress plays an integral role in the progression of atherosclerosis and subsequently acute coronary syndromes, culminating in significant morbidity and mortality of the diabetic patient [18].
In recent years, it is known that diabetes may influence heart muscles independently in addition to early atherosclerosis of the coronary artery, which causes ischemic heart disease [19]. This condition is referred as diabetic cardiomyopathy, a disease process that affects the myocardium in diabetic patients, causing a wide range of structural abnormalities that eventually lead to left ventricular hypertrophy and diastolic and systolic dysfunction [20]. The cardiomyopathy associated with diabetes is a unique myopathic state that appears to be independent of macrovascular/microvascular disease and contributes significantly to CVD morbidity and mortality in diabetic patients, especially those with coexistent hypertension or coronary artery disease with resulting synergistic adverse effects. At the onset of diabetes, the heart undergoes short-term physiological adaptation to metabolic alterations, but prolonged hyperglycemic conditions induce degenerative changes that eventually culminate in irreversible pathological remodeling. Morphological changes include thickening of capillary basement membrane, proliferation of small arterioles, myocyte atrophy, accumulation of ground matrix, and cardiac fibrosis [21, 22] in the diabetic myocardium. Clinically, diabetic patients manifest abnormalities in diastolic left ventricular function, which is caused by changes such as interstitial fibrosis process, collagen formation, reduced ventricular elasticity, and hypertrophy of heart muscle cells. At the cellular level, disruption of calcium release from cytoplasm, changes of troponin T structure, and increased activity of pyruvate kinase appear [19]. These changes cause distraction of heart muscle contraction and relaxation, as well as elevation of end-diastolic pressure, that cause diabetic cardiomyopathy [20, 23].
The pathogenesis of CVD in diabetes is multifactorial, including increased oxidative stress, disturbances in glucose and fatty acid metabolism, mitochondrial dysfunction, alterations of vasoactive factors such as endothelin-1 and NO, abnormalities in intracellular calcium homeostasis, altered transcription of genes encoding for contractile and structural proteins, autonomic dysfunction, and abnormal expression of growth factors and their receptors [24, 25]. Moreover, there is increase in inflammatory cytokines, apoptosis of cardiac muscles, dysregulation of the renin-angiotensin system, hyperglycemia-induced activation of protein kinase C isoforms, and alterations in gene expression induced by miRNAs [26–28]. Additionally, diabetic conditions are known to impede key cardioprotective signaling pathways and blunt a number of cardioprotective modalities [29–31].
3 Role of NO-cGMP Signaling in Diabetes-Induced Cardiovascular Damage
High levels of glucose cause biochemical changes in the endothelial cells that are reminiscent of early molecular alterations in the target organs of diabetes [32, 33]. In vitro studies revealed that acute exposure of endothelial cells to high glucose significantly lowers NO production [34]. In addition to its reduced level under hyperglycemic conditions, NO may also be sequestered by glucose-induced oxidative stress, which generates an imbalance in the counter-activity of NO and endothelins, which are major vasoactive factors produced by endothelial cells [32, 33, 35]. Endothelium-derived NO activates soluble guanylyl cyclase in vascular smooth muscle cells, resulting in enhanced cyclic guanosine monophosphate (cGMP) concentrations and vasorelaxation. Downregulation of the NO-cGMP pathway has been implicated in the pathogenesis of diabetes-induced cardiovascular complications [36, 37]. Consistent with these observations, type 2 diabetic patients have impaired NO synthesis and decreased expression of eNOS and iNOS in skeletal muscle [38]. Furthermore, nitric oxide synthase (NOS) enzymes have been suggested to play important roles in acetylcholine-induced paradoxical vasoconstriction in atherosclerotic coronary arteries [39]. Recent animal studies with NOS inhibitors and eNOS gene deficiency suggest that the NO signaling pathway may regulate and promote glucose uptake in myocytes and enhance muscle glucose utilization as well [40–43]. Moreover, insulin-stimulated muscle glucose uptake and endothelial NO production was blocked using the eNOS inhibitor, L-NMMA (NG-monomethyl-L-arginine) [44]. eNOS knockout mice had decreased oxygen consumption, increased weight gain, and were resistant to insulin [45]. Furthermore, genetic variations of eNOS gene influenced energy expenditure, severity of glucose intolerance, and risk of developing type 2 diabetes [46]. A recent epidemiological study provided evidence of a strong correlation between the risk factors associated with metabolic syndrome (i.e., obesity, elevated fasting glucose levels, dyslipidemia, hypertension) and urinary cGMP excretion, suggesting that a reduction of NO bioactivity concurs with these cardiovascular risk factors [47].
4 PDE-5 Inhibitors in the Treatment of Diabetes-Associated Complications
Phosphodiesterase 5 (PDE-5) inhibitors including sildenafil (Viagra), vardenafil (Levitra), and tadalafil (Cialis) are erectile dysfunction (ED) drugs that reduce damage to the penile vascular endothelium by upregulating eNOS, iNOS, and increased NO production [48–50]. PDE-5 inhibitors are commonly used among the diabetic patient population because ED is a major and prevalent vascular complication in diabetes [51]. ED is present in 32 % of insulin-dependent diabetics and 46 % of non-insulin-dependent diabetics. Chronic administration of PDE-5 inhibitors has been associated with increased persistent vascular and endothelial function by increasing the level of endothelial cGMP generated by activation of eNOS [52]. Sildenafil dilates epicardial coronary arteries, improves endothelial dysfunction, and inhibits platelet activation in patients with coronary artery disease [53] and acutely enhances flow-mediated vasodilation in patients with heart failure [54]. Therefore, inhibiting cGMP degradation by sildenafil might be a rational approach to treat patients with diabetes, coronary artery disease, or heart failure [55]. In support of this notion, previous reports demonstrated that sildenafil and vardenafil improved vasorelaxation through enhanced endogenous NO signaling in streptozotocin-induced diabetic rats [56, 57]. In streptozotocin-induced type 1 diabetic rats, 14 days of treatment with sildenafil improved vasorelaxation, and long-term administration of the PDE-5 inhibitor DA-8159 prevented ED and preserved endothelial function through enhanced endogenous NO signaling [56, 58]. The PDE-5 inhibitor vardenafil improved cardiovascular dysfunction in experimental diabetes mellitus [57]. Diabetic rats treated with vardenafil showed a tendency toward higher dP/dtmax and dP/dtmin, without reaching statistical significance. The load-independent, PV-loop-derived contractility indices (Emax, PRSW, and dP/dtmax-EDV) were significantly improved in the vardenafil treatment group. In a clinical study, chronic (alternate-day) administration of tadalafil in men with ED of any etiology had improved endothelial function as indicated by marked changes in serum markers of endothelial function [59]. Furthermore, both acute and chronic administration of sildenafil improved endothelial function in patients with type 2 diabetes [60, 61].
Pioneering studies from our laboratory have shown that PDE-5 inhibitors restore NO signaling and protect against myocardial ischemia–reperfusion (I/R) injury in normoglycemic mice [52, 62–64]. Several other investigators have also demonstrated the cardioprotective effects of PDE-5 inhibitors in different models of ischemic injury [65–67]. Moreover, PDE-5 inhibitors attenuate cardiac dysfunction following myocardial infarction and doxorubicin-induced cardiomyopathy [68–71]. Using proteomic analysis, our laboratory has recently demonstrated that chronic tadalafil treatment (28-day treatment) modulates cardiac proteins, specifically those associated with cytoskeletal rearrangement such as myosin light chain-2, myosin light chain-4, myosin heavy chain-α, and myosin binding protein-C, which contribute to contractile dysfunction [72]. These data suggest that tadalafil therapy may downregulate cytoskeletal contractile proteins associated with cardiac remodeling and heart failure. A recent study from our laboratory has shown that tadalafil, similar to other PDE-5 inhibitors in nondiabetic models, significantly reduces infarct size following I/R in the diabetic heart and attenuates necrosis and apoptosis following simulated ischemia and reoxygenation in isolated ventricular cardiomyocytes [73]. Moreover, tadalafil therapy in type 2 diabetic mice ameliorated circulating inflammatory cytokines and chemokines while improving fasting glucose levels and reducing infarct size following I/R injury in the diabetic heart [73]. Tadalafil treatment also improved PKG activity in the cardiomyocytes of diabetic mice compared to vehicle-treated controls [73]. These studies provided evidence that PDE-5 inhibitors restore NO signaling and induce cardioprotective effects through several cellular and molecular mechanisms.
5 PDE-5 Inhibitors in Attenuation of Oxidative Stress in the Diabetic Myocardium
In diabetes, multiple hyperglycemia-induced pathways causing oxidative stress including oxidized low lipoprotein, AGEs/RAGEs, and heme oxygenase pathways [74, 75] have been reported. In addition, increased mitochondrial superoxide and ROS production lead to apoptosis of cardiomyocytes. Depletion of proteins involved in electron-chain transport and increased expression of proteins involved in β-oxidation causing increased superoxide production, mitochondrial uncoupling, and reduced ATP generation in diabetic myocardium were reported earlier [76, 77]. Enhanced oxidative stress can modify cellular components (including proteins) to elicit damage and alter gene transcription of specific vasoactive and cardioprotective factors, leading to structural and functional defects in the diabetic myocardium [35, 78]. Oxidative stress is a major cause of reduced endothelial NO bioavailability in diabetes and is involved in the pathogenesis and progression of diabetic tissue damage. Increased ROS generation and impaired antioxidant defenses could both contribute to oxidative stress. Many studies have shown that ROS generation increases in both type 1 and type 2 diabetes [79–82]. PDE-5 inhibitors may suppress oxidative stress. Importantly, we previously demonstrated that PDE-5 inhibitors sildenafil and vardenafil reduced myocardial infarct size when administered at reperfusion following ischemia [83], a well-established model in which ROS have been widely implicated in causing reperfusion injury. Recent studies have also shown that sildenafil inhibits superoxide formation in cultured corpus cavernosal smooth muscle cells derived from rabbit penis [84, 85]. Chronic treatment with tadalafil in diabetic mice was shown to improve redox signaling by enhancing the antioxidant enzyme glutathione-S-transferase kappa-1 (GSKT-1) and downregulating redox regulatory chaperones, heat shock protein 8, and 75-kDa glucose regulatory protein [72]. Moreover, tadalafil-treated diabetic mice had significantly lowered plasma levels of GSSG/GSH, suggesting reduction of oxidative stress [72]. Using a combined physiological and biochemical approach we recently demonstrated that chronic treatment with tadalafil attenuated oxidative stress induced in type 2 diabetic hearts [86]. Tadalafil treatment protected the diabetic mice hearts from I/R injury, consistent with our previous studies that demonstrated PDE-5 inhibitors induce powerful cardioprotective effects against in vivo myocardial I/R injury in normoglycemic mice [64, 71]. Tadalafil also exhibited beneficial effects on the systemic abnormalities induced by diabetes. Tadalafil treatment improved the metabolic status of mice as evidenced by slight decrease in body weight and blood glucose coupled with significant reductions in hyperinsulinemia and hypertriglyceridemia. Furthermore, tadalafil treatment decreased ROS production in isolated ventricular myocytes following simulated ischemia and reoxygenation (Fig. 1).

Effect of tadalafil on reactive oxygen species (ROS) generation in adult cardiomyocytes following simulated ischemia and reoxygenation: representative images with chlorofluorescein (DCF) staining in isolated cardiomyocytes. (a) Bright-field images (top) and green fluorescent images (bottom). ×200. (b) ROS production in cardiomyocytes quantified and expressed as percent (%) for DCF-positive cells among total cells. Data are mean ± SEM (n = 4/group). *P < 0.05 versus db/db mouse cardiomyocytes
Multiple sources of endogenously generated ROS have been implicated in oxidative damage of the diabetic vasculature and heart [87–90]. Importantly, increased expression of NAD(P)H oxidase proteins has been observed in the vasculature from animal models of diabetes or from diabetic patients [87, 91, 92]. In this recent study, we also tested the effect of tadalafil treatment on NAD(P)H oxidase, a major enzyme involved in oxidative stress [86]. The NAD(P)H oxidase enzyme complex consists of the membrane subunit cytochrome b 558 (p22 phox and gp91 phox), multiple cytoplasmic subunits (p67 phox and p47phox), and the small G protein Rac-1. We showed that tadalafil treatment attenuated expression of NAD(P)H oxidase subunits pRac-1 and gp91phox in type 2 diabetic hearts (Fig. 2) and NAD(P)H oxidase activity (Fig. 3a). Enhanced lipid peroxidation in diabetes is an autocatalytic mechanism leading to oxidative destruction of cellular membranes in the heart. Cardiac lipid peroxidation activity in the db/db mice was increased by 41.2 % as compared to the control group (n = 6/group, P < 0.05, Fig. 3b). Tadalafil treatment significantly attenuated the enhanced lipid peroxidation in the diabetic mice. Moreover, tadalafil treatment enhanced the GSH/GSSG ratio in the myocardium of db/db mice (Fig. 3c). Reduced glutathione is a major intracellular redox buffer and functions as a direct free radical scavenger.

Effect of tadalafil on myocardial expression of NAD(P)H oxidase subunits. (a) Western blots show myocardial expression levels of pRac1, Rac1, gp91phox p47phox p67phox, and representative actin bands. (b) Densitometric quantification of protein expression of pRac1 normalized against Rac1. (c) Densitometric quantification of protein expression of gp91phox normalized against actin. (d) Densitometric quantification of protein expression of p47phox normalized against actin. (e) Densitometric quantification of protein expression of p67phox normalized against actin. Data are mean ± SEM (n = 6/group). *P < 0.05 versus control; # P < 0.05 versus db/db

Effect of tadalafil on NAD(P)H oxidase activity, lipid peroxidation and glutathione levels in diabetic hearts. NAD(P)H-dependent activity represented as percent increases in ethidium fluorescence compared to control (a), myocardial lipid peroxidation (b), and GSH/GSSG ratio (c) in control, db/db, and tadalafil (TAD)-treated db/db groups. Data are mean ± SEM (n = 6/group).*P < 0.05 versus control; # P < 0.05 versus db/db
In type 2 diabetes, ROS are involved in insulin resistance, via its regulatory effects on mitochondrial function [93]. Maintenance of mitochondrial membrane potential (ΔΨm) is necessary for production of energy (ATP) and preservation of cellular homeostasis. It has been demonstrated that maintenance of ΔΨm is a critical primary determinant of myocyte survival [94]. We measured dissipation of Δψm of ventricular cardiomyocytes following simulated ischemia and reoxygenation injury by JC-1 staining. Cardiomyocytes from untreated diabetic mice exhibited loss of Δψm whereas control and tadalafil-treated diabetic mice demonstrated preserved Δψm and intact mitochondrial membrane. Similar preservation of Δψm was observed following simulated ischemia/reoxygenation in nondiabetic cardiomyocytes treated with sildenafil [49]. The mechanism by which tadalafil preserves Δψm in diabetic cardiomyocytes is not clear, although it may be mediated in part through opening of mitochondrial ATP-sensitive potassium channels, which appear to be the effectors of cardioprotection with PDE-5 inhibitors as reported previously [63, 83]. The protective effects of tadalafil in diabetic heart may be caused by maintenance of balance in oxidant–antioxidant status, particularly considering that increased ROS generation augments impairment of mitochondrial function in diabetic hearts [95]. Moreover, tadalafil treatment enhanced cGMP and PKG levels in mouse models of cardiac injury [64, 69]. As mentioned earlier, recent studies from our laboratory demonstrated that tadalafil reversed detrimental remodeling of myocardial proteins [72], ameliorated pro-inflammatory cytokines, and reduced infarct size following I/R injury while upregulating PKG activity in isolated cardiomyocytes of diabetic mice [73]. In this respect, tadalafil is attractive because this drug can modulate multiple molecular targets of cardioprotection as compared to antioxidants, which only reduce oxidative stress.
6 Conclusions
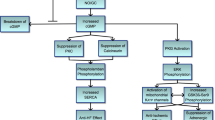
PDE-5 inhibitors have been successfully used by millions of people worldwide for treatment of male erectile dysfunction. Several basic science studies now demonstrate that sildenafil and other PDE-5 inhibitors have a protective effect in clinical scenarios including ischemia/reperfusion injury, myocardial infarction, heart transplantation, cardiac hypertrophy, heart failure, doxorubicin-induced cardiotoxicity, Duchenne muscular dystrophy, and stem-cell preconditioning [96]. Today, nearly 100 clinical trials with PDE-5 inhibitors have been completed or are ongoing that focus on the potential cardiovascular benefits [97]. Our studies provide new insights into the potential role of PDE-5 inhibitors in type 2 diabetic heart (Fig. 4). We believe these drugs may protect the diabetic heart through multiple redundant mechanisms. First, PDE-5 inhibitors could attenuate ROS generation and decrease the accumulation of oxidized glutathione (GSSG) in the diabetic through inhibition of NADPH oxidase [86]. Second, initiation of cGMP-dependent PKG signaling by PDE-5 inhibitors may increase PI3/Akt phosphorylation, which could increase eNOS phosphorylation leading to enhanced NO bioavailability and activation of PKG through soluble guanylate cyclase (sGC)-dependent formation of cGMP. The ROS-dependent inhibition of AMPK and PI3/Akt phosphorylation may reduce NO formation through disruption of eNOS phosphorylation. The increased NO bioavailability and formation of cGMP/PKG may reverse or attenuate mitochondrial ROS by increased synthesis of the putative mitochondrial antioxidant enzyme GSTK1. Finally, PDE-5 inhibition could increase PKG-dependent phosphorylation of glycogen synthase 3β (GSK-3β), which has a role in inhibition of MPTP [96] and therefore apoptosis in the diabetic heart following I/R. Our results have also shown that PDE-5 inhibition attenuated detrimental alterations in proteins involved in the cytoskeletal structure, cardiac contractility, and redox regulatory mechanisms of the diabetic myocardium. We propose that tadalafil treatment could be pursued as a novel therapeutic approach in protection against diabetes-induced cardiomyopathy. In fact, a recent clinical study suggested that chronic treatment with sildenafil caused an anti-remodeling effect in patients with early features of diabetic cardiomyopathy, such as left ventricular concentric hypertrophy associated with altered myocardial contraction dynamics [98]. Moreover, it is tempting to speculate the use of PDE-5 inhibitors may be therapeutically beneficial in protection against other oxidative stress-induced organ damage during diabetes, including nephropathy and retinopathy. These findings have potential clinical significance in the current scenario as PDE-5 inhibitors are now serving as first-line therapeutics in the treatments of ED in diabetic patients.

Proposed cardioprotective pathways by phosphodiesterase 5 (PDE-5) inhibition in diabetes
References
King H, Aubert RE, Herman WH (1998) Global burden of diabetes, 1995–2025: prevalence, numerical estimates, and projections. Diabetes Care 21:1414–1431
Zimmet P, Alberti KG, Shaw J (2001) Global and societal implications of the diabetes epidemic. Nature 414:782–787
Garcia MJ, McNamara PM, Gordon T et al (1974) Morbidity and mortality in diabetics in the Framingham population. Sixteen year follow-up study. Diabetes 23:105–111
Tschoepe D, Roesen P (1998) Heart disease in diabetes mellitus: a challenge for early diagnosis and intervention. Exp Clin Endocrinol Diabetes 106:16–24
Kannel WB, Hjortland M, Castelli WP (1974) Role of diabetes in congestive heart failure: the Framingham study. Am J Cardiol 34:29–34
Malmberg K, Ryden L (1988) Myocardial infarction in patients with diabetes mellitus. Eur Heart J 9:259–264
Fein FS (1990) Diabetic cardiomyopathy. Diabetes Care 13:1169–1179
Iltis I, Kober F, Dalmasso C et al (2005) Noninvasive characterization of myocardial blood flow in diabetic, hypertensive, and diabetic-hypertensive rats using spin-labeling MRI. Microcirculation 12:607–614
Lopaschuk GD, Rebeyka IM, Allard MF (2002) Metabolic modulation: a means to mend a broken heart. Circulation 105:140–142
Maier C, Clodi M, Neuhold S et al (2009) Endothelial markers may link kidney function to cardiovascular events in type 2 diabetes. Diabetes Care 32:1890–1895
Caro JJ, Ward AJ, O’Brien JA (2002) Lifetime costs of complications resulting from type 2 diabetes in the U.S. Diabetes Care 25:476–481
Grossman E, Messerli FH (1996) Diabetic and hypertensive heart disease. Ann Intern Med 125:304–310
Ayala JE, Bracy DP, Julien BM et al (2007) Chronic treatment with sildenafil improves energy balance and insulin action in high-fat-fed conscious mice. Diabetes 57:1025–1033
Deyoung L, Chung E, Kovac JR et al (2012) Daily use of sildenafil improves endothelial function in men with type 2 diabetes. J Androl 33:176–180
Lerman A, Zeiher AM (2005) Endothelial function: cardiac events. Circulation 111:363–368
Wen Y, Skidmore JC, Porter-Turner MM et al (2002) Relationship of glycation, antioxidant status and oxidative stress to vascular damage in diabetes. Diabetes Obes Metab 4:305–308
Soloman H, Man JW, Jackson G (2003) Erectile dysfunction and the cardiovascular patient: endothelial dysfunction is the common denominator. Heart 89:251–254
Versari A (2009) Endothelial dysfunction as a target for prevention of cardiovascular disease. Diabetes Care 32:S314–S321
Shahab A (2006) Why does diabetes mellitus increase the risk of cardiovascular disease? Acta Med Indones 38:33–41
Hayat SA, Patel B, Khattar RS et al (2004) Diabetic cardiomyopathy: mechanisms, diagnosis and treatment. Clin Sci (Lond) 107:539–557
Bella JN, Devereux RB, Roman MJ et al (2001) Separate and joint effects of systemic hypertension and diabetes mellitus on left ventricular structure and function in American Indians (the Strong Heart Study). Am J Cardiol 87:1260–1265
Fein FS, Sonnenblick EH (1985) Diabetic cardiomyopathy. Prog Cardiovasc Dis 27:255–270
Bauters C, Lamblin N, Mc Fadden EP et al (2003) Influence of diabetes mellitus on heart failure risk and outcome. Cardiovasc Diabetol 2:1
Aronow WS, Ahn C (1999) Incidence of heart failure in 2,737 older persons with and without diabetes mellitus. Chest 115:867–868
Liu JE, Palmieri V, Roman MJ et al (2001) The impact of diabetes on left ventricular filling pattern in normotensive and hypertensive adults: the Strong Heart Study. J Am Coll Cardiol 37:1943–1949
Spector KS (1998) Diabetic cardiomyopathy. Clin Cardiol 21:885–887
Tziakas DN, Chalikias GK, Kaski JC (2005) Epidemiology of the diabetic heart. Coron Artery Dis 16:S3–S10
Feng B, Chen S, George B et al (2010) miR133a regulates cardiomyocyte hypertrophy in diabetes. Diabetes Metab Res Rev 26:40–49
Zhu SG, Xi L, Kukreja RC (2011) Type 2 diabetic obese db/db mice are refractory to myocardial ischemic postconditioning in vivo: potential role for Hsp20, F(1)-ATPase δ, and Echs1. J Cell Mol Med 15:2512–2524
Ye Y, Perez-Polo JR, Aguilar D et al (2011) The potential effects of anti-diabetic medications on myocardial ischemia-reperfusion injury. Basic Res Cardiol 106:925–952
Ghaboura N, Tamareille S, Ducluzeau PH et al (2011) Diabetes mellitus abrogates erythropoietin-induced cardioprotection against ischemic-reperfusion injury by alteration of the RISK/GSK-3 beta signaling. Basic Res Cardiol 106:147–162
Khan ZA, Chakrabarti S (2003) Endothelins in chronic diabetic complications. Can J Physiol Pharmacol 81:622–634
Khan ZA, Chakrabarti S (2006) Therapeutic targeting of endothelial dysfunction in chronic diabetic complications. Recent Pat Cardiovasc Drug Discov 1:167–175
Giugliano D, Marfella R, Coppola L et al (1997) Vascular effects of acute hyperglycemia in humans are reversed by L-arginine. Evidence for reduced availability of nitric oxide during hyperglycemia. Circulation 95:1783–1790
Khan ZA, Farhangkhoee H, Chakrabarti S (2006) Towards newer molecular targets for chronic diabetic complications. Curr Vasc Pharmacol 4:45–57
Lin KY, Ito A, Asagami T et al (2002) Impaired nitric oxide synthase pathway in diabetes mellitus: role of asymmetric dimethylarginine and dimethylarginine dimethylaminohydrolase. Circulation 106:987–992
Munzel T, Daiber A, Ullrich V (2005) Vascular consequences of endothelial nitric oxide synthase uncoupling for the activity and expression of the soluble guanylyl cyclase and the cGMP-dependent protein kinase. Arterioscler Thromb Vasc Biol 25:1551–1557
Torres SH, De Sanctis JB, de L Briceño M (2004) Inflammation and nitric oxide production in skeletal muscle of type 2 diabetic patients. J Endocrinol 181:419–427
Ludmer PL, Selwyn AP, Shook TL et al (1986) Paradoxical vasoconstriction induced by acetylcholine in atherosclerotic coronary arteries. N Engl J Med 315:1046–1051
Duplain H, Burcelin R, Sartori C et al (2001) Insulin resistance, hyperlipidemia, and hypertension in mice lacking endothelial nitric oxide synthase. Circulation 104:342–345
Shankar RR, Wu Y, Shen HQ et al (2000) Mice with gene disruption of both endothelial and neuronal nitric oxide synthase exhibit insulin resistance. Diabetes 49:684–687
Roy D, Perreault M, Marette A (1998) Insulin stimulation of glucose uptake in skeletal muscles and adipose tissue in vivo is NO dependent. Am J Physiol 274:E692–E699
Baron AD, Zhu JS, Marshall S et al (1995) Insulin resistance after hypertension induced by the nitric oxide synthase inhibitor L-NMMA in rats. Am J Physiol 269:E709–E715
Higaki Y, Hirshman MF, Fujii N et al (2001) Nitric oxide increases glucose uptake through a mechanism that is distinct from the insulin and contraction pathways in rat skeletal muscle. Diabetes 50:241–247
Cook S, Hugli O, Egli M et al (2003) Clustering of cardiovascular risk factors mimicking the human metabolic syndrome X in eNOS null mice. Swiss Med Wkly 133:360–363
Franks PW, Luan J, Barroso I et al (2005) Variation in the eNOS gene modifies the association between total energy expenditure and glucose intolerance. Diabetes 54:2795–2801
Cui R, Iso H, Pi J et al (2007) Metabolic syndrome and urinary cGMP excretion in the general population. Atherosclerosis 190:423–428
Behr-Roussel D, Gorny D, Mevel K et al (2005) Chronic sildenafil improves erectile function and endothelium-dependent cavernosal relaxations in rats: lack of tachyphylaxis. Eur Urol 47:87–91
Das A, Xi L, Kukreja RC (2005) Phosphodiesterase-5 inhibitor sildenafil preconditions adult cardiac myocytes against necrosis and apoptosis. Essential role of nitric oxide signaling. J Biol Chem 280:12944–12955
Wallis RM (1999) The pharmacology of sildenafil, a novel and selective inhibitor of phosphodiesterase (PDE) type 5. Nippon Yakurigaku Zasshi 114:22P–26P
Vickers MA, Satyanarayana R (2002) Phosphodiesterase type 5 inhibitors for the treatment of erectile dysfunction in patients with diabetes mellitus. Int J Impot Res 14:466–471
Salloum F, Yin C, Xi L (2003) Sildenafil induces delayed preconditioning through inducible nitric oxide synthase-dependent pathway in mouse heart. Circ Res 92:595–597
Halcox JP, Nour KR, Zalos G et al (2002) The effect of sildenafil on human vascular function, platelet activation, and myocardial ischemia. J Am Coll Cardiol 40:1232–1240
Hryniewicz K, Dimayuga C, Hudaihed A et al (2005) Inhibition of angiotensin-converting enzyme and phosphodiesterase type 5 improves endothelial function in heart failure. Clin Sci (Lond) 108:331–338
Gross GJ (2005) Sildenafil and endothelial dysfunction in humans. Circulation 111:721–723
Schafer A, Fraccarollo D, Pfortsch S et al (2008) Improvement of vascular function by acute and chronic treatment with the PDE-5 inhibitor sildenafil in experimental diabetes mellitus. Br J Pharmacol 153:886–893
Radovits T, Bomicke T, Kokeny G et al (2009) The phosphodiesterase-5 inhibitor vardenafil improves cardiovascular dysfunction in experimental diabetes mellitus. Br J Pharmacol 156:909–919
Choi SM, Kim JE, Kang KK (2006) Chronic treatment of DA-8159, a new phosphodiesterase type V inhibitor, attenuates endothelial dysfunction in stroke-prone spontaneously hypertensive rat. Life Sci 78:1211–1216
Aversa A, Greco E, Bruzziches R et al (2007) Relationship between chronic tadalafil administration and improvement of endothelial function in men with erectile dysfunction: a pilot study. Int J Impot Res 19:200–207
DeSouza C, Parulkar A, Lumpkin D et al (2002) Acute and prolonged effects of sildenafil on brachial artery flow-mediated dilatation in type 2 diabetes. Diabetes Care 25:1336–1339
Aversa A, Bruzziches R, Vitale C et al (2007) Chronic sildenafil in men with diabetes and erectile dysfunction. Expert Opin Drug Metab Toxicol 3:451–464
Kukreja RC, Ockaili R, Salloum F et al (2003) Sildenafil-induced cardioprotection in rabbits. Cardiovasc Res 60:700–701
Ockaili R, Salloum F, Hawkins J et al (2002) Sildenafil (Viagra) induces powerful cardioprotective effect via opening of mitochondrial K(ATP) channels in rabbits. Am J Physiol Heart Circ Physiol 283:H1263–H1269
Salloum FN, Chau VQ, Hoke NN et al (2009) Phosphodiesterase-5 inhibitor, tadalafil, protects against myocardial ischemia/reperfusion through protein-kinase g-dependent generation of hydrogen sulfide. Circulation 120:S31–S36
Elrod JW, Greer JJ, Lefer DJ (2007) Sildenafil-mediated acute cardioprotection is independent of the NO/cGMP pathway. Am J Physiol Heart Circ Physiol 292:H342–H347
Nagy O, Hajnal A, Parratt JR et al (2004) Sildenafil (Viagra) reduces arrhythmia severity during ischaemia 24 h after oral administration in dogs. Br J Pharmacol 141:549–551
Sesti C, Florio V, Johnson EG et al (2007) The phosphodiesterase-5 inhibitor tadalafil reduces myocardial infarct size. Int J Impot Res 19:55–61
Fisher PW, Salloum F, Das A et al (2005) Phosphodiesterase-5 inhibition with sildenafil attenuates cardiomyocyte apoptosis and left ventricular dysfunction in a chronic model of doxorubicin cardiotoxicity. Circulation 111:1601–1610
Koka S, Das A, Zhu SG et al (2010) Long-acting phosphodiesterase-5 inhibitor tadalafil attenuates doxorubicin-induced cardiomyopathy without interfering with chemotherapeutic effect. J Pharmacol Exp Ther 334:1023–1030
Koka S, Kukreja RC (2010) Attenuation of doxorubicin-induced cardiotoxicity by Tadalafil: a long acting phosphodiesterase-5 inhibitor. Mol Cell Pharmacol 2:173–178
Salloum FN, Abbate A, Das A et al (2008) Sildenafil (Viagra) attenuates ischemic cardiomyopathy and improves left ventricular function in mice. Am J Physiol Heart Circ Physiol 294:H1398–H1406
Koka S, Xi L, Kukreja RC (2012) Chronic treatment with long acting phosphodiesterase-5 inhibitor tadalafil alters proteomic changes associated with cytoskeletal rearrangement and redox regulation in type II diabetic hearts. Basic Res Cardiol 107:249
Varma A, Das A, Hoke NN et al (2012) Anti-inflammatory and cardioprotective effects of tadalafil in diabetic mice. PLoS One 7:e45243
Bucala R, Tracey KJ, Cerami A (1991) Advanced glycosylation products quench nitric oxide and mediate defective endothelium-dependent vasodilatation in experimental diabetes. J Clin Invest 87:432–438
Farhangkhoee H, Khan ZA, Mukherjee S et al (2003) Heme oxygenase in diabetes-induced oxidative stress in the heart. J Mol Cell Cardiol 35:1439–1448
Boudina S, Abel ED (2007) Diabetic cardiomyopathy revisited. Circulation 115:3213–3223
Shen X, Zheng S, Metreveli NS et al (2006) Protection of cardiac mitochondria by overexpression of MnSOD reduces diabetic cardiomyopathy. Diabetes 55:798–805
Zanetti M, Sato J, Katusic ZS et al (2000) Gene transfer of endothelial nitric oxide synthase alters endothelium-dependent relaxations in aortas from diabetic rabbits. Diabetologia 43:340–347
Barouch LA, Berkowitz DE, Harrison RW et al (2003) Disruption of leptin signaling contributes to cardiac hypertrophy independently of body weight in mice. Circulation 108:754–759
Cai L, Li W, Wang G et al (2002) Hyperglycemia-induced apoptosis in mouse myocardium: mitochondrial cytochrome C-mediated caspase-3 activation pathway. Diabetes 51:1938–1948
Wold LE, Ren J (2004) Streptozotocin directly impairs cardiac contractile function in isolated ventricular myocytes via a p38 MAP kinase-dependent oxidative stress mechanism. Biochem Biophys Res Commun 318:1066–1071
Zhou YT, Grayburn P, Karim A et al (2000) Lipotoxic heart disease in obese rats: implications for human obesity. Proc Natl Acad Sci U S A 97:1784–1789
Salloum FN, Takenoshita Y, Ockaili RA et al (2007) Sildenafil and vardenafil but not nitroglycerin limit myocardial infarction through opening of mitochondrial K(ATP) channels when administered at reperfusion following ischemia in rabbits. J Mol Cell Cardiol 42:453–458
Hotston MR, Jeremy JY, Bloor J et al (2007) Sildenafil inhibits the up-regulation of phosphodiesterase type 5 elicited with nicotine and tumour necrosis factor-alpha in cavernosal vascular smooth muscle cells: mediation by superoxide. BJU Int 99:612–618
Koupparis AJ, Jeremy JY, Muzaffar S et al (2005) Sildenafil inhibits the formation of superoxide and the expression of gp47 NAD[P]H oxidase induced by the thromboxane A2 mimetic, U46619, in corpus cavernosal smooth muscle cells. BJU Int 96:423–427
Koka S, Das A, Salloum FN et al (2013) Phosphodiesterase-5 inhibitor tadalafil attenuates oxidative stress and protects against myocardial ischemia/reperfusion injury in type 2 diabetic mice. Free Radic Biol Med 60:80–88
Guzik TJ, Mussa S, Gastaldi D et al (2002) Mechanisms of increased vascular superoxide production in human diabetes mellitus: role of NAD(P)H oxidase and endothelial nitric oxide synthase. Circulation 105:1656–1662
Kuroda J, Ago T, Matsushima S et al (2010) NADPH oxidase 4 (Nox4) is a major source of oxidative stress in the failing heart. Proc Natl Acad Sci U S A 107:15565–15570
Kuroda J, Sadoshima J (2010) NADPH oxidase and cardiac failure. J Cardiovasc Transl Res 3:314–320
Shao D, Oka S, Brady CD et al (2012) Redox modification of cell signaling in the cardiovascular system. J Mol Cell Cardiol 52:550–558
Hink U, Li H, Mollnau H et al (2001) Mechanisms underlying endothelial dysfunction in diabetes mellitus. Circ Res 88:E14–E22
Kim YK, Lee MS, Son SM et al (2002) Vascular NADH oxidase is involved in impaired endothelium-dependent vasodilation in OLETF rats, a model of type 2 diabetes. Diabetes 51:522–527
Newsholme P, Haber EP, Hirabara SM et al (2007) Diabetes associated cell stress and dysfunction: role of mitochondrial and non-mitochondrial ROS production and activity. J Physiol 583:9–24
Akao M, O’Rourke B, Teshima Y et al (2003) Mechanistically distinct steps in the mitochondrial death pathway triggered by oxidative stress in cardiac myocytes. Circ Res 92:186–194
Sack MN (2009) Type 2 diabetes, mitochondrial biology and the heart. J Mol Cell Cardiol 46:842–849
Kukreja RC, Salloum FN, Das A (2012) Cyclic guanosine monophosphate signaling and phosphodiesterase-5 inhibitors in cardioprotection. J Am Coll Cardiol 59:1921–1927
Giannetta E, Isidori AM, Galea N et al (2012) Chronic inhibition of cGMP phosphodiesterase 5A improves diabetic cardiomyopathy: a randomized, controlled clinical trial using magnetic resonance imaging with myocardial tagging. Circulation 125:2323–2333
Acknowledgments
This work was supported by grants from the National Institutes of Health (HL51045, HL79424, HL93685) to Dr. Rakesh C Kukreja. Dr. Saisudha Koka is supported by postdoctoral fellowship (11POST7400028) from American Heart Association.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2014 Springer Science+Business Media New York
About this chapter
Cite this chapter
Koka, S., Kukreja, R.C. (2014). PDE-5 Inhibitors in Protection of Diabetic Heart. In: Turan, B., Dhalla, N. (eds) Diabetic Cardiomyopathy. Advances in Biochemistry in Health and Disease, vol 9. Springer, New York, NY. https://doi.org/10.1007/978-1-4614-9317-4_20
Download citation
DOI: https://doi.org/10.1007/978-1-4614-9317-4_20
Published:
Publisher Name: Springer, New York, NY
Print ISBN: 978-1-4614-9316-7
Online ISBN: 978-1-4614-9317-4
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)