
Abstract
The development and optimization of CNS drug is hampered by the inaccessibility of the human brain and the difficulty to quantify human CNS drug effects. The use of serial CSF sampling in animals and mathematical modeling of plasma pharmacokinetics, in conjunction with CNS effects, provided only useful information for drugs that distribute to the brain target site by simple diffusion and having direct and reversible CNS effects. Active transport processes across blood–brain barriers and brain cell membranes may be applicable for many drugs and should be taken into account. Also, context dependencies of the rates and extents of all transport processes should be included. This indicates the need for cross-compare designed preclinical experimental approaches and mathematical modeling to provide information on contributions of the (main) individual processes, in terms of rate and extent, as well as their interplay, to be able to predict human CNS drug effects.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others


Keywords
- Intranasal Administration
- PKPD Model
- Target Occupancy
- Prolactin Plasma Concentration
- Total Brain Concentration
These keywords were added by machine and not by the authors. This process is experimental and the keywords may be updated as the learning algorithm improves.
1 Introduction
Despite enormous advances in CNS research, CNS disorders remain the world’s leading cause of disability and account for more hospitalizations and prolonged care than almost all other diseases combined. This indicates a high unmet need for good CNS drugs and drug therapies. For a proper CNS effect the drug should have the ability to access the CNS “at the right place, at the right time, and at the right concentration.” To that end a number of key issues need to be considered.
-
Only the unbound drug is able to pass the BBB and to interact with its target to drive the effect (Urien et al. 1987; Jolliet et al. 1997; Tanaka and Mizojiri 1999; Liu et al. 2005; Hammarlund-Udenaes et al. 2008; Hammarlund-Udenaes 2009; Stevens et al. 2012).
-
Transport across the blood–brain barrier (BBB) needs to take place for adequate drug delivery to the CNS. It is of great importance to understand the mechanisms involved in uptake into and efflux from the brain, on one hand being governed by BBB functionality (in terms of passive paracellular and transcellular diffusion), facilitated diffusion, active influx, active efflux, and absorptive or receptor-mediated endocytosis and on the other hand by drug physicochemical properties and structure (Mayer et al. 1959; Oldendorf 1974; Betz and Goldstein 1986; Suzuki et al. 1997; Kalvass and Maurer 2002; Danhof et al. 2005, 2007; Westerhout et al. 2011).
-
Not only BBB transport is of importance but also plasma pharmacokinetics and intrabrain distribution, the latter indicating spatial and temporal exchange of a drug between brain ECF, brain cells, and CSF (De Lange et al. 1995c; Kalvass and Maurer 2002; Liu et al. 2005; Westerhout et al. 2011, 2012).
-
Mechanisms that underlie BBB functionality and brain tissue characteristics all have their specific rate and extent, being dynamically regulated. Therefore, heterogeneity in species, gender, genetic background, tissue, age, diet, disease conditions, drug treatment, etc., (Letrent et al. 1999; Karssen et al. 2001; Kooij et al. 2010; De Lange et al. 2005; Mulder et al. 2001; Danhof et al. 2007; Ravenstijn et al. 2007, 2012; Syvänen et al. 2009; Westerhout et al. 2011, 2012) contributes to context-dependent variability in CNS target site PK.
-
Then, not only CNS target site distribution is context dependent so is the observed effect or the biomarker(s) of the effect. Context-dependent PKPD relationships of CNS drugs most of all underlies the relative high failure of CNS drug candidates. Therefore the link between target concentration and CNS response should preferentially be obtained within the same subject (De Lange 2013a).
-
Information on time-dependency is crucial (De Lange et al. 2005; Hammarlund-Udenaes et al. 2008).
The inaccessibility of the human brain for sampling hampers obtaining relevant human target site concentrations of CNS drugs, while also it is often difficult to quantify human CNS drug effects. This indicates that CNS drug distribution and effects in humans should be predicted by other measures.
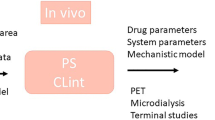
To decipher and learn more on the factors that govern plasma pharmacokinetics, BBB transport intrabrain distribution, as well as their interrelationships and consequences for CNS effects in the different settings, systematic preclinical research on CNS drugs will be of help. To that end, investigations should be performed such that variables are systematically varied (e.g., inhibition of an efflux transporter, or induction of pathological state) in which time-dependency is explicitly included. As our brains do not have the capacity to integrate all these data and determine contributions of individual mechanisms in PKPD relationships, we need to organize, condense, and store knowledge in mathematical frameworks, by the use of advanced mathematical modeling. This provides the links to the human situation.
This chapter deals with more classical, current, and future approaches to PKPD aspects of brain drug delivery in a translational perspective.
2 History
2.1 CSF Concentrations to Predict CNS Target Site Concentration
For a long time monitoring approaches have been searched for to obtain information that could be used to predict human target site kinetics and CNS effects. As it is the free drug that is available for target binding, in the early 1980s it was anticipated that the drug concentration in the cerebrospinal fluid (CSF) could serve as a biomarker of the free brain target site concentrations, because in CSF, at least under physiological conditions, no binding of drugs to proteins occurs (Bonati et al. 1984).
A step forward was then made by the development of the methodology of serial CSF sampling. Experiments were designed to obtain the time-course of concentrations in relation to parallelly obtained CNS drug effects, and many samples could be obtained from single animals providing the possibility of within-subject cross-over designed studies, and minimizing the number of animals to be used. Serial CSF sampling in conjunction with measuring CNS effects was applied for pento- and heptabarbital, ethanol, and pentylenetetrazole, and by varying the rate and duration of an intravenous infusion of a single dose, it could be clearly demonstrated that the CSF compartment was pharmacokinetically indistinguishable from the site of action of this drug (Danhof and Levy 1984; Dingemanse et al. 1988; Ramzan and Levy 1986). On that basis CSF concentrations were considered to be of key value for studying PKPD relationships of CNS active drugs, and methodologies for sequential CSF sampling in human became available (Bruce and Oldfield 1988).
2.2 Predictions of CNS Drug Response by Compartmental PKPD Modeling
In the early 1990s, as an alternative to serial CSF sampling, mathematical modeling techniques were developed to describe the effect–time course of a CNS drug on the basis of its plasma pharmacokinetics (Campbell 1990). Concentration–response profiles often have a sigmoidal shape when the percentage of the maximal response (Emax) is plotted against the logarithm of the drug concentration (Fig. 9.1). Therefore, the sigmoid Emax model is (still) most generally used to fit a plasma concentration–effect profiles to provide estimates of EC50 and Emax values of drugs. The sigmoid Emax equation (9.1) is the following:

Plasma concentration–effect profiles often have a sigmoidal shape when the percentage of the maximal response (Emax) of a drug (agonist) is plotted against the logarithm of the drug concentration
With E0, the baseline response, E the response observed for a given drug concentration [A] at time t, Emax, the maximal effect of the drug, EC50 is the plasma concentration of the drug that produces 50 % of Emax, and h, the Hill coefficient, which determines the steepness of the concentration–effect relationship. The EC50 (“potency”) is simply the concentration of agonist required to provoke a response halfway between the baseline and maximum responses. It is usually not the same as the dissociation equilibrium constant (KA) for the binding of agonist to its receptor. If drug concentrations at the target site are in equilibrium with those in plasma (site of measurement) and drug effects are direct and in case the effect is (assumed to be) direct and reversible this is a very useful approach, especially as it can also be applied to humans.
However, when the drug concentration in plasma is not in equilibrium with its site of action, hysteresis occurs and drug levels are out of phase with activity. So-called counter-clockwise hysteresis (Fig. 9.2) is observed when the effect increases with time for a given drug concentration in plasma. Such situation can be caused by pharmacokinetic processes such as slow diffusion of the drug towards the target site taking time, active influx of the drug towards the target site, formation of active agonistic metabolites, but also by pharmacodynamic processes like relatively slow signal transduction processes and sensitization. Clockwise hysteresis, in which the effect decreases with time for a given drug concentration, can be caused by tolerance, active antagonistic metabolites, learning effects, and feedback regulation.

Time developments of plasma concentration and effect are not usually in phase. A number of processes may cause a delay in effect relative to plasma concentrations of the drug. This will result in a so-called hysteresis loop for the effect versus drug concentration in the climbing and falling phase of drug concentrations in plasma
Hysteresis can be mathematically dealt with by incorporating an “effect site compartment” using the COLLAPS algorithm (Sheiner et al. 1979; Veng-Pedersen et al. 1991), also called the “link model,” that contains a compartment with hypothetical target site concentrations (the effect site compartment), being linked to the plasma concentration by a first-order rate constant for influx k 1e and a rate constant k eo for drug efflux from the hypothetical effect compartment.
In which Ce is the concentration of the drug (agonist) in the effect compartment.
With inclusion of the effect compartment, simultaneous PKPD modeling may provide estimates of EC50, Emax, and Hill factor, as well as the rate of CNS target site equilibration as has been shown for benzodiazepines, baclofen, antiepileptic drugs, and adenosine receptor agonists and antagonists (Mandema and Danhof 1992; Mandema et al. 1992; Danhof et al. 1993). Therewith it was demonstrated that by using PKPD modeling in preclinical investigations, useful quantitative information on the pharmacodynamics of new drugs in vivo could be obtained (Breimer and Danhof 1997a, b).
However, compartmental direct effect PKPD modeling cannot distinguish between slow diffusion and other active pharmacokinetic processes determining the concentration of the drug at the target site, nor does it allow the discrimination between drug affinity (binding of the drug to its receptor) and efficacy (ability of the drug to cause an effect after binding to the receptor) (De Lange et al. 2005). It, therefore, lacks the power to predict drug responses under different physiologic or pathologic conditions, where active transport processes are involved, or where both affinity and efficacy may be affected. This means that for prediction of CNS drug effects a more in depth investigation on PKPD relationships is needed on one hand by incorporating information on target site distribution and on the other hand by including information on target site interaction and signal transduction.
3 More in Depth Investigation on PKPD Relationships Is Needed
3.1 Drug Transport Processes Between Blood and CNS Target Site
In the last decades it has become clear that exchange of drugs between blood and brain (Fig. 9.3; Davson and Segal 1996; Fenstermacher et al. 1974) is to a high extent governed by active transport processes, and may therefore affect CNS target site pharmacokinetics (Greig et al. 1987; Hammarlund-Udenaes et al. 1997, 2008; Bouw et al. 2001a, b; De Lange et al. 2005; Girardin 2006; Westerhout et al. 2011), as depicted in Fig. 9.4. This indicates that for building a proper PKPD model for CNS drugs it is of importance to determine BBB transport as that will help to reveal the mechanisms that play a role in the relation between plasma concentrations and CNS drug effects, in other words to distinguish target site distribution from target interaction and signal transduction processes (Fig. 9.5).

Exchange of drugs between blood and brain may occur via different transport processes, among which multiple active transport mechanisms (Influx transport, efflux transport, pinocytosis, and transcytosis). Arrows indicate possible direction and circles indicate drugs with concentration gradient

Using simulations on a simple plasma and brain compartment model in which only unbound drug concentrations are present, one can clearly see that plasma and brain pharmacokinetics may be considerably different, depending on the (virtual) values of CLin and Clout. Left: For CLin = Clout, both varying from high (1.0) to low (0.1) values, with that showing greater discrepancy between plasma and brain PK. Right: For a fixed value for CLout = 0.5, varying of Clin from high (0.5) to low (0.01) shows a decrease of the PK in brain in parallel to plasma PK. Adapted from (Hammarlund-Udenaes et al. 1997)

Factors between drug dose and CNS effect. For building a proper PKPD model for CNS drugs it is of importance to determine BBB transport and target site distribution to be distinguished from target interaction and signal transduction processes
3.1.1 Blood–Brain Barrier (BBB) Transport
Among the transporters present at the BBB the P-glycoprotein efflux transporter (P-gp) is the earliest discovered best characterized one. By the development of the so-called P-gp knockout or mdr1a(−/−) mice, Schinkel et al. (1994) demonstrated the importance of the efflux by P-gp for brain distribution of many clinically important drugs and revolutionized research on active transport mechanisms at the level of the BBB. Later, also the multidrug-related transport proteins (MRP’S; Borst et al. 2000; Wijnholds et al. 2000) and the breast cancer-resistance protein (BCRP; Enokizono et al. 2008) were found to play a role in the brain disposition of many drugs, with partial overlap on substrates with each other.
For quantitative determination of P-gp efflux at the level of the BBB unbound concentrations at either side of the BBB are needed, i.e. unbound plasma and brain extracellular (brain ECF) concentrations. Microdialysis is widely considered to be the best technique to monitor concentrations in the brain ECF over time (Cremers et al. 2009), which combined with simultaneous serial blood sampling from the same animal is a powerful approach to study pharmacokinetic properties related to BBB transport and intracerebral distribution mechanisms (Wang and Welty 1996; De Lange et al. 1994, 1997; Hammarlund-Udenaes et al. 1997). Rate and extent of BBB transport for the unbound drug can be determined using this technique, without confounding influence of binding in plasma or brain (Hammarlund-Udenaes et al. 2008).
Furthermore, in many cases, CNS targets are membrane bound receptors facing the brain ECF, or enzymes within the brain ECF. This makes information on brain ECF concentrations highly valuable (De Lange and Danhof 2002; Watson et al. 2009; Jeffrey and Summerfield 2010; Westerhout et al. 2011). For intracellular targets, however, obtaining in vivo information is more complicated. There are no means to directly monitor brain intracellular concentration–time profiles. At best, (at equilibrium), brain intracellular concentrations can be derived by combining different experimental approaches (Fridén et al. 2007; Hammarlund-Udenaes et al. 2008).
The use of the microdialysis technique showed that even if drugs cross the BBB by passive diffusion, important differences may exist between brain ECF and plasma concentration profiles (Wong et al. 1992; Malhotra et al. 1994; De Lange et al. 1994, 1995a, b, c-critical factors; Yang et al. 1997; Wang et al. 1997; Bouw et al. 2000, 2001a, b) which are influenced upon (induced) changes in BBB properties (de Lange et al. 1995a, b). That has led to a more general theoretical framework on the rate and extent of BBB transport and influences thereof on the relationship between plasma and brain ECF concentration profiles (Hammarlund-Udenaes et al. 1997, 2008).
Also active transport processes could be determined by microdialysis, with P-gp-mediated efflux at the BBB being addressed first. De Lange et al. (1998) studied the BBB transport and P-gp functionality in mdr1a(−/−) mice and wild-type for the model P-gp substrate rhodamine-123 (R123), and Xie et al. (1999) studied the effect of P-gp functionality at the BBB for morphine in these mice, indicating that P-gp participates in regulating morphine transport across the BBB, with an approximately twofold higher extent of brain distribution in the absence of P-gp efflux transport. Likewise, for the fluoroquinolone sparfloxacin, a clear effect of P-gp functionality on BBB transport was found, with about a fivefold increase in brain ECF distribution in the absence of P-gp efflux (De Lange et al. 2000). Another example is the increase of imipramine brain distribution by inhibition of P-gp (O’Brien et al. 2012). Other active transporters at the BBB were indicated by the use of intracerebral microdialysis with probenecid as inhibitor of other active transport at the level of the BBB. Xie studied the BBB transport characteristics of morphine-3-glucuronide (M3G) in the rat and found that its extent of BBB transport increased about twofold upon coadministration of probenecid (Xie et al. 2000). The possible influence of probenecid on morphine transport across the BBB was studied by Tunblad with a ~1.3-fold increase of extent of BBB transport of morphine (Tunblad et al. 2005). As final example, microdialysis studies by Sun in rats indicated that multidrug-resistance-related proteins (MRPs) or MRP-like transport system(s) play a role in fluorescein distribution across both BBB and BCSFB, formerly considered as a marker for passive paracellular transport (Sun et al. 2001).
Actually, apart from P-gp and MRP’s many more active transporters have been found at the level of the BBB (Begley 2004; de Boer et al. 2003; Kusuhara and Sugiyama 2004, 2005; Löscher and Potschka 2005; Boström et al. 2006; Uchida et al. 2011, 2012)
3.1.2 Intracerebral Distribution
Apart from plasma pharmacokinetics and BBB transport also other factors processes may be important determinants for actual target site concentrations. These factors may include extracellular metabolism, extra-intracellular distribution, and exchange of the drug between ECF and CSF (Cserr and Bundgaard 1984; Wong et al. 1992; Malhotra et al. 1994; Williams et al. 1995; De Lange et al. 1995c; Yang et al. 1997; Shen et al. 2004; Westerhout et al. 2012; Syvänen et al. 2012). As an example, for a drug with low BBB permeability but fast accumulation into brain cells, the ECF concentrations will be lower than in case no intracellular accumulation takes place. Therefore, it is a prerequisite to take total brain concentrations into account, because otherwise the extent and rate of transport into the brain will be underestimated. Moreover, intra-extracellular exchange may include active transport mechanisms (Lee et al. 2001) and potential change in this transport by coadministration of transport inhibitors, intended to modify BBB transport, could as well modify extra-intracellular exchange. The effect of brain ECF to parenchymal exchange has been clearly demonstrated by the microdialysis study by Scism on valproate in rabbits. Coadministration of probenecid via the probe increased the intracellular concentrations without affecting brain ECF concentrations, indicating the presence of a probenecid-sensitive efflux transporter at the brain parenchymal cells (Scism et al. 1997).
3.1.3 Blood–Cerebrospinal Fluid-Barrier (BCSFB) Transport
With time, the potential contribution of the BCSFB in drug transport into and out of the brain has become clear. The BCSFB is based in the epithelial cells of the choroid plexus in which also transporters are expressed (Nishino et al. 1999; Wijnholds et al. 2001; De Lange 2004). To date there is no full consensus on the transport direction and subcellular localization of all the different transporters. As presented above, it has been well established that P-gp functions as an efflux transporter at the BBB, either by efflux enhancement or by influx hindrance (Tunblad et al. 2004b; Syvänen et al. 2006). However, the transport direction of P-gp at the level of the BCSFB is unclear. There have been some indications that P-gp functions as an influx transporter at the BCSFB. Noninvasive single-photon-emission computed tomography (SPECT) studies with 99mTc-sestamibi, a membrane-permeant radiopharmaceutical that is a substrate of both P-gp and MRP, were performed by Rao et al. (1999). It was concluded that P-gp localizes subapically at the choroid plexus epithelium, with transport into the direction of the CSF. Also Kassem et al. (2007) came to the same conclusion based on their studies on thyroxine transfer from CSF to choroid plexus and ventricular brain regions in rabbit. A recent study of the detailed kinetics of the strong P-gp substrate quinidine in different sites of the brain (brain extracellular, lateral ventricle, and cistern magna) could not confirm active influx of quinidine from blood into the ventricles (Westerhout et al. 2012). This is in line with findings of only minimal expression of P-gp at the choroid plexus cells of the lateral and fourth ventricle by Gazzin et al. (2008).
Interestingly, the BBB and the BCSFB have many similarities but also differences with regard to location and surface, but also both qualitatively and quantitatively between the plethora of active transport properties being expressed. It can be seen that this may impact on drug distribution at different sites/parts of the brain (Fig. 9.6).

Simplified and schematic representation of the brain, with passive and active transport processes, and metabolism, that all govern the concentration–time profile of the free drug at different locations in the CNS and therewith CNS drug effects
3.1.4 Pathologic Conditions
An important feature is that the BBB is under continuous physiologic control by surrounding astrocytes, pericytes, neurons, and plasma components. All together, these factors determine the delicate homeostasis of the brain environment. This dynamic regulation of the BBB indicates that different situations may result in different BBB functionalities and changes in pathological conditions (Zlokovic et al. 1989; Oztaş and Küçük 1995; Oztaş et al. 2004; Oztas et al. 2007; Mulder et al. 2001; Ederoth et al. 2004; Langford et al. 2004; De Lange et al. 2005; Bell and Zlokovic 2009; Bengtsson et al. 2009; Zlokovic 2010). BBB functionality changes may influence drug transport across the BBB and, therefore, they may have important implications for the target site kinetics.
3.1.5 Impact of Blood–Brain Transport and Brain Distribution on PKPD Relationships
Wang and Welty (1993) studied the concentration–time profile of gabapentin in plasma and brain ECF by microdialysis, and end-of-experiment whole brain tissue in rats, and determined the anticonvulsant effects of gabapentin by maximal electroshock. Brain ECF concentrations of gabapentin were very small (~5 %) in comparison with those in plasma, while brain tissue concentrations were equal to or greater than those in plasma. Wang and Welty were the first to introduce the term “volume of distribution in brain” (Ve,app) as the extent of drug distribution between brain unbound to brain tissue. For gabapentin a large Ve,app was found (5.5 mL/g-brain). Furthermore, the maximal anticonvulsant effect of gabapentin lagged behind both plasma and brain ECF gabapentin concentrations indicating that the anticonvulsant effect of gabapentin is delayed relative to plasma concentrations by time-dependent events in distribution from blood to brain and even deeper into the brain.
Stain-Texier et al. (1999) showed that M6G brain ECF concentrations were ~125-fold (!) higher than the calculated intracellular levels, showing that M6G is almost exclusively distributed into brain ECF, which is highly favorable for exposure to the opioid receptors. Bouw et al. (2001a, b) further investigated the contribution of the blood–brain barrier (BBB) transport to the delay in antinociceptive effect of morphine-6-glucuronide (M6G), and studied the equilibration of M6G in vivo across the BBB with microdialysis measuring unbound concentrations. They found a significant longer half-live of M6G in brain ECF than in blood. Active efflux in BBB transport of M6G was indicated by the extent of BBB transport being far below unity (~25 %). Ve,app of M6G was ~20 % of the brain, corresponding with the brain ECF space, in line with the data of Stain-Texier et al. (1999). Furthermore, it was found that about half of the delay between blood concentrations and antinociceptive effect of M6G was attributed to slow transport across the BBB. For morphine the contribution of BBB transport of to the delay in antinociceptive effect was even larger (Bouw et al. 2000). Lötsch et al. (2002) further assessed the relationship between spinal concentrations and antinociceptive effects of M6G in rats, and showed that pharmacological inhibition of P-gp resulted in approximately twofold increase in the M6G spinal cord/plasma concentration ratio while also the antinociceptive effects of M6G were significantly enhanced.
For the antiepileptic drug oxcarbamazepine Clinckers et al. (2008) studied simultaneously the concentration–time profile of oxcarbamazepine and effects on hippocampal monoamines as pharmacodynamic markers for the anticonvulsant activity, in absence or presence of locally administered P-gp inhibitors in a rat model of epilepsy. Although systemic oxcarbazepine administration alone failed in preventing the animals from developing seizures, coadministration with verapamil (as P-gp blocker) or probenecid (as MRP’s blocker) offered complete protection. Concomitantly, significant increases in extracellular hippocampal dopamine and serotonin levels were observed.
All together these studies clearly demonstrate that (active) transport processes at the BBB and brain distribution beyond have an impact on the response and should be taken into account to better understand PKPD relationships of CNS drugs.
3.2 Target Site Interaction and Signal Transduction
Many times brain distribution is studied without measuring associated (biomarkers of the) effects. Actually, it would be of great added value if PK and associated PD would be obtained in a single experimental subject or at least single experimental context. So, therefore, it is of importance to learn more about factors in target activation and signal transduction, as addressed in this chapter.
Here we assume the target being a receptor. At equilibrium, the relationship between agonist concentration ([A]) and agonist-occupied receptor ([AR]) is described by (9.3):
in which [RT] represents total receptor concentration and KA represents the agonist-receptor equilibrium dissociation constant.
3.2.1 Operational Model of Agonism
Receptor theory as included in the operational model of agonism assigns mathematical rules to biological systems in order to quantify drug effects and define what biological systems can and cannot do, leading to the design of experiments that may further modify the model. For the relation between agonist-occupied receptors [AR] and receptor activation Black and Leff (1983) derived a practical or “operational” equation. If agonist binding to the target is hyperbolic and the concentration–response curve has a Hill slope of 1.0, the equation linking the concentration of “agonist-occupied receptors” to the response must also be hyperbolic. This leads to the “transducer function,” as the mathematical representation of the transduction of receptor occupation into a response, in (9.4):
The parameter, Em, is the maximum response possible in the system (tissue). It is important to note that this is not necessarily equal to the maximum response that a particular agonist actually produces (Fig. 9.7). The parameter KE is the concentration of [AR] that elicits half the maximal tissue response, Em. The efficacy of an agonist is determined by both KE and the total receptor density of the tissue ([RT]). Black and Leff (1983) combined those two parameters into a ratio ([RT]/KE) and called this parameter tau (τ), the “transducer constant.”

The maximal tissue response is termed Em. It is important to note that this is not necessarily equal to maximal response, Emax, that can be produced by a particular agonist
It actually indicates that two agents in a setting with equivalent sets of receptors may not produce equal degrees of effect even if both agents are given in maximally effective doses. This is due to differences in “Intrinsic activity” (or efficacy) that can be defined as the property of a drug that determines the amount of biological effect produced per unit of drug–receptor complex formed. Thus, the drug that produces the greater maximum effect has the greater intrinsic activity. It is important to note that intrinsic activity is not the same as “potency” and may be completely independent of it.
Activation of the receptor should be “transduced” to elicit the response. Combining the hyperbolic occupancy equation with the hyperbolic transducer function yields an explicit equation (9.5) describing the effect at any concentration of agonist:
in which E = effect, Em = maximum response achievable in system, KA = agonist dissociation equilibrium constant, and n = slope index of the receptor occupancy effect function. It actually describes a 3-dimensional interrelationship as can be seen in Fig. 9.8.

A certain agonist concentration [A] leads to a certain occupancy of the receptor (concentration of the receptor–agonist complex [AR]). Then, receptor occupancy should be “transduced” to elicit the response E. The relation between agonist concentration, receptor occupancy, and elicited effect can be described by a 3-dimensional interrelationship. Em = maximum response achievable in system, K A = agonist dissociation equilibrium constant, n = slope index of the occupancy effect function, R 0 = total number of available receptors, Ke = concentration occupied receptors [AR] that produces 50 % of maximal effect, τ = transducer constant or efficacy parameter (=R 0/Ke)
Intrinsic activity—like affinity—depends on the characteristics of both the drug and the receptor, but intrinsic activity and affinity apparently can vary independently. This means that the EC50 does not equal KA but rather KA/(1 + τ). As an example, having a strong agonist that reaches a 50 % response upon binding fewer than half the available receptors, its EC50 will be much less than KA.
Receptor affinity and intrinsic activity are “drug-specific” properties and can be estimated in in vitro bioassays, with the maximal response of the drug being determined, not from single dose–response curves but from using pairs of dose–response curves (usually treatment and control) for a particular tissue, here CNS, sharing some parameters.
Subsequent simultaneous analysis of the resulting different PKPD relationships must be performed to build a mechanism-based model that explicitly distinguishes between the drug-specific and the system-specific properties to allow prediction of the intrinsic activity and potency of another drug for a particular pharmacological effect or response. These different PKPD relationships may be obtained in different ways.
-
Studying one agonist under control conditions and conditions in which the number of receptors available for binding is reduced (Furchgott 1966; Garrido et al. 2000).
-
Studying series of chemically similar drugs with varying degrees of agonism for the specific receptor and simultaneous analysis of the PKPD relationships (Cox et al. 1998; Groenendaal et al. 2008).
The operational model of agonism has been successfully applied in numerous in vitro studies and later also in mechanisms-based PKPD analysis of in vivo drug effects (Kenakin 2004; Danhof et al. 2005, 2007). For adenosine A1 receptor agonists a good correlation was observed between the in vivo pK A and the in vitro pK i and also between the in vivo efficacy parameter (τ) and the in vitro GTP shift (as measure for intrinsic activity), thus enabling the prediction of in vivo concentration–effect relationships (Van der Graaf and Danhof 1997a, b; Van der Graaf et al. 1999). In addition, excellent in vitro–in vivo correlations have also been observed for benzodiazepines (Tuk et al. 1999, 2002; Visser et al. 2003) and neuroactive steroids (Visser et al. 2002).
Taken together, incorporation of receptor theory into PKPD models on in vivo concentration–effect relationships could provide information on:
-
Tissue selectivity of drug effects (Van Schaick et al. 1998)
-
Interspecies differences in concentration–effect relationships
-
Tolerance and sensitization (Cleton et al. 2000)
-
Intra- and interindividual variability
Of course, life is not that simple that in all cases the incorporation of receptor theory in mechanism-based PKPD models was successful. For the opioids alfentanil, fentanyl, and sufentanil, it was shown by simulation that the concentration–effect relationships could be explained by the operational model of agonism under the assumption of a considerable receptor reserve (Cox et al. 1998), while also, a shift in the concentration–effect relationship of alfentanil was observed following pretreatment with the irreversible μ-opioid receptor antagonist β-funaltrexamine, which was consistent with the 40–60 % reduction in the available number of specific μ-opioid binding sites as shown in an in vitro receptor bioassay (Garrido et al. 2000). However, a proper incorporation of the receptor theory in a mechanism-based PKPD model of the opioid receptor agonists could not been accomplished.
Also, for the 5-HT1A receptor agonists, a rather poor correlation was found between the in vivo pK A and the in vitro pK i, despite a good correlation between in vivo and the in vitro GTP shift (Zuideveld et al. 2007). Failure of successful inclusion of the receptor theory in the PKPD models of the opioid and 5-HT1A agonists could be due to complexities at the level of blood–brain transport and intracerebral distribution which was not addressed in these studies, as estimates of hypothetical target site concentrations were made using the link model.
When solving shortcomings in knowledge on target site distribution of drugs, the principles of the operational model will provide the basis for future developments in drug development by classifying drugs and predicting their mechanism of action in pharmacology (Kenakin and Christopoulos 2011)
3.3 Mechanism-Based PKPD Modeling Including Complex Target Site Distribution
As indicated above, the mechanism-based PKPD analysis of the EEG effects of the opioids alfentanil, fentanyl, and sufentanil, using the operational model of agonism (Cox et al. 1998) did not predict in vivo efficacies of these opioids. Moreover, alfentanil, fentanyl, and sufentanil all appeared to behave as high-efficacy (full) agonists. However, for the development of a mechanism-based PKPD model for the central effects of opioids, additional PKPD data on low-efficacy (partial) agonists were needed, as well as information on the target site equilibration. Therefore, also in vivo PKPD studies on the EEG effects of nalbuphine, butorphanol, and morphine were included to contribute to further data analysis (Groenendaal et al. 2007a, b, 2008). In addition, in vitro studies on passive permeability rates of membrane transport and P-gp interaction of all opioids were performed using cell systems comprised of epithelial cells transfected with either the human MDR1 or the rodent MDR1a gene. The results of these investigations confirmed that morphine is a P-gp substrate and that its transport could be inhibited by the P-gp inhibitor GF120918 (elacridar). Alfentanil, fentanyl, and sufentanil were found to be inhibitors of P-gp, but could not be identified as substrates for this efflux transporter. No interaction with P-gp was observed for butorphanol. For alfentanil, fentanyl, sufentanil, and butorphanol, the passive permeability across the monolayers was very high, whereas for morphine and nalbuphine the passive permeability was low. For morphine more information was needed on its BBB transport in conjunction to its EEG effect. To quantitatively determine the influence of BBB transport on the PKPD relationship of morphine, including P-gp-mediated efflux, the combined EEG/microdialysis technique was developed and used. For morphine the functionality of transporters at the BBB was found to be a major determinant of the time-course of brain ECF concentrations as well as on the EEG effect though brain ECF concentrations could not be used to directly predict EEG effects. Still, the data of all opioids could not be condensed into one mechanism-based model on the central effects of opioids using the operational model of agonism. So, this indicates that lots of insights on PKPD relationships of opioids have been gained, but remaining parts between brain unbound morphine concentration and EEG effect remaining to be determined.
4 Current Status
4.1 Quantitative Translational Systems Approach in PKPD Modeling
Since biological systems operate at different set points in the body under different conditions, the ability to predict drug effects under a variety of circumstances is important (Ingss 1990; Van der Graaf and Danhof 1997a, b; Kenakin 2008; Gabrielsson and Green 2009; Van Steeg et al. 2007, 2009, 2010). Moreover, as biological system mechanisms are concurrently working, there is a need for integrated in vivo experiments, e.g., that the experiments address multiple mechanisms (/biomarkers) at the same time. Using animals, we can learn more on the interrelationship of the different pharmacokinetic processes, by performing integrative studies in which variables are systematically varied (e.g., inhibition of an efflux transporter or induction of pathological state, or using a different drug or route of administration). By these are so-called integrative cross-compare designed studies (Westerhout et al. 2011, 2012; De Lange 2013a, b) we can dissect contributions of individual mechanisms in animals using mechanism-based mathematical modeling. This provides the links to the human situation based on the parsimony of the biological system.
4.2 Classification of Biomarkers
In translational models, specific expressions are needed that quantitatively characterize processes on the causal path between drug administration and effect. These include target site distribution, target binding and activation, transduction, PD interactions, and homeostatic feedback mechanisms (Mandema et al. 1991; Cox et al. 1998; Van der Graaf and Danhof 1997a, b; van Steeg et al. 2009). Ultimately also the effects on and of disease processes and disease progression have to be considered. These can be characterized by biomarkers according to the following classification of biomarkers (Danhof et al. 2005; Fig. 9.9):

In translational models, specific expressions are needed that quantitatively characterize processes on the causal path between drug administration and effect. These can be characterized by different types of biomarkers according to the presented classification
-
Type 0 biomarkers refer to the genotype or phenotype as determinant of the drug response, that influences target site exposure or response due to variation in the expression of e.g., enzymes or receptors. They are commonly used as covariates in PKPD models.
-
Type 1 biomarkers refer to drug concentrations in general and at the target site in particular. As previously pointed out, quantitative biomarkers that represent the target site distribution of drugs and metabolites for compounds that act in the CNS are difficult to obtain in man, but readily available in vivo in animals (De Lange et al. 2005).
-
Type 2 biomarkers refer to the degree of target occupancy. In theory, effects may occur at different degrees of target occupancy and may be species dependent. The relationship between target occupancy and effect is therefore important for the understanding of inter- and intraindividual variability. Information on target occupancy is available by bioassays in vitro and can also be noninvasively measured in humans by positron emission tomography (Kapur et al. 2000; Kvernmo et al. 2006, 2008).
-
Type 3 biomarkers refer to quantification of the target site activation. By means of in vitro bioassays information can be obtained on receptor activation in animal and man. Techniques like electroencephalograms (EEG) (Kropf and Kuschinsky 1993; Vorobyov et al. 2003) and functional-magnetic resonance imaging (fMRI) can obtain specific receptor activation in preclinical and clinical in vivo setting.
-
Type 4 biomarkers refer to physiological measures in the integral biological system, which are often controlled by homeostatic feedback mechanisms (Bagli et al. 1999). Such measures can for example be on pituitary hormones that play a very important role in communication between CNS and periphery (Freeman et al. 2000).
-
Type 5 biomarkers characterize disease processes and are particularly useful in clinical settings. (However, an important question is whether type 5 biomarkers can be identified in animal models of disease; Holford and Nutt 2008).
-
Type 6 biomarkers refer to clinical endpoints, such as occurrence of a disease, symptom, sign, or laboratory abnormality that links to target outcomes (Holford and Nutt 2008).
Obtaining combined information on a number of biomarker types (preferable in parallel, within a single biological system) will allows the development of better models, with increased accuracy and predictability. The better we will be able to develop predictive models in preclinical studies, the more the number of often extremely costly clinical studies can be reduced.
The focus should therefore be on the design of quantitative in vivo animal studies such that translational pharmacology approaches can be applied (Boxenbaum 1982; Danhof et al. 2008; Fridén et al. 2009). Especially, to that end, in refined animal models the biomarkers of the effect that can be measured in both animals and human will be useful.
4.3 Development of a Translational PKPD Model on D2 Receptor Inhibition
Investigations on drugs that interact with the dopaminergic system in the brain are of interest as many diseases, including Parkinson’s disease, schizophrenia, and depression, are related to dysfunctions in the dopaminergic system. Since dopamine is an important neurotransmitter in hypothalamic control, pituitary hormones have high potential as type 4 biomarkers for dopaminergic activity in the brain (Freeman et al. 2000), as these are secreted into blood and blood levels can be assessed in both animal and human. One of these hormones is prolactin. Prolactin is synthesized in the lactotrophs of the pituitary, and its release into plasma will occur upon dopaminergic inhibition (specifically the D2 receptor; Fig. 9.10).

Cartoon of transversal cross section of human brain with dopaminergic areas dopaminergic receptor-type distributions
Remoxipride is a weak, but selective, dopamine-D2 receptor antagonist (Farde and Von Bahr 1990; Köhler 1990) and was prescribed as an atypical antipsychotic (Roxiam®) at the end of the 1980s. Due to a few cases of aplastic anemia, the drug was withdrawn from the market (Philpott 1993). The data that have been obtained before that time in clinical setting can still be used and extensive clinical PKPD datasets are available for remoxipride and prolactin plasma data (Movin-Osswald et al. 1995). In recent studies, remoxipride was used as a paradigm compound in rats, finally enabling investigation of animal to human extrapolation of dopaminergic drug effects (Stevens et al. 2012) and the development of the translational model, first for intravenous to intranasal administration, then from rat to human, is presented below.
4.3.1 Development of the PKPD Model Following Intravenous Administration in Rats
Following intravenous administration of remoxipride as a model drug for dopaminergic D2 receptor inhibition, using the levels of the pituitary hormone prolactin in plasma as a pharmacodynamic readout (Fitzgerald and Dinan 2008; Stevens et al. 2011, 2012). Remoxipride pharmacokinetics was determined in plasma, and in brain ECF by microdialysis, as the latter was anticipated to allow better prediction of pharmacodynamic effects in a PKPD model. After assessment of baseline variation in prolactin plasma concentrations, the prolactin response (increase in plasma concentrations) upon intravenous administration of three different single doses of remoxipride was obtained. Also, the prolactine response was measured following double low dosing of remoxipride with different time intervals to get information on the synthesis of prolactin in the lactotrophs of the pituitary similar to the data obtained in human (Movin-Osswald and Hammarlund-Udenaes 1995; Fig. 9.11)

Information on the synthesis rate of prolactin in the lactotrophs can be obtained by the double dosing approach (Movin-Osswald and Hammarlund-Udenaes 1995). After the first dose of the D2 antagonist the lactotroph is depleted from prolactin. The time to the second dose (interval) will determine how much prolactin has been newly synthesized and can be released by the D2 antagonist at that time. (a) Fully filled lactotroph with dopaminergic inhibition (b) release of prolactin content from the lactotroph upon antagonizing dopaminergic inhibition by the D2 antagonist (c) partly filled lactotroph after some time and (d) release of the newly synthesized prolactin by the second dose of the D2 antagonist
The PKPD model was developed in multiple steps, comparing different structural models and model quality testing. The final mechanistic PKPD model consisted of a:
-
Pharmacokinetic model for plasma and brain unbound remoxipride concentrations.
-
Pool model for prolactin synthesis and storage and its release into- and elimination from plasma.
-
Positive feedback of prolactin plasma concentrations on prolactin synthesis.
-
(Not unbound plasma but specifically) the unbound brain concentrations of remoxipride for the inhibition of the D2 receptor, and resulting stimulation of prolactin release into plasma.
It is of interest that plasma prolactin concentrations had a positive feedback on prolactin synthesis in the lactotrophs and that brain unbound remoxipride concentrations were indistinguishable from target site concentrations to drive the release of prolactin into plasma.
Although the strong positive feedback by plasma prolactin suggested in this study is not consistent with a few previous findings (Movin-Osswald et al. 1995; Friberg et al. 2008; Ma et al. 2010), other literature seems to support our current observation of a positive feedback (Freeman et al. 2000; Phelps 1986; Ben Jonathan et al. 2008). Since prolactin receptors are present in the cell membranes of lactotrophs and their activation can result in synthesis of prolactin, this demonstrates that lactotrophs can perceive and respond to prolactin concentrations in a paracrine manner by which release of prolactin by lactotrophs (depletion) increases the prolactin synthesis to “refill” the lactotrophs.
4.3.2 Extension of the PKPD Model for Intranasal Administration in Rats
Next to rapid systemic uptake of compounds, intranasal administration may provide a direct way for delivery of therapeutics into the CNS (Hanson and Frey et al. 2008; Baker and Spencer 1986; Bagger and Bechgaard 2004; Constantino et al. 2007). If direct transport into the brain would be possible, intranasal administration could enhance the CNS target site bioavailability and therewith a more selective effect of CNS drugs (Graff and Pollack 2004; Illum 2000, 2004; Jansson and Bjork 2002). Intranasal administration could be a promising alternative for dopaminergic drugs because oral administration is often limited due to active first-pass clearance by the liver while also frequently restricted BBB transport of dopaminergic drugs has been reported (Dhuria et al. 2009).
Using a previously reported minimum stress, freely moving rat model for intranasal drug administration (Stevens et al. 2009), plasma- and brain ECF samples were obtained over time, after giving remoxipride intranasally at the same dosages as in the intravenous study, and measuring the resulting prolactine levels in plasma within the same rats.
The remoxipride PKPD model as developed on intravenous data was extended by adding an absorption compartment to allow simultaneous fitting of the intravenous and intranasal datasets. However, for proper description of the intranasal data by the model, a second absorption compartment with transport of remoxipride direct from nose to brain had to be included. The visual predictive check of the final model showed good prediction of the plasma- and brain ECF observations after intravenous and intranasal administration (Stevens et al. 2011; Fig. 9.12). Thus, a multicompartment pharmacokinetic model with two distinct absorption compartments, nose-to-systemic and direct nose-to-brain was found to best describe the observed pharmacokinetic data. Absorption was described in terms of bioavailability and rate. Total bioavailability following intranasal administration was ~90 % of which ~75 % was attributed to direct nose-to brain transport. The advanced mathematical model and appropriate data allowed further for having information not only on the extent of brain distribution but also on the rates of transport. The direct nose-to-brain absorption did not turn out to be a rapid route to the brain. The rate was slow, explaining prolonged brain ECF exposure after intranasal compared to intravenous administration. Thus, by the experimental combined with mathematical modeling approach explicit separation and quantitation of systemic- and direct nose-to-brain transport after intranasal administration of remoxipride in the rat could be made.

Visual predictive check of the PK model for plasma and brainECF data (dots) (95 % percentiles inclusion as gray areas) for remoxipride following intravenous (IV) and intranasal (IN) administration
An important finding was that brain ECF (brain unbound) concentrations could directly be linked to the observed effect on prolactin plasma concentrations following intranasal administration, while the model did not converge with using plasma concentration data of remoxipride. It shows the importance of having kinetic information of unbound concentrations as close to the receptor as possible, as was indicated by study of Watson et al. (2009), in which brain unbound concentrations were found to be a better predictor of dopamine D2 receptor occupancy than total brain concentration, CSF concentration, or blood unbound concentration.
4.3.3 Use of the Structural Preclinical PKPD Model to Finally Predict Human PKPD
When drug-specific and biological system-specific parameters are quantified in a PKPD model it provides the opportunity to scale the system-specific parameters from animal to human to translate PKPD relationship to man. Allometric scaling of drug pharmacokinetic properties and biological system-specific parameters has been used in translational investigations, with reasonable degree of success, to predict drug effects in humans (Yassen et al. 2007; Zuideveld et al. 2007). But, pharmacodynamic properties are more difficult to scale compared to pharmacokinetic properties, since pharmacodynamic parameters are often not related to bodyweight (e.g., receptor occupancy). Such information may be available by in vitro bioassays. For many drugs and endogenous compounds, clinical information is often readily available, like for target binding characteristics of dopaminergic compounds (Kvernmo et al. 2006, 2008) and on prolactin in animals and human (Ben Jonathan et al. 2008). BBB transport of remoxipride in humans was assumed to be comparable to that in rat (in essence based on passive diffusion). The preclinical translational human PKPD model successfully predicted the system prolactin response in humans, indicating that positive feedback on prolactin synthesis and allometric scaling thereof could be a new feature in describing complex homeostatic mechanisms (Fig. 9.13).

Preclinical-derived translational PKPD model for remoxipride and its effect on plasma concentrations of prolactin
5 Future Directions
We have to accept that CNS drug delivery and CNS disease research is complex, and we need to (continue to) put efforts in performing the type of investigations that provide data that we learn from in having a CNS drug “at the right place, at the right time, and at the right concentration.”
Since biological systems operate at different set points in the body under different conditions, the ability to predict drug effects under a variety of circumstances is important and more advanced experimental designs are needed to decipher and learn more on the factors that govern plasma pharmacokinetics, BBB transport intrabrain distribution, as well as their interrelationships and consequences for CNS effects in the different settings (Garrido et al. 2000; Grime and Riley 2006; Gabrielsson and Green 2009; Danhof et al. 2007, 2008; Ploeger et al. 2009; Kenakin and Christopoulos 2011). Therefore, individual processes on the causal path between drug dose and CNS effect should be systematically varied (e.g., inhibition of an efflux transporter, or induction of pathological state, and so on and so forth) to study the impact on the PKPD relationships are measured in a time-dependent manner. Therefore, in the design of future experiments we always need to consider the following.
-
Combining different levels of biomarkers (see all types Sect. 9.4.2) in single subjects as outcomes are context dependent (most important!).
-
Measuring unbound drug concentrations as it is the unbound drug that is able to interact with its target and therefore drives the effect.
-
Involving time dependencies.
-
Understanding the mechanisms involved in uptake into and efflux from the brain, but also plasma pharmacokinetics and intrabrain distribution and their mutual interrelationships.
-
Including drug receptor theory as a tool for quantifying the activity of drugs in a system-independent manner.
-
Identifying the heterogeneity in rate and extent of mechanisms on the causal chain between dose and CNS effects (including challenges/disease conditions).
-
Using advanced mathematical modeling to integrate all these data to build preclinical mathematical models (generalized frameworks).
-
Using human data to test validity of the models after tuning this to human conditions.
-
Improving interspecies extrapolation of pharmacokinetics, by using a more physiologically based pharmacokinetic modeling approach.
Such approach may be referred to as the “Mastermind Research Approach,” in analogy of the game (Fig. 9.14, De Lange 2013a). It makes that the predictive value of the models on PKPD relationships of CNS drugs will increase significantly and the outlook is therefore that clinical studies would suffice with fewer individuals and less samples per individual, for proof of concept in man.

The approach of using integrative cross-compared designed studies, literature data, and advanced mathematical modeling to dissect contributions of rate and extent of individual mechanisms in dose–CNS effect relationships in different conditions, as the basis for translation between conditions. This approach may be shortly abbreviated as the “Mastermind Research Approach,” in analogy of the strategic approach that is needed to “decipher the code” in the game called “Mastermind.” This approach makes that the predictive value of the models on PKPD relationships of CNS drugs will increase significantly
6 Challenges
Performing integrative studies is not without big challenges.
-
From the perspective of the subject, it is of course impossible to integrate research at all biomarker levels within a single subject, but a combination of a subset of major aspects will do.
-
From a technical perspective, there are limitations to what level subjects can be instrumented, and development, improvement, and refinement of techniques remain important.
-
When it comes down to challenges applied to the subject, there are limitations, for humans much more than for animals. Integrity of the physiology of subjects remains to be a high priority. Then, intentionally inducing a disease state can be performed only in animals, and animal disease may at best partly reflect that in human.
-
With increasing complexity of experiments, the chance of failures will increase. Thus, from the people perspective, performing advanced surgeries, complex experimentation, and the use of apparatus needed for monitoring techniques, advanced mathematical data analysis, and model development can be performed only by well-trained and skilled persons.
-
In an overall perspective, Good Academic (/Clinical) Research Practice in terms of preparation of experiments, administration of files, data storage and use in building mathematical models, and last but not least communication, should be effectuated.
7 Conclusions
For a proper CNS effect the drug should have the ability to access the CNS “at the right place, at the right time, and at the right concentration.” To that end a number of key issues need to be considered:
-
To develop treatments with improved safety and efficacy, one of the scientific challenges is to understand the biological mechanisms underlying the PKPD relationships of CNS drugs. Knowledge on an only one individual processes is worthless and its role should be investigated in multiple contexts.
-
PKPD modeling is the golden standard to investigate such complex mechanisms. Often, these models include plasma drug concentration–effect relationships. However, when the target site is in a tissue, a discrepancy between unbound plasma concentrations and unbound tissue concentrations should be considered.
-
Investigations on the kinetics of the unbound drug are indispensable. These are the concentrations seen by the target and a more mechanistic approach should be aimed at understanding the factors that control unbound drug concentrations at the target site.
-
To have information on what concentrations can actually represent target site concentrations, measurement of concomitant effects of the drug is needed.
-
Advanced mathematical modeling techniques are needed to reveal complex relationships of body processes and interactions of the body and the drug, to be ultimately settled down in mathematical models.
8 Points for Discussion
-
Body is a total system in which processes are interdependent. Studies need to be designed such that mutual dependence gets clear. How can studies be best designed to have the most valuable data collected?
-
What concentrations in human can be assessed and used best to predict CNS target site concentrations?
-
Can we address sources of variability between drug responses in human populations, aiming at personalized CNS medicine?
Abbreviations
- AR:
-
Agonist receptor complex density
- BBB:
-
Blood–brain barrier
- BCSFB:
-
Blood–cerebrospinal fluid barrier
- Ce:
-
Concentration of the drug in the effect compartment
- CNS:
-
Central nervous system
- CSF:
-
Cerebrospinal fluid
- E :
-
Effect
- E0:
-
Effect in the absence of the agonist
- EC50 :
-
Concentration of agonist at half-maximal effect
- ECF:
-
Extracellular fluid
- Em:
-
Maximal effect in the biological system
- Emax:
-
Maximal effect of the agonist
- KA:
-
Agonist-receptor binding dissociation equilibrium constant
- Ke:
-
Density of agonist receptor complex that elicits the half maximal effect
- K1e:
-
First-order rate constant for influx K1e
- Keo:
-
Rate constant for drug efflux from the hypothetical effect compartment
- P-gp:
-
P-glycoprotein
- PD:
-
Pharmacodynamics
- PK:
-
Pharmacokinetics
- PKPD:
-
Pharmacokinetic-pharmacodynamic
- RT:
-
Total receptor density
- τ :
-
Transducer constant (efficacy parameter)
- Ve,app:
-
Apparent volume of distribution in the brain
References
Bagger M, Bechgaard E (2004) A microdialysis model to examine nasal drug delivery and olfactory absorption in rats using lidocaine hydrochloride as a model drug. Int J Pharm 269:311–322
Bagli M, Suverkrup R, Quadflieg R, Hoflich G, Kasper S, Moller HJ, Langer M, Barlage U, Rao ML (1999) Pharmacokinetic-pharmacodynamic modeling of tolerance to the prolactin-secreting effect of chlorprothixene after different modes of drug administration. J Pharmacol Exp Ther 291:547–554
Baker H, Spencer RF (1986) Transneuronal transport of peroxidase-conjugated wheat germ agglutinin (WGA-HRP) from the olfactory epithelium to the brain of the adult rat. Exp Brain Res 63(3):461–473
Begley DJ (2004) ABC transporters and the blood-brain barrier. Curr Pharm Des 10:1295–1312
Bell RD, Zlokovic BV (2009) Neurovascular mechanisms and blood-brain barrier disorder in Alzheimer’s disease. Acta Neuropathol 118(1):103–113
Ben Jonathan N, LaPensee CR, LaPensee EW (2008) What can we learn from rodents about prolactin in humans? Endocr Rev 29:1–41
Bengtsson J, Ederoth P, Ley D, Hansson S, Amer-Wåhlin I, Hellström-Westas L, Marsál K, Nordström CH, Hammarlund-Udenaes M (2009) The influence of age on the distribution of morphine and morphine-3-glucuronide across the blood-brain barrier in sheep. Br J Pharmacol 157(6):1085–1096
Betz AL, Goldstein GW (1986) Specialized properties and solute transport in brain capillaries. Annu Rev Physiol 48:241–250
Black J, Leff P (1983) Operational model of pharmacological agonism. Proc R Soc Lond B 220:141–162
Bonati M, Latini R, Tognini G, Young JF, Garattini S (1984) Interspecies comparison of in vivo caffeine pharmacokinetics in man, monkey, rabbit, rat and mouse. Drug Metab Rev 15:1355–1383
Borst P, Zelcer N, van Helvoort A (2000) ABC transporters in lipid transport. Biochim Biophys Acta 1486(1):128–144
Borst P, Elferink RO (2002) Mammalian ABC transporters in health and disease. Annu Rev Biochem 71:537–592
Boström E, Simonsson US, Hammarlund-Udenaes M (2006) In vivo blood-brain barrier transport of oxycodone in the rat: indications for active influx and implications for PK/PD. Drug Metab Dispos 34(9):1624–1631
Bouw MR, Ederoth P, Lundberg J, Ungerstedt U, Nordstrom CH, Hammarlund-Udenaes M (2001a) Increased blood-brain barrier permeability of morphine in a patient with severe brain lesions as determined by microdialysis. Acta Anesthesiol Scand 45:390–392
Bouw MR, Gardmark M, Hammarlund-Udenaes M (2000) PK-PD modelling of morphine transport across the blood-brain barrier as a cause of the antinociceptive effect delay in rats—a microdialysis study. Pharm Res 17:1220–1227
Bouw MR, Xie R, Tunblad K, Hammarlund-Udenaes M (2001b) Blood-brain barrier transport and brain distribution of morphine-6-glucuronide in relation to the antinociceptive effect in rats—pharmacokinetic/pharmacodynamic modelling. Br J Pharmacol 134:1796–1804
Boxenbaum H (1982) Interspecies scaling, allometry, physiological time, and the ground plan of pharmacokinetics. J Pharmacokinet Biopharm 10:201–227
Breimer DD, Danhof M (1997a) Prediction of the time course of drug effects in vivo in health and disease (intensity and duration). Clin Pharmacokinet 32:259–267
Breimer DD, Danhof M (1997b) Relevance of the application of pharmacokinetic-pharmacodynamic modelling concepts in drug development. The ‘wooden shoe’ paradigm. Clin Pharmacokinet 32:259–267
Bruce JN, Oldfield EH (1988) Method for sequential sampling of cerebrospinal fluid in humans. Neurosurgery 23:788–790
Campbell DB (1990) The use of kinetic-dynamic interactions in the evaluation of drugs. Psychopharmacology (Berl) 100(4):433–450
Cleton A, Odman J, Van der Graaf PH, Ghijsen W, Voskuyl R, Danhof M (2000) Mechanism-based modeling of functional adaptation upon chronic treatment with midazolam. Pharm Res 17:321–327
Clinckers R, Smolders I, Meurs A, Ebinger G, Michotte Y (2005) Quantitative in vivo microdialysis study on the influence of multidrug transporters on the blood-brain barrier passage of oxcarbazepine: concomitant use of hippocampal monoamines as pharmacodynamic markers for the anticonvulsant activity. J Pharmacol Exp Ther 14(2):725–731
Clinckers R, Smolders I, Michotte Y, Ebinger G, Danhof M, Voskuyl RA, Della Pasqua O (2008) Impact of efflux transporters and of seizures on the pharmacokinetics of oxcarbazepine metabolite in the rat brain. Br J Pharmacol 155(7):1127–1138
Costantino HR, Illum L, Brandt G, Johnson PH and Quay SC (2007) Intranasal delivery: physicochemical and therapeutic aspects. Int J Pharm 337:1–24
Cox EH, Kerbusch T, van der Graaf PH, Danhof M (1998) Pharmacokinetic-pharmacodynamic modeling of the electroencephalogram effect of synthetic opioids in the rat. Correlation with binding at the μ-opioid receptor. J Pharmacol Exp Ther 284:1095–1103
Cremers TI, de Vries MG, Huinink KD, Loon JP, Hart MV, Ebert B, Westerink BH, De Lange EC (2009) Quantitative microdialysis using modified ultraslow microdialysis: direct rapid and reliable determination of free brain concentrations with the MetaQuant technique. J Neurosci Methods 178(2):249–254
Cserr HF, Bundgaard M (1984) Blood-brain interfaces in vertebrates: a comparative approach. Am J Physiol 246:R277–R288
Danhof M, Alvan G, Dahl SG, Kuhlmann J, Paintaud G (2005) Mechanism-based pharmacokinetic-pharmacodynamic modeling-a new classification of biomarkers. Pharm Res 22:1432–1437
Danhof M, de Jongh J, de Lange ECM, Della Pasqua OE, Ploeger BA, Voskuyl RA (2007) Mechanism-based pharmacokinetic-pharmacodynamic modeling: biophase distribution, receptor theory, and dynamical systems analysis. Annu Rev Pharmacol Toxicol 47:357–400
Danhof M, de Lange EC, Della Pasqua OE, Ploeger BA, Voskuyl RA (2008) Mechanism-based pharmacokinetic-pharmacodynamic (PKPD) modeling in translational drug research. Trends Pharmacol Sci 29:186–191
Danhof M, Levy G (1984) Kinetics of drug action in disease states. I. Effect of infusion rate on phenobarbital concentrations in serum, brain and cerebrospinal fluid of normal rats at onset of loss of righting reflex. J Pharmacol Exp Ther 229(1):44–50
Danhof M, Mandema JW, Hoogerkamp A, Mathot RA (1993) Pharmacokinetic-pharmacodynamic modeling in pre-clinical investigations: principles and perspectives. Eur J Drug Metab Pharmacokinet 18(1):41–47
Davson H, Segal MB (1996) Physiology of the CSF and blood-brain barriers. CRC, Boca Raton, FL
De Boer AG, van der Sandt I, Gaillard PJ (2003) The role of drug transporters at the blood-brain barrier. Annu Rev Pharmacol Toxicol 43:629–656
De Lange EC, Danhof M (2002) Considerations in the use of cerebrospinal fluid pharmacokinetics to predict brain target concentrations in the clinical setting: implications of the barriers between blood and brain. Clin Pharmacokinet 41:691–703
De Lange EC, Danhof M, de Boer AG, Breimer DD (1994) Critical factors of intracerebral microdialysis as a technique to determine the pharmacokinetics of drugs in rat brain. Brain Res 666:1–8
De Lange EC, de Bock G, Schinkel AH, de Boer AG, Breimer DD (1998) BBB transport and P-glycoprotein functionality using MDR1A (−/−) and wild-type mice. Total brain versus microdialysis concentration profiles of rhodamine-123. Pharm Res 15(11):1657–1665
De Lange EC, Marchand S, van den Berg D, van der Sandt IC, de Boer AG, Delon A, Bouquet S, Couet W (2000) In vitro and in vivo investigations on fluoroquinolones; effects of the P-glycoprotein efflux transporter on brain distribution of sparfloxacin. Eur J Pharm Sci 12(2):85–93
De Lange EC, Ravenstijn PGM, Groenendaal D, van Steeg TS (2005) Toward the prediction of CNS drug effect profiles in physiological and pathological conditions using microdialysis and mechanism-based pharmacokinetic-pharmacodynamic modeling. AAPS J 7(3), 54
De Lange EC (2004) Potential role of ABC transporters as a detoxification system at the blood-cerebrospinal fluid-barrier. Adv Drug Deliv Rev 56(12):1793–1809
De Lange EC (2013a) Utility of CSF in translational neuroscience. J Pharmacokinet Pharmacodyn 40(3):315–326
De Lange ECM, Bouw MR, Danhof M, De Boer AG, Breimer DD (1995a) Application of intracerebral microdialysis to study regional distribution kinetics of drugs in rat brain. Br J Pharmacol 116:2538–2544
De Lange ECM, Danhof M, De Boer AG, Breimer DD (1997) Methodological considerations of intracerebral microdialysis in pharmacokinetic studies on blood-brain barrier transport of drugs. Brain Res Rev 25:27–49
De Lange ECM, de Vries JD, Zurcher C, Danhof M, De Boer AG, Breimer DD (1995b) The use of intracerebral microdialysis to study blood-brain barrier transport of anticancer drugs in tumor-bearing rat brain. Pharm Res 12:1924–1931
De Lange ECM, Hesselink MB, Danhof M, De Boer AG, Breimer DD (1995c) The use of intracerebral microdialysis to determine changes in blood-brain barrier transport characteristics. Pharm Res 12:129–133
De Lange ECM (2013b) The use of the mastermind research approach: factors in brain distribution and prediction of human brain target site kinetics and CNS drug effects. Fluids Barriers CNS 10:12
Dhuria SV, Hanson LR, Frey WH (2009) Intranasal delivery to the central nervous system: Mechanisms and experimental considerations. J Pharm Sci 99:1654–1673
Dingemanse J, Hutson PH, Langemeijer MW, Curzon G, Danhof M (1988) Application of serial sampling of cerebrospinal fluid in pharmacodynamic studies with a drug active in the CNS: heptabarbital concentrations at onset and offset of loss of righting reflex in rats. Neuropharmacology 27(5):467–474
Ederoth P, Tunblad K, Bouw R, Lundberg CJ, Ungerstedt U, Nordström CH, Hammarlund-Udenaes M (2004) Blood-brain barrier transport of morphine in patients with severe brain trauma. Br J Clin Pharmacol 57(4):427–435
Enokizono J, Kusuhara H, Ose A, Schinkel AH, Sugiyama Y (2008) Quantitative investigation of the role of breast cancer resistance protein (Bcrp/Abcg2) in limiting brain and testis penetration of xenobiotic compounds. Drug Metab Dispos 36(6):995–1002
Faraci FM (1993) Endothelium-derived vasoactive factors and regulation of the cerebral circulation. Neurosurgery 33(4):648–659
Farde L, von Bahr C (1990) Distribution of remoxipride to the human brain and central D2-dopamine receptor binding examined in vivo by PET. Acta Psychiatr Scand Suppl 358:67–71
Fenstermacher JD, Patlak CS, Blasberg RG (1974) Transport of material between brain extracellular fluid, brain cells and blood. Fed Proc 33:2070–2074
Fenstermacher JD, Wei L, Acuff V, Lin SZ, Chen JL, Bereczki D, Otsuka T, Nakata H, Tajima A, Hans FJ, Ghersi-Egea JF, Finnegan W, Richardson G, Haspel H, Patlak C (1995) The dependency of influx across the blood-brain barrier on blood flow and the apparent flow-independence of glucose influx during stress. In: Greenwood J et al (eds) New concepts of a blood-brain barrier. Plenum, New York, pp 89–101
Fitzgerald P, Dinan TG (2008) Prolactin and dopamine: what is the connection? A review article. J Psychopharmacol 22(2 Suppl):12–19
Freeman ME, Kanyicska B, Lerant A, Nagy G (2000) Prolactin: structure, function, and regulation of secretion. Physiol Rev 80:1523–1631
Frey WH (2002) Intranasal delivery: bypassing the blood-brain barrier to deliver therapeutic agents to the brain and spinal cord. Drug Deliv Technol 2:46–49
Friberg LE, Vermeulen AM, Petersson KJF, Karlsson MO (2008) An agonist-antagonist interaction model for prolactin release following risperidone and paliperidone treatment. Clin Pharmacol Ther 85:409–417
Fridén M, Gupta A, Antonsson M, Bredberg U, Hammarlund-Udenaes M (2007) In vitro methods for estimating unbound drug concentrations in the brain interstitial and intracellular fluids. Drug Metab Dispos 35:1711–1719
Fridén M, Winiwarter S, Jerndal G, Bengtsson O, Wan H, Bredberg U, Hammarlund-Udenaes M, Antonsson M (2009) Structure—brain exposure relationships in rat and human using a novel data set of unbound drug concentrations in brain interstitial and cerebrospinal fluids. J Med Chem 52:6233–6243
Furchgott RF (1966) The use of β-haloalkylamines in the differentiation of receptors and in the determination of dissociation constants of receptor-agonist complexes. Adv Drug Res 3:21–55
Gabrielsson J, Green AR (2009) Quantitative pharmacology or pharmacokinetic pharmacodynamic integration should be a vital component in integrative pharmacology. J Pharmacol Exp Ther 331:767–774
Garrido M, Gubbens-Stibbe J, Tukker E, Cox E, von Frijtag J, Künzel DM, Ijzerman A, Danhof M, Van der Graaf PH (2000) Pharmacokinetic-pharmacodynamic analysis of the EEG effect of alfentanil in rats following beta-funaltrexamine-induced mu-opioid receptor “knockdown” in vivo. Pharm Res 17:653–659
Gazzin S, Strazielle N, Schmitt C, Fevre-Montange M, Ostrow JD, Tiribelli C, Ghersi-Egea JF (2008) Differential expression of the multidrug resistance-related proteins ABCb1 and ABCc1 between blood-brain interfaces. J Comp Neurol 510(5):497–507
Ghersi-Egea JF, Leininger-Muller B, Cecchelli R, Fenstermacher JD (1995) Blood-brain interfaces: relevance to cerebral drug metabolism. Toxicol Lett 82–83:645–653
Girardin F (2006) Membrane transporter proteins: a challenge for CNS drug development. Dialogues Clin Neurosci 8:311–321
Gjedde A, Crone C (1983) Biochemical modulation of blood-brain barrier permeability. Acta Neuropathol Suppl 8:59–74
Graff CL, Pollack G (2004) Drug transport at the blood-brain barrier and the choroid plexus. Curr Drug Metab 5:95–108
Greig NH, Momma S, Sweeney DJ, Smith QR, Rapoport SI (1987) Facilitated transport of melphalan at the rat blood-brain barrier by the large neutral amino acid carrier system. Cancer Res 47:1571–1576
Grime K, Riley RJ (2006) The impact of in vitro binding on in vitro-in vivo extrapolations, projections of metabolic clearance and clinical drug-drug interactions. Curr Drug Metab 7(3):251–264
Groenendaal D, Freijer J, de Mik D, Bouw MR, Danhof M, De Lange EC (2007a) PK-PD modelling of the electroencephalogram effects of morphine: the influence of biophase equilibration and P-glycoprotein interaction. Br J Pharmacol 151(5):713–720
Groenendaal D, Freijer J, de Mik D, Bouw MR, Danhof M, De Lange EC (2007b) Population pharmacokinetic modelling of non-linear brain distribution of morphine: influence of active saturable influx and P-glycoprotein mediated efflux. Br J Pharmacol 151(5):701–712
Groenendaal D, Freijer J, Rosier A, de Mik D, Nicholls G, Hersey A, Ayrton AD, Danhof M, de Lange EC (2008) Pharmacokinetic/pharmacodynamic modelling of the EEG effects of opioids: the role of complex biophase distribution kinetics. Eur J Pharm Sci 34(2–3):149–163
Hammarlund-Udenaes M, Fridén M, Syvänen S, Gupta A (2008) On the rate and extent of drug delivery to the brain. Pharm Res 25(8):1737–1750
Hammarlund-Udenaes M, Paalzow LN, De Lange ECM (1997) Drug equilibration across the blood-brain-barrier—pharmacokinetic considerations based on the microdialysis method. Pharm Res 14:128–134
Hammarlund-Udenaes M (2009) Active-site concentrations of chemicals—are they a better predictor of effect than plasma/organ/tissue concentrations? Basic Clin Pharmacol Toxicol 106:215–220
Hanson LR, Frey WH (2008) 2nd. Intranasal delivery bypasses the blood-brain barrier to target therapeutic agents to the central nervous system and treat neurodegenerative disease. BMC Neurosci 2008 Dec 10;9 Suppl 3:S5
Holford N, Nutt JG (2008) Disease progression, drug action and Parkinson’s disease: why time cannot be ignored. Eur J Clin Pharmacol 64(2):207–216
Illum L (2004) Is nose-to-brain transport of drugs in man a reality? J Pharm Pharmacol 56:3–17
Illum L (2000) Transport of drugs from the nasal cavity to the central nervous system. Eur J Pharm Sci 11:1–18
Ings RMJ (1990) Interspecies scaling and comparisons in drug development and toxicogenetics. Xenobiotica 20:1201–1231
Jansson B, Bjork E (2002) Visualization of in vivo olfactory uptake and transfer using fluorescein dextran. J Drug Target 10:379–386
Jeffrey P, Summerfield S (2010) Assessment of the blood-brain barrier in CNS drug discovery. Neurobiol Dis 37:33–37
Jolliet P, Simon N, Bree F, Brée F, Urien S, Pagliara A, Carrupt PA, Testa B, Tillement JP (1997) Blood-to-brain transfer of various oxicams: effects of plasma binding on their brain delivery. Pharm Res 14:650–656
Kalvass JC, Maurer TS (2002) Influence of nonspecific brain and plasma binding of CNS exposure: implications for rational drug discovery. Biopharm Drug Dispos 23:327–338
Kapur S, Zipursky R, Jones C, Remington G, Houle S (2000) Relationship between dopamine D2 occupancy, clinical response, and side effects: a double-blind PET study of first-episode schizophrenia. Am J Psychiatry 157:514–520
Karssen AM, Meijer OC, van der Sandt ICJ, Lucassen PJ, de Lange ECM, de Boer AG, de Kloet ER (2001) Multidrug resistance P-glycoprotein hampers the access of cortisol but not of corticosterone to mouse and human brain. Endocrinology 142:2686–2694
Kassem NA, Deane R, Segal MB, Chen RL, Preston JE (2007) Thyroxine (T4) transfer from CSF to choroid plexus and ventricular brain regions in rabbit: contributory role of P-glycoprotein and organic anion transporting polypeptides. Brain Res 1181:44–50
Kenakin T, Christopoulos A (2011) Analytical pharmacology: the impact of numbers on pharmacology. Trends Pharmacol Sci 32(4):189–196
Kenakin T (2004) Principles: receptor theory in pharmacology. Trends Pharmacol Sci 25(4):186–192
Kenakin T (2008) Receptor theory. Curr Protoc Pharmacol. Chapter 1:Unit 1.2
Kooij G, van Horssen J, de Lange EC, Reijerkerk A, van der Pol SM, van Het Hof B, Drexhage J, Vennegoor A, Killestein J, Scheffer G, Oerlemans R, Scheper R, van der Valk P, Dijkstra CD, de Vries HE (2010) T lymphocytes impair P-glycoprotein function during neuroinflammation. J Autoimmun 34(4):416–425
Köhler C, Hall H, Magnusson O, Lewander T, Gustafsson K (1990) Biochemical pharmacology of the atypical neuroleptic remoxipride. Acta Psychiatr Scand Suppl. 358:27–36
Kropf W, Kuschinsky K (1993) Effects of stimulation of dopamine D1 receptors on the cortical EEG in rats: different influences by a blockade of D2 receptors and by an activation of putative dopamine autoreceptors. Neuropharmacology 32:493–500
Kusuhara H, Sugiyama Y (2004) Efflux transport systems for organic anions and cations at the blood-CSF barrier. Adv Drug Deliv Rev 56:1741–1763
Kusuhara H, Sugiyama Y (2005) Active efflux across the blood-brain barrier: role of the solute carrier family. NeuroRx 2:73–85
Kvernmo T, Hartter S, Burger E (2006) A review of the receptor-binding and pharmacokinetic properties of dopamine agonists. Clin Ther 28:1065–1078
Kvernmo T, Houben J, Sylte I (2008) Receptor-binding and pharmacokinetic properties of dopaminergic agonists. Curr Top Med Chem 8(12):1049–1067
Langford D, Grigorian A, Hurford R, Adame A, Ellis RJ, Hansen L, Masliah E (2004) Altered P-gp expression in AIDS patients with HIV encephalitis. J Neuropathol Exp Neurol 63:1038–1047
Lee G, Dallas S, Hong M, Bendayan R (2001) Drug transporters in the central nervous system: brain barriers and brain parenchyma considerations. Pharmacol Rev 53:569–596
Letrent SP, Pollack GM, Brouwer KR, Brouwer KL (1999) Effects of a potent and specific P-glycoprotein inhibitor on the blood-brain barrier distribution and antinociceptive effect of morphine in the rat. Drug Metab Dispos 27:827–834
Liu X, Smith BJ, Chen C, Callegari E, Becker SL, Chen X, Cianfrogna J, Doran AC, Doran SD, Gibbs JP, Hosea N, Liu J, Nelson FR, Szewc MA, Van Deusen J (2005) Use of a physiologically based pharmacokinetic model to study the time to reach brain equilibrium: an experimental analysis of the role of blood–brain barrier permeability, plasma protein binding, and brain tissue binding. J Pharmacol Exp Ther 313:1254–1262
Löscher W, Potschka H (2005) Blood-brain barrier active efflux transporters: ATP-binding cassette gene family. Nat Rev Neurosci 6:591–602
Lötsch J, Schmidt R, Vetter G, Schmidt H, Niederberger E, Geisslinger G, Tegeder I (2002) Increased CNS uptake and enhanced antinociception of morphine-6-glucuronide in rats after inhibition of P-glycoprotein. J Neurochem 83(2):241–248
Ma G, Friberg LE, Movin-Osswald G, Karlsson MO (2010) Comparison of the agonist-antagonist interaction model and the pool model for the effect of remoxipride on prolactin. Br J Clin Pharmacol 70:815–824
Malhotra BK, Lemaire M, Sawchuk RJ (1994) Investigation of the distribution of EAB 515 to cortical ECF and CSF in freely moving rats utilizing microdialysis. Pharm Res 11:1223–1231
Mandema JW, Sansom LN, Dios-Viéitez MC, Hollander-Jansen M, Danhof M (1991) Pharmacokinetic-pharmacodynamic modelling of the EEG effects of benzodiazepines. Correlation with receptor binding and anticonvulsant activity. J Pharmacol Exp Ther 257:472–478
Mandema JW, Danhof M (1992) Electroencephalogram effect measures and relationships between pharmacokinetics and pharmacodynamics of centrally acting drugs. Clin Pharmacokinet 23(3):191–215
Mandema JW, Kuck MT, Danhof M (1992) Differences in intrinsic efficacy of benzodiazepines are reflected in their concentration-EEG effect relationship. Br J Pharmacol 105(1):164–170
Mayer S, Maickel RP, Brodie BB (1959) Kinetics of penetration of drugs and other foreign compounds into cerebrospinal fluid and brain. J Pharmacol Exp Ther 127:205–211
Movin-Osswald G, Hammarlund-Udenaes M (1995) Prolactin release after remoxipride by an integrated pharmacokinetic-pharmacodynamic model with intra- and interindividual aspects. J Pharmacol Exp Ther 274:921–927
Movin-Osswald G, Hammarlund-Udenaes M, Von Bahr C, Eneroth P, Walton-Bowen K (1995) Influence of the dosing interval on prolactin release after remoxipride. Br J Clin Pharmacol 39:503–510
Mulder M, Blokland A, van den Berg DJ, Schulten H, Bakker AH, Terwel D, Honig W, de Kloet ER, Havekes LM, Steinbusch HW, de Lange EC (2001) Apolipoprotein E protects against neuropathology induced by a high-fat diet and maintains the integrity of the blood-brain barrier during aging. Lab Invest 81(7):953–960
Nishino J, Suzuki H, Sugiyama D, Kitazawa T, Ito K, Hanano M, Sugiyama Y (1999) Transepithelial transport of organic anions across the choroid plexus: possible involvement of organic anion transporter and multidrug resistance-associated protein. J Pharmacol Exp Ther 290(1):289–294
O’Brien FE, Clarke G, Fitzgerald P, Dinan TG, Griffin BT, Cryan JF (2012) Inhibition of P-glycoprotein enhances transport of the antidepressant imipramine across the blood-brain barrier: microdialysis studies in the conscious freely moving rat. Br J Pharmacol. doi:10.1111/j.1476-5381.2012.01858.x
Oldendorf WH (1974) Lipid solubility and drug penetration of the blood-brain barrier. Proc Soc Exp Biol Med 14:813–816
Oztaş B, Akgül S, Arslan FB (2004) Influence of surgical pain stress on the blood-brain barrier permeability in rats. Life Sci 74(16):1973–1979
Oztas B, Akgul S, Seker FB (2007) Gender difference in the influence of antioxidants on the blood-brain barrier permeability during pentylenetetrazol-induced seizures in hyperthermic rat pups. Biol Trace Elem Res 118(1):77–83
Oztaş B, Küçük M (1995) Influence of acute arterial hypertension on blood-brain barrier permeability in streptozocin-induced diabetic rats. Neurosci Lett 188(1):53–56
Phelps CJ (1986) Immunocytochemical analysis of prolactin cells in the adult rat adenohypophysis: distribution and quantitation relative to sex and strain. Am J Anat 176:233–242
Philpott NJ (1993) Aplastic anaemia and remoxipride. Lancet 342:1244
Ploeger BA, van der Graaf PH, Danhof M (2009) Incorporating receptor theory in mechanism-based pharmacokinetic-pharmacodynamic (PK-PD) modeling. Drug Metab Pharmacokinet 24:3–15
Ramzan IM, Levy G (1986) Chronic theophylline administration has no apparent effect on theophylline concentrations required to produce seizures in rats. Proc Soc Exp Biol Med 182(2):176–180
Rao VV, Dahlheimer JL, Bardgett ME, Snyder AZ, Finch RA, Sartorelli AC, Piwnica-Worms D (1999) Choroid plexus epithelial expression of MDR1 P-glycoprotein and multidrug resistance-associated protein contribute to the blood-cerebrospinal-fluid drug-permeability barrier. Proc Natl Acad Sci U S A 96:3900–3905
Ravenstijn PG, Merlini M, Hameetman M, Murray TK, Ward MA, Lewis H, Ball G, Mottart C, de Ville de Goyet C, Lemarchand T, van Belle K, O’Neill MJ, Danhof M, De Lange EC (2007) The exploration of rotenone as a toxin for inducing Parkinson’s disease in rats, for application in BBB transport and PKPD experiments. J Pharmacol Toxicol Methods 57(2):114–130
Ravenstijn PGM, Drenth H, Baatje MS, O’Neill MJ, Danhof M, de Lange ECM (2012) Evaluation of BBB transport and CNS drug metabolism in diseased and control brain after intravenous L-DOPA in a unilateral rat model of Parkinson’s disease. Fluids Barriers CNS 9:4
Schinkel AH, Smit JJM, van Tellingen O, Beijnen JH, Wagenaar E, van Deemter L, Mol CAAM, van der Valk MA, Robanus-Maandag EC, te Riele HPJ, Berns AJM, Borst P (1994) Disruption of the Mouse mdr1a P-glycoprotein gene leads to a deficiency in the blood-brain barrier and to increased sensitivity to drugs. Cell 77:491–502
Scism JL, Powers KM, Artru AA, Chambers AC, Lewis L, Adkison KK, Kalhorn TF, Shen DD (1997) Effects of probenecid on brain-cerebrospinal fluid-blood distribution kinetics of E-Delta(2)-valproic acid in rabbits. Drug Metab Dispos 25:1337–1346
Scism JL, Powers KM, Artru AA, Lewis L, Shen DD (2000) Probenecid-inhibitable efflux transport of valproic acid in the brain parenchymal cells of rabbits: a microdialysis study. Brain Res. 884(1–2):77–86
Sheiner LB, Stanski DR, Vozeh S, Miller RD, Ham J (1979) Simultaneous modeling of pharmacokinetics and pharmacodynamics: application to d-tubocurarine. Clin Pharmacol Ther 25(3):358–371
Shen DD, Artru AA, Adkison KK (2004) Principles and applicability of CSF sampling for the assessment of CNS drug delivery and pharmacodynamics. Adv Drug Deliv Rev 56:1825–1857
Stain-Texier F, Boschi G, Sandouk P, Scherrmann JM (1999) Elevated concentrations of morphine 6-beta-D-glucuronide in brain extracellular fluid despite low blood-brain barrier permeability. Br J Pharmacol 128:917–924
Stevens J, Ploeger B, Hammarlund-Udenaes M, Osswald G, van der Graaf PH, Danhof M, de Lange ECM (2012) Mechanism-based PK–PD model for the prolactin biological system response following an acute dopamine inhibition challenge: quantitative extrapolation to humans. J Pharmacokinet Pharmacodyn 39(5):463–477
Stevens J, Ploeger B, van der Graaf PH, Danhof M, de Lange ECM (2011) Systemic- and direct nose-to-brain transport in the rat; a mechanistic pharmacokinetic model for remoxipride after intravenous and intranasal administration. Drug Metab Dispos 39(12):2275–2282
Stevens J, Suidgeest E, van der Graaf PH, Danhof M, de Lange EC (2009) A new minimal-stress freely-moving rat model for preclinical studies on intranasal administration of CNS drugs. Pharm Res 26(8):1911–1917
Sun H, Miller DW, Elmquist WF (2001) Effect of probenecid on fluorescein transport in the central nervous system using in vitro and in vivo models. Pharm Res 18(11):1542–1549
Suzuki H, Terasaki T, Sugiyama Y (1997) Role of efflux transport across the blood-brain barrier and blood-cerebrospinal fluid barrier on the disposition of xenobiotics in the central nervous system. Adv Drug Deliv Rev 25:257–285
Syvänen S, Lindhe Ö, Palner M, Kornum BR, Rahman O, Långström B, Knudsen GM, Hammarlund-Udenaes M (2009) Species differences in blood-brain barrier transport of three positron emission tomography radioligands with emphasis on P-glycoprotein transport. Drug Metab Dispos 37:635–643
Syvänen S, Schenke M, van den Berg D-J, Voskuyl RA, de Lange ECM (2012) Alteration in P-glycoprotein functionality affects intrabrain distribution of quinidine more than brain entry—a study in rats subjected to status epilepticus by kainate. AAPS J 14:87–96
Syvänen S, Xie R, Sahin S, Hammarlund-Udenaes M (2006) Consequences of active drug efflux at the blood–brain barrier. Pharm Res 23:705–717
Tanaka H, Mizojiri K (1999) Drug-protein binding and blood-brain barrier permeability. J Pharmacol Exp Ther 288:912–918
Tuk B, van Gool T, Danhof M (2002) Mechanism-based pharmacodynamic modeling of the interaction of midazolam, bretazenil, and zolpidem with ethanol. J Pharmacokinet Pharmacodyn 29(3):235–250
Tuk B, van Oostenbruggen MF, Herben VM, Mandema JW, Danhof M (1999) Characterization of the pharmacodynamic interaction between parent drug and active metabolite in vivo: midazolam and alpha-OH-midazolam. J Pharmacol Exp Ther 289(2):1067–1074
Tunblad K, Ederoth P, Gardenfors A, Hammarlund-Udenaes M, Nordstrom CH (2004a) Altered brain exposure of morphine in experimental meningitis studied with microdialysis. Acta Anaesthesiol Scand 48:294–301
Tunblad K, Hammarlund-Udenaes M, Jonsson E (2004b) An integrated model for the analysis of pharmacokinetic data from microdialysis experiments. Pharm Res 21:1698–1707
Tunblad K, Hammarlund-Udenaes M, Jonsson EN (2005) Influence of probenecid on the delivery of morphine-6-glucuronide to the brain. Eur J Pharm Sci 24:49–57
Tunblad K, Jonsson EN, Hammarlund-Udenaes M (2003) Morphine blood-brain barrier transport is influenced by probenecid co-administration. Pharm Res 20:618–623
Uchida Y, Ohtsuki S, Kamiie J, Terasaki T (2012) Blood-brain barrier (BBB) pharmacoproteomics (PPx): reconstruction of in vivo brain distribution of 11 P-glycoprotein substrates based on the BBB transporter protein concentration, in vitro intrinsic transport activity, and unbound fraction in plasma and brain in mice. J Pharmacol Exp Ther 339(2):579–588
Uchida Y, Ohtsuki S, Katsukura Y, Ikeda C, Suzuki T, Kamiie J, Terasaki T (2011) Quantitative targeted absolute proteomics of human blood-brain barrier transporters and receptors. J Neurochem 117(2):333–345
Urien S, Pinquier JL, Paquette B, Chaumet RP, Kiechel JR, Tillement JP (1987) Effect of the binding of isradipine and darodipine to different plasma proteins on their transfer through the blood-brain barrier. J Pharmacol Exp Ther 242:349–353
Van der Graaf PH, Danhof M (1997a) Analysis of drug-receptor interactions in vivo: a new approach in pharmacokinetic-pharmacodynamic modelling. Int J Clin Pharmacol Ther 35:442–446
Van der Graaf PH, Van Schaick EA, Visser SA, De Greef HJ, Ijzerman AP, Danhof M (1999) Mechanism-based pharmacokinetic-pharmacodynamic modeling of antilipolytic effects of adenosine A(1) receptor agonists in rats: prediction of tissue-dependent efficacy in vivo. J Pharmacol Exp Ther 290:702–709
Van der Graaf PH, Danhof M (1997b) On the reliability of affinity and efficacy estimates obtained by direct operational model fitting of agonist concentration-effect curves following irreversible receptor inactivation. J Pharmacol Toxicol Methods 38(2):81–85
Van Schaick EA, Tukker HE, Roelen HCPF, IJzerman AP, Danhof M (1998) Selectivity of action of 8-alkylamino analogues of N6-cyclopentyladenosine in vivo: haemodynamic versus anti-lipolytic responses in rats. Br J Pharmacol 124(3):607–618
Van Steeg T, Krekels EHJ, Danhof M, de Lange ECM (2007) Experimental alteration of serum AGP and albumin concentrations in the rat; an approach to assess the impact of changes in serum protein binding on pharmacodynamics. J Pharmacol Toxicol Methods 56:72–78
Van Steeg T, Krekels EHJ, Freijer J, Danhof M, de Lange ECM (2010) Effect of altered AGP plasma binding on heart rate changes by S(-)-propranolol in rats using mechanism-based estimations of in vivo receptor affinity (KB, vivo). J Pharm Sci 99(5):2511–2520
Van Steeg TJ, Boralli VB, Krekels EHJ, Slijkerman P, Freijer J, Danhof M, de Lange EC (2009) Influence of plasma protein binding on pharmacodynamics: estimation of in vivo receptor affinities of b blockers using a new mechanism-based PK–PD modelling approach. J Pharm Sci 98(10):3816–3828
Veng-Pedersen P, Mandema JW, Danhof M (1991) A system approach to pharmacodynamics. III: An algorithm and computer program, COLAPS, for pharmacodynamic modeling. J Pharm Sci 80(5):488–495
Visser SA, Wolters FL, Gubbens-Stibbe JM, Tukker E, Van Der Graaf PH, Peletier LA, Danhof M (2003) Mechanism-based pharmacokinetic/pharmacodynamic modeling of the electroencephalogram effects of GABAA receptor modulators: in vitro-in vivo correlations. J Pharmacol Exp Ther 304(1):88–101
Visser SAG, Gladdines WWFT, van der Graaf PH, Peletier LA, Danhof M (2002) Neuroactive steroids differ in potency but not in intrinsic efficacy at the GABAA receptor in vivo. J Pharmacol Exp Ther 303(6):616–626
Vorobyov VV, Schibaev NV, Morelli M, Carta AR (2003) EEG modifications in the cortex and striatum after dopaminergic priming in the 6-hydroxydopamine rat model of Parkinson’s disease. Brain Res 97(2):177–185
Wang Y, Wei Y, Sawchuk RJ (1997) Zidovudine transport within the rabbit brain during intracerebroventricular administration and the effect of probenecid. J Pharm Sci 86(12):1484–1490
Wang YF, Welty DF (1996) The simultaneous estimation of the influx and efflux blood-brain barrier permeabilities of gabapentin using a microdialysis-pharmacokinetic approach. Pharm Res 13:398–403
Watson J, Wright S, Lucas A, Clarke KL, Viggers J, Cheetham S, Jeffrey P, Porter R, Read KD (2009) Receptor occupancy and brain free fraction. Drug Metab Dispos 37:753–760
Westerhout J, Danhof M, de Lange EC (2011) Preclinical prediction of human brain target site concentrations: considerations in extrapolating to the clinical setting. J Pharm Sci 100(9): 3577–3593
Westerhout J, Ploeger B, Smeets J, Danhof M, de Lange ECM (2012) Physiologically based pharmacokinetic modeling to investigate regional brain distribution kinetics in rats. AAPS J 14(3):543–553
Westerhout J, Smeets J, Danhof M, de Lange ECM (2013) The impact of P-gp functionality on non-steady state relationships between CSF and brain extracellular fluid. J Pharmacokinet Pharmacodyn 40(3):327–342
Wijnholds J, de Lange ECM, Scheffer GL, van den Berg D-J, Mol CAAM, van der Valk M, Schinkel AH, Scheper RJ, Breimer DD, Borst P (2000) Multidrug resistance protein 1 protects the choroid plexus epithelium and contributes to the blood-cerebrospinal fluid barrier. J Clin Invest 105:279–285
Williams SA, Davson H, Segal MB (1995) Transport of the nucleoside thymidine, in the central nervous system: the blood-cerebrospinal fluid and blood-brain barriers. In: Greenwood J, Begley DJ, Segal MB (eds) New concepts of a blood-brain barrier. Plenum, New York
Wong SL, Van Belle K, Sawchuk RJ (1993) Distributional transport kinetics of zidovudine between plasma and brain extracellular fluid/cerebrospinal fluid in the rabbit: investigation of the inhibitory effect of probenecid utilizing microdialysis. J Pharmacol Exp Ther 264(2):899–909
Wong SL, Wang Y, Sawchuk RJ (1992) Analysis of zidovudine distribution to specific regions in rabbit brain using microdialysis. Pharm Res 9(3):332–338
Xie R, Bouw MR, Hammarlund-Udenaes M (2000) Modelling of the blood-brain barrier transport of morphine-3-glucuronide studied using microdialysis in the rat: involvement of probenecid-sensitive transport. Br J Pharmacol 131(8):1784–1792
Xie R, Hammarlund-Udenaes M, de Boer AG, de Lange ECM (1999) The role of P-glycoprotein in blood-brain barrier transport of morphine: transcortical microdialysis studies in mdr1a (−/−) and mdr1a (+/+) mice. Br J Pharmacol 128:563–568
Yang Z, Brundage RC, Barbhaiya RH, Sawchuk RJ (1997) Microdialysis studies of the distribution of stavudine into the central nervous system in the freely-moving rat. Pharm Res 14(7):865–872
Yassen A, Olofsen E, Kan J, Dahan A, Danhof M (2007) Animal-to-human extrapolation of the pharmacokinetic and pharmacodynamic properties of buprenorphine. Clin Pharmacokinet 46:433–447
Zhang Y, Schuetz JD, Elmquist WF, Miller DW (2004) Plasma membrane localization of multidrug resistance-associated protein homologs in brain capillary endothelial cells. J Pharmacol Exp Ther 311:449–455
Zlokovic BV, Skundric DS, Segal MB, Colover J, Jankov RM, Pejnovic N, Lackovic V, Mackic J, Lipovac MN, Davson H et al (1989) Blood-brain barrier permeability changes during acute allergic encephalomyelitis induced in the guinea pig. Metab Brain Dis 4(1):33–40
Zlokovic BV (2010) Neurodegeneration and the neurovascular unit. Nat Med 16(12):1370–1371
Zuideveld KP, van der Graaf PH, Peletier LA, Danhof M (2007) Allometric scaling of pharmacodynamic responses: application to 5-Ht1A receptor mediated responses from rat to man. Pharm Res 24:2031–2039
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2014 American Association of Pharmaceutical Scientists
About this chapter
Cite this chapter
de Lange, E.C.M. (2014). PKPD Aspects of Brain Drug Delivery in a Translational Perspective. In: Hammarlund-Udenaes, M., de Lange, E., Thorne, R. (eds) Drug Delivery to the Brain. AAPS Advances in the Pharmaceutical Sciences Series, vol 10. Springer, New York, NY. https://doi.org/10.1007/978-1-4614-9105-7_9
Download citation
DOI: https://doi.org/10.1007/978-1-4614-9105-7_9
Published:
Publisher Name: Springer, New York, NY
Print ISBN: 978-1-4614-9104-0
Online ISBN: 978-1-4614-9105-7
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)