
Abstract
Cells encounter stress on a daily basis that can damage their DNA and promote malignant transformation, yet the latter rarely occurs. The DNA damage response (DDR) is a highly coordinated signaling pathway that functions to detect and repair DNA damage in cells, inhibiting transformation. However, when DNA damage is so severe that it cannot be repaired, the DDR promotes apoptosis, thus preventing the propagation of abnormal cells. The tumor suppressor protein, p53, is one of the most essential molecules keeping DNA damage in check. Here, we discuss the signaling cascades that activate p53 upon DNA damage and the molecular mechanisms that mediate p53-dependent and -independent apoptosis. Moreover, we discuss the signals that trigger the DDR during malignant propagation and the importance of DNA damage-mediated apoptosis in preventing tumorigenesis.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others

Keywords
7.1 DNA Damage Induced Cell Death
This chapter is aimed at understanding the molecular processes that drive cell death in response to DNA damage. We briefly explore the DNA damage response (DDR) pathway, and how cells translate DNA damage signals into appropriate biological outcomes. The mechanism by which the tumor suppressor, p53, induces apoptosis in response to DNA damage is identified, as are other p53-independent pathways. Finally the role of DNA-damage induced apoptosis in the prevention of cancer is explored.
7.2 The DNA Damage Response
Every day, cells encounter numerous stresses and toxins that damage DNA, yet these cells rarely become tumorigenic. This is primarily due to the DDR which functions to detect and repair DNA damage in cells, to arrest the cell cycle, and to remove the cells if the damage cannot be repaired, through senescence or apoptosis [1–4].
DNA lesions can be generated through normal physiological processes such as DNA replication which can result in DNA mismatches and DNA double-strand breaks (DSB) as well as free oxygen radicals generated as a result of cellular metabolism [5, 6]. In addition, DNA can also be damaged by exposure to environmental stresses such as ultraviolet (UV) light and ionizing radiation (IR) [7]. Lesions are repaired through repair mechanisms such as homologous recombination (HR), nonhomologous end joining (NHEJ), or mismatch repair (MMR) [8]. During the process, cells undergo cycle arrest to allow time for optimal repair and to prevent the damage from being passed on to future generations [8]. Should the extent of DNA damage be too severe, the cells undergo permanent arrest (senescence) or death (apoptosis). A tightly coordinated molecular signaling cascade determines these cellular responses (Fig. 7.1). This section will focus primarily of the signaling events of the DDR.

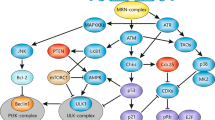
The DNA Damage Response (DDR): DNA stress in the form of double-strand breaks (DSB) or stalled replication forks trigger ATM and ATR kinases. DNA lesions promote the auto-phosphorylation of ATM and a shift from an inactive dimer to active monomer. ATM is recruited to the site of damage by MRN complex (Mre11–Rad50–Nbs1). A complex between ATR and ATRIP renders ATR inactive, and it is activated through RPA recruitment to the site of a stalled replication fork. The kinase activities of ATM and ATR are amplified in DNA damage associated foci formed by the phosphorylation of H2AX. This scaffold promotes the recruitment of checkpoint mediator proteins that amplify the kinase activity of ATM (Mdc1, p53BP1 and Brca1) and ATR (Claspin, RSR, and the 9-1-1 complex). Active ATM and ATR phosphorylate numerous target proteins; the most commonly recognized are Chk1, Chk2, and p53 that function in regulating the cell cycle and inducing apoptosis or senescence. ATM and ATR can also phosphorylate other proteins that mediate apoptosis in a p53-independent manner
The signal transduction pathway of the DDR is controlled by the PI(3)K (phosphatidyl-inositol-3-OH kinase)-related kinases (PIKKs): ATM (ataxia telangiectasia mutated) and ATR (ATM- and Rad3-related) [9]. ATM kinase is induced after DNA damage, whereas ATR kinase is induced as a result of stalled replication forks during DNA replication [10, 11]. Here we will discuss the three different stages in the ATM/ATR-dependent signal transduction cascade: (1) Identification of DNA damage results in the activation of the ATM /ATR kinases and their recruitment to the site of damage. (2) Checkpoint mediator proteins enhance ATM/ATR kinase function. (3) ATM/ATR-phosphorylated effector proteins mediate cellular responses to damage.
Sensing DNA damage: In unstressed cells ATM kinase activity is low since the protein exists as an inactive dimer, blocking the active kinase domain [12]. DNA damage-induced DSBs mediate a conformational change in ATM, resulting in its auto-phosphorylation and dissociation of the dimer complex to an active monomeric form [12]. Recently, a complex of the proteins Mre11–Rad50–Nbs1 (MRN) has been shown to act as a sensor of DSBs, aiding in the recruitment of ATM to the site of DNA damage [13]. ATR does not require a conformational change to mediate its kinase activity. Instead ATR remains inactive through its association with ATR interacting protein (ATRIP), and becomes activated when recruited to the site of a stalled replication fork through its interaction with the single-strand DNA-binding protein, replication protein A (RPA) [14, 15]. Once activated and recruited to the site of DNA damage, ATM and ATR can phosphorylate their relative substrates, which include hundreds of proteins [16]. Effective ATM/ATR function is dependent on checkpoint mediator proteins, which create a scaffold for amplification of kinase function.
Checkpoint mediator proteins enhance the kinase activity of ATM/ATR: The variant histone, H2AX, is phosphorylated at the site of DNA damage by ATM and ATR, creating a scaffold for the recruitment and binding of numerous checkpoint mediator proteins, resulting in the formation of DNA damage associated foci [17, 18]. The exact function of these damage-associated foci are unclear, but they may serve to enhance the signal transduction pathway [19]. Instead, phosphorylated H2AX and the associated damage foci are most commonly used to identify cells with damaged DNA. The ATM-related mediator proteins include: Mdc1 (mediator of DNA damage checkpoint 1), p53BP1 (p53 binding protein 1), and Brac1 (breast cancer type 1 susceptibility protein) [20–25]. ATR-related mediator Claspin, RSR (RAD17-containing complex), and the 9-1-1-complex (RAD9-RAD1-HUS1) all serve to amplify ATR kinase activity in DNA-damage foci [26–29]. Cells lacking mediator proteins show defective DDR and cancer susceptibility, indicating their importance for propagating ATM/ATR kinase activity [2].
Effector proteins mediate the cellular response to DNA damage: More than 700 proteins are phosphorylated in an ATM/ATR-dependent manner; however, the checkpoint-transducer serine/threonine kinases, Chk2 and Chk1, are by far the most prominent downstream targets of ATM and ATR respectively [16, 30]. The major function of Chk1 and Chk2 is to induce cell cycle arrest at the G1–S, S, and G2–M phases of the cell cycle by inhibiting the CDC25 (cell division cycle) family of phosphatases (CDC25a, CDC25b, and CDC25c) that normally activate the cyclin-dependent kinases (CDKs) [2, 31]. Chk1 and Chk2 also mediate the regulation of various proteins directly involved in DNA repair through transcriptional and posttranslational mechanisms. Thus, these two very important functions of the Chk proteins facilitate DNA repair in a rapid and efficient manner. Another key target of both ATM and Chk2 is the tumor suppressor protein, p53 and its negative regulator MDM2 (murine double minute) [32, 33]. p53 functions as a transcription factor that mediates the expression of proteins involved in cell cycle arrest, senescence, and apoptosis (Fig. 7.3) [34, 35]. Expression of p21/WAF1 is induced by p53, and this cyclin-dependent kinase inhibitor 1 (or CDK-interacting protein 1) functions to mediate G1 cell cycle arrest by binding to and inhibiting the activity of cyclins [36]. This process takes longer, but aids in maintaining the G1 arrest initiated by CDC25 inhibition, further facilitating DNA repair. If the damage to the DNA is irreparable, cells can undergo permanent cell cycle arrest (senescence) or apoptosis. P53 can facilitate these responses, but other p53-independent yet ATM-modulated proteins can also be involved.
7.3 Death in the Absence of DNA Damage
Interestingly, although most chemotherapeutic agents induce a DDR and apoptosis in tumor cells, cisplatin can also induce apoptosis independently of DNA damage. In this context, cisplatin induces endoplasmic reticulum (ER) stress-mediated apoptosis [37, 38]. Moreover, Poly(ADP-ribose) polymerase (PARP) proteins which detect single-strand DNA breaks and recruit repair machineries, may also promote an ER stress response independently of DNA [39, 40]. ER stress promotes an accumulation of unfolded proteins, inducing the unfolded protein response (UPR) [41]. The UPR is a complex and dynamic network of signaling pathways that function to maintain cellular homeostasis by reducing the levels of unfolded proteins. If ER stress is prolonged the UPR triggers apoptosis. Below, we will focus primarily on cellular responses to DNA damage per se.
The role of p53-dependent and independent mechanisms of apoptosis in response to DNA damage will be discussed in the section below.
7.4 DNA-Damage Mediated Apoptosis
Apoptosis is an active cell death process that functions during normal development and cellular homeostasis and is important in tumor suppression, maintaining host defense to pathogens, and the response to cellular stress [42]. A family of cysteine proteases called caspases orchestrates apoptosis, and a subset of these, the executioner caspases, cleaves hundreds of substrate proteins, resulting in cell shrinkage, chromatin condensation, and membrane blebbing [43, 44].
Caspases can be activated by several pathways of apoptosis; the best characterized of these are the death receptor pathway and the mitochondrial pathway of apoptosis. Although the exact molecular signals are not entirely clear, DNA damage can trigger both of these pathways of apoptosis through p53-dependent and independent means. This section will focus on the molecular pathways of p53-dependent and -independent apoptosis induced by the DDR.
The death receptor pathway of apoptosis: This is a tightly regulated signal transduction pathway that is mediated by a subset of the tumor necrosis factor receptor (TNFR) super family, the death receptors [45]. Signals to die are generated once membrane-bound death receptors such as TNFR1, CD95 (Fas), and TRAIL-R are recognized by their specific death ligands (TNF, CD95L, TRAIL) [46]. Trimerization of the receptor follows, with accompanying recruitment of adaptor molecules such as Fas associated death domain (FADD) through a DD–DD homotypic interaction [47]. Adaptor molecules recruit the initiator caspases (8 and 10) through their shared death effector domains (DED), thus forming the death inducing signaling complex (DISC) [48, 49]. The DISC functions to activate the initiator capsases through their respective dimerization and intramolecular cleavage [50]. Once active, initiator caspases cleave and activate the executioner caspases (3 and 7) as well as BID, which stimulates the mitochondrial cell death pathway (Fig. 7.2) [44].

The Death Receptor and Mitochondrial pathways of Apoptosis: DNA damage triggers the death receptor and mitochondrial apoptotic pathways. Death ligands that bind to their respective receptors trigger the recruitment of adaptor molecules (FADD), and subsequent recruitment and activation of caspases to mediate apoptosis. Activated caspases can also cleave Bid to promote the mitochondrial pathway of apoptosis. This pathway is defined by mitochondrial outer membrane permeabilization (MOMP) and is regulated by the BCL-2 proteins. Pro-apoptotic proteins such as BID or BIM promote BAX or BAK homo oligomerization in the mitochondrial membrane, whereas anti-apoptotic proteins such as BCL-2 and BCL-xL inhibit this process. De-repressor proteins BAD, PUMA or NOXA bind the anti-apoptotic proteins and reduce the threshold for BAX/BAK activation. MOMP results in cytochrome c release into the cytosol, which promotes APAF-1 oligomerization, caspase activation, and apoptosis
The mitochondrial pathway of apoptosis: This pathway is defined by the permeabilization of mitochondria and release of proteins, including cytochrome c, from the inner mitochondrial space (IMS) into the cytosol [51, 52]. Cytochrome c binds to and triggers the oligomerization of apoptotic protease activating factor-1 (APAF-1) [53, 54]. This provides a signaling platform called the apoptosome for the activation of the initiator caspase-9 and consequent executioner caspases-3 and -7 [55, 56]. The BCL-2 (B cell CLL/lymphona-2) family consists of both pro- and anti-apoptotic proteins that regulate the integrity of the outer mitochondrial membrane (OMM) [57, 58]. The anti-apoptotic proteins such as BCL-2, BCL-xL (BCL-2-related gene, long isoform), MCL-1 (myeloid cell leukemia 1), and A1 (BCL-2 related gene A1) are generally localized on the OMM to promote membrane integrity by inhibiting the pro-apoptotic family members [59]. The pro-apoptotic proteins consist of two groups that promote cell death. Upon activation, the effector proteins BAX (BCL-2 associated x proteins) and BAK (BCL-2 antagonist killer 1), homo-oligomerize and cause mitochondrial outer membrane permeabilization (MOMP) [60, 61]. The BH3-only “direct activator” pro-apoptotic proteins, such as BID or BIM, function to directly activate BAX or BAK [58, 62, 63]. In contrast, the BH3-only “de-repressor or sensitizer” pro-apoptotic proteins such as Bad, PUMA (p53-upregulated modulator of apoptosis), and NOXA bind to anti-apoptotic proteins and lower the threshold required for BAX or BAK activation [58, 64]. Thus, the specific interactions between the pro- and anti-apoptotic proteins directly mediate MOMP (Fig. 7.2).
7.5 p53-Dependendent Pathways of Apoptosis in Response to DNA Damage
p53 has been termed “guardian of the genome” because it prevents the malignant transformation of cells by inducing a variety of cellular effects such as cell cycle arrest, senescence, and apoptosis (Fig. 7.3) [65, 66]. P53 loss predisposes the host to a variety of spontaneous and induced tumors [67]. Moreover, tumor cells routinely induce p53 instability and loss with more than 50 % of tumors lacking p53 function [68]. This highlights the importance of p53 as a tumor suppressor protein. P53 is a transcription factor whose targets are largely responsible for these effects.

DNA damage induced p53 promotes apoptosis in a transcription-dependent and -independent manner. In unstressed cells p53 levels are low due to its interaction with MDM2 where MDM2 polyubiquitylates p53, promoting its proteasomal degradation. Upon DNA damage residues within p53 and MDM2 are posttranslationally modified generating a fully functional, stabilized p53 protein. P53 binds to the promoter regions and transactivates numerous genes that control the cell cycle, senescence, and apoptosis. P53 can have transcription-independent functions in the cytosol and act analogously to a pro-apoptotic BH3 only protein by promoting MOMP
In unstressed cells, p53 levels remain low due to its constant degradation through the ubiquitin–proteasome pathway. MDM2 is an E3 ubiquitin ligase that regulates p53 levels via ubiquitylation [69]. MDMX/MDM4 (murine double minute x/4) is also an E3 ubiquitin ligase, and an additional negative regulator of p53, although this function is not via p53 ubiquitylation but through MDM2 stabilization [70]. MDM2 is a transcriptional target of p53, and thus controls its protein level by a simple feedback loop (Fig. 7.3) [71, 72]. Upon DNA damage, p53 becomes active in two ways: the inhibition of the interaction of p53 with its negative regulators, and through various posttranslational modifications that promote its transcription function (Fig. 7.4) [32, 35]. The specific regulation of p53 function will be discussed in greater detail below. Stabilized and active p53 binds to the promoter region of numerous pro-apoptotic target genes of the mitochondrial cell death pathway such as BAX, PUMA, NOXA, and BID and represses the expression of both BCL-2 and BCL-xL [35, 73]. P53 also mediates the expression of CD95 and TRAIL-R1, to facilitate the death receptor pathway of apoptosis (Fig. 7.3) [35, 73].

DNA damage results in posttranslational modifications of p53 and its negative regulators, MDM2 and MDMX, to promote apoptosis: After DNA damage the N-terminus of p53 is phosphorylated by ATM, ATR, CHK1, and CHK2 at different serine residues. This serves to inhibit the interaction of p53 with its negative regulators MDM2 and MDMX and promote its transcription function. MDM2 and MDMX are also phosphorylated by ATM resulting in the stabilization of p53 levels through its inability to be exported to the cytosol and polyubiquitylated at its C-terminus. Upon DNA damage, residues in the C-terminus of p53 that are normally ubiquitylated by MDM2 are now acetylated, sumoylated, or neddylated to induce sequence-specific DNA binding and promote p53-dependent apoptosis. Acetylation of lysines (K120/K164) in the DBD of p53 can potentiate the apoptotic function of p53 after DNA damage
In addition to its transcription function, studies have shown that p53 can mediate death through transcription-independent means (Fig. 7.3) [34, 74, 75]. In this setting, p53 functions analogously to a BH3-only pro-apoptotic member of the BCL-2 family, although the exact mechanism by which it mediates MOMP remains unclear. Cytosolic p53 can directly activate and oligomerize BAX [76, 77]. Additionally, mitochondrially localized p53 can promote BAK oligomerization to induce MOMP and cytochrome c release [78, 79]. Other studies suggest that cytosolic p53 can bind and neutralize the anti-apoptotic function of BCL-2 and BCL-xL, promoting MOMP in an indirect manner [80, 81]. Further studies suggest that the cytoplasmic function of p53 may also require its transcription function [82]. BCL-xL binds and inhibits cytosolic p53, which can only be released by PUMA [83]. PUMA-BCL-xL binding promotes p53 release and subsequent MOMP via BAX oligomerization, due to a very precise interaction between a tryptophan residue near the PUMA BH3 region (W71) and a histidine residue within the BCL-2 groove of BCL-xL (H113). The biological significance of cytoplasmic p53-mediated death remains unknown. Experimental evidence suggesting a role in tumor suppression is still lacking, but remains a very interesting topic to be investigated. The exact mechanism by which transcription-independent p53 is regulated by DNA damage is also unclear and will be discussed in detail below.
7.6 The Regulation of p53 Levels in Response to DNA Damage
One of the most interesting topics regarding p53 function is how the cell determines whether to undergo repair, cell cycle arrest, senescence, or apoptosis in response to DNA damage. There is some suggestion that the strength and duration of the damage signal is important, as is the tissue type or compartment undergoing stress. However, what is probably the clearest requirement for a differential p53 response are the posttranslational modifications of various residues within the protein.
P53 is a 393-amino-acid protein (390 in mouse) consisting of distinct functional domains (Fig. 7.4) [84]. The N-terminus contains the transactivation domain (TAD) where transcription co-activators p300 and CBP (CREB-binding protein) bind to promote the transcription function of p53 [85]. MDM2 and MDMX also bind in this region to regulate p53 levels [69, 86]. A proline-rich domain, of unclear function, follows this region [87]. The core region of p53 consists of its DNA-binding domain (DBD) and is also the region in which many cancer-associated mutations are found [88]. The tetramerization/oligomerization (OG) domain promotes oligomerization and the C-terminal region is the regulatory domain that can be either ubiquitylated to mediate proteasomal degradation of p53 or undergo a variety of posttranslational modifications such as acetylation, phosphorylation, neddylation, sumoylation, and methylation to regulate the cellular function of p53 [86, 89, 90].
N-terminal p53 regulation: N-terminal phosphorylation is necessary to inhibit MDM2 binding and promote p53 stabilization [91–93]. Following DNA damage, ATM, ATR, Chk1, and Chk2 all contribute to phosphorylate a number of serine and threonine residues in the N-terminus (Fig. 7.4) [33, 94, 95]. It remains unclear whether an individual kinase is responsible for phosphorylating one or more residues upon DNA damage, or whether numerous kinases are required. It also remains unclear whether one or more residues must be phosphorylated to inhibit MDM2 interaction. A knockin mouse containing individual phosphorylation mutants S18A (human S15) and S23A (human S20) or combined S18/23A mutations was generated to address this question. Individual mutations showed little difference in p53 stabilization upon DNA damage in numerous tissues [96, 97]. However, p53 was unable to be stabilized in response to DNA damage in the combined S18/23A mutant mouse [98]. Although there are other serine residues that are phosphorylated in the N-terminal region upon DNA damage, their specific function in vivo remains unclear. It is possible that in addition to stabilizing p53, N-terminal phosphorylation may be important for the recruitment and binding of transcription co-factors to specific gene promoter regions, thus influencing cellular response to stress.
Modification of MDM2 and MDMX by ATM stabilizes p53: DNA damage induced kinases stabilize p53 not only through modification of p53 residues, but also through posttranslational modification of residues within the negative regulators, MDM2 and MDMX (Fig. 7.4). Numerous residues near the C-terminal RING domain of HDM2 are modified by ATM in vitro including; S395, T419, S425, and S429 [99]. A knockin mouse containing an S394A (S395 human) mutation, demonstrated that phosphorylation of this residue is important for p53 stabilization after DNA damage in vivo [100]. This mechanism of p53 stabilization remains unclear; some experiments suggest that S395 phosphorylation prevents the nuclear export of p53 and its subsequent ubiquitylation and degradation, while other experiments suggest that ring domain oligomerization is inhibited, thus inhibiting the ubiquitylation function of MDM2 [101, 102]. Interestingly another study proposed that ATM-dependent phosphorylation of MDM2 switches its function from a negative to positive regulator of p53 by binding p53 mRNA and promoting the translation of p53 [103].
Recently ATM has also been shown to decrease the levels of the deubiquitylation enzyme USP7/HAUSP (Ubiquitin-specific-processing protease 7/herpesvirus-associated ubiquitin-specific protease) [104]. Since MDM2 levels are determined through self-ubiquitylation, diminished USP7 can stabilize p53 [105, 106]. It has long been thought that MDM2 regulates the levels of MDMX through ubiquitylation; however, it is becoming more apparent that DNA damage induced kinases can also directly modify residues within the C-terminal region of MDMX, thus inhibiting its function and promoting p53 stabilization. MDMX can be phosphorylated by ATM and Chk2 kinase on S342, S367 and S403, enhancing its specificity for MDM2-mediated binding and degradation [107, 108]. Other E3 ligases such as COP-1 (constitutive photomorphogenetic 1), Pirh2 (p53-induced RING-H2 protein), and HUWE1 (HECT-domain ubiquitin ligase) can mediate low p53 levels through ubiquitylation [109–111]. DNA damage can also regulate these proteins and stabilize p53; for instance ATM can phosphorylate COP-1 on S387 and stimulate its rapid degradation [109].
C-terminal p53 regulation: The C-terminal domain of p53 contains numerous lysine residues that are heavily modified upon DNA damage through acetylation, sumoylation, methylation, and neddylation (Fig. 7.4). In non-stressed cells, the lysine residues in this region are constantly ubiquitylated by MDM2 to promote p53 degradation [69, 86]. In stressed cells, modification of these same residues may help to promote sequence-specific DNA binding, thus influencing the cellular response to stress [90]. However, recent studies using p53 knockin mice where 6 or 7 of the extreme C-terminal lysines were modified, showed little difference in p53 stability or response to stress [112, 113]. This suggests that perhaps other residues in the DNA-binding site (K120 or K164) can contribute to p53 stability and function [114, 115]. Either way, the exact role of C-terminal modifications after DNA damage in mediating p53 cellular effects remains unclear. Perhaps the most interesting new development in this area of research pertains to the idea that p53 is bound to DNA and fully active in the absence of DNA damage and is held in a “repressed state” by it negative regulators MDM2/MDMX [116]. This idea is based on the observation that MDM2 and MDMX deficient mice are embryonic lethal due to p53-mediated death and this lethality can be rescued upon p53 deletion [117–119]. Furthermore, a knockin mouse, p53QS, that retains DNA-binding potential but cannot interact with its negative regulators MDM2/MDMX is also embryonic lethal [120]. The various posttranslational modifications that are required to release p53 from this repressed state are currently being elucidated.
Posttranslational modifications of p53 that mediate its transcription-independent function: The posttranslational modifications of p53 that mediate its cytosolic function remain unclear. MDM2 and the E3 ligase MSL2 (male specific lethal 2) are both proposed to monoubiquitylate p53 to promote mitochondrial localization [121–123]. These E3 ligases do not directly shuttle p53 to the mitochondria; instead this function is performed by the mitochondrial chaperone, Tid1 [124]. Once localized to the mitochondrial membrane, HAUSP deubiquitylates p53 to promote MOMP [125]. A recent study has identified that K351 mutation in p53 cannot be monoubiquitylated by MDM2 or MSL2 and cannot be localized to the mitochondria, suggesting that this is the critical residue for modification and subsequent cytoplasmic p53 function [126]. Furthermore, MDM2-mediated monoubiquitylation can promote other modifications of the p53 C-terminal lysine residues, such as sumoylation of K386 by PIASy (protein inhibitor of activated STAT protein gamma) [101]. These modifications not only expose the p53 nuclear export signal (NES) but also release MDM2, further promoting nuclear export. The acetylation of K120 by Tip60/hMOF histone acetyl transferases (HATs) in the p53 DNA-binding domain may promote transcription-independent cell death by displacing Mcl-1 from BAK [127]. However, another study proposed that acetylation of C-terminal K320/K373/K382 mediates p53 binding to the DNA repair protein, Ku, thus inhibiting its interaction with BAX and inducing cell death [128]. Many tumors exhibit p53 mutations within the DNA-binding domain resulting in a transcription-deficient p53. Identifying the mechanism by which DNA damage induces and promotes p53 nuclear export and cytoplasmic/mitochondrial function may be of great value for future neoplastic therapeutics.
7.7 p53-Independendent Pathways of Apoptosis in Response to DNA Damage
Although p53 clearly plays an important role in mediating death in response to DNA damage, there are many other proteins that can promote the mitochondrial and death receptor apoptotic pathways in the absence of p53. This is particularly important during tumor therapy since many tumors have mutated or nonfunctional p53. Some of these proteins will be discussed below.
p63 and p73: These proteins are part of the p53 family of transcription factors and they share similar homology, and some transcriptional targets and functions with p53 [129]. Following DNA damage all three family members have the potential to mediate cell cycle arrest and apoptosis [129]. Therefore, it is probable that in response to DNA damage p63 and p73 can compensate for lack of p53 function and mediate death in some settings. Similar to p53, p63 and p73 contain an N-terminal TAD, a central DBD and an OG domain. Unlike p53, they contain a sterile alpha motif (SAM) in their C-terminal regions that promotes protein binding. Two different promoters at the N-terminus of the gene generate two different isoforms; Transcriptional activator (TA)-containing isoform (TAp63 and TAp73) and N-terminal truncated isoform (ΔNp63 and ΔNp73) [130]. Alternative splicing at the C-terminus gives further rise to numerous isoforms of p63 and p73 [130]. TAp63 and TAp73 mediate their cellular effects primarily through transcription-dependent means, whereas ΔNp63 and ΔNp73 function as dominant negative proteins since they can bind to the promoter binding sites but are unable to transactivate gene expression [131]. Like p53, levels of TAp63 and TAp73 increase upon DNA damage and their transactivation function is mediated primarily by posttranslational modifications, although the exact nature and regulation of these modifications still remain to be fully elucidated [132–134]. p63 and p73 transactivate and/or increase the expression of BAX, PUMA, NOXA, BAD, APAF1, caspase-3, -8, and -9, CD95, TNFR, and TRAIL-R1 to influence cell death pathways [135–137]. In addition, a transcription-independent function has been attributed to p73, but not p63. Full length TAp73 or a caspase-cleaved p73 fragment were found localized to the mitochondria, and mediated apoptosis [138, 139]. Interestingly, purified p73 could also induce MOMP on isolated mitochondrial fractions [138]. This function of p73 is still quite controversial and requires further validation in vivo.
TAp63-induced death may be tissue specific since TAp63 is expressed strongly in oocytes and mice deficient for TAp63 were resistant to DNA damage mediated oocyte death [140]. In response to DNA damage, TAp63 induced primordial follicle oocyte death through the induction of PUMA and NOXA [141]. Therefore, p63 may play an important role in female reproduction and oocyte maintenance independently of p53 function.
p63 and p73 may also display tumor suppressor functions. Mice heterozygous for both proteins display spontaneous tumor formation at a rate that is only slightly longer than that seen with p53 heterozygous mice [142]. Furthermore, mice lacking only the TAp73 isoforms displayed a high incidence of spontaneous tumor formation [143]. Unlike p53, however, p63 and p73 are rarely mutated in cancers, but the N-terminal truncated version of both proteins is very commonly over-expressed [144]. These results all indicate a role in tumor suppression for p63 and p73.
NF-κB transcription factor is induced by DNA damage: NF-κB (nuclear factor kappa-light-chain-enhancer of activated B cells) mediates a large array of biological processes such as immune regulation and cellular growth but it can also be activated upon DNA damage to promote apoptosis [145–148]. NF-κB is a dimeric transcription factor that belongs to the Rel family of proteins that consists of RelA (p65), c-Rel, or RelB as one half, and p50 (NFKB1) or p52 (NFKB2) as the other [149]. NF-κB is inactive in the cytoplasm by virtue of its interaction with inhibitory IkB proteins, such as IkBα. The IkB kinase complex (IKK) is made up of three proteins; two catalytically active kinases IKKα and IKKβ and the regulatory IKKγ (NEMO) protein that functions to phosphorylate IkBα, inducing its ubiquitylation and subsequent proteasomal degradation, thus allowing the now unbound NF-kB to translocate to the nucleus and mediate is transcription function (Fig. 7.5) [150, 151].

DNA damage promotes NF-κB mediated apoptosis: DNA damage results in ATM phosphorylation of nuclear localized NEMO. The ATM/NEMO complex is subsequently translocated to the cytosol resulting in TAK1 phosphorylation and activation. TAK1 phosphorylates IKKβ resulting in IκB degradation and NF-κB activation. Persistent DNA damage promotes an NF-κB mediated TNFα feed-forward loop, resulting in conversion of complex I to the apoptotic complex II. DNA damage reduces the levels of IAPs, promoting the formation of a ripoptosome complex that promotes either apoptosis or programmed necrosis
NF-κB can be activated through one of two pathways, the canonical or noncanonical pathways [149]. Here we will briefly discuss the canonical pathway, since NF-κB activation by DNA damage converges with the canonical pathway of NF-kB activation at the point of IKK activation and Ikβ degradation [148, 149].
Bacterial or viral infections or pro-inflammatory cytokines such as TNFα engage the TNFR1, triggering the recruitment of TRADD (TNF receptor associated death domain), FADD, and RIP1 (receptor interacting protein 1) all through their respective death domains (DD). TRADD also binds the E3 ubiquitin ligase, TRAF2 (TNF receptor associated factor 2) that subsequently binds the cellular inhibitors of apoptosis 1 and 2 (cIAP1 and cIAP2). Together, this group of proteins makes the core of the TNFR1 signaling complex, also called complex-I. RIP1 ubiquitylation by cIAP1 recruits the kinase TAK1 (transforming growth factor β-activated kinase 1), which phosphorylates IKKβ, activating the IKK complex, resulting in the degradation of IκBα and an active NF-κB, composed mainly of p50/p65 dimers.
The exact mechanism by which nuclear ATM can activate a complex localized in the cytoplasm is unclear, but IKK-independent functions of NEMO may provide the key to this conundrum [152]. Posttranslational modifications of NEMO mediate its role in transducing DNA damage signals. NEMO is firstly SUMOylated by PIASY-1 (protein inhibitor of activated STAT protein gamma) to promote its nuclear localization and is subsequently phosphorylated by ATM on serine 85 [153, 154]. NEMO is then monoubiquitylated by cIAP1 and exported to the cytoplasm along with a fraction of ATM, upon which TAK1 becomes phosphorylated, thus activating IKK [155–157]. Nuclear translocated NF-κB transcribes genes involved in cell survival such as the anti-apoptotic genes, cIAP1&2, XIAP, FLIPL, BCL-2, A1, and BCL-xL, presumably to allow the cells time to mediate DNA repair [150, 151, 158]. In some cases NF-κB can induce the expression of genes to promote cell death, such as CD95L in T cells exposed to stress [159].
DNA damage can promote cell death through the formation of two cytoplasmically localized protein complexes, complex-II and the ripoptosome (Fig. 7.5). TNF-mediated signaling induces the formation of complex-II. This complex consists of RIP1, FADD, caspase-8, and FLIPL and forms secondary to the membrane-associated complex-I, hours after initial TNF ligation to the TNF-R1 [160]. The molecular mechanism for transition from complex-I to complex-II remains unclear, but a recent study suggests that that strength and duration of DNA damage promotes apoptosis through a TNFα-feed forward mechanism [161]. Low levels of DNA damage induced ATM-dependent NF-κB induction of pro-survival genes, including the expression of TNFα. Newly synthesized TNFα engaged TNFR1 resulting in the formation of complex-II. Here, RIP1 is autophosphorylated resulting in FADD recruitment and caspase 8 activation and apoptosis [161]. Another study has identified the formation NEMO, RIP1, and PIDD (p53-induced death domain) complex upon DNA damage. PIDD promotes NF-κB activation through NEMO sumoylation and ubiquitylation [162]. Caspase-2 can also bind PIDD to form the “PIDDosome” and induce apoptosis [163]. Whether or not a feed forward mechanism of caspase-2 dependent apoptotic signaling can also exist remains unclear, but caspase-2 deficient cells show aberrant death after DNA damage, suggesting that maybe it does [164].
DNA damage can also mediate death independently of TNF signaling through the down regulation of cIAP1, cIAP2, and XIAP [165, 166]. In the absence of IAPs a spontaneous complex is formed in the cytosol called the ripoptosome that can induce both apoptotic and programmed necrotic cell death pathways [166]. The ripoptosome is made up of RIP1/FADD/caspase-8 with additional association of FLIPL and RIP3. The ripoptosome requires the kinase activity of RIP1 and is negatively regulated by the IAP’s through the ubiquitin-mediated proteasomal degradation of RIP1 [166]. Although FLIPL is also a negative regulator of the ripoptosome, DNA damage can significantly decrease the levels of FLIPL in a cell [167]. Since FLIP can form heterodimers with caspase 8 and inhibit both apoptosis and programmed necrosis, decreased FLIP levels allow both caspase 8 homodimerization and RIP1/RIP3 association to induce apoptosis and necrosis, respectively, upon DNA damage and ripoptosome formation [168, 169].
This form of cell death may be extremely important in cancer therapy since it was shown to kill tumor cells that were resistant to apoptosis [166].
7.8 DNA Damage Response and p53-Mediated Apoptosis in the Prevention of Tumorigenesis
Aberrant proliferation is a feature commonly seen in tumor cells and is the combined result of active oncogenes and dysfunctional tumor suppressor genes [170]. p53-mediated apoptosis is one the most important mechanisms by which cells prevent oncogene dependent malignant transformation [171, 172]. The DDR pathway is crucial to control initial damage signals to the DNA that can induce genomic instability, but importantly, there is evidence that the DDR is also induced by oncogenic transformation of a cell, thus preventing propagation of early malignant cells.
In this section we discuss the signals that trigger the DDR during malignant propagation and the importance of the DDR in preventing tumorigenesis. Finally we will determine whether tumor-suppression by p53 is mediated through the DDR pathway or whether ARF (alternate reading frame of the INK4a/ARF locus)-dependent mechanisms are preferentially utilized.
Oncogene-mediated DDR: The idea that the DDR acts as a barrier against genomic instability and cancer development came about after histochemical observations in very early neoplastic lung, colon, bladder, and skin tissue. Pre-neoplastic tissue was associated with DNA damage foci and high levels of apoptosis and senescence [173, 174]. Markers of an active DDR, such as phosphorylated H2AX, accumulation of p53BP foci and Chk2 phosphorylation were visible in pre-neoplastic and neoplastic tissue but not in normal tissue [173, 175]. Thus, DNA damage checkpoints could be activated in response to oncogenes during the early stages of malignant progression to promote apoptosis.
The mechanism by which oncogenes promote the DDR in these precancerous tissues remains unclear. Oncogenes promote cellular hyper-proliferation that is accompanied by DNA hyper-replication, resulting in replication stress [176]. Oncogene expressing cells displayed high cyclin E levels, DNA hyper-replication, increased numbers of active replicons, increased replication fork instability and LOH in common fragile sites [175, 177, 178]. These DNA damage signals promoted a specific ATR/Chk1 response with ATR and RPA found to be co-localized in H2AX foci in cells [15, 173].
Another mechanism by which oncogenes can trigger a DDR is through telomere erosion [179, 180]. Telomeres are repetitive nucleotide sequences that protect the very ends of chromosomes from instability. Neoplastic cells exhibit telomere erosion and unless lengthened by telomerase, are recognized as DSBs, promoting a DDR [181]. The shelterin complex caps telomeres for further chromosomal protection and facilitates telomerase function. Oncogene-induced mutations in this complex or telomerase can also contribute to telomere erosion and promote a DDR [180, 182].
Although it still remains unclear exactly how the DDR is induced in tumor cells, DNA damage dependent signaling pathways seem to be a crucial factor in inhibiting tumorigenesis.
DDR suppresses tumorigenesis: Mutations in DNA-damage responses allow the continued cell growth and proliferation of cells with damaged DNA and genomic alterations, thus enhancing the propensity for oncogenic transformation [183]. This is evident in cells over-expressing oncogenic Ras. Cells lacking ATM or Chk2 did not stop proliferating, were more susceptible to transformation, and formed tumors in recipient mice, whereas DDR competent cells arrested [178].
The importance of the DDR in preventing malignancy is further highlighted in mouse genetic models and human conditions in which proteins of the DDR are nonfunctional. Patients displaying defects in genes that mediate NHEJ and HR repair have syndromes such as LIG4 syndrome and FANCD1 (Fanconi Anemia) and a predisposition to developing lymphoma and leukemia [184, 185]. By the same token, genetic defects in proteins that mediate DNA damage signaling pathways also predispose individuals to tumors. Patients with mutated ATM have a disorder called ataxia telangiectasia (AT) [186]. These patients are sensitive to γ-irradiation (IR) and are predisposed to developing leukemias and lymphomas. Furthermore, mutations in genes that encode downstream targets of ATM also predispose patients to cancers, such as Mre11 (A-T like disorder, ATLD), NBS1 (Nijmegen breakage syndrome, NBS), Brca1 and Brca2 (familial breast, ovarian carcinoma syndrome), Chk2 and p53 (Li–Fraumeni syndrome) [187–192]. In contrast, patients with an ATR mutation (Seckel syndrome) are not predisposed to cancer but display dwarfism, developmental delay, and microencephaly [193]. Similarly, mouse models of a defective DDR also show susceptibility to tumor development. Mice lacking ATM are born viable but are strongly predisposed to developing T cell lymphoma at 2–4 months of age [194]. Mice lacking the checkpoint mediator proteins H2AX and p53BP1 are also predisposed to spontaneous tumor development [195, 196].
DNA damage as an effective therapy against cancer: It may seem paradoxical that the initiation of tumorigenesis may be due to a defective DDR, yet the most common therapy used to treat malignancies involves chemotherapeutic drugs that induce DNA damage. Tumor cells usually have defective repair pathways and proliferate much faster than nonmalignant cells, making them more susceptible to treatment [197]. Without a fully functional repair process, DNA damage preferentially promotes apoptosis. Since effective DNA repair can promote resistance to therapy, inhibitors of DNA repair are currently being explored in the clinic as treatment against cancers resistant to traditional chemotherapy [198, 199].
Another reason why tumor cells may respond to better to chemotherapy is because they are “primed to die” [200]. Most normal cells that encounter unregulated oncogene expression induce an apoptotic response, but some cells survive and grow because they express high levels of anti-apoptotic proteins such as BCL-2 [201]. Importantly, the anti-apoptotic proteins in these cells are occupied with pro-apoptotic proteins such as BIM [202, 203]. These tumor cells are therefore “primed” for death with pro-apoptotic molecules and are more sensitive to conventional chemotherapy that tips the balance of this fragile equilibrium towards cell death [204].
Is the tumor suppressor function of p53 due solely to the DNA damage response? Tumorigenesis in animals lacking a functional DDR may be primarily due to a lack of p53 function. Cells deficient for ATM generally show an inability to stabilize and induce p53-mediated cellular effects after IR, suggesting the importance of ATM-mediated p53 effects in tumor suppression [205]. Furthermore p53 knock in mice that are unable to be stabilized and activated by ATM (combined S18/23A mutations in p53) develop late-onset B cell lymphomas [98, 206]. However, these lymphomas do not develop at the same rate and intensity as is seen with p53 knockout (KO) mice. This suggests that other mechanisms are still mediating p53 activation and function.
Oncogene-mediated hyper-proliferation signals can also stabilize p53 through ARF (p14ARF in human and p19ARF in mouse), and this occurs in a DNA damage-independent manner [207]. ARF is transcribed through the alternate reading frame of the INK4a/ARF locus, which also encodes p16INK4a, a cyclin-dependent kinase inhibitor. ARF stabilizes p53 by direct binding and sequestration of its negative regulator MDM2 [208]. Mice lacking ARF are prone to developing tumors, and tumors that have high levels of ARF have silenced p53, whereas tumors that retain WT p53 function have nonfunctional ARF [209, 210]. Furthermore, mice null for both p53 and ATM show dramatic acceleration of tumor formation relative to mice deficient for one gene only, thus indicating an alternative mechanism of p53 induction in tumors [211].
Elegant studies performed in knock in mice containing a p53 estrogen receptor fusion protein (p53ERTam) that can restore p53 function upon 4-hydroxytamoxifen (4OHT) administration, have supported the idea that p53-dependent tumor suppression is attributed to ARF and not the DDR [212, 213]. In this model, p53 was required to prohibit radiation-induced lymphoma; however, the time of p53 restoration after the acute radiation response determined its tumor-suppressor function [213]. When p53 was restored in mice before IR, the mice displayed all the features of an effective DDR coinciding with high levels of apoptosis, but this did not prevent lymphoma development. However, when p53 was restored after the acute DDR was cleared, a significant delay in lymphomagenesis was observed. This effect was lost in an ARF null background, suggesting that ARF engaged the tumor suppressor function of p53 in response to the outgrowth of malignant cells after IR [213].
Furthermore, mice that contain an additional copy of the p53 gene (p53Super) show a superior response to DNA damage inducing high levels of apoptosis and are highly resistant to tumors [214]. These mice do not confer protection against tumor development the absence of ARF.
It is possible however that the tumor suppressor function of p53 stabilized through DNA damage or ARF is not mutually exclusive. ARF-deficient cells are unable to mount an effective DDR and ARF levels can be increased upon DNA damage [215]. Therefore, additional studies are require to further elucidate the mechanism by which p53 can mediate its tumor-suppressor capabilities.
p53-mediated apoptosis in tumor suppression: The apoptotic function of p53 is primarily mediated through PUMA and NOXA. Mice deficient for PUMA have thymocytes that are highly resistant to IR, yet mice do not develop spontaneous tumors [216, 217]. However, in an Eμ-MYC model of lymphomagenesis, PUMA deficiently accelerated tumor formation [218]. NOXA-deficient mice also show a defect in DNA damage induced apoptosis, although this is specific for mouse embryonic fibroblasts (MEFs) and not thymocytes [217]. They are also resistant to spontaneous tumor formation and fail to accelerate Eμ-MYC tumorigenesis [219]. Mice lacking both PUMA and NOXA phenocopy mice deficient for PUMA alone and are resistant to tumors [220].
Mice deficient for p53-target genes that mediate the death receptor cell death pathway such as TRAIL-R1 and CD95 are also not prone to spontaneous tumors, but may accelerate tumorigenesis under some conditions [221, 222]. These experiments suggest that there may be a redundancy for p53-mediated apoptosis for tumor suppression and that the cell cycle arrest or senescent functions of p53 can compensate in the absence of apoptosis and mediate tumor suppression. Mice deficient for p21 are generally not tumor prone, but p21 deficiency can accelerate tumorigenesis in some settings [223, 224]. Importantly, however, mice lacking PUMA, NOXA, and p21 display no spontaneous tumor formation although these studies have not been performed in the presence of a cellular stress [225]. This study indicates that the tumor suppressor function of p53 is not dependent on apoptosis or cell cycle arrest. Mice that contain mutations within the TAD of p53 are unable to mediate apoptosis or cell cycle arrest in response to DNA damage. However, p53 with a mutated TAD maintains some transactivation function and can mediate the expression of genes involved in senescence. Importantly, the expression of these genes is enough to mediate tumor suppression in these mice [225, 226]. Another study generated mice with mutations in the lysines of the DBD of p53. In this scenario, p53 was unable to express proteins involved in apoptosis, cell cycle arrest or senescence, yet these mice were also resistant to tumors [227]. A metabolic function of p53 was proposed to be tumor-suppressive in this model, representing a very interesting theme that is being explored in the p53 field at present [228].
It remains unclear why some tumor cells respond to DNA damage by mediating p53 dependent-apoptosis, senescence, or even metabolic effects. Whether it depends on the type or strength of DNA damage, or the tissue type, or the posttranslational modifications of p53 is unknown but requires further identification [229].
7.9 DNA Damage Response Promotes p53-Mediated Cell Competition in HSCs and May Promote Tumorigenesis
Recent evidence suggests that the DDR can not only mediate a cellular response to stress, such apoptosis or cell cycle arrest, but can also regulate the self-renewal and differentiation function of stem cells in a p53-dependent manner. DNA damage mediated cellular effects and DNA damage mediated stem cell effects represent two independent functions of p53 that may have opposing roles in the development of cancer. Here we will discuss how a DDR can mediate a form of cellular competition in the hematopoietic compartment by regulating HSC population. We will also discuss the contribution that the modified stem cell compartment may have in cancer progression.
Cell competition describes the clonal survival of developmentally identical cells in a compartment that has been exposed to a type of stress, such as DNA damage [230]. The “fittest” cells survive the stress and repopulate the cellular compartment. For example, when bone marrow was combined in equal measures from IR and non-IR mice and injected into lethally irradiated mice, the non-irradiated HSCs repopulated the bone marrow and therefore represent the fittest population [231]. In short, undamaged cells outcompete the damaged counterparts.
Reports suggest that DDR mediated cellular competition in the hematopoietic compartment is p53-dependent [231, 232]. HSCs with lower p53 levels displayed greater self-renewal capacity and outcompete high p53 expressing HSCs after DNA damage. Cell competition is distinct from the p53-mediated DDR, since cells become out-competed at a much later time point after all the DNA damage has been cleared. The outcompeted cells in this scenario did not die by apoptosis but instead saw decreased proliferative capacity and a senescent phenotype, whereas the fitter cells showed better proliferation. Since high levels of DNA damage can deplete stem cell number, lower levels of p53 may be beneficial for long-term stem cell survival. However, increased self-renewal in stem cells is also associated in tumors [233]. Moreover, HSCs with mutated p53 also displayed lower levels of p53 and better competition and this may also contribute to the outgrowth of cells that contribute to malignancy [234, 235].
Another report proposed that survival of HSCs after DNA damage was dependent on ATM-mediated phosphorylation of BID [236]. ATM-dependent phosphorylation of BID at residues S61 and S78 resulted in cell cycle arrest in the S and G2 phase suggesting that BID may have a pro-survival function in response to low levels of DNA damage [237, 238]. Phosphorylated BID was unable to localize to the mitochondria and promote oxidative stress and apoptosis, thus maintaining the quiescence of HSCs [236].
Therefore, although the DDR may be crucial to inhibit cancer progression and induce tumor suppression in tissues, it may however promote the survival of tumor-initiating stem cells that repopulate the organism [239]. A clearer understanding of the mechanisms by which p53 contributes to cell competition in the hematopoietic compartment in response to DNA damage is necessary.
7.10 Concluding Remarks
We have discussed the many ways by which the DDR can trigger both mitochondrial and death receptor cell death pathways in a p53-dependent and -independent manner. The kinases of the DDR mediate detailed posttranslational modifications of p53 and its negative regulators to promote its transcription-dependent and -independent functions. DNA damage kinases can also modulate other pathways to induce apoptosis in a p53-independent manner. This represents an important “backup” for tumor suppression when p53 is deleted or dysfunctional. Although p53-mediated apoptosis is crucial for the prevention of tumorigenesis, whether this is induced through the DDR or ARF-dependent means is still unclear. Interestingly, the apoptotic effects of p53 may not be solely responsible for tumor suppression after DNA damage—senescence or even metabolic effects may play a part, thus presenting a redundancy in mechanism of p53-dependent tumor suppression. Finally, DNA damage mediated tumor suppression in cells may actually promote cellular competition in the hematopoietic compartment and induce the differentiation of stem cells to a more malignant phenotype.
References
Harper JW, Elledge SJ. The DNA damage response: ten years after. Mol Cell. 2007;28:739–45. doi:S1097-2765(07)00783-6 [pii]10.1016/j.molcel.2007.11.015.
Kastan MB, Bartek J. Cell-cycle checkpoints and cancer. Nature. 2004;432:316–23. doi:nature03097 [pii]10.1038/nature03097.
Jackson SP, Bartek J. The DNA-damage response in human biology and disease. Nature. 2009;461:1071–8. doi:nature08467 [pii]10.1038/nature08467.
Ciccia A, Elledge SJ. The DNA damage response: making it safe to play with knives. Mol Cell. 2010;40:179–204. doi:S1097-2765(10)00747-1 [pii]10.1016/j.molcel.2010.09.019.
Lindahl T, Barnes DE. Repair of endogenous DNA damage. Cold Spring Harb Symp Quant Biol. 2000;65:127–33.
Guo Z, Kozlov S, Lavin MF, Person MD, Paull TT. ATM activation by oxidative stress. Science. 2010;330:517–21. doi:330/6003/517 [pii]10.1126/science.1192912.
Doll R, Peto R. The causes of cancer: quantitative estimates of avoidable risks of cancer in the United States today. J Natl Cancer Inst. 1981;66:1191–308.
Hoeijmakers JH. Genome maintenance mechanisms for preventing cancer. Nature. 2001;411:366–74. doi:10.1038/3507723235077232 [pii].
Smith J, Tho LM, Xu N, Gillespie DA. The ATM-Chk2 and ATR-Chk1 pathways in DNA damage signaling and cancer. Adv Cancer Res. 2010;108:73–112. doi:B978-0-12-380888-2.00003-0 [pii]10.1016/B978-0-12-380888-2.00003-0.
Lavin MF, Kozlov S. ATM activation and DNA damage response. Cell Cycle. 2007;6:931–42. doi:4180 [pii].
Cimprich KA, Cortez D. ATR: an essential regulator of genome integrity. Nat Rev Mol Cell Biol. 2008;9:616–27. doi:nrm2450 [pii]10.1038/nrm2450.
Bakkenist CJ, Kastan MB. DNA damage activates ATM through intermolecular autophosphorylation and dimer dissociation. Nature. 2003;421:499–506. doi:10.1038/nature01368[pii].
Lee JH, Paull TT. ATM activation by DNA double-strand breaks through the Mre11-Rad50-Nbs1 complex. Science. 2005;308:551–4. doi:1108297 [pii]10.1126/science.1108297.
Zou L, Elledge SJ. Sensing DNA damage through ATRIP recognition of RPA-ssDNA complexes. Science. 2003;300:1542–8. doi:10.1126/science.1083430300/5625/1542 [pii].
Cortez D, Guntuku S, Qin J, Elledge SJ. ATR and ATRIP: partners in checkpoint signaling. Science. 2001;294:1713–6. doi:10.1126/science.1065521294/5547/1713 [pii].
Matsuoka S, et al. ATM and ATR substrate analysis reveals extensive protein networks responsive to DNA damage. Science. 2007;316:1160–6. doi:316/5828/1160 [pii]10.1126/science.1140321.
Rogakou EP, Pilch DR, Orr AH, Ivanova VS, Bonner WM. DNA double-stranded breaks induce histone H2AX phosphorylation on serine 139. J Biol Chem. 1998;273:5858–68.
Paull TT, et al. A critical role for histone H2AX in recruitment of repair factors to nuclear foci after DNA damage. Curr Biol. 2000;10:886–95. doi:S0960-9822(00)00610-2 [pii].
Lukas C, Falck J, Bartkova J, Bartek J, Lukas J. Distinct spatiotemporal dynamics of mammalian checkpoint regulators induced by DNA damage. Nat Cell Biol. 2003;5:255–60. doi:10.1038/ncb945 [pii].
Stewart GS, Wang B, Bignell CR, Taylor AM, Elledge SJ. MDC1 is a mediator of the mammalian DNA damage checkpoint. Nature. 2003;421:961–6. doi:10.1038/nature01446 [pii].
Wang B, Matsuoka S, Carpenter PB, Elledge SJ. 53BP1, a mediator of the DNA damage checkpoint. Science. 2002;298:1435–8. doi:10.1126/science.1076182 [pii].
Lou Z, et al. MDC1 maintains genomic stability by participating in the amplification of ATM-dependent DNA damage signals. Mol Cell. 2006;21:187–200. doi:S1097-2765(05)01813-7 [pii]10.1016/j.molcel.2005.11.025.
Mochan TA, Venere M, DiTullio Jr RA, Halazonetis TD. 53BP1 and NFBD1/MDC1-Nbs1 function in parallel interacting pathways activating ataxia-telangiectasia mutated (ATM) in response to DNA damage. Cancer Res. 2003;63:8586–91.
Huyen Y, et al. Methylated lysine 79 of histone H3 targets 53BP1 to DNA double-strand breaks. Nature. 2004;432:406–11. doi:nature03114 [pii]10.1038/nature03114.
Cortez D, Wang Y, Qin J, Elledge SJ. Requirement of ATM-dependent phosphorylation of brca1 in the DNA damage response to double-strand breaks. Science. 1999;286:1162–6. doi:7959 [pii].
Zou L, Cortez D, Elledge SJ. Regulation of ATR substrate selection by Rad17-dependent loading of Rad9 complexes onto chromatin. Genes Dev. 2002;16:198–208. doi:10.1101/gad.950302.
Wang X, et al. Rad17 phosphorylation is required for claspin recruitment and Chk1 activation in response to replication stress. Mol Cell. 2006;23:331–41. doi:S1097-2765(06)00449-7 [pii]10.1016/j.molcel.2006.06.022.
Bao S, et al. ATR/ATM-mediated phosphorylation of human Rad17 is required for genotoxic stress responses. Nature. 2001;411:969–74. doi:10.1038/35082110 [pii].
Lee J, Kumagai A, Dunphy WG. Claspin, a Chk1-regulatory protein, monitors DNA replication on chromatin independently of RPA, ATR, and Rad17. Mol Cell. 2003;11:329–40. doi:S1097276503000455 [pii].
Liu Q, et al. Chk1 is an essential kinase that is regulated by Atr and required for the G(2)/M DNA damage checkpoint. Genes Dev. 2000;14:1448–59.
Sanchez Y, et al. Conservation of the Chk1 checkpoint pathway in mammals: linkage of DNA damage to Cdk regulation through Cdc25. Science. 1997;277:1497–501.
Kruse JP, Gu W. Modes of p53 regulation. Cell. 2009;137:609–22. doi:S0092-8674(09)00511-X [pii]10.1016/j.cell.2009.04.050.
Hirao A, et al. DNA damage-induced activation of p53 by the checkpoint kinase Chk2. Science. 2000;287:1824–7. doi:8333 [pii].
Chipuk JE, Green DR. Dissecting p53-dependent apoptosis. Cell Death Differ. 2006;13:994–1002. doi:4401908 [pii]10.1038/sj.cdd.4401908.
Riley T, Sontag E, Chen P, Levine A. Transcriptional control of human p53-regulated genes. Nat Rev Mol Cell Biol. 2008;9:402–12. doi:nrm2395 [pii]10.1038/nrm2395.
Abbas T, Dutta A. p21 in cancer: intricate networks and multiple activities. Nat Rev Cancer. 2009;9:400–14. doi:nrc2657 [pii]10.1038/nrc2657.
Mandic A, Hansson J, Linder S, Shoshan MC. Cisplatin induces endoplasmic reticulum stress and nucleus-independent apoptotic signaling. J Biol Chem. 2003;278:9100–6. doi:10.1074/jbc.M210284200 [pii].
Tabas I, Ron D. Integrating the mechanisms of apoptosis induced by endoplasmic reticulum stress. Nat Cell Biol. 2011;13:184–90. doi:ncb0311-184 [pii]10.1038/ncb0311-184.
Gibson BA, Kraus WL. New insights into the molecular and cellular functions of poly(ADP-ribose) and PARPs. Nat Rev Mol Cell Biol. 2012;13:411–24. doi:nrm3376 [pii]10.1038/nrm3376.
Jwa M, Chang P. PARP16 is a tail-anchored endoplasmic reticulum protein required for the PERK- and IRE1alpha-mediated unfolded protein response. Nat Cell Biol. 2012;14:1223–30. doi:ncb2593 [pii]10.1038/ncb2593.
Gorman AM, Healy SJ, Jager R, Samali A. Stress management at the ER: regulators of ER stress-induced apoptosis. Pharmacol Ther. 2012;134:306–16. doi:S0163-7258(12)00043-5 [pii]10.1016/j.pharmthera.2012.02.003.
Green DR. Means to an end: apoptosis and other cell death mechanisms. Cold Spring Harbor, NY: Cold Spring Harbor Laboratory Press; 2011.
Kerr JF, Wyllie AH, Currie AR. Apoptosis: a basic biological phenomenon with wide-ranging implications in tissue kinetics. Br J Cancer. 1972;26:239–57.
Taylor RC, Cullen SP, Martin SJ. Apoptosis: controlled demolition at the cellular level. Nat Rev Mol Cell Biol. 2008;9:231–41. doi:nrm2312 [pii]10.1038/nrm2312.
Dickens LS, Powley IR, Hughes MA, MacFarlane M. The ‘complexities’ of life and death: death receptor signalling platforms. Exp Cell Res. 2012;318:1269–77. doi:S0014-4827(12)00188-7 [pii]10.1016/j.yexcr.2012.04.005.
Locksley RM, Killeen N, Lenardo MJ. The TNF and TNF receptor superfamilies: integrating mammalian biology. Cell. 2001;104:487–501. doi:S0092-8674(01)00237-9 [pii].
Scott FL, et al. The Fas-FADD death domain complex structure unravels signalling by receptor clustering. Nature. 2009;457:1019–22. doi:nature07606 [pii]10.1038/nature07606.
Kischkel FC, et al. Cytotoxicity-dependent APO-1 (Fas/CD95)-associated proteins form a death-inducing signaling complex (DISC) with the receptor. EMBO J. 1995;14:5579–88.
Peter ME, Krammer PH. The CD95(APO-1/Fas) DISC and beyond. Cell Death Differ. 2003;10:26–35. doi:10.1038/sj.cdd.4401186 [pii].
Oberst A, et al. Inducible dimerization and inducible cleavage reveal a requirement for both processes in caspase-8 activation. J Biol Chem. 2010;285:16632–42. doi:M109.095083 [pii]10.1074/jbc.M109.095083.
Goldstein JC, Waterhouse NJ, Juin P, Evan GI, Green DR. The coordinate release of cytochrome c during apoptosis is rapid, complete and kinetically invariant. Nat Cell Biol. 2000;2:156–62. doi:10.1038/35004029.
Tait SW, Green DR. Mitochondria and cell death: outer membrane permeabilization and beyond. Nat Rev Mol Cell Biol. 2010;11:621–32. doi:nrm2952 [pii]10.1038/nrm2952.
Zou H, Henzel WJ, Liu X, Lutschg A, Wang X. Apaf-1, a human protein homologous to C. elegans CED-4, participates in cytochrome c-dependent activation of caspase-3. Cell. 1997;90:405–13. doi:S0092-8674(00)80501-2 [pii].
Zou H, Li Y, Liu X, Wang X. An APAF-1.cytochrome c multimeric complex is a functional apoptosome that activates procaspase-9. J Biol Chem. 1999;274:11549–56.
Schafer ZT, Kornbluth S. The apoptosome: physiological, developmental, and pathological modes of regulation. Dev Cell. 2006;10:549–61. doi:S1534-5807(06)00169-9 [pii]10.1016/j.devcel.2006.04.008.
Riedl SJ, Salvesen GS. The apoptosome: signalling platform of cell death. Nat Rev Mol Cell Biol. 2007;8:405–13. doi:nrm2153 [pii]10.1038/nrm2153.
Chipuk JE, Moldoveanu T, Llambi F, Parsons MJ, Green DR. The BCL-2 family reunion. Mol Cell. 2010;37:299–310. doi:S1097-2765(10)00079-1 [pii]10.1016/j.molcel.2010.01.025.
Llambi F, et al. A unified model of mammalian BCL-2 protein family interactions at the mitochondria. Mol Cell. 2011;44:517–31. doi:S1097-2765(11)00760-X [pii]10.1016/j.molcel.2011.10.001.
Cheng EH, et al. BCL-2, BCL-X(L) sequester BH3 domain-only molecules preventing BAX- and BAK-mediated mitochondrial apoptosis. Mol Cell. 2001;8:705–11. doi:S1097-2765(01)00320-3 [pii].
Wei MC, et al. Proapoptotic BAX and BAK: a requisite gateway to mitochondrial dysfunction and death. Science. 2001;292:727–30. doi:10.1126/science.1059108292/5517/727 [pii].
Willis SN, et al. Apoptosis initiated when BH3 ligands engage multiple Bcl-2 homologs, not Bax or Bak. Science. 2007;315:856–9. doi:315/5813/856 [pii]10.1126/science.1133289.
Wei MC, et al. tBID, a membrane-targeted death ligand, oligomerizes BAK to release cytochrome c. Genes Dev. 2000;14:2060–71.
Eskes R, Desagher S, Antonsson B, Martinou JC. Bid induces the oligomerization and insertion of Bax into the outer mitochondrial membrane. Mol Cell Biol. 2000;20:929–35.
Kuwana T, et al. BH3 domains of BH3-only proteins differentially regulate Bax-mediated mitochondrial membrane permeabilization both directly and indirectly. Mol Cell. 2005;17:525–35. doi:S1097276505010774 [pii]10.1016/j.molcel.2005.02.003.
Lane DP. Cancer. p53, guardian of the genome. Nature. 1992;358:15–6. doi:10.1038/358015a0.
Levine AJ. p53, the cellular gatekeeper for growth and division. Cell. 1997;88:323–31. doi:S0092-8674(00)81871-1 [pii].
Donehower LA, et al. Mice deficient for p53 are developmentally normal but susceptible to spontaneous tumours. Nature. 1992;356:215–21. doi:10.1038/356215a0.
Lang GA, et al. Gain of function of a p53 hot spot mutation in a mouse model of Li-Fraumeni syndrome. Cell. 2004;119:861–72. doi:S0092867404010487 [pii]10.1016/j.cell.2004.11.006.
Kubbutat MH, Jones SN, Vousden KH. Regulation of p53 stability by Mdm2. Nature. 1997;387:299–303. doi:10.1038/387299a0.
Linares LK, Hengstermann A, Ciechanover A, Muller S, Scheffner M. HdmX stimulates Hdm2-mediated ubiquitination and degradation of p53. Proc Natl Acad Sci U S A. 2003;100:12009–14. doi:10.1073/pnas.2030930100 [pii].
Momand J, Zambetti GP, Olson DC, George D, Levine AJ. The mdm-2 oncogene product forms a complex with the p53 protein and inhibits p53-mediated transactivation. Cell. 1992;69:1237–45. doi:0092-8674(92)90644-R [pii].
Wu X, Bayle JH, Olson D, Levine AJ. The p53-mdm-2 autoregulatory feedback loop. Genes Dev. 1993;7:1126–32.
Vousden KH, Prives C. Blinded by the light: the growing complexity of p53. Cell. 2009;137:413–31. doi:S0092-8674(09)00459-0 [pii]10.1016/j.cell.2009.04.037.
Haupt Y, Rowan S, Shaulian E, Vousden KH, Oren M. Induction of apoptosis in HeLa cells by trans-activation-deficient p53. Genes Dev. 1995;9:2170–83.
Speidel D. Transcription-independent p53 apoptosis: an alternative route to death. Trends Cell Biol. 2010;20:14–24. doi:S0962-8924(09)00240-2 [pii]10.1016/j.tcb.2009.10.002.
Chipuk JE, Maurer U, Green DR, Schuler M. Pharmacologic activation of p53 elicits Bax-dependent apoptosis in the absence of transcription. Cancer Cell. 2003;4:371–81. doi:S1535610803002721 [pii].
Chipuk JE, et al. Direct activation of Bax by p53 mediates mitochondrial membrane permeabilization and apoptosis. Science. 2004;303:1010–4. doi:10.1126/science.1092734303/5660/1010 [pii].
Pietsch EC, et al. The tetramerization domain of p53 is required for efficient BAK oligomerization. Cancer Biol Ther. 2007;6:1576–83. doi:4719 [pii].
Leu JI, Dumont P, Hafey M, Murphy ME, George DL. Mitochondrial p53 activates Bak and causes disruption of a Bak-Mcl1 complex. Nat Cell Biol. 2004;6:443–50. doi:10.1038/ncb1123 [pii].
Petros AM, Gunasekera A, Xu N, Olejniczak ET, Fesik SW. Defining the p53 DNA-binding domain/Bcl-x(L)-binding interface using NMR. FEBS Lett. 2004;559:171–4. doi:10.1016/S0014-5793(04)00059-6S0014579304000596 [pii].
Mihara M, et al. p53 has a direct apoptogenic role at the mitochondria. Mol Cell. 2003;11:577–90. doi:S1097276503000509 [pii].
Schuler M, Green DR. Transcription, apoptosis and p53: catch-22. Trends Genet. 2005;21:182–7. doi:S0168-9525(05)00015-6 [pii]10.1016/j.tig.2005.01.001.
Chipuk JE, Bouchier-Hayes L, Kuwana T, Newmeyer DD, Green DR. PUMA couples the nuclear and cytoplasmic proapoptotic function of p53. Science. 2005;309:1732–5. doi:309/5741/1732 [pii]10.1126/science.1114297.
Joerger AC, Fersht AR. Structural biology of the tumor suppressor p53. Annu Rev Biochem. 2008;77:557–82. doi:10.1146/annurev.biochem.77.060806.091238.
Scolnick DM, et al. CREB-binding protein and p300/CBP-associated factor are transcriptional coactivators of the p53 tumor suppressor protein. Cancer Res. 1997;57:3693–6.
Haupt Y, Maya R, Kazaz A, Oren M. Mdm2 promotes the rapid degradation of p53. Nature. 1997;387:296–9. doi:10.1038/387296a0.
Walker KK, Levine AJ. Identification of a novel p53 functional domain that is necessary for efficient growth suppression. Proc Natl Acad Sci U S A. 1996;93:15335–40.
Cho Y, Gorina S, Jeffrey PD, Pavletich NP. Crystal structure of a p53 tumor suppressor-DNA complex: understanding tumorigenic mutations. Science. 1994;265:346–55.
Wang Y, et al. p53 domains: identification and characterization of two autonomous DNA-binding regions. Genes Dev. 1993;7:2575–86.
Gu W, Roeder RG. Activation of p53 sequence-specific DNA binding by acetylation of the p53 C-terminal domain. Cell. 1997;90:595–606. doi:S0092-8674(00)80521-8 [pii].
Siliciano JD, et al. DNA damage induces phosphorylation of the amino terminus of p53. Genes Dev. 1997;11:3471–81.
Shieh SY, Taya Y, Prives C. DNA damage-inducible phosphorylation of p53 at N-terminal sites including a novel site, Ser20, requires tetramerization. EMBO J. 1999;18:1815–23. doi:10.1093/emboj/18.7.1815.
Ashcroft M, Kubbutat MH, Vousden KH. Regulation of p53 function and stability by phosphorylation. Mol Cell Biol. 1999;19:1751–8.
Tibbetts RS, et al. A role for ATR in the DNA damage-induced phosphorylation of p53. Genes Dev. 1999;13:152–7.
Unger T, et al. Critical role for Ser20 of human p53 in the negative regulation of p53 by Mdm2. EMBO J. 1999;18:1805–14. doi:10.1093/emboj/18.7.1805.
Wu Z, et al. Mutation of mouse p53 Ser23 and the response to DNA damage. Mol Cell Biol. 2002;22:2441–9.
Sluss HK, Armata H, Gallant J, Jones SN. Phosphorylation of serine 18 regulates distinct p53 functions in mice. Mol Cell Biol. 2004;24:976–84.
Chao C, Herr D, Chun J, Xu Y. Ser18 and 23 phosphorylation is required for p53-dependent apoptosis and tumor suppression. EMBO J. 2006;25:2615–22. doi:7601167 [pii]10.1038/sj.emboj.7601167.
Maya R, et al. ATM-dependent phosphorylation of Mdm2 on serine 395: role in p53 activation by DNA damage. Genes Dev. 2001;15:1067–77. doi:10.1101/gad.886901.
Gannon HS, Woda BA, Jones SN. ATM phosphorylation of Mdm2 Ser394 regulates the amplitude and duration of the DNA damage response in mice. Cancer Cell. 2012;21:668–79. doi:S1535-6108(12)00163-8 [pii]10.1016/j.ccr.2012.04.011.
Carter S, Bischof O, Dejean A, Vousden KH. C-terminal modifications regulate MDM2 dissociation and nuclear export of p53. Nat Cell Biol. 2007;9:428–35. doi:ncb1562 [pii]10.1038/ncb1562.
Cheng Q, Chen L, Li Z, Lane WS, Chen J. ATM activates p53 by regulating MDM2 oligomerization and E3 processivity. EMBO J. 2009;28:3857–67. doi:emboj2009294 [pii]10.1038/emboj.2009.294.
Gajjar M, et al. The p53 mRNA-Mdm2 interaction controls Mdm2 nuclear trafficking and is required for p53 activation following DNA damage. Cancer Cell. 2012;21:25–35. doi:S1535-6108(11)00439-9 [pii]10.1016/j.ccr.2011.11.016.
Khoronenkova SV, et al. ATM-dependent downregulation of USP7/HAUSP by PPM1G activates p53 response to DNA damage. Mol Cell. 2012;45:801–13. doi:S1097-2765(12)00087-1 [pii]10.1016/j.molcel.2012.01.021.
Cummins JM, et al. Tumour suppression: disruption of HAUSP gene stabilizes p53. Nature. 2004;428:1 p following 486. doi:10.1038/nature02501.
Meulmeester E, et al. Loss of HAUSP-mediated deubiquitination contributes to DNA damage-induced destabilization of Hdmx and Hdm2. Mol Cell. 2005;18:565–76. doi:S1097-2765(05)01287-6 [pii]10.1016/j.molcel.2005.04.024.
Chen L, Gilkes DM, Pan Y, Lane WS, Chen J. ATM and Chk2-dependent phosphorylation of MDMX contribute to p53 activation after DNA damage. EMBO J. 2005;24:3411–22. doi:7600812 [pii]10.1038/sj.emboj.7600812.
Okamoto K, et al. DNA damage-induced phosphorylation of MdmX at serine 367 activates p53 by targeting MdmX for Mdm2-dependent degradation. Mol Cell Biol. 2005;25:9608–20. doi:25/21/9608 [pii]10.1128/MCB.25.21.9608-9620.2005.
Dornan D, et al. ATM engages autodegradation of the E3 ubiquitin ligase COP1 after DNA damage. Science. 2006;313:1122–6. doi:313/5790/1122 [pii]10.1126/science.1127335.
Leng RP, et al. Pirh2, a p53-induced ubiquitin-protein ligase, promotes p53 degradation. Cell. 2003;112:779–91. doi:S0092867403001934 [pii].
Chen D, et al. ARF-BP1/Mule is a critical mediator of the ARF tumor suppressor. Cell. 2005;121:1071–83. doi:S0092-8674(05)00356-9 [pii]10.1016/j.cell.2005.03.037.
Krummel KA, Lee CJ, Toledo F, Wahl GM. The C-terminal lysines fine-tune P53 stress responses in a mouse model but are not required for stability control or transactivation. Proc Natl Acad Sci U S A. 2005;102:10188–93. doi:0503068102 [pii]10.1073/pnas.0503068102.
Feng L, Lin T, Uranishi H, Gu W, Xu Y. Functional analysis of the roles of posttranslational modifications at the p53 C terminus in regulating p53 stability and activity. Mol Cell Biol. 2005;25:5389–95. doi:25/13/5389 [pii]10.1128/MCB.25.13.5389-5395.2005.
Sykes SM, et al. Acetylation of the p53 DNA-binding domain regulates apoptosis induction. Mol Cell. 2006;24:841–51. doi:S1097-2765(06)00821-5 [pii]10.1016/j.molcel.2006.11.026.
Tang Y, Zhao W, Chen Y, Zhao Y, Gu W. Acetylation is indispensable for p53 activation. Cell. 2008;133:612–26. doi:S0092-8674(08)00441-8 [pii]10.1016/j.cell.2008.03.025.
Brooks CL, Gu W. New insights into p53 activation. Cell Res. 2010;20:614–21. doi:cr201053 [pii]10.1038/cr.2010.53.
Montes de Oca Luna R, Wagner DS, Lozano G. Rescue of early embryonic lethality in mdm2-deficient mice by deletion of p53. Nature. 1995;378:203–6. doi:10.1038/378203a0.
Jones SN, Roe AE, Donehower LA, Bradley A. Rescue of embryonic lethality in Mdm2-deficient mice by absence of p53. Nature. 1995;378:206–8. doi:10.1038/378206a0.
Parant J, et al. Rescue of embryonic lethality in Mdm4-null mice by loss of Trp53 suggests a nonoverlapping pathway with MDM2 to regulate p53. Nat Genet. 2001;29:92–5. doi:10.1038/ng714 [pii].
Johnson TM, Hammond EM, Giaccia A, Attardi LD. The p53QS transactivation-deficient mutant shows stress-specific apoptotic activity and induces embryonic lethality. Nat Genet. 2005;37:145–52. doi:ng1498 [pii]10.1038/ng1498.
Li M, et al. Mono-versus polyubiquitination: differential control of p53 fate by Mdm2. Science. 2003;302:1972–5. doi:10.1126/science.1091362302/5652/1972 [pii].
Marchenko ND, Wolff S, Erster S, Becker K, Moll UM. Monoubiquitylation promotes mitochondrial p53 translocation. EMBO J. 2007;26:923–34. doi:7601560 [pii]10.1038/sj.emboj.7601560.
Kruse JP, Gu W. MSL2 promotes Mdm2-independent cytoplasmic localization of p53. J Biol Chem. 2009;284:3250–63. doi:M805658200 [pii]10.1074/jbc.M805658200.
Ahn BY, et al. Tid1 is a new regulator of p53 mitochondrial translocation and apoptosis in cancer. Oncogene. 2010;29:1155–66. doi:onc2009413 [pii]10.1038/onc.2009.413.
Li M, et al. Deubiquitination of p53 by HAUSP is an important pathway for p53 stabilization. Nature. 2002;416:648–53. doi:10.1038/nature737 [pii].
Muscolini M, et al. The cancer-associated K351N mutation affects the ubiquitination and the translocation to mitochondria of p53 protein. J Biol Chem. 2011;286:39693–702. doi:M111.279539 [pii]10.1074/jbc.M111.279539.
Sykes SM, Stanek TJ, Frank A, Murphy ME, McMahon SB. Acetylation of the DNA binding domain regulates transcription-independent apoptosis by p53. J Biol Chem. 2009;284:20197–205. doi:M109.026096 [pii]10.1074/jbc.M109.026096.
Yamaguchi H, et al. p53 acetylation is crucial for its transcription-independent proapoptotic functions. J Biol Chem. 2009;284:11171–83. doi:M809268200 [pii]10.1074/jbc.M809268200.
Pietsch EC, Sykes SM, McMahon SB, Murphy ME. The p53 family and programmed cell death. Oncogene. 2008;27:6507–21. doi:onc2008315 [pii]10.1038/onc.2008.315.
Murray-Zmijewski F, Lane DP, Bourdon JC. p53/p63/p73 isoforms: an orchestra of isoforms to harmonise cell differentiation and response to stress. Cell Death Differ. 2006;13:962–72. doi:4401914 [pii]10.1038/sj.cdd.4401914.
Pozniak CD, et al. An anti-apoptotic role for the p53 family member, p73, during developmental neuron death. Science. 2000;289:304–6.
Okada Y, et al. p53 gene family p51(p63)-encoded, secondary transactivator p51B(TAp63alpha) occurs without forming an immunoprecipitable complex with MDM2, but responds to genotoxic stress by accumulation. Exp Cell Res. 2002;276:194–200. doi:10.1006/excr.2002.5535S0014482702955357 [pii].
Katoh I, Aisaki KI, Kurata SI, Ikawa S, Ikawa Y. p51A (TAp63gamma), a p53 homolog, accumulates in response to DNA damage for cell regulation. Oncogene. 2000;19:3126–30. doi:10.1038/sj.onc.1203644.
Urist M, Tanaka T, Poyurovsky MV, Prives C. p73 induction after DNA damage is regulated by checkpoint kinases Chk1 and Chk2. Genes Dev. 2004;18:3041–54. doi:18/24/3041 [pii]10.1101/gad.1221004.
Gressner O, et al. TAp63alpha induces apoptosis by activating signaling via death receptors and mitochondria. EMBO J. 2005;24:2458–71. doi:7600708 [pii]10.1038/sj.emboj.7600708.
Zhu J, Jiang J, Zhou W, Chen X. The potential tumor suppressor p73 differentially regulates cellular p53 target genes. Cancer Res. 1998;58:5061–5.
Melino G, et al. p73 Induces apoptosis via PUMA transactivation and Bax mitochondrial translocation. J Biol Chem. 2004;279:8076–83. doi:10.1074/jbc.M307469200 [pii].
Sayan AE, et al. P73 and caspase-cleaved p73 fragments localize to mitochondria and augment TRAIL-induced apoptosis. Oncogene. 2008;27:4363–72. doi:onc200864 [pii]10.1038/onc.2008.64.
John K, Alla V, Meier C, Putzer BM. GRAMD4 mimics p53 and mediates the apoptotic function of p73 at mitochondria. Cell Death Differ. 2011;18:874–86. doi:cdd2010153 [pii]10.1038/cdd.2010.153.
Suh EK, et al. p63 protects the female germ line during meiotic arrest. Nature. 2006;444:624–8. doi:nature05337 [pii]10.1038/nature05337.
Kerr JB, et al. DNA damage-induced primordial follicle oocyte apoptosis and loss of fertility require TAp63-mediated induction of Puma and Noxa. Mol Cell. 2012;48:343–52. doi:S1097-2765(12)00735-6 [pii]10.1016/j.molcel.2012.08.017.
Flores ER, et al. Tumor predisposition in mice mutant for p63 and p73: evidence for broader tumor suppressor functions for the p53 family. Cancer Cell. 2005;7:363–73. doi:S1535-6108(05)00092-9 [pii]10.1016/j.ccr.2005.02.019.
Tomasini R, et al. TAp73 knockout shows genomic instability with infertility and tumor suppressor functions. Genes Dev. 2008;22:2677–91. doi:gad.1695308 [pii]10.1101/gad.1695308.
Deyoung MP, Ellisen LW. p63 and p73 in human cancer: defining the network. Oncogene. 2007;26:5169–83. doi:1210337 [pii]10.1038/sj.onc.1210337.
Karin M, Lin A. NF-kappaB at the crossroads of life and death. Nat Immunol. 2002;3:221–7. doi:10.1038/ni0302-221 [pii].
Rosette C, Karin M. Ultraviolet light and osmotic stress: activation of the JNK cascade through multiple growth factor and cytokine receptors. Science. 1996;274:1194–7.
McCool KW, Miyamoto S. DNA damage-dependent NF-kappaB activation: NEMO turns nuclear signaling inside out. Immunol Rev. 2012;246:311–26. doi:10.1111/j.1600-065X.2012.01101.x.
Miyamoto S. Nuclear initiated NF-kappaB signaling: NEMO and ATM take center stage. Cell Res. 2011;21:116–30. doi:cr2010179 [pii]10.1038/cr.2010.179.
Hayden MS, Ghosh S. Shared principles in NF-kappaB signaling. Cell. 2008;132:344–62. doi:S0092-8674(08)00120-7 [pii]10.1016/j.cell.2008.01.020.
Pahl HL. Activators and target genes of Rel/NF-kappaB transcription factors. Oncogene. 1999;18:6853–66. doi:10.1038/sj.onc.1203239.
Hayden MS, Ghosh S. Signaling to NF-kappaB. Genes Dev. 2004;18:2195–224. doi:10.1101/gad.122870418/18/2195 [pii].
Huang TT, Wuerzberger-Davis SM, Wu ZH, Miyamoto S. Sequential modification of NEMO/IKKgamma by SUMO-1 and ubiquitin mediates NF-kappaB activation by genotoxic stress. Cell. 2003;115:565–76. doi:S009286740300895X [pii].
Mabb AM, Wuerzberger-Davis SM, Miyamoto S. PIASy mediates NEMO sumoylation and NF-kappaB activation in response to genotoxic stress. Nat Cell Biol. 2006;8:986–93. doi:ncb1458 [pii]10.1038/ncb1458.
Wu ZH, Shi Y, Tibbetts RS, Miyamoto S. Molecular linkage between the kinase ATM and NF-kappaB signaling in response to genotoxic stimuli. Science. 2006;311:1141–6. doi:311/5764/1141 [pii]10.1126/science.1121513.
Jin HS, et al. cIAP1, cIAP2, and XIAP act cooperatively via nonredundant pathways to regulate genotoxic stress-induced nuclear factor-kappaB activation. Cancer Res. 2009;69:1782–91. doi:0008-5472.CAN-08-2256 [pii]10.1158/0008-5472.CAN-08-2256.
Hinz M, et al. A cytoplasmic ATM-TRAF6-cIAP1 module links nuclear DNA damage signaling to ubiquitin-mediated NF-kappaB activation. Mol Cell. 2010;40:63–74. doi:S1097-2765(10)00710-0 [pii]10.1016/j.molcel.2010.09.008.
Wu ZH, et al. ATM- and NEMO-dependent ELKS ubiquitination coordinates TAK1-mediated IKK activation in response to genotoxic stress. Mol Cell. 2010;40:75–86. doi:S1097-2765(10)00712-4 [pii]10.1016/j.molcel.2010.09.010.
Janssens S, Tschopp J. Signals from within: the DNA-damage-induced NF-kappaB response. Cell Death Differ. 2006;13:773–84. doi:4401843 [pii]10.1038/sj.cdd.4401843.
Kasibhatla S, et al. DNA damaging agents induce expression of Fas ligand and subsequent apoptosis in T lymphocytes via the activation of NF-kappa B and AP-1. Mol Cell. 1998;1:543–51. doi:S1097-2765(00)80054-4 [pii].
Micheau O, Tschopp J. Induction of TNF receptor I-mediated apoptosis via two sequential signaling complexes. Cell. 2003;114:181–90. doi:S009286740300521X [pii].
Biton S, Ashkenazi A. NEMO and RIP1 control cell fate in response to extensive DNA damage via TNF-alpha feedforward signaling. Cell. 2011;145:92–103. doi:S0092-8674(11)00175-9 [pii]10.1016/j.cell.2011.02.023.
Janssens S, Tinel A, Lippens S, Tschopp J. PIDD mediates NF-kappaB activation in response to DNA damage. Cell. 2005;123:1079–92. doi:S0092-8674(05)01042-1 [pii]10.1016/j.cell.2005.09.036.
Tinel A, Tschopp J. The PIDDosome, a protein complex implicated in activation of caspase-2 in response to genotoxic stress. Science. 2004;304:843–6. doi:10.1126/science.1095432 [pii].
Dorstyn L, et al. Caspase-2 deficiency promotes aberrant DNA-damage response and genetic instability. Cell Death Differ. 2012;19:1288–98. doi:cdd201236 [pii]10.1038/cdd.2012.36.
Yang Y, Fang S, Jensen JP, Weissman AM, Ashwell JD. Ubiquitin protein ligase activity of IAPs and their degradation in proteasomes in response to apoptotic stimuli. Science. 2000;288:874–7. doi:8502 [pii].
Tenev T, et al. The Ripoptosome, a signaling platform that assembles in response to genotoxic stress and loss of IAPs. Mol Cell. 2011;43:432–48. doi:S1097-2765(11)00420-5 [pii]10.1016/j.molcel.2011.06.006.
Ganten TM, et al. Enhanced caspase-8 recruitment to and activation at the DISC is critical for sensitisation of human hepatocellular carcinoma cells to TRAIL-induced apoptosis by chemotherapeutic drugs. Cell Death Differ. 2004;11 Suppl 1:S86–96. doi:10.1038/sj.cdd.4401437 [pii].
Pop C, et al. FLIP(L) induces caspase 8 activity in the absence of interdomain caspase 8 cleavage and alters substrate specificity. Biochem J. 2011;433:447–57. doi:BJ20101738 [pii]10.1042/BJ20101738.
Oberst A, et al. Catalytic activity of the caspase-8-FLIP(L) complex inhibits RIPK3-dependent necrosis. Nature. 2011;471:363–7. doi:nature09852 [pii]10.1038/nature09852.
Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell. 2011;144:646–74. doi:S0092-8674(11)00127-9 [pii]10.1016/j.cell.2011.02.013.
Schmitt CA, et al. Dissecting p53 tumor suppressor functions in vivo. Cancer Cell. 2002;1:289–98. doi:S1535610802000478 [pii].
Junttila MR, Evan GI. p53—a Jack of all trades but master of none. Nat Rev Cancer. 2009;9:821–9. doi:nrc2728 [pii]10.1038/nrc2728.
Bartkova J, et al. DNA damage response as a candidate anti-cancer barrier in early human tumorigenesis. Nature. 2005;434:864–70. doi:nature03482 [pii]10.1038/nature03482.
Bartkova J, et al. Oncogene-induced senescence is part of the tumorigenesis barrier imposed by DNA damage checkpoints. Nature. 2006;444:633–7. doi:nature05268 [pii]10.1038/nature05268.
Gorgoulis VG, et al. Activation of the DNA damage checkpoint and genomic instability in human precancerous lesions. Nature. 2005;434:907–13. doi:nature03485 [pii]10.1038/nature03485.
Dominguez-Sola D, et al. Non-transcriptional control of DNA replication by c-Myc. Nature. 2007;448:445–51. doi:nature05953 [pii]10.1038/nature05953.
Casper AM, Nghiem P, Arlt MF, Glover TW. ATR regulates fragile site stability. Cell. 2002;111:779–89. doi:S0092867402011133 [pii].
Di Micco R, et al. Oncogene-induced senescence is a DNA damage response triggered by DNA hyper-replication. Nature. 2006;444:638–42. doi:nature05327 [pii]10.1038/nature05327.
d’Adda di Fagagna F, et al. A DNA damage checkpoint response in telomere-initiated senescence. Nature. 2003;426:194–8. doi:10.1038/nature02118 [pii].
Martinez P, Blasco MA. Telomeric and extra-telomeric roles for telomerase and the telomere-binding proteins. Nat Rev Cancer. 2011;11:161–76. doi:nrc3025 [pii]10.1038/nrc3025.
Maser RS, DePinho RA. Connecting chromosomes, crisis, and cancer. Science. 2002;297:565–9. doi:10.1126/science.297.5581.565297/5581/565 [pii].
Sfeir A, de Lange T. Removal of shelterin reveals the telomere end-protection problem. Science. 2012;336:593–7. doi:336/6081/593 [pii]10.1126/science.1218498.
O’Driscoll M, Jeggo PA. The role of double-strand break repair—insights from human genetics. Nat Rev Genet. 2006;7:45–54. doi:nrg1746 [pii]10.1038/nrg1746.
O’Driscoll M, et al. DNA ligase IV mutations identified in patients exhibiting developmental delay and immunodeficiency. Mol Cell. 2001;8:1175–85. doi:S1097-2765(01)00408-7 [pii].
Wang W. Emergence of a DNA-damage response network consisting of Fanconi anaemia and BRCA proteins. Nat Rev Genet. 2007;8:735–48. doi:nrg2159 [pii]10.1038/nrg2159.
Savitsky K, et al. A single ataxia telangiectasia gene with a product similar to PI-3 kinase. Science. 1995;268:1749–53.
Bell DW, et al. Heterozygous germ line hCHK2 mutations in Li-Fraumeni syndrome. Science. 1999;286:2528–31. doi:8128 [pii].
Stewart GS, et al. The DNA double-strand break repair gene hMRE11 is mutated in individuals with an ataxia-telangiectasia-like disorder. Cell. 1999;99:577–87. doi:S0092-8674(00)81547-0 [pii].
Carney JP, et al. The hMre11/hRad50 protein complex and Nijmegen breakage syndrome: linkage of double-strand break repair to the cellular DNA damage response. Cell. 1998;93:477–86. doi:S0092-8674(00)81175-7 [pii].
Miki Y, et al. A strong candidate for the breast and ovarian cancer susceptibility gene BRCA1. Science. 1994;266:66–71.
Wooster R, et al. Identification of the breast cancer susceptibility gene BRCA2. Nature. 1995;378:789–92. doi:10.1038/378789a0.
Srivastava S, Zou ZQ, Pirollo K, Blattner W, Chang EH. Germ-line transmission of a mutated p53 gene in a cancer-prone family with Li-Fraumeni syndrome. Nature. 1990;348:747–9. doi:10.1038/348747a0.
O’Driscoll M, Ruiz-Perez VL, Woods CG, Jeggo PA, Goodship JA. A splicing mutation affecting expression of ataxia-telangiectasia and Rad3-related protein (ATR) results in Seckel syndrome. Nat Genet. 2003;33:497–501. doi:10.1038/ng1129 [pii].
Barlow C, et al. Atm-deficient mice: a paradigm of ataxia telangiectasia. Cell. 1996;86:159–71. doi:S0092-8674(00)80086-0 [pii].
Bassing CH, et al. Histone H2AX: a dosage-dependent suppressor of oncogenic translocations and tumors. Cell. 2003;114:359–70. doi:S009286740300566X [pii].
DiTullio Jr RA, et al. 53BP1 functions in an ATM-dependent checkpoint pathway that is constitutively activated in human cancer. Nat Cell Biol. 2002;4:998–1002. doi:10.1038/ncb892 [pii].
Bouwman P, Jonkers J. The effects of deregulated DNA damage signalling on cancer chemotherapy response and resistance. Nat Rev Cancer. 2012;12:587–98. doi:nrc3342 [pii]10.1038/nrc3342.
Helleday T, Petermann E, Lundin C, Hodgson B, Sharma RA. DNA repair pathways as targets for cancer therapy. Nat Rev Cancer. 2008;8:193–204. doi:nrc2342 [pii]10.1038/nrc2342.
Martin SA, Lord CJ, Ashworth A. DNA repair deficiency as a therapeutic target in cancer. Curr Opin Genet Dev. 2008;18:80–6. doi:S0959-437X(08)00021-X [pii]10.1016/j.gde.2008.01.016.
Letai AG. Diagnosing and exploiting cancer’s addiction to blocks in apoptosis. Nat Rev Cancer. 2008;8:121–32. doi:nrc2297 [pii]10.1038/nrc2297.
Vaux DL, Cory S, Adams JM. Bcl-2 gene promotes haemopoietic cell survival and cooperates with c-myc to immortalize pre-B cells. Nature. 1988;335:440–2. doi:10.1038/335440a0.
Certo M, et al. Mitochondria primed by death signals determine cellular addiction to antiapoptotic BCL-2 family members. Cancer Cell. 2006;9:351–65. doi:S1535-6108(06)00113-9 [pii]10.1016/j.ccr.2006.03.027.
Del Gaizo Moore V, et al. Chronic lymphocytic leukemia requires BCL2 to sequester prodeath BIM, explaining sensitivity to BCL2 antagonist ABT-737. J Clin Invest. 2007;117:112–21. doi:10.1172/JCI28281.
Ni Chonghaile T, et al. Pretreatment mitochondrial priming correlates with clinical response to cytotoxic chemotherapy. Science. 2011;334:1129–33. doi:science.1206727 [pii]10.1126/science.1206727.
Kastan MB, et al. A mammalian cell cycle checkpoint pathway utilizing p53 and GADD45 is defective in ataxia-telangiectasia. Cell. 1992;71:587–97. doi:0092-8674(92)90593-2 [pii].
Armata HL, Garlick DS, Sluss HK. The ataxia telangiectasia-mutated target site Ser18 is required for p53-mediated tumor suppression. Cancer Res. 2007;67:11696–703. doi:67/24/11696 [pii]10.1158/0008-5472.CAN-07-1610.
Quelle DE, Zindy F, Ashmun RA, Sherr CJ. Alternative reading frames of the INK4a tumor suppressor gene encode two unrelated proteins capable of inducing cell cycle arrest. Cell. 1995;83:993–1000. doi:0092-8674(95)90214-7 [pii].
Weber JD, Taylor LJ, Roussel MF, Sherr CJ, Bar-Sagi D. Nucleolar Arf sequesters Mdm2 and activates p53. Nat Cell Biol. 1999;1:20–6. doi:10.1038/8991.
Kamijo T, et al. Tumor suppression at the mouse INK4a locus mediated by the alternative reading frame product p19ARF. Cell. 1997;91:649–59. doi:S0092-8674(00)80452-3 [pii].
Eischen CM, Weber JD, Roussel MF, Sherr CJ, Cleveland JL. Disruption of the ARF-Mdm2-p53 tumor suppressor pathway in Myc-induced lymphomagenesis. Genes Dev. 1999;13:2658–69.
Westphal CH, et al. atm and p53 cooperate in apoptosis and suppression of tumorigenesis, but not in resistance to acute radiation toxicity. Nat Genet. 1997;16:397–401. doi:10.1038/ng0897-397.
Martins CP, Brown-Swigart L, Evan GI. Modeling the therapeutic efficacy of p53 restoration in tumors. Cell. 2006;127:1323–34. doi:S0092-8674(06)01597-2 [pii]10.1016/j.cell.2006.12.007.
Christophorou MA, Ringshausen I, Finch AJ, Swigart LB, Evan GI. The pathological response to DNA damage does not contribute to p53-mediated tumour suppression. Nature. 2006;443:214–7. doi:nature05077 [pii]10.1038/nature05077.
Garcia-Cao I, et al. “Super p53” mice exhibit enhanced DNA damage response, are tumor resistant and age normally. EMBO J. 2002;21:6225–35.
Khan SH, Moritsugu J, Wahl GM. Differential requirement for p19ARF in the p53-dependent arrest induced by DNA damage, microtubule disruption, and ribonucleotide depletion. Proc Natl Acad Sci U S A. 2000;97:3266–71. doi:10.1073/pnas.050560997 [pii].
Jeffers JR, et al. Puma is an essential mediator of p53-dependent and -independent apoptotic pathways. Cancer Cell. 2003;4:321–8. doi:S1535610803002447 [pii].
Villunger A, et al. p53- and drug-induced apoptotic responses mediated by BH3-only proteins puma and noxa. Science. 2003;302:1036–8. doi:10.1126/science.1090072 [pii].
Garrison SP, et al. Selection against PUMA gene expression in Myc-driven B-cell lymphomagenesis. Mol Cell Biol. 2008;28:5391–402. doi:MCB.00907-07 [pii]10.1128/MCB.00907-07.
Michalak EM, et al. Puma and to a lesser extent Noxa are suppressors of Myc-induced lymphomagenesis. Cell Death Differ. 2009;16:684–96. doi:cdd2008195 [pii]10.1038/cdd.2008.195.
Michalak EM, Villunger A, Adams JM, Strasser A. In several cell types tumour suppressor p53 induces apoptosis largely via Puma but Noxa can contribute. Cell Death Differ. 2008;15:1019–29. doi:cdd200816 [pii]10.1038/cdd.2008.16.
Finnberg N, et al. DR5 knockout mice are compromised in radiation-induced apoptosis. Mol Cell Biol. 2005;25:2000–13. doi:25/5/2000 [pii]10.1128/MCB.25.5.2000-2013.2005.
Chen L, et al. CD95 promotes tumour growth. Nature. 2010;465:492–6. doi:nature09075 [pii]10.1038/nature09075.
Deng C, Zhang P, Harper JW, Elledge SJ, Leder P. Mice lacking p21CIP1/WAF1 undergo normal development, but are defective in G1 checkpoint control. Cell. 1995;82:675–84. doi:0092-8674(95)90039-X [pii].
Jackson RJ, et al. p21Cip1 nullizygosity increases tumor metastasis in irradiated mice. Cancer Res. 2003;63:3021–5.
Brady CA, et al. Distinct p53 transcriptional programs dictate acute DNA-damage responses and tumor suppression. Cell. 2011;145:571–83. doi:S0092-8674(11)00312-6 [pii]10.1016/j.cell.2011.03.035.
Jiang D, et al. Full p53 transcriptional activation potential is dispensable for tumor suppression in diverse lineages. Proc Natl Acad Sci U S A. 2011;108:17123–8. doi:1111245108 [pii]10.1073/pnas.1111245108.
Li T, et al. Tumor suppression in the absence of p53-mediated cell-cycle arrest, apoptosis, and senescence. Cell. 2012;149:1269–83. doi:S0092-8674(12)00533-8 [pii]10.1016/j.cell.2012.04.026.
Gottlieb E, Vousden KH. p53 regulation of metabolic pathways. Cold Spring Harb Perspect Biol. 2010;2:a001040. doi:cshperspect.a001040 [pii]10.1101/cshperspect.a001040.
Ventura A, et al. Restoration of p53 function leads to tumour regression in vivo. Nature. 2007;445:661–5. doi:nature05541 [pii]10.1038/nature05541.
Green DR. Cell competition: pirates on the tangled bank. Cell Stem Cell. 2010;6:287–8. doi:S1934-5909(10)00103-7 [pii]10.1016/j.stem.2010.03.006.
Bondar T, Medzhitov R. p53-mediated hematopoietic stem and progenitor cell competition. Cell Stem Cell. 2010;6:309–22. doi:S1934-5909(10)00099-8 [pii]10.1016/j.stem.2010.03.002.
Marusyk A, Porter CC, Zaberezhnyy V, DeGregori J. Irradiation selects for p53-deficient hematopoietic progenitors. PLoS Biol. 2010;8:e1000324. doi:10.1371/journal.pbio.1000324.
Sperka T, Wang J, Rudolph KL. DNA damage checkpoints in stem cells, ageing and cancer. Nat Rev Mol Cell Biol. 2012;13:579–90. doi:nrm3420 [pii]10.1038/nrm3420.
Zhao Z, et al. p53 loss promotes acute myeloid leukemia by enabling aberrant self-renewal. Genes Dev. 2010;24:1389–402. doi:24/13/1389 [pii]10.1101/gad.1940710.
Cicalese A, et al. The tumor suppressor p53 regulates polarity of self-renewing divisions in mammary stem cells. Cell. 2009;138:1083–95. doi:S0092-8674(09)00840-X [pii]10.1016/j.cell.2009.06.048.
Maryanovich M, et al. The ATM-BID pathway regulates quiescence and survival of haematopoietic stem cells. Nat Cell Biol. 2012;14:535–41. doi:ncb2468 [pii]10.1038/ncb2468.
Zinkel SS, et al. A role for proapoptotic BID in the DNA-damage response. Cell. 2005;122:579–91. doi:S0092-8674(05)00641-0 [pii]10.1016/j.cell.2005.06.022.
Kamer I, et al. Proapoptotic BID is an ATM effector in the DNA-damage response. Cell. 2005;122:593–603. doi:S0092-8674(05)00597-0 [pii]10.1016/j.cell.2005.06.014.
Ito K, et al. Regulation of oxidative stress by ATM is required for self-renewal of haematopoietic stem cells. Nature. 2004;431:997–1002. doi:nature02989 [pii]10.1038/nature02989.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2014 Springer Science+Business Media New York
About this chapter
Cite this chapter
Baran, K., Rodriguez, D., Green, D. (2014). The DNA Damage Response Mediates Apoptosis and Tumor Suppression. In: Wu, H. (eds) Cell Death. Springer, New York, NY. https://doi.org/10.1007/978-1-4614-9302-0_7
Download citation
DOI: https://doi.org/10.1007/978-1-4614-9302-0_7
Published:
Publisher Name: Springer, New York, NY
Print ISBN: 978-1-4614-9301-3
Online ISBN: 978-1-4614-9302-0
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)