
Abstract
Proteases play a fundamental role in multiple biological and pathological conditions including cancer. They contribute to cancer development and promotion by regulating the activities of growth factors/cytokines and signalling receptors, as well as the composition of the extracellular matrix, thereby suppressing cell death pathways and activating cell survival pathways. With strong evidence of protease involvement in cancer, proteases serve an important role in anticancer drug development. In this review we will first introduce key proteases along with their function in tumorigenesis. Finally we will discuss the key proteases as viable therapeutic targets for anticancer drug development. Further elucidation of the role of proteases in cancer will allow us to design more effective inhibitors and novel protease-based drugs for clinical use.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others


Keywords
- Caspases
- Cysteine cathepsins
- Urokinase-type plasminogen activator
- Kallikreins
- Matrix metalloproteinases
- A disintegrin and metalloproteinases
- A disintegrin and metalloproteinase with thrombospondin motifs
- Protease-activated prodrugs
1 Introduction
Proteases refer to a group of enzymes whose catalytic function is to hydrolyze peptide bonds of proteins. Till recent times, they were essentially considered to be protein degrading enzymes having nonspecific functions in protein catabolism. Recent developments in this field indicate that proteases can cleave specific substrates, thereby having an influence on the varied vital processes and pathological conditions. An insight into the genome of human and other model organisms has revealed the impressive diversity existing in protease functions. There are at least 569 proteases and homologues produced by human cells there by forming a human degradome. These proteases and their homologues are further classified into five classes: 194 metalloproteinases, 176 serine, 150 cysteine, 28 threonine, and 21 aspartic proteases (Fig. 12.1) [1].

Classification of human degradome. On the basis of the mechanism of catalysis, the human proteases are classified into 5 different classes: metallo, serine, cysteine, threonine, and aspartic proteases
The number as well as diversity of these proteases is indicative of their importance in the biological processes. Proteases have been found to regulate the fate, localization, and activity of many proteins, modulate protein–protein interactions, create new bioactive molecules thereby contributing to the processing of cellular information, and generate, transduce, and amplify molecular signals. A direct result of these multiple actions is that proteases influence DNA replication and transcription, cell proliferation and differentiation, adhesion, tissue morphogenesis and remodelling, angiogenesis, stem cell mobilization, autophagy, senescence, necrosis, apoptosis, and evasion of immune system [2]. Therefore, proteases have an influence on cell behavior, survival, and death of all organisms [3]. Alterations in proteolytic systems can lead to multiple pathological conditions such as neurodegenerative disorders, inflammatory and cardiovascular diseases, and cancer.
Given the role of proteases in protein degradation and tissue remodelling, they have been suggested to be involved in cancer invasion and metastasis, which accounts for a majority of lethal outcomes related to cancer [4]. This was first proposed by Fisher in 1946. Following this, many individual proteases were identified to have a role in cellular invasion. Intracellular proteases like cysteine and aspartyl proteases take part in removing damaged or undesirable products and degradation of endocytosed proteins [5]. Other intracellular proteases like cysteine proteases (of caspase family of proteins) and autophagins regulate proteolytic activities which lead to apoptosis and autophagy, respectively [6, 7]. All the intracellular proteases, including the deubiquitinases, confer protection to the cell via proteolytic cascades, and loss of function mutations in these proteases leads to various human cancers [6, 8]. In contrast to the protective mechanism of intracellular proteases, extracellular proteases take part in facilitating tumorigenesis. Activation of oncogenic transcriptional pathways often leads to over expression of these enzymes in the tumor tissues [9].
The ability of cancer cells to invade normal tissues and cross physiological barriers depends on proteolytic function of these proteins. In order to metastasize, tumor cells must cross the basement membrane which comprises a continuous and dense network of collagen, glycoproteins, and proteoglycans. Recent data suggest that the action of proteases present within and on the surface of cells results in local proteolysis which helps in the movement of the cells from their primary location to a distant location. Metastasis basically involves a sequence of events wherein a cell or group of cancer cells attach to the underlying basement membrane, intravasate into the vasculature, survive in the circulation, arrest at a distant vasculature bed, extravasate into surrounding tissues, and proliferate into a secondary tumor where proteases play a major role [10]. In the pages that follow, you will be introduced to some of the important proteases which have been shown to play key roles at different stages of cancer progression.
2 Caspases
Caspases (cysteine-dependent aspartyl-specific protease) belong to a family of cysteine proteases that mediate proteolytic events indispensable for biological phenomena such as cell death and inflammation. To date, a number of caspases have been identified in various vertebrate and invertebrate species. In humans, 11 caspases including caspase 1 to caspase 10 and caspase 14 have been identified. Several additional caspases, including caspase 11, caspase 12, and caspase 13, have been detected in other mammals such as rodents and the cow Bos taurus. To date 14 mammalian caspases have been broadly categorized into initiators (caspases 2, 8, 9, and 10), effectors (caspases 3, 6, and 7), and inflammatory caspases (caspases 1, 4, 5, 11, 12 and 13). Caspase 14 is a unique caspase as it belongs to neither apoptotic caspases nor inflammatory caspases [11]. Recently, caspases 15, 16, 17, and 18 have also been identified as new members of the caspase family in vertebrates, although their function has not yet been identified [12–14]. In addition, fish-specific caspases have been found: caspy, caspy2, and caspase recruitment domain (CARD)-casp8 [15, 16]. Not limited to vertebrates, caspases have been identified in a wide variety of animals such as sponge Geodia cydonium, Hydra vulgaris, sea anemone Aiptasia pallida, nematode Caenorhabditis elegans, fruit fly Drosophila melanogaster, sea urchin Strongylocentrotus purpuratus, and ascidians Ciona intestinalis and Ciona savignyi [17–25].
Since apoptosis is central to tumorigenesis, the role of caspases has been a subject of great interest. Literature reveals that caspase alterations are not rare in a variety of tumors which might result from mutations, promoter methylation, alternative splicing, and posttranslational modifications. Some of these defects lead to loss of function, but in other cases mutated caspases act as dominant negatives preventing the activation of wild-type protein [26]. The importance of caspase 8 was brought to light by showing that knockout of caspase 8 leads to embryonic lethality. In contrast, another study pointed out that the deletion or silencing of Caspase 8 gene promotes cell survival and metastasis in neuroblastoma [6, 27]. Somatic mutations leading to inactivation of Caspase 8 gene have also been seen in tumors of head and neck, lung, colorectal, and gastric tumors [28, 29]. Following this, genetic alterations in other caspases have also been observed, and it has been pointed out that Caspase 10 is also frequently mutated in tumors [30, 31]. Presence of mutation in Caspase 8, Caspase 10, and other caspase family members suggests their involvement in progression of malignant tumors.
Literature further reveals that altered caspase function can also be a consequence of modified expression of their specific inhibitors, for example, cFLIPs that competes with caspase 8 for FADD binding, thereby preventing its activation. cFLIPs is often elevated in tumors, while its downregulation has been shown to sensitize tumor cells towards therapy [32]. Among caspase inhibitors an important role is played by inhibitors of apoptosis (IAPs). While initially described as caspase inhibitors, IAPs are now recognized to regulate a multitude of other cellular functions including regulation of the immune response, cell migration, mitosis, and proliferation [33]. Alterations of IAPs are found in a variety of human cancers and are associated with poor prognosis and resistance to therapy. In some cases however, loss of IAPs correlates with tumor progression complicating the issue and suggesting that the role of IAPs has to be carefully evaluated based on cell context.
3 Cysteine Cathepsins
Cathepsins were originally identified as endopeptides that are located in the lysosomes, while recent reports have uncovered nontraditional roles for cathepsins in the extracellular space as well as in the cytosol and nucleus [34, 35]. Cysteine cathepsins specifically are capable of efficiently cleaving a wide variety of substrates and thought to participate in protein turnover. They comprise 11 proteases that show increased expression in tumors and are referred to as clan CA, family C1a: cathepsins B, C (also known as cathepsin J and dipeptidyl-peptidase 1), F, H, K (also known as cathepsin O2), L, O, S, W, V (also known as cathepsin L2), and Z (also known as cathepsin X and cathepsin P) in humans.
Cysteine cathepsins are highly upregulated in a wide variety of cancers by mechanism which includes gene amplification, transcript variation (arises from the use of alternate promoters and alternative splicing), transcriptional regulation, posttranscriptional regulation, and epigenetic regulation to name a few [5]. Presence of diversity in the expression of specific cysteine cathepsins in tumor cells and tumor-associated cells at different times during neoplastic progression indicates that individual enzymes have distinct roles during progression in the various cell types that comprise the tumor microenvironment and in the tumor cells. The pattern of expression often varies with tumor type and the cellular composition of the tumors. Increase in the expression of cysteine cathepsins occurs in premalignant or early lesions, for example, cathepsin B in Barrett’s esophagus and stage I esophageal tumors [36, 37], cathepsin H in node-negative lung tumors [38], and cathepsin S and cathepsin X in high-grade prostatic intraepithelial neoplasias. In addition to these in ductal carcinoma in situ of the breast, there is increased expression of the cysteine cathepsins F, K, and L [39]. Cathepsin S, which is increased in stage IV astrocytomas, is found in both tumor cells and tumor-associated macrophages [40], as has been reported for cathepsin B in colon carcinomas and observed for cathepsin B in transgenic mouse mammary tumors. Moreover, overexpression of Cathepsin B gene has also been found in esophageal carcinoma and transformed rat ovarian cells. Recent studies also show that increased expression of Cathepsin B is associated with breast, lung, gastric, colorectal, and prostate carcinomas, melanomas, gliomas, and osteoclastomas and with low survival rates in patients with colorectal cancer [10]. Cathepsin B can cleave a wide variety of substrates including extracellular matrix proteins, proteinases, as well as proteinase inhibitors. Reports suggest that Cathepsin B is involved in detachment of migrating cancer cells, and inhibitors of cysteine cathepsins can reduce the proteolysis and migration of oral squamous cell carcinoma cells. Inhibitors against intracellular Cathepsin B have been shown to reduce the invasiveness of human melanoma and prostate carcinoma cells [5]. Cysteine cathepsins are also upregulated during HPV16-induced cervical carcinogenesis, further encouraging consideration of this protease family as a therapeutic target in human cancers. In contrast to these studies, deletion of Cathepsin L in HPV16-induced skin carcinogenesis mouse model leads to formation of early onset aggressive tumors. Keratinocytes with homozygous deletion of Cathepsin L show increased proliferation rates, suggesting a tumor suppressor function of Cathepsin L with respect to squamous cell skin carcinoma [41]. Table 12.1 shows the distribution of cysteine cathepsins in tumor and tumor-associated cells that express cysteine cathepsins.
4 Urokinase-Type Plasminogen Activator
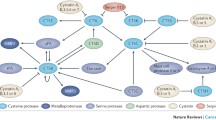
Urokinase-type plasminogen activator (uPA) is an extracellular proteolytic enzyme belonging to serine protease family of enzymes. The uPA system comprises urokinase-type plasminogen activator, 2 inhibitors of plasminogen activators, namely, PAI-1 and PAI-2, and the urokinase receptor (uPAR) [42, 43]. The binding of uPA to the cell surface receptor uPAR leads to activation of uPA, and it further cleaves the surface-associated plasminogen into the serine protease called plasmin which in turn is involved in a number of pathophysiological processes requiring basement membrane (BM) or extracellular matrix (ECM) remodelling, including tumor progression and metastasis (Fig. 12.2) [44, 45]. Plasmin can also activate specific growth factors like FGF2, VEGF, IGF-2, and HGF that stimulate cell proliferation/mitogenesis [46–48]. FGF-2 and VEGF the two well-known stimulators of endothelial cell growth have also been found to play a role in angiogenesis [49].

Schematic representation of the PAS system. Given illustration represents the proteolytic cascade which leads to generation of plasmin
Preliminary evidences support the role of uPA system in early stages of tumorigenesis. The expression of both uPAR and uPA is significantly upregulated during cancer progression and is primarily confined to the tumor-associated stromal compartment. uPA, plasminogen, and plasmin have also been shown to play roles in cell migration and adhesion [42]. Increased expression of uPAR in virtually all human cancers suggests possible clinical applications as diagnostic marker, predictive tool of survival or clinical response, and as a target for therapy and imaging. In fact, increased expression of uPA/uPAR and PAI-1 in tumors shows strong correlation with metastatic potential and lower rates of patient prognosis [50] and indicates poor survival. uPA/PAI-1 has been designated as a prognostic markers associated with poor disease outcome for early stage breast cancer and has been recommended by the American Society of Clinical Oncology for screening in routine clinical practice [51]. The cleaved forms of uPAR are also prognostic markers and a potential diagnostic, and predictive impact of the different uPAR forms has been reported [52, 53].
Literature further reveals that in comparison to wild-type mice, uPA-deficient mice which have been chemically induced with blue nevi failed to progress to melanomas [54]. In vivo studies have shown that uPA and matrix-metalloproteases (MMPs) work together for the degradation of the ECM thereby facilitating metastasis. Recent data have also provided new insights into the role of uPAR in gastric cancer progression, and in addition to mediating proteolysis, this receptor also appears to mediate cell signalling, proliferation, and survival, and these observations have revealed novel ways to target uPAR. Dual role has also been suggested for uPA in angiogenesis depending on the stage of angiogenesis. In the beginning, uPA helps by degrading the ECM and promoting the proliferation of endothelial cells, while later on it might activate angiostatin (inhibitor of angiogenesis) formation [55].
5 Kallikreins
Kallikrein (Greek synonym for pancreas: kallikreas) was named by the three German scientists H. Kraut, E.K. Frey, and E. Werle, who in 1930 reported that the pancreas is a rich source of this endogenous hypotensive substance. The human kallikrein gene locus spans a region of 2, 61,558 bp on chromosome 19q13.4. It is formed of 15 tandemly localized kallikrein genes with no intervention from other genes and is the largest cluster of serine proteases within the human genome.
Kallikreins are basically serine proteases which are responsible for the coordination of various physiological functions including blood pressure, semen liquefaction, and skin desquamation [56]. The expression of kallikreins has also been found to be altered in hormonally regulated human carcinomas, and there have been reports in the literature that suggest that kallikreins might function as tumor-promoting or tumor-suppressing enzymes on the basis of hormonal balances and tissue type. Prostate-specific antigen (PSA) kallikrein 3 is proposed to promote tumorigenesis by initiation of growth factors and proteolytic degradation of the ECM. Kallikrein 3, the most commonly known kallikrein, is a useful biomarker that aids in the diagnosis, staging, and follow-up of prostate cancer. Apart from KLK3 several other kallikreins, including kallikreins 2 (KLK2) and 11 (KLK11), are also emerging as complementary prostate cancer biomarkers [57, 58]. Kallikrein 4 has also been found to be overexpressed in prostate cancer [59, 60]. Along with these kallikreins, several others have been implicated in the other cancers. For example, KLK5, KLK6, KLK7, KLK10, KLK11, and KLK14 are emerging biomarkers for ovarian cancer [61–63], and kallikrein 1, also known as tissue kallikrein, cleaves kininogen to release the vasoactive kinin peptide, bradykinin, or lysyl bradykinin. Kallikrein 5 is widely expressed but found at high levels in skin, breast, brain, and testis, and its overexpression is an indicator of aggressive ovarian tumors which result in poor patient prognosis [64, 65]. Kallikrein 8 is expressed in the brain and is a novel marker of ovarian and cervical cancer. It is worth mentioning here that upregulation of 12 kallikrein genes has been found in ovarian cancer. KLK3, KLK8, and KLK10 have been shown to have tumor suppressive functions [66, 67]. Owing to their interaction with other serine proteases like uPA and its receptor uPAR, human kallikreins have been implicated in tissue invasion, angiogenesis, and metastasis. Furthermore, KLKs can activate MMPs like collagenase IV and thereby promote tumorigenesis. KLK2 and KLK4 can inactivate PAI-1 and in turn activate the uPA pathway [68]. Controversies still exist on the role of KLK2 in angiogenesis. While KLK2 can activate tumor growth factor β (TGF-β) and promote angiogenesis, it is also known to block FGF 2 and hence inhibit angiogenesis. Furthermore, KLKs can activate MMPs like collagenase IV and thereby promote tumorigenesis. Due to their role as biomarkers, KLKs are used for the screening, diagnosis, prognosis, and monitoring of various cancers, e.g., prostate, ovarian, breast, testicular, and lung; human tissue kallikreins (KLKs) are attracting increased attention these days.
6 Matrix Metalloproteinases
This family consists of 23 zinc-dependent endopeptidases which are expressed during processes involving tissue remodelling like embryonic development, wound healing, uterine and mammary involution, cartilage to bone transition during ossification, and trophoblast invasion into endometrial stoma during placenta development [69–72]. This extracellular remodelling property of MMPs has implications in pathological processes like periodontitis and rheumatoid arthritis. Recent findings further provide evidence that MMPs can modulate the different stages of tumor formation which include tumor growth, invasion, metastasis, and angiogenesis.
MMP-9 has also been found to enhance endothelial cell growth in vitro [73, 74], and MMP-8 when overexpressed in breast cancer cells reduces their metastatic potential. Literature also reveals that MMP-8-deficient mice show increased incidence of skin carcinomas [75]. Furthermore, MMP-12, which is mainly expressed in macrophages, has been shown to reduce tumor growth rates in mice [76]. However, the precise role of MMP-12 in human cancers is not yet clear. This stems from studies supporting its dual role: its expression in colon and hepatocellular carcinomas has been found to have favorable outcomes, while its expression in other tumors correlates with poor prognosis of patients [77–79]. Dual roles have been reported for other MMPs like MMP-3, MMP-9, and MMP-11 which have been associated with tumor progression and in some instances with antitumor effect [1].
MMPs facilitate metastasis and angiogenesis by degrading the physical barriers and allowing increased signalling by signalling molecules like growth factors and cytokines. MMPs mediate cleavage of the ectodomain of VE-cadherins which leads to loss of cell–cell adhesions. In vivo studies show that MMP-7 (matrilysin) is necessary for endothelial cell proliferation, and upregulation of other MMPs like MMP-1 and MMP-2 causes induction of angiogenesis [80]. In comparison to quiescent vessels, angiogenic and tumor blood vessels contain exposed cryptic binding sites for αvβ3 which is brought by cleavage of type IV collagenase by MMPs. This also correlates with increased expression of MMP-2 which binds to αvβ3 and facilitates angiogenesis [81, 82]. Further studies on these enzymes showed that loss of MMP-8 causes abnormalities in inflammatory response induced by carcinogens leading to sustained inflammation thereby generation of a favorable environment for tumor development.
Another zinc-dependent metalloprotease family is the disintegrin and metalloproteinase which include two subgroups: the membrane-bound ADAM and a disintegrin and metalloproteinase with thrombospondin motifs (ADAMTS) [83, 84]. This family of proteases bears structural relationships with other MMPs. ADAMs and ADAMTS have been implicated in different stages of cancer progression. ADAMs mediate the shedding of cytokines and growth factors and regulate the fusion of membranes and motility of cells as well as muscle development, fertilization, and cell fate determination [85]. ADAM17 plays a major role in inflammatory processes by facilitating the shedding of TNF-α [86]. ADAM10 and ADAM12 have been found to be overexpressed in a number of carcinoma tissues and cell lines consistent with their ability to regulate adhesion and motility of cells [87]. Reports show that ADAMTS1, the first identified member of ADAMTS family, inhibits angiogenesis and reduces growth of tumor and metastasis [88–90]. Table 12.2 shows role of different MMPs and their implications in tumorigenesis.
7 Proteases as Viable Therapeutic Targets for Anticancer Drug Development
7.1 Caspases as Drug Targets
Cell death inhibition is a very successful strategy that cancer cells employ to combat the immune system and various anticancer therapies. An alteration in the apoptotic signalling pathway is one of the main reasons for tumorigenesis. Hence, components as well as triggers and regulators of this pathway are among the most promising targets for pharmacological interventions with respect to cancer. Currently chemotherapeutic treatments aim to promote cellular toxicity and damage which in turn induces apoptosis either directly or indirectly via caspases. Approaches are being developed that will activate death receptor pathways, synthetically activate caspases, restore the activity of tumor suppressor genes such as p53, and counteract the effects of antiapoptotic factors. Among these approaches, small molecules are in clinical trials against several antiapoptotic players, namely, IAP proteins and the Bcl-2 protein. Cellular IAP proteins regulate expression of antiapoptotic molecules and prevent assembly of proapoptotic protein-signalling complexes. In addition, amplifications, mutations, and chromosomal translocations of IAP genes are associated with various malignancies. Several therapeutic strategies have been designed to target IAP proteins, including a small-molecule approach that is based on mimicking the IAP-binding motif of an endogenous IAP antagonist—the second mitochondrial activator of caspases (Smac). Other strategies involve antisense nucleotides and transcriptional repression. Inhibitors of IAPs like second mitochondria-derived activator of caspase/direct IAP-binding protein with low pI (Smac/DIABLO) and heat-inducible serine protease (A2HtrA2) have increased the interest of the pharmacological industry in them. Omi/HtrA2 is a mitochondrial serine protease that is released from mitochondria during apoptosis. It binds to IAP and antagonizes its binding to caspase 9, thereby modulating the caspase activity. Another member of the IAP family is survivin, which plays a role in apoptosis as well as cell cycle regulation. Caspase 3 and cyclin-dependent kinase inhibitor p21 have been found to colocalize with survivin. Regardless of its function as an inhibitor of caspase 3 or as a regulator of cell cycle, downregulation of survivin affects the growth of transformed cells. As many of these processes are often modified in cancer, it is clear how alteration of IAPs can play a role in tumorigenesis. The most important pathway regulated by IAPs that contributes to cancer development is the NF-kB signalling pathway, and XIAP, cIAP1, and cIAP2 have been shown to regulate this pathway and in turn regulate inflammation, immunity, and cell survival. Moreover evidence is there in the literature that IAPs protect from TNF-α killing. In addition, recent findings show a role for IAPs in metastasis also as XIAP/survivin complex has been found to trigger the NF-kB pathway leading to activation of cell motility kinases [91]. This however is still a controversial issue, and other studies show a suppressive effect of IAPs on cell mobility, therefore raising the need for further investigations. Experiments with administration of antisense nucleotides and ribozymes against survivin have also been shown to induce apoptosis in various cell lines. This finding has triggered the development of antisense-based strategies which target the expression of survivin. In any case due to their involvement in cancer progression and their ability to suppress apoptosis IAPs have become an attractive therapeutically target, leading to the development of IAP inhibitors, some of which are based on natural inhibitors such as Smac/DIABLO [91–93]. These drugs appear to be able to directly kill cancer cells or at least sensitize them to other killing agents while sparing normal cells. A number of these compounds are currently entering clinical trials; Table 12.3 [94].
Bcl-2 is an antiapoptotic molecule and overexpression of Bcl-2 protein has been reported in many types of cancers, including leukemia, lymphomas, and carcinomas. Bcl-2 has also been associated with chemotherapy resistance in various human cancers. Thus targeted inhibition of Bcl2 can be used as a tool for the treatment of different cancers. Several classes of drugs have been found to regulate gene expression of antiapoptotic Bcl-2 members, and several of these are in different phases of clinical trials. Until recently, most research efforts aimed at developing anticancer tools were focusing on small molecules. Alternative compounds are now being increasingly assessed for their potential anticancer properties, including peptides and their derivatives. Most anticancer peptidic compounds induce apoptosis of tumor cells by modulating the activity of Bcl-2 family members that control the release of death factors from the mitochondria. Some of these peptides have been shown to inhibit the growth of tumors in mouse models. Several agents targeting antiapoptotic Bcl-2 family of proteins are also in preclinical/clinical trials (Table 12.4).
7.2 Cysteine Cathepsins as Drug Targets
An increased cell proliferation rate represents a key aspect of tumor biology, and cysteine cathepsins have been discovered to influence the regulation of cell proliferation by several means. Upregulation of cysteine cathepsins has been reported in many human tumors, including breast, lung, brain, gastrointestinal, prostate, and melanoma. Knockout of specific cathepsins in mice has confirmed that targeting of individual cysteine cathepsins can prove to be a beneficial strategy for cancer treatment. Avascular tumors are severely restricted in their growth potential because of the lack of blood supply, and it is well known that angiogenesis is required for invasive tumor growth and metastasis and constitutes an important point in the control of cancer progression. Therefore, inhibition of angiogenesis is a valuable approach to cancer therapy. There is also increasing evidence that cysteine cathepsins promote invasion and metastasis by remodelling the extracellular matrix (ECM) in the tumor microenvironment. Active cathepsins have been shown to be able to degrade the protein components of basement membranes and the interstitial connective matrix including laminin, fibronectin, elastin, tenascin, and various types of collagen. Following the report by Szpaderska et al. that inhibitors of intracellular cathepsins can reduce the invasiveness of human melanoma and prostate carcinomas, they are being studied for anticancer therapies. Endogenous inhibitors of these enzymes, known as cystatins, are also being used to reduce tumor growth, invasion, metastasis, and angiogenesis. Administration of small molecule inhibitors of cathepsins or increasing the expression of endogenous inhibitors is suggested to be of therapeutic benefit. Presently, only one small molecule inhibitor has been successful which is a broad-spectrum inhibitor and targets the intracellular and extracellular pool of cathepsins. Therapeutic agents that can be activated by subsequent cleaving at the tumor cell surface by cathepsins have also proven to be efficacious. Such therapeutic agents generally contain a pore forming toxins conjugated to cathepsin B cleavable linkers. Prodrugs which can be cleaved by cathepsins are also being developed, for example, prodrugs of doxorubicin [5].
7.3 uPA System as Drug Targets
Breast cancer is one of the most common malignancies and is responsible for many deaths. The plasminogen activation system (PAS) has been found to be frequently upregulated in metastatic breast cancer and also correlates with poor prognosis of patients. Many antimetastatic prophylactic drugs are being developed which target the PAS. Inhibitors of uPA and the interaction of uPA with its cell surface receptor (uPAR) are promising molecules for drug development. Known inhibitors bind to the S1 subsite of uPA which forms a salt bridge with the negatively charged Asp189 residue by incorporation of positive charges. Essentially all uPA inhibitors which are being used as antimetastatic drugs retain the crucial interaction with improvisations in the pharmacokinetics of the positively charged molecules. An example is amiloride that is a potassium-sparring diuretic that has been reported to prevent lung metastasis in a rat adenocarcinoma model. It has also been found to significantly reduce metastasis of MATB rat mammary cancer cells. Amiloride as well as B428, another uPA inhibitor, have showed the potential to reduce the invasive capacity of two breast cancer cell lines, namely, MDA-MB-231 and MDA-MB-436. Other small molecule inhibitors of uPA are tranexamic acid, aprotinin, and leupeptin. Arginine mimetics which bears either an N-tri-isopropyl-phenylsulfonamide group or 4-amidinobenzylamine group are also being considered as potent inhibitors. Besides this cyclic peptide antagonists who have the potential to displace the uPA molecules bound to the surface receptors and hence inhibit the tumor cell associated activation of plasminogen and fibrin degradation [50]. The interaction between uPA and its receptor uPAR has also been investigated to find possible strategies of intervention. This has devised the development of linear peptide antagonists which compete with uPA for the binding site/epitope on its receptor.
7.4 MMPs as Drug Targets
Given the positive correlation between tumor aggressiveness and the levels of proteases, these enzymes have become a subject of interest for development of anticancer drugs. MMPs regulate the destruction of the extracellular matrix which facilitates malignant invasion making them a suitable target for drug development. One such strategy involves the use of tissue inhibitor of metalloproteinases (TIMPs) or TIMP fragments as direct inhibitors of MMP activation or activity. Another strategy involves using peptide inhibitors which mimic the amino terminal motif of the MMPs which contain the latent state enzyme. A third strategy involves using synthetic compounds as competitive inhibitors of substrates which bind to the active site of the enzymes. In vivo studies have shown that TIMP-1, TIMP-2, and synthetic substrate inhibitors can be used to block angiogenesis. In vivo studies in animal models provide evidence that TIMP-1 inhibits invasion and metastasis. Simultaneously, TIMP-2 has also been shown to inhibit cell invasiveness in in vitro and in vivo studies. An example of synthetic inhibitors of MMPs is BB94 whose administration to athymic nude mice-bearing fragments of colon cancer inhibited the growth of primary tumor and a reduction in incidence of tumor invasion and spontaneous metastasis. In a separate study it has also been shown to delay growth of B16-BL6 transplanted primary melanoma. Chemically modified tetracycline derivatives have also been shown to inhibit the enzyme activity as well as the synthesis of MMPs by blocking its transcription. These compounds inhibit MMPs by chelating the metal ions like zinc and calcium. A few examples of these compounds are metastat, minocycline, and doxycycline. Doxycycline is currently the only approved drug which is being used against periodontitis, brain tumor, and Kaposi sarcoma [86].
8 Development of Protease-Activated Prodrugs
As mentioned above, proteases are molecules important for the development of anticancer drugs. Besides being target molecules for therapeutic drugs, proteases are also instrumental molecules for development of prodrugs. Prodrugs are derivatives of therapeutic agents which upon chemical or environmental stimuli release the parent drug. They are inactive molecules in their native state and are transition to active drugs upon being acted by a stimulus. Given that proteases lead to selective cleavage of their substrates, they have been considered for development of protease-activated prodrugs (PAP), ensuring an efficient delivery and efficacy of the drug with minimum toxic effects on healthy cells. They are made up of a parent drug conjugated to a polymer or peptide substrate via a cleavable linkage and/or a targeting moiety for specific delivery, which in the case of PAPs should be stable in blood stream till it reaches target protease [95]. Examples are prodrugs made by conjugating single amino acids or dipeptides to cancer drugs like doxorubicin and daunorubicin. For further information please go through reference [95].
9 Conclusions
Proteases are involved in a wide variety of functions like immunological responses, degradation of the articular cartilage matrix, and other pathological processes, playing a major role in both intra- and extracellular protein turnover. Apart from their role in various physiological and pathological processes, proteases have also been found to be involved in tumor growth, invasion, and migration. A number of proteases have been associated with various stages of cancer and also serve as biomarkers which can predict the prognosis of patients. Given their role in tumorigenesis, proteases are being considered as highly relevant targets. Endogenous inhibitors of proteases have been found to be suitable molecules for drug development. Synthetic peptides mimicking the inhibitors of pro-tumorigenic proteases are also being developed. Research is also going on to identify selective inhibitors of proteases rather than broad-spectrum inhibitors. In addition to this, RNA interference can also prove to be instrumental in silencing protease genes which show aberrant expression in tumors. Such therapies can prove to be useful not only in cancer but several pathological states, such as immune disorders, osteoporosis, rheumatoid, and osteoarthritis, where proteases are known to be involved. In addition to being target molecules for different anticancer drugs, proteases are now also being used for the development of protease-activated prodrugs which will ensure the efficient delivery and action of conventional anticancer drugs.
References
Lopez-Otin C, Matrisian LM (2007) Emerging roles of proteases in tumour suppression. Nat Rev Cancer. 7(10): 800-808
Lopez-Otin C, Overall CM (2002) Protease degradomics: a new challenge for proteomics. Nat Rev. Mol. Cell Biol. 3(7): 509-519
Turk B (2006) Targeting proteases: successes, failures and future prospects. Nature Rev. Drug Discov. 5(9): 785-799
Fisher A (1946) Mechanism of proteolytic activity of malignant tissue cells. Nature. 157: 442
Mohamed MM, Sloane BF (2006) Cysteine cathepsins: multifunctional enzymes in cancer. Nat Rev Cancer. 6(10): 764-775
Teitz T, Wei T, Valentine MB et al (2000) Caspase 8 is deleted or silenced preferentially in childhood neuroblastomas with amplification of MYCN. Nature Med. 6(5): 529-535
Marino G, Uría JA, Puente XS et al (2003) Human autophagins, a family of cysteine proteases potentially implicated in cell degradation by autophagy. J. Biol. Chem. 278(6): 3671-3678
Hoeller D, Hecker CM, Dikic I (2006) Ubiquitin and ubiquitin-like proteins in cancer pathogenesis. Nature Rev. Cancer. 6(10): 776-788
Egeblad M, Werb Z (2002) New functions for matrix metalloproteinases in cancer progression. Nature Rev. Cancer. 2(3): 161-174
Koblinskia JE, Ahrama M, Sloane BF (2000) Unraveling the role of proteases in cancer. Clinica Chimica Acta. 291(2): 113-135
Lippens S, Kockx M, Knaapen M et al (2000) Epidermal differentiation does not involve the pro-apoptotic executioner caspases, but is associated with caspase-14 induction and processing. Cell Death and Differentiation. 7(12): 1218–1224
Eckhart L, Ballaun C, Uthman A et al (2005) Identification and characterization of a novel mammalian caspase with proapoptotic activity. J Biol Chem. 280(42): 35077-80
Sakata S, Yan Y, Satou Y et al (2007) Conserved function of caspase-8 in apoptosis during bony fish evolution. Gene. 396(1): 134-48
Eckhart L, Ballaun C, Hermann M et al (2008) Identification of novel mammalian caspases reveals an important role of gene loss in shaping the human caspase repertoire. Mol Biol Evol. 25(5): 831-41
Masumoto J, Zhou W, Chen FF et al (2003) Caspy, a zebrafish caspase, activated by ASC oligomerization is required for pharyngeal arch development. J Biol Chem. 278(6): 4268-76
Sakamaki K, Nozaki M, Kominami K et al (2007) The evolutionary conservation of the core components necessary for the extrinsic apoptotic signaling pathway, in Medaka fish. BMC Genomics. 8: 141
Shaham S (1998) Identification of multiple Caenorhabditis elegans caspases and their potential roles in proteolytic cascades. J Biol Chem. 273(52): 35109-17
Cikala M, Wilm B, Hobmayer E et al (1999) Identification of caspases and apoptosis in the simple metazoan Hydra. Curr Biol. 9:959-62
Lamkanfi M, Declercq W, Kalai M et al (2002) Alice in caspase land. A phylogenetic analysis of caspases from worm to man. Cell Death Differ. 9(17): 358-61
Terajima D, Shida K, Takada N et al (2003) Identification of candidate genes encoding the core components of the cell death machinery in the Ciona intestinalis genome. Cell Death Differ. 10(6): 749-53
Wiens M, Krasko A, Perovic S et al (2003) Caspase-mediated apoptosis in sponges: cloning and function of the phylogenetic oldest apoptotic proteases from Metazoa. Biochim Biophys Acta. 1593(2-3): 179-89
Weill M, Philips A, Chourrout D et al (2005) The caspase family in urochordates: distinct evolutionary fates in ascidians and larvaceans. Biol Cell. 97(11): 857-66
Dunn SR, Phillips WS, Spatafora JW et al (2006) Highly conserved caspase and Bcl-2 homologues from the sea anemone Aiptasia pallida: lower metazoans as models for the study of apoptosis evolution. J Mol Evol. 63(1): 95-107
Robertson AJ, Croce J, Carbonneau S et al (2006) The genomic underpinnings of apoptosis in Strongylocentrotus purpuratus. Dev Biol. 300(1): 321-34
Kumar S (2007) Caspase function in programmed cell death. Cell Death Differ. 14(1): 32-43
Olsson M and Zhivotovsky B (2011) Caspases and cancer. Cell Death Differ. 18(9): 1441-1449
Stupack DG, Teitz T, Potter MD et al (2006) Potentiation of neuroblastoma metastasis by loss of caspase-8. Nature. 439(7072): 95-99
Mandruzzato S, Brasseur F, Andry G et al (1997) CASP-8 mutation recognized by cytosolic T lymphocytes on a human head and neck carcinoma. J. Exp. Med. 186: 785-793
Soung YH, Lee JW, Kim SY et al (2005) CASPASE-8 gene is inactivated by somatic mutations in gastric carcinomas. Cancer Res. 65(3): 815-821
Shin MS, Kim HS, Kang CS et al (2002) Inactivating mutations of CASP 10 gene in non-Hodgkin lymphomas. Blood. 99(11): 4094-4099
Park WS, Lee JH, Shin MS et al (2002) Inactivating mutations of caspase- 10 gene in gastric cancer. Oncogene. 21(18): 2919-2925
Kataoka T (2005) The caspase-8 modulator c-FLIP. Crit Rev Immunol. 25(1): p. 31-58
Gyrd-Hansen M, Meier P (2010) IAPs: from caspase inhibitors to modulators of NF-kappaB, inflammation and cancer. Nat Rev Cancer. 10(8): 561-574
Rawlings ND, Morton FR, Kok CY et al (2008) MEROPS: the peptidase database. Nucleic Acids Res. 36(Database issue): D320–D325
Xia L, Kilb J, Wex H et al (1999) Localization of rat cathepsin K in osteoclasts and resorption pits: inhibition of bone resorption and cathepsin K-activity by peptidyl vinyl sulfones. Biol. Chem. 380(6): 679–687
Shuja S, Murnane MJ (1996) Marked increases in cathepsin B and L activities distinguish papillary carcinoma of the thyroid from normal thyroid or thyroid with non-neoplastic disease. Int J Cancer. 66(4): 420–6
Hughes SJ, Glover TW, Zhu XX et al (1998) A novel amplicon at 8p22–23 results in overexpression of cathepsin B in esophageal adenocarcinoma. Proc. Natl Acad. Sci. USA. 95(21): 12410–12415
Linnerth NM, Sirbovan, K, Moorehead RA (2005) Use of a transgenic mouse model to identify markers of human lung tumors. Int. J. Cancer. 114(6): 977–982
Allinen, M, Beroukhim R, Cai L et al (2004) Molecular characterization of the tumor microenvironment in breast cancer. Cancer Cell. 6(1): 17–32
Flannery T, Gibson D, Mirakhur M et al (2003) The clinical significance of cathepsin S expression in human astrocytomas. Am. J. Pathol. 163(1): 175–182
Reinheckel T, Hagemann S, Dollwet-Mack S et al (2005) The lysosomal cysteine protease cathepsin L regulates keratinocyte proliferation by control of growth factor recycling. J. Cell Sci. 118(Pt 15): 3387-3395
Andreasen PA, Kjoller L, Christensen L et al (1997) The urokinase type plasminogen activator system in cancer metastasis: a review. Int J Cancer. 72(1): 1-22
Dano K, Andreasen PA, Grondahl-Hansen K et al (1985) Plasminogen activators, tissue degradation and cancer. Adv Cancer Res. 44: 139-266
Plough M, Ellis V, Dano K (1994) Ligand interaction between urokinase type plasminogen activator and its receptor probed with 8-anilino- 1-naphthalenesulfonate: evidence for a hydrophobic binding site exposed only on the intact receptor. Biochemistry. 33(30): 8991- 8997
Zhou HM, Nichols A, Meda P et al (2000) Urokinase-type plasminogen activator and its receptor synergize to promote pathogenic proteolysis. EMBO J. 19(17): 4817-4826
Rifkin DB (1997) Cross-talk among proteases and matrix in the control of growth factor action. Fibrinol Proteolysis. 11: 3-9
Plouet J, Moro F, Bertagnolli S et al (1997) Extracellular cleavage of the vascular endothelial growth factor 189-amino acid form by urokinase is required for its mitogenic effect. J Biol Chem. 272: 13390-13396
Mars WM, Zarnegar R, Michalopoulos GK (1993) Activation of hepatocyte growth factor by the plasminogen activators uPA and tPA. Am J Path. 143(3): p. 949-958
Cross MJ, Claesson-Welsh L (2001) FGF and VEGF function in angiogenesis: signalling pathways, biological responses and therapeutic inhibition. Trends Pharmacol Sci. 22(4): 201-207.
Tyndall JDA, Kelso MJ, Clingan P et al (2008) Peptides and Small Molecules Targeting the Plasminogen Activation System: Towards Prophylactic Anti-Metastasis Drugs for Breast Cancer. Recent Patents on Anti-Cancer Drug Discovery. 3(1): 1-13
Hayes DF, Bast RC, Desch CE et al (1996) Tumor marker utility grading system: a framework to evaluate clinical utility of tumor markers. J Natl Cancer Inst. 88(20): 1456-66
Shapiro RL, Duquette JG, Roses DF et al (1996) Induction of primary cutaneous melanocytic neoplasms in urokinase-type plasminogen activator (uPA)-deficient and wild type mice: cellular blue nevi invade but do not progress to malignant melanoma in uPA-deficient animals. Cancer Res. 56: 3597-3604
Cao R, Wu HL, Veitonmaki N et al (1996) Suppression of angiogenesis and tumor growth by the inhibitor K1-5 generated by plasmin-mediated proteolysis. Proc Natl Acad Sci USA. 96(10): 5728-5733
Duggan C, Maguire T, McDermott E et al (1995) Urokinase plasminogen activator and urokinase plasminogen activator receptor in breast cancer. Int J Cancer. 61(5): 597-600
Grondahl-Hansen J, Peters HA, van Putten WLJ et al (1995) Prognostic significance of the receptor for urokinase type plasminogen activator in breast cancer. Clin Cancer Res. 1: 1079-1087
Paliouras M, Borgono C, Diamandis EP (2007) Human tissue kallikreins: The cancer biomarker family. Cancer Lett. 249: 61-79
Scorilas A, Gregorakis AK (2006) mRNA expression analysis of human kallikrein 11 (KLK11) may be useful in the discrimination of benign prostatic hyperplasia from prostate cancer after needle prostate biopsy. Biol. Chem. 387(6): 789–793
Stavropoulou P, Gregorakis AK, Plebani M et al (2005) Expression analysis and prognostic significance of human kallikrein 11 in prostate cancer. Clin. Chim. Acta 357(2): 190–195
Obiezu CV, Soosaipillai A, Jung K et al (2002) Detection of human kallikrein 4 in healthy and cancerous prostatic tissues by immunofluorometry and immunohistochemistry. Clin. Chem. 48(8): 1232–1240
Obiezu CV, Shan SJ, Soosaipillai A et al (2005) Human kallikrein 4: quantitative study in tissues and evidence for its secretion into biological fluids. Clin. Chem. 51(8): 1432–1442
Borgono CA, Michael IP, Diamandis EP (2004) Human tissue kallikreins: physiologic roles and applications in cancer. Mol. Cancer Res. 2: 257–280
Borgono CA, Diamandis EP (2004) The emerging roles of human tissue kallikreins in cancer. Nat. Rev. Cancer 4: 876–890
Obiezu CV, Diamandis EP (2005) Human tissue kallikrein gene family: applications in cancer. Cancer Lett. 224(1): 1–22
Obiezu CV, Scorilas A, Katsaros D et al (2001) Higher human kallikrein gene 4 (KLK4) expression indicates poor prognosis of ovarian cancer patients. Clin. Cancer Res. 7: 2380–2386
Kim H, Scorilas A, Katsaros D et al (2001) Human kallikrein gene 5 (KLK5) expression is an indicator of poor prognosis in ovarian cancer. Br. J. Cancer 84(5): 643–650
Sher YP, Chou CC, Chou RH et al (2006) Human kallikrein 8 protease confers a favourable clinical outcome in non-small cell lung cancer by suppressing tumor cell invasiveness. Cancer Res. 66: 11763-11770
Goyal J, Smith KM, Cowan JM et al (1998) The role of NES1 serine protease as a novel tumor suppressor. Cancer Res. 58: 4782-4786
Beaufort N, Debela M, Creutzburg S et al (2006) Interplay of human tissue kallikrein 4 (hK4) with the plasminogen activation system: hK4 regulates the structure and functions of the urokinase-type plasminogen activator receptor (uPAR). Biol Chem. 387(2): 217-22
Page-McCaw A, Ewald AJ, Werb Z (2007) Matrix metalloproteinases and the regulation of tissue remodelling. Nat Rev Mol Cell Biol. 8: 221–233
Parks WC, Wilson CL, Lopez-Boado YS (2004) Matrix metalloproteinases as modulators of inflammation and innate immunity. Nat Rev Immunol. 4: 617–629
Nagase H, Visse R, Murphy G (2006) Structure and function of matrix metalloproteinases and TIMPs. Cardiovasc Res. 69(3): 562–573
Egeblad M, Werb Z (2002) New functions for the matrix metalloproteinases in cancer progression. Nat Rev Cancer. 2: 161–174
Genersch E, Haye K, Neuenfeld Y et al (2000) Sustained ERK phosphorylation is necessary but not sufficient for MMP-9 regulation in endothelial cells: involvement of Ras-dependent and independent pathways. J. Cell Sci. 113: 4319-4330
Stetler-Stevenson WG (1999) Matrix metalloproteinases in angiogenesis: a moving target for therapeutic intervention. J Clin Invest. 103(9): 1237–1241
Balbín M, Fueyo A, Tester AM et al (2003) Loss of collagenase-2 confers increased skin tumor susceptibility to male mice. Nature Genet. 35: 252-257
Gorrin-Rivas MJ, Arii S, Furutani M et al (2000) Mouse macrophage metalloelastase gene transfer into a murine melanoma suppresses primary tumor growth by halting angiogenesis. Clin. Cancer Res. 6: 1647-1654
Gorrin-Rivas MJ, Arii S, Mori A et al (2001) Implications of human macrophage metalloelastase and vascular endothelial growth factor gene expression in angiogenesis of hepatocellular carcinoma. Ann. Surg. 231(1): 67-73
Hofmann HS, Hansen G, Richter G et al (2005) Matrix metalloproteinase-12 expression correlates with local recurrence and metastatic disease in non-small cell lung cancer patients. Cancer Res. 11: 1086-1092
Sternlicht MD, Lochter A, Sympson CJ et al (1999) The stromal proteinase MMP3/stromelysin-1 promotes mammary carcinogenesis. Cell. 98(2): 137-146
Ichikawa Y, Ishikawa T, Momiyama N et al (2006) Matrilysin (MMP-7) degrades VE-cadherin and accelerates accumulation of beta-catenin in the nucleus of human umbilical vein endothelial cells. Oncol. Rep. 15: 311-315
Stetler-Stevenson WG (1999) Matrix metalloproteinases in angiogenesis: a moving target for therapeutic intervention. J Clin Invest. 103: 1237–1241
Rojiani MV, Alidina J, Esposito N et al (2010) Expression of MMP-2 correlates with increased angiogenesis in CNS metastasis of lung carcinoma. Int J Clin Exp Pathol. 3: 775–781
Noël A, Jost M, Maquoi E (2008) Matrix metalloproteinases at cancer tumor-host interface. Semin Cell Dev Biol. 19(1): 52–60
Murphy G (2008) The ADAMs: signalling scissors in the tumour microenvironment. Nat Rev Cancer. 8: 932– 941
Rocks N, Paulissen G, El Hour M et al (2008) Emerging roles of ADAM and ADAMTS metalloproteinases in cancer. Biochimie. 90(2): 369–379
Boutet P, Agüera-González S, Atkinson S et al (2009) Cutting edge: the metalloproteinase ADAM17/TNF-alpha-converting enzyme regulates proteolytic shedding of the MHC class I-related chain B protein. J Immunol. 1: 182(1): p. 49-53
Gialeli C, Theocharis AD, Karamanos NK (2011) Roles of matrix metalloproteinases in cancer progression and their pharmacological targeting. FEBS J. 278(1): 16-27
Iruela-Arispe ML, Carpizo D, Luque A (2003) ADAMTS1: a matrix metalloprotease with angioinhibitory properties. Ann. NY. Acad. Sci. 995: 183-190
Kuno K, Bannai K, Hakozaki M et al (2004) The carboxy-terminal half region of ADAMTS-1 suppresses both tumorigenicity and experimental tumor metastatic potential. Biochem. Biophys. Res. Commun. 319: 1327-1333
Masui T, Hosotani R, Tsuji S et al (2001) Expression of METH-1 and METH-2 in pancreatic cancer. Clin. Cancer Res. 7: 3437-3443
Cheung HH, St Jean M, Beug ST et al (2011) SMG1 and NIK regulate apoptosis induced by Smac mimetic compounds. Cell Death Dis. 2: e146
Hengartner MO (2000) The biochemistry of apoptosis. Nature. 407: 770-776
MacKenzie SH, Schipper JL, Clark AC (2010) The potential for caspases in drug discovery. Curr Opin Drug Discov Devel. 13(5): 568–576
Los M, Burek CJ, Stroh C, Benedyk K et al (2003) Anticancer drugs of tomorrow: apoptotic pathways as targets for drug design. Drug Discov Today. 8(2): 67-77
Choi KY, Swierczewska M, Lee S et al (2012) Protease-Activated Drug Development. Theranostics. 2(2): 156-178
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2014 Springer Science+Business Media New York
About this chapter
Cite this chapter
Upadrasta, S., Saini, N. (2014). Proteases and Their Role in Drug Development with an Emphasis in Cancer. In: Dhalla, N., Chakraborti, S. (eds) Role of Proteases in Cellular Dysfunction. Advances in Biochemistry in Health and Disease, vol 8. Springer, New York, NY. https://doi.org/10.1007/978-1-4614-9099-9_12
Download citation
DOI: https://doi.org/10.1007/978-1-4614-9099-9_12
Published:
Publisher Name: Springer, New York, NY
Print ISBN: 978-1-4614-9098-2
Online ISBN: 978-1-4614-9099-9
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)