
Abstract
Sickle cell disease (SCD) is a hereditary disorder that is so named for the presence of characteristic malformed sickle-shaped red blood cells (RBCs). These abnormal RBCs are short-lived and can also cause vaso-occlusion throughout the body, which may lead to ischemia and infarcts in multiple organ systems. Neurovascular complications of SCD include cerebral vasculopathy, such as stroke and neurocognitive deficits, which are significant causes of morbidity and mortality in both children and adults. In current clinical practice, various conventional imaging tools, such as computed tomography (CT), magnetic resonance imaging (MRI), and catheter angiogram, still play an important role in diagnosis and follow-up of neurovascular complications of SCD. Ultrasounds, including transcranial Doppler (TCD) ultrasound, are useful in screening for intra- and extracranial vasculopathy. Advanced imaging techniques, such as diffusion tensor imaging (DTI), perfusion imaging, MR spectroscopy, quantitative MRI (qMRI), and nuclear medicine such as single-photon emission computed tomography (SPECT) and positron emission tomography (PET), are emerging as diagnostic tools in the evaluation of SCD.
In this chapter, a brief overview of the etiology, classification, pathophysiology, epidemiology, clinical manifestation, and treatment of SCD is provided. The complications of SCD seen across the spectrum of conventional to advanced imaging modalities are also illustrated and discussed. The focus is on neurovascular complications and its management, but there will also be a discussion of posterior reversible encephalopathy syndrome, craniofacial osseous complications, and inner ear complications, which may be encountered in the daily neuroradiology practice.
Access provided by Autonomous University of Puebla. Download reference work entry PDF
Similar content being viewed by others


Keywords
- Sickle cell disease (SCD)
- Quantitative MRI (qMRI)
- Diffusion tensor imaging (DTI)
- Perfusion imaging
- MR spectroscopy (MRS)
- Vasculopathy
- Vaso-occlusion
- Hemolytic anemia
- Stroke
- Ischemic stroke
- Hemorrhagic stroke
- Silent cerebral infarct (SCI)
- Cerebral fat embolism
- Moyamoya syndrome
- Posterior reversible encephalopathy syndrome (PRES)
- Bone infarction
- Osteomyelitis
- Labyrinthine hemorrhage (LH)
- Labyrinthitis ossificans (LO)
- Arterial spin labeling (ASL)
- Positron emission tomography (PET)
- Single-photon emission computed tomography (SPECT)
- Transcranial Doppler (TCD)
1 Introduction
Sickle cell disease (SCD) is an inherited hematologic disorder characterized by the formation of misshapen red blood cells (RBCs) when sickle hemoglobin becomes deoxygenated and polymerizes. SCD causes a broad spectrum of clinical manifestations in various organ systems and is associated with significant morbidity and mortality in children and adults. Imaging plays a crucial role in the detection and description of the extent of involvement of SCD in the brain, head and neck, and spine.
Neurovascular complications, such as cerebrovascular ischemia/infarcts, hemorrhage, various intra- and extracranial vasculopathy, and cognitive impairment, are among the most significant complications in SCD. SCD also affects the osseous structures of the head and neck, including the inner ears. Neuroimaging plays an important role in diagnosing neurological complications of SCD as well as in monitoring the effects of medical and surgical treatments. Currently, neuroimaging of SCD largely relies on various conventional imaging modalities such as computed tomography (CT), magnetic resonance imaging (MRI), Doppler ultrasound, and catheter angiogram. Recently, quantitative MRI (qMRI) and functional MRI techniques, such as diffusion tensor imaging (DTI), perfusion imaging, and MR spectroscopy, have been introduced to assess and monitor patients with SCD. In this chapter, the pathophysiology, epidemiology, clinical features, and characteristic neuroimaging manifestations of SCD with conventional as well as advanced imaging techniques are reviewed. Current therapeutic approaches and management of SCD and the potential role of imaging in management will also be discussed.
1.1 Etiology, Classification, and Pathophysiology
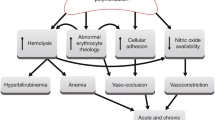
The term SCD refers to a variety of β-globin (HBB) genotypes that all result in abnormally shaped RBCs. The mutation underlying SCD is an A-to-T transversion in the codon for amino acid position 6 in the β-globin gene. Because of this mutation, a valine residue replaces the normal glutamic acid residue (glu6val) and HbS β-globin chains are substituted for normal β-globin chains [1, 2]. Each hemoglobin molecule contains two β-globin and two α-globin subunits. Hemoglobin with two normal β-globin chains is referred to as hemoglobin A (HbA) and is the major hemoglobin of adults. Fetal hemoglobin (HbF), a molecule of two γ-globin and two α-globin chains, is most prevalent in the fetus and newborns up to about three months of age. Other variant hemoglobin that can be present as compound heterozygotes with HbS are hemoglobin C (HbC), hemoglobin E (HbE), and hemoglobin D (HbD) [1, 2]. SCD is a recessive trait, which means that patients must carry two abnormal alleles of HBB in order for the disease to fully manifest. Homozygosity for the HbS gene or sickle cell anemia is the most common genotype of SCD [1, 2]. HbSC disease is the second most common genotype. SCD can also be caused by compound heterozygosity for HbS and one of the many forms of β-thalassemia. Deoxygenation causes HbS molecules to polymerize, causing RBCs to assume various abnormal shapes, some of which resemble a sickle. The change in shape causes RBC membrane damage and is associated with a decrease in RBC elasticity, rendering them unable to pass through small capillaries. Hemolytic anemia or a shortened lifespan of the RBC is also a feature of this cellular damage [1, 2]. Sickled cells can become irreversibly fixed in the “sickle” shape because of permanent membrane damage and fail to resume normal shape after oxygen tension is restored. These sickled cells and other RBCs can be sequestered in the spleen and other reticuloendothelial cell-containing organs and also lyse in the circulation, leading to anemia.
Patients who carry one abnormal HBB allele and a normal β-globin gene are said to have sickle cell trait (HbAS) and have 60 % HbA and 40 % HbS. HbAS is considered a benign entity with only rare complications. In carriers of HbAS, because each RBC contains 40 % HbS, polymerization of HbS occurs only under special conditions, like in the hypoxic, acidotic, and hyperosmolar conditions of the renal medulla and in the spleen of some people who are hypoxic. One prospective study in patients with HbAS showed that even otherwise asymptomatic patients had a high percentage of vasculopathy, such as arterial tortuosity, but the pathophysiologic consequences remain unclear [3]. Patients with HbAS may develop hyposthenuria and occasional hematuria and have a higher incidence of thromboembolic disease.
The two major pathophysiological processes in SCD are vaso-occlusion and hemolytic anemia that is both intra- and extravascular [1, 2, 4]. 3Vaso-occlusion can occur at both the micro- and macrovascular levels. At the microvascular level, the abnormal adherence of sickled RBCs to the capillary and venule endothelium can lead to intravascular stasis of blood flow. Stasis attracts leukocytes and fosters platelet adherence and degranulation, as well as fibrin deposition. This microvascular occlusion can lead to tissue ischemia and infarct. Inflammation can enhance the expression of adhesion molecules, further increasing the tendency of sickled erythrocytes to adhere to the vascular endothelium, worsening vaso-occlusion [1, 2, 4]. At the macrovascular level, abnormal adhesion of sickled RBCs and white blood cells (WBCs) to vessel sites of high flow turbulence may damage the vascular wall, resulting in intimal hyperplasia of large vessels, stenosis, and progressive occlusion of the large cerebral arteries [5]. Chronic hemolytic anemia from the accelerated breakdown of sickled RBCs may also result in macrovascular occlusion from intimal hyperplasia secondary to an inflammatory cascade triggered by increased free hemoglobin levels [1, 2, 4]. In addition, sickled RBCs may adhere to damaged vessel walls, which can result in secondary thrombosis and distal branch emboli [5–7]. In the past, small vessel occlusion by intravascular sickling and slugging of RBCs was considered to be the primary cause of strokes in SCD. It is now understood, however, that occlusion of small vessels accounts for about 25 % of cerebral infarct in SCD while macrovascular complications account for 75 % [8].
1.2 Epidemiology
SCD is most common in people from sub-Saharan Africa, South and Central America, the Caribbean islands, the Mediterranean countries, India, Saudi Arabia, and their descendants worldwide [1, 2]. It is now estimated that 200,000 children affected with SCD are born every year, approximately 2,000 children in the USA. The estimated US prevalence ranges from 70,000 to 140,000 [4]. About 8 % of African Americans have HbAS. In some regions of Africa, more than a quarter of the populations are carriers; very high gene frequencies are also found in some parts of Saudi Arabia and Greece, in tribal populations of India, and in Brazil [1, 2, 4]. HbAS confers some protection from Plasmodium falciparum malaria; the underlying protective mechanism is still subject of scientific investigation [9].
Mortality in patients with SCD has significantly improved in high-resource countries. The universal newborn screening for hemoglobinopathies has become standard practice in such countries, enabling early diagnosis and patient management [1, 4]. In 2000, the universal administration of the pneumococcal vaccine has further decreased the mortality and morbidity of childhood invasive pneumococcal disease, including in children with SCD. As a result of these programs, SCD-related death fell by 42 % from 1999 through 2002 [10]. It is estimated that 94 % of children with SCD now live to adulthood, and more than 98 % of children with SCD in high-resource countries are living into adulthood [11]. This has caused a mortality shift toward older ages and SCD is considered a chronic condition requiring comprehensive, lifelong management [4]. However, in low-resource countries, more than 50 % of children younger than five years of age die due to complication of SCD [4].
2 Clinical Manifestations
2.1 Painful Episodes
Pain, both acute and chronic, is a hallmark of SCD. Acute painful episodes are the most common vaso-occlusive events with inflammatory and ischemic consequences in patients with SCD of all ages, although they are more common in teenagers and young adults [1, 2, 4]. In young children, one of the first manifestations of SCD is the so-called hand-foot syndrome (i.e., sickle cell dactylitis), a painful swelling of the hands and feet due to inflammation of the metacarpal and metatarsal periosteum. Painful episodes may involve any bones, muscles, mesentery, and other organs. Although acute vaso-occlusive pain is generally self-limiting and does not result in permanent organ damage, increased frequency of pain is associated with early death in patients with SCD who are older than 20 years [12]. The cause of chronic pain is poorly understood, but it can be debilitating and may result from leg ulcers, avascular necrosis of bone, and neuropathic complications [4]. An important consideration for the neuroradiologist is that the skull and maxillofacial bones may be affected by acute pain. Typical symptoms are headaches or localized pain and swelling. Frequently, these are caused by bone infarcts with subperiosteal hemorrhage [13].
2.2 Sickle Cell Acute Events
Besides the painful episodes described above, patients with SCD may also experience splenic and hepatic sequestration crisis, aplastic crisis, acute chest syndrome (ACS), and stroke. In addition, SCD patients are more susceptible to infection and its complications, such as sepsis, pulmonary embolism, pulmonary hypertension, cardiomyopathy, and hepatic disease [1, 2, 4].
Infection with B19 parvovirus can suppress the production of RBCs in patients with SCD for 2 to 3 days, causing an acute, life-threatening aplastic anemia with pallor, tachycardia, and fatigue. Severe anemia can also result acutely from sequestration of blood in the spleen or liver [2]. ACS is the second most common cause of hospitalization among patients with SCD and a leading cause of mortality [14]. ACS is an acute lung injury that causes development of a new alveolar pulmonary infiltrate involving at least one lung segment. ACS is caused by a combination of pulmonary infection, fat embolism, and intravascular pulmonary sequestration of sickled erythrocytes, resulting in lung injury and infarct. Recurrent ACS can lead to chronic respiratory insufficiency. Occasionally, ACS culminates with multi-organ failure [2].
2.3 Infarcts
Infarcts are the consequences of vaso-occlusive crises. Infarcts may involve the brain, liver, spleen, kidney, and bones. Infections often precede vaso-occlusive episodes in children, suggesting that fever, dehydration, and acidosis may be contributing factors [6]. Bone infarction can occur anywhere in the skeleton. It results directly from the sickling of RBCs in the bone marrow, which causes stasis of blood and sequestration of cells [13, 15].
Stroke is one of the most severe complications of SCD because it may result in permanent neurological deficits or death. The incidence of stroke varies depending on the SCD genotype. The prevalence of cerebral vascular accident (CVA) was 11 % in patients under 20 years of age with homozygous HBSS [16]. Ischemic strokes are more common in the children, adolescent, and old adult SCD patients, while the incidence of hemorrhagic strokes peaks between the ages of 20–29 years [16]. Silent cerebral infarcts (SCIs) occur more commonly than overt strokes, with a prevalence of 21.8 % in children with HbSS ages 6–19 years [17]. SCIs are defined as a lesion on MRI, without corresponding neurological deficits lasting longer than 24 h. SCIs are clinically significant because they indicate a higher likelihood of subsequent overt stroke [18]. Although SCD is recognized as a hypercoagulable state, the incidence of venous infarction due to thrombosis is unknown [19].
2.4 Cognitive Deficits
As more SCD patients survive to adulthood, the neurocognitive dysfunction caused by SCD is becoming an emerging problem in young adults [20]. Studies correlating MRI evidence of brain damage have come to controversial conclusions, with some stating that neurocognitive deficits in SCD patients are associated with focal brain injury and silent strokes in children while others state that these deficits are associated more with anemia and age. A recent study suggests that volume reduction of the basal ganglia and thalamus may correlate with cognitive deficits in adult with SCD [21]. These reports suggest that the cause of the neurocognitive impairment is multifactorial and that further correlating studies using functional imaging for cerebral blood flow and hypoxia may be needed [20].
3 Treatment
3.1 Hydroxyurea (HU)
Hydroxyurea (HU) is a drug that promotes the production of HbF, the normal hemoglobin which is more prevalent in newborns. HU is currently the only established preventive pharmacologic treatment for both children and adults with SCD. The presence of HbF inhibits HbS polymerization and higher levels of HbF are associated with reduced mortality in SCD patients [2]. In a randomized controlled trial, HU was shown to decrease the frequency of painful episodes and acute chest syndrome and the need for blood transfusion and hospital admissions in SCD patients [22].
3.2 Blood Transfusion
Chronic blood transfusions have been demonstrated to reduce the risk of both primary and secondary strokes and SCIs [23] and prevent repeated ACS [2, 4]. There is currently no specific treatment for acute stroke in SCD. However, blood transfusion within 48 hours after acute stroke may decrease the incidence of subsequent strokes [24]. The aim of transfusion therapy is to reduce HbS to below 30 %, which effectively prevents overt stroke and SCIs [25]. However, it can result in morbidity related to iron overload and iron deposition in multiple organs, including the liver, heart, and endocrine systems, and often necessitates iron chelation therapy [2, 4].
3.3 Hematopoietic Cell Transplantation
Hematopoietic cell transplantation is the only curative treatment for SCD thus far. However, there is a lack of suitable allogeneic marrow donors (usually HLA-matched siblings) for about 90 % of eligible patients [2]. There is a 5–10 % mortality rate associated with this treatment, mostly due to graft failure, lack of response to first-line immunosuppressive therapy, graft-versus-host disease (GVHD), relapse after the initial successful treatment, and secondary clonal and malignant disease [26]. There are especially poor outcomes in patients over the age of 16 years who are subjected to myeloablative conditioning regimens, making the use of non-myeloablative regimens standard in older patients [2].
4 Neurovascular Complications Concept and Imaging Features
4.1 Stroke
Stroke or CVA is defined as a sudden-onset neurological deficit that persists for more than 24 hours. SCD is one of the most common causes of stroke in children and young adults. The Cooperative Study of Sickle Cell Disease (CSSCD) classified stroke in SCD patients as ischemic, hemorrhagic, or transient ischemic attack [16]. The special forms of ischemic stroke in SCD patients are the so-called silent cerebral infarcts (SCIs) and stroke caused by cerebral fat embolism [27].
Imaging approaches for stroke in SCD patients are the same as for non-SCD patients. The purpose of acute stroke imaging in SCD is to determine the extent of brain damage, to differentiate ischemic stroke from hemorrhagic stroke, and to help identify and delineate underlying vasculopathy.
CT of the brain without contrast is usually the first imaging modality used in the acute setting, which allows for quick assessment of hemorrhagic versus ischemic stroke, for detection of other causes to explain new neurological deficits, and for evaluation of the extent of damage and complications. Other benefits of CT include its easier accessibility and 24/7 availability at most institutions, as well as short exam times [28]. Disadvantages of CT include ionizing radiation exposure, low sensitivity for detection of small infarcts, and low sensitivity for stroke detection in the first several hours [29].
MRI with diffusion-weighted imaging (DWI) is very useful in detecting acute infarcts as well as determining the exact location and extent of ischemic lesions [30, 31]. Apparent diffusion coefficient (ADC) maps in conjunction with DWI help to pinpoint regions of early ischemia and infarct [32]. T2-weighted (T2-WI) imaging and fluid-attenuated inversion recovery (FLAIR) imaging are used for detecting T2 high signal intensity lesions [33], such as acute stroke and silent infarcts. Gradient echo T2*-weighted (T2*WI) images and susceptibility-weighted images (SWI) are used to detect hemorrhagic lesions [34]. While MRI offers the benefits of increased sensitivity for pathology compared to CT and avoidance of radiation exposure, it requires considerable patient cooperation and in many pediatric patients, may require sedation. The other disadvantages of MRI are limited access for acutely ill patients at many institutions and relatively long duration of imaging studies.
Vascular imaging studies in the head and neck may be considered in all patients with SCD, irrespective of whether they had previous episodes of stroke [30, 35]. CTA and MRA can be used to detect intracranial arterial occlusion or stenosis and aneurysms. CTA exposes patients to high levels of ionizing radiations and contrast media; therefore, MRA is preferred in children [30]. MR venography can be useful when venous thrombosis is suspected. Conventional catheter angiogram is an invasive procedure but yields higher anatomical detail of vascular anatomy and pathology than other imaging modalities, including information on hemodynamic function. Catheter angiogram is usually indicated prior to surgery or endovascular interventions [31]. A careful risk–benefit evaluation should be performed prior to subjecting an SCD patient to catheter angiogram. If anesthesia is necessary, special care should be taken in the perioperative care of patients with SCD [36].
4.1.1 Ischemic Stroke
The majority of stroke in SCD is caused by large vessel disease, which may involve the proximal cerebral arteries and the neck arteries [8, 27]. Macrovascular stroke in SCD has imaging features of cortical involvement and is more commonly unilateral [8]. DWI with ADC map is essential for diagnosing acute ischemic infarct and for differentiating it from chronic ischemic changes. Both acute and chronic changes can have high intensity on T2-WI images. With FLAIR imaging, abnormal T2 high signal may be more easily detected than conventional T2-WI imaging, particularly in the periventricular and subcortical regions (Figs.1, 2, 3, 4, and 5).


A 38-year-old woman with SCD, presenting with altered mental status. (a) Axial FLAIR image shows multiple high signal intensity lesions of the bilateral periventricular white matter (arrows). (b, c) Corresponding axial DWI (b = 1,000) (b) with ADC map (c) shows multiple foci of restricted diffusion in the left periventricular white matter consistent with acute infarcts (arrows). Right periventricular lesions suggest chronic ischemia or prior infarcts


A 6-year-old girl presenting with right-sided weakness. (a, b) Axial FLAIR images show multiple high-signal-intensity lesions at the left frontal border zone (watershed) areas (arrows). (c, d) Corresponding axial DWI (b = 1,000) shows restricted diffusion in the left MCA territory and border zone larger than that shown on FLAIR images consistent with acute infarct (arrows). (e) Axial T2-WI image shows loss of flow voids in the left proximal MCA (arrow). (f) MRA shows obvious stenosis of left MCA (arrow) and decrease of flow-related signal distally


A 16-year-old woman presenting with headache and syncope. (a) Axial FLAIR image demonstrates encephalomalacia (asterisks) with surrounding gliosis (arrows) in the right frontoparietal ACA-MCA border zone (watershed) distribution indicating old ischemic changes. (b) MRA shows decreased caliber and flow-related signal with wall irregularity of the visualized right extracranial ICA (arrows)


A 22-year-old woman presenting with seizure. Axial T2-WI image shows bilateral frontal high signal intensity, indicating chronic ischemic changes and encephalomalacia (asterisks), predominantly in ACA-MCA border zone (watershed) distribution


A 14-year-old girl presenting with past episode of right hemiplegia. Axial FLAIR image shows high signal (arrows) and prominent sulci (asterisks), indicating volume loss and gliosis with cortical involvement in the left cerebral hemisphere
Arterial stenosis or occlusion is often seen in conjunction with infarcts of the cerebral gray matter (the cortex, basal ganglia, and thalamus) (Fig. 2). However, white matter infarcts can be seen in the setting of arterial stenosis or occlusion as well, particularly in border (watershed) zones [8] (Fig. 3). The anterior circulation is more often involved than the posterior circulation. The radiologist should screen for loss of vascular flow voids on T2-WI images, which may indicate vasculopathy (Fig. 2e). FLAIR imaging can show high signal in cortical vessels in the setting of altered flow velocities, such as slow distal flow of a stenotic arterial branch or leptomeningeal collaterals [37] (Fig. 6). In these cases, the radiologist may decide to add brain MRA or recommend other angiographic imaging such as CTA or catheter angiogram for further evaluations regarding diagnosis, treatment, and management.


A 10-year-old girl presenting with left facial droop, drooling, and hand tingling. (a) (b) Axial FLAIR images show multiple high signal intensity vessels in the Sylvian fissure and frontoparietal sulci (arrows), indicating slow flow or thrombosis. (c) MRA shows loss of flow-related signal in the right MCA distribution (asterisks) and bilateral proximal ACAs (arrows)
4.1.2 Hemorrhagic Stroke
Hemorrhagic stroke in SCD is relatively more common in adults than in children. Although it is rare, primary hemorrhagic stroke, such as subarachnoid and parenchymal hemorrhage, is frequently fatal in SCD patients. Aneurysms are the most common identified cause of subarachnoid hemorrhage in adult patients with SCD [27], although their exact prevalence is unknown. Compared with patients without SCD, aneurysms in SCD patients are often multiple, have an increased propensity for the posterior cerebral circulation, and may be prone to rupture at smaller sizes [38]. Moyamoya syndrome is a common cause of parenchymal hemorrhage in SCD in children [8], which will be discussed later in more detail.
Non-contrast CT is the first imaging modality of choice in the diagnosis of acute hemorrhagic stroke (Figs. 7 and 8). In the current clinical practice, MRI may occasionally be obtained prior to CT when there is high suspicion for acute stroke based on clinical symptoms. Radiologists should be familiar with the MR imaging features of acute hemorrhage that are the same as non-SCD patients, to avoid delayed diagnosis of hemorrhagic strokes. The accuracy of MRI in detecting acute parenchymal hemorrhage is similar to CT, especially when gradient echo sequences, such as T2*-WI imaging and SWI, are used [39]. Catheter angiogram using three-dimensional (3D) digital subtraction angiography is still the gold standard for the diagnosis and preoperative delineation of intracranial aneurysms. Noninvasive techniques such as MRA and CTA are more commonly used, and recently their accuracy is considered satisfactory [40, 41]. CTA is often the first-line modality to search for underlying aneurysms in the setting of acute subarachnoid hemorrhage, while MRA is commonly used in the non-acute setting (Figs. 9 and 10).


A 20-year-old man presenting with altered mental status. Axial non-contrast CT shows a high density area in the right frontal lobe white matter with surrounding low density indicating edema (arrow), compatible with acute hemorrhage


A 14-year-old boy presenting with severe headache. Axial non-contrast CT shows high density in the right Sylvian fissure consistent with acute subarachnoid hemorrhage (arrows)


A 34-year-old man presenting with headache. (a) MRA shows an ovoid focus of flow-related signal arising from the junction of the cavernous–clinoid segments of the left ICA extending inferiorly, consistent with an aneurysm (arrow). (b) Catheter angiogram of the left ICA lateral view confirmed the aneurysm (arrow)


A 22-year-old man presenting with headache and vertigo. (a) Axial FLAIR image shows multiple high signal foci (arrows) in the left frontal white matter without abnormal diffusion restriction (not shown) consistent with silent cerebral infarcts. (b) MRA shows a 3 mm aneurysm at the medial portion of the left IC-ophthalmic segment (arrow). Tortuous extracranial right internal carotid artery and bilateral vertebral arteries are also noted (open arrows)
4.1.3 Silent Cerebral Infarct (SCI)
SCI is the most common neurovascular complication in both pediatric and adult patients with SCD. SCIs have been seen in children under the age of 6 years [42]. They may represent a different pathogenesis from either ischemic or hemorrhagic strokes as they involve small vessels in watershed distributions instead of the larger cerebral vessels [43]. Measurement of arterial velocities with transcranial Doppler (TCD) ultrasound are not predictive of SCI risk, unlike with overt ischemic stroke, which supports a different pathophysiology between overt stroke and SCI [44]. Several studies showed that SCIs are more frequent in patients with arterial stenosis and intracranial arterial tortuosity, suggesting that large vessel disease may play a role in the pathophysiology of these lesions [42, 45, 46]. Children with arterial stenosis are at higher risk of brain parenchymal injury as they have more SCIs [47] (Fig. 10).
The presence of SCI is considered a predictor of future strokes [17, 48, 49]. SCIs have been shown to progress in both number and size over time [49]. Associated neurocognitive abnormalities also may be progressive [50]. Small foci of acute SCI may be found in asymptomatic children with SCD [42]. SCIs may continue to progress in spite of initiation of chronic transfusion therapy after a stroke [51]; however, a recent study showed that in patients without a stroke and not at high risk of a stroke, chronic transfusions reduced the occurrence of new SCIs by 56 % over a median observation period of 3 years [23].
SCIs are defined as T2 high signal intensity lesions of at least 3 mm in the greatest linear dimension in children and 5 mm in adults, visible on at least two planes of T2-WI images [20, 52] (Figs. 11 and 12). Because of their small size, detection of SCI depends largely on the MR imaging technique, such as the use of FLAIR, slice thickness, multiplanar acquisitions, and the magnetic field strength [53]. With further advances in imaging and more utilization of 3.0-T or higher magnet field units, it is likely that more cases of SCI will be detected in individuals with SCD [49].


A 6-year-old girl presenting with SCD for screening MRI. Coronal FLAIR image shows high signal foci in the right frontal subcortical white matter (arrow), compatible with silent cerebral infarct


A 42-year-old woman presenting with left facial numbness. (a) Axial FLAIR image shows multiple high signal intensity lesions in the right frontoparietal white matter (arrows). (b) DWI (b = 1,000) shows corresponding restricted diffusion in the postcentral gyrus consistent with acute infarct (arrow)
4.1.4 Cerebral Fat Embolism
Fat embolism syndrome is a very rare but potentially lethal complication of SCD that is not widely recognized [54]. In non-SCD patients, fat embolism syndrome is usually a complication of long bone fractures. The pathogenesis of fat embolism syndrome in SCD remains controversial. It is thought to be caused by bone marrow infarcts and necrosis, with subsequent embolization through osseous venous channels into the venous circulation. Neurologic dysfunction in the setting of fat embolism syndrome may result directly from vessel occlusion by fat emboli, from disruption of the blood–brain barrier by toxic free fatty acids, or both [54, 55].
Typical MR imaging findings of cerebral fat embolism include tiny foci of restricted diffusion and corresponding T2 high signal intensities representing microinfarcts. Lesions can involve the cerebral white matter, basal ganglia, thalamus, brain stem, and cerebellum. This MR imaging pattern, sometimes called a “starfield” pattern, is not very specific and can also be seen in cardiogenic or septic emboli, vasculitis, and tiny hemorrhagic metastases [54]. T2*-WI images and SWI frequently demonstrate microhemorrhages in cerebral fat embolism [55].
4.2 Moyamoya Syndrome
Vascular pathology in SCD includes arterial tortuosity, stenosis, occlusion, and aneurysm formation. A special form of cerebrovascular manifestation in SCD patients is “moyamoya syndrome,” a progressive stenosis of major intracranial arteries with formation of numerous collateral circulations, typically via lenticulostriate and thalamoperforating arteries (Fig. 13). Moyamoya syndrome typically involves the distal ICA (internal carotid artery) and proximal ACA (anterior cerebral artery) and MCA, with relative sparing of the posterior circulation. The name moyamoya syndrome is derived from moyamoya disease, a rare, presumed genetic, progressive cerebrovascular disorder. The term “moyamoya” means “puff of smoke” in Japanese and refers to the appearance of collateral vessels on cerebral angiogram [56]. The pathophysiology of moyamoya syndrome in SCD is poorly understood. It is hypothesized that sickle-shaped RBCs occlude the vasa vasorum of the carotid arteries [31], leading to intimal hyperplasia [57]. The prevalence of moyamoya syndrome in SCD patients has been reported for 35 % of conventional catheter angiogram studies [7] and in 20–40 % of MRA studies [8, 58]. It was shown that 75 % of patients with moyamoya syndrome demonstrated radiographic progression of arteriopathy and 65 % of patients showed clinical progression during follow-up [59] (Fig. 14).


A 24-year-old man presenting with multiple episodes of ischemic infarct. (a) Axial FLAIR image shows bilateral periventricular high signal intensity with cystic foci compatible with old ischemic change (arrows). (b) MRA shows occlusion of the right ICA terminus (asterisk) and severe stenosis of the left ACA (arrow) and distal MCA (open arrow). (c) Catheter angiogram of the right ICA shows occlusion of the supraclinoid ICA (arrow). Posterior communicating artery (PComm) is markedly prominent (asterisk) and diffuse moyamoya-like collateral vessels are noted (open arrows). (d) Subsequent image of the ICA catheter angiogram shows more apparent “moyamoya” (puff-of-smoke) appearance of the collateral vessels (open arrows)


A 15-year-old man presenting with recurrent headache. (a, b) In 2005, Axial FLAIR image (a) shows normal brain and MRA (b) shows possible stenosis of the right proximal ACA and bilateral proximal MCA (asterisk). (c, d) In 2006, axial FLAIR image (c) still shows normal brain, but MRA (d) shows obvious occlusion of the right terminal ICA (asterisk). The patient received bilateral pial synangiosis for cerebral revascularization in 2007. (e, f) In 2013, axial FLAIR image (e) shows bilateral frontal old infarcts with encephalomalacia and gliosis (arrows). MRA (f) shows bilateral occlusion of the terminal ICAs (asterisks) and bilateral prominent superficial temporal arteries (arrowheads) after revascularization. Serial FLAIR images and MRA images over 8 years demonstrate progression of arterial stenosis, bilateral frontal lobe infarcts, and atrophy in spite of revascularization
Moyamoya syndrome can be detected on MRA as progressive large vessel occlusions and major collateral flow patterns, the so-called moyamoya vessels. On T2-WI images, moyamoya syndrome presents with loss of flow voids in the circle of Willis (Fig. 15) and on time-of-flight MRA as a loss of flow-related signal (Fig. 16). Collaterals typically present as multiple flow voids on T2-WI and T1-WI images in the thalamoperforate and lenticulostriate territories. High signal intensity vessels within the cortical sulci on FLAIR images can indicate a decreased flow of cortical arteries [37] (Fig. 6).


A 22-year-old woman presenting with seizure. Axial T2-WI image shows loss of normal flow voids of the bilateral MCA and ACA (arrows). In contrast, prominent posterior circulation flow voids are noted (arrowheads)


A 38-year-old woman with SCD presenting with altered mental status. MRA shows multiple stenoses and occlusions of the right ICA terminus, bilateral ACAs, bilateral PCAs (posterior cerebral artery), and left MCA (arrows)
Incidentally discovered asymptomatic moyamoya syndrome in children has the potential to progress, both radiographically and clinically. Monitoring and screening with imaging in high-risk patients facilitates early referral to neurosurgery and allows for timely revascularization therapy [59]. Patients with moyamoya syndrome may have a particularly poor prognosis and may benefit from revascularization of ischemic brain parenchyma by establishing collaterals from the external carotid artery (ECA) branches to the ICA territories [60]. Superficial temporal artery (STA) to middle cerebral artery (MCA) bypass is a direct revascularization procedure, which is technically difficult. Indirect revascularization procedures such as encephaloduroarteriosynangiosis (EDAS) and pial synangiosis are becoming popular for the surgical treatment of moyamoya syndrome [61]. Pial synangiosis is a technique in which the dura and the arachnoid are opened and the STA adventitia is sutured directly to the pia [62], and it has a relatively low risk of complications [63]. Pial synangiosis typically results in an increase in collaterals from the STA or middle meningeal artery (MMA) to the brain [62]. MRA or CTA can be used to monitor the patency of synangiosis between STA branches and pial arterial branches after cerebral revascularization (Fig. 14) .
4.3 Extracranial Vasculopathy
Extracranial ICA stenosis is rare, but it has been reported that there is an association between the presence of extracranial internal carotid arteriopathy and stroke risk in SCD [64–66]. Doppler ultrasound screening studies found stenosis or occlusion of the cervical ICA in 3–6 % of the children, and the majority of these also had strokes [65]. One MRA study reported that patients with an acute neurologic event, TCD abnormalities, and previous history of stroke also showed cervical ICA stenosis and/or occlusion. The most commonly involved artery is the proximal ICA, within 1 cm from its origin from the CCA [66] (Fig. 17). In addition, a recent study showed that the high velocity of extracranial ICA detected with submandibular Doppler ultrasound is highly predictive of intracranial arterial stenosis seen on MRA [67]. The differential diagnosis of extracranial ICA stenosis and/or occlusion includes atherosclerosis, arteritis, and fibromuscular dysplasia. Age, location, and lack of systemic inflammation or hypercoagulability help differentiate SCD-related extracranial internal carotid arteriopathy from other conditions [68].


A 30-year-old woman with SCD, evaluation for revascularization. (a) Catheter angiogram of the right CCA (lateral view) shows complete occlusion of the extracranial ICA near its origin (arrow). (b) Catheter angiogram of the left CCA (lateral view) shows stenosis of the ICA at the ophthalmic segment (arrow). (c) Catheter angiogram of the left subclavian artery shows tortuosity of the left VA (arrows)
Arterial tortuosity is considered to be an adaptive response to chronic anemia in patients with SCD. Chronic anemia is associated with increased cardiac output and high blood flow velocity due to the increased need for blood supply, which can lead to arterial tortuosity and can occur in all cervical arteries [68]. Cerebrovascular tortuosity may serve as a predictor of chronic brain hypoxia [53]. On imaging, tortuosity can be diagnosed when the following features are present: dilatation or ectasia of an artery segment, abnormal increase in length of an arterial segment, and obvious bowing of an artery [53, 69] (Figs. 17, 18, and 19) .


A 34-year-old woman with SCD, presenting for annual MRI follow-up. MRA shows no significant stenosis or occlusion of the intracranial arteries; however, dilatation and elongation of the extracranial left ICA are seen (arrows)


A 20-year-old man with SCD. MRA shows tortuous left extracranial ICA (arrows) and aneurysmal dilatation of the extracranial segment of the left VA (open arrow)
5 Posterior Reversible Encephalopathy Syndrome (PRES)
There have been several case reports of posterior reversible encephalopathy syndrome (PRES), also known as reversible posterior leukoencephalopathy syndrome (RPLS), in patients with SCD [70–74]. The vast majority of the patients are in the pediatric population [71]. It is believed that there are two major pathomechanisms in PRES: a cytotoxic event after an increase in blood pressure that causes vasoconstriction and cerebral ischemia or a vasogenic etiology where hypertension in combination with endothelial dysfunction and failure of cerebral autoregulation causes vasodilation [70]. Studies have identified PRES as a common neurological complication of SCD, and this entity needs to be considered when evaluating a child with acute neurological deterioration, especially if the patient is hypertensive, has history of ACS, and has received systemic steroid therapy or blood transfusion [72–74]. A diagnosis of PRES may predict a high risk for morbidity and mortality [73]. On MR imaging, PRES usually presents as symmetrical areas of high signal intensities on T2-WI and FLAIR images without associated restricted diffusion, suggesting vasogenic edema, which is the same as non-SCD patients [73] (Fig. 20) .


A 5-year-old girl with SCD, presenting with headache and hypertensive status. Axial FLAIR image shows high signal intensity lesions in the bilateral occipital lobes (arrows). Given the distribution, clinical course, and documented hypertension, the findings are compatible with PRES
6 Other Sickle Cell Disease Imaging Manifestations
There are several osseous complications related to SCD. These include chronic bone marrow changes in response to long-standing anemia and acute bone infarcts as well as osteomyelitis. Osteoporosis and iron deposition in the bone marrow due to repeated transfusions can also be seen [13, 15, 68, 75, 76].
6.1 Craniofacial Bone
6.1.1 Bone Marrow Hyperplasia
As a result of chronic anemia in SCD patients, the red marrow in bony erythrocyte production never undergoes the conversion to fatty yellow marrow [68, 75]. Furthermore, as the anemia becomes more severe and long-standing, the bone marrow spaces expand secondary to increased RBC production [68, 75]. This marrow expansion is most frequently noted clinically in the skull and facial bones. The non-conversion of the bone marrow is demonstrated by diffuse low signal intensity on T1-WI images relative to the signal in the intervertebral disks [15] (Figs. 21 and 22).


An 18-year-old man with SCD. Sagittal T1-WI image shows the diffuse expansion of the marrow space of the skull (arrows)


A 10-year-old boy with SCD, presenting with back pain. Sagittal T1-WI image of the lumbar spine shows low signal in the vertebral bodies (asterisks) relative to the intervertebral disk, compatible with an activated bone marrow triggered by chronic anemia. Note also the “H”-shaped deformity of vertebral bodies, compatible with end-plate infarcts and subsequent height loss
6.1.2 Bone Infarct
Bone infarcts are more common than osteomyelitis among children with SCD. Ischemia causes pain, even before infarct occurs. For SCD patients presenting with headache or localizing facial pain, acute craniofacial bone infarct should be a major diagnostic consideration [13, 15]. The orbital walls, mandible, cranial vault, and skull base are commonly affected by bone infarcts in SCD patients [13, 15] (Fig. 23). In the spine, it is hypothesized that bone infarcts contribute to the characteristic “letter H” deformity of vertebral bodies that is typical of patients with SCD [77] (Fig. 22).


A 6-year-old boy presenting with temporomandibular joint pain and headache. Axial fat-suppressed T2-WI image demonstrates high signal intensity in the left mandibular ramus (arrow), compatible with bone infarct
MR imaging is more sensitive than CT in detecting bone infarcts. MR imaging findings of acute bone infarcts include increased signal on T2-WI images, particularly with fat-suppression techniques such as STIR, and associated high signal intensity on DWI with corresponding low signal intensity on ADC map (Fig. 24). There may also be subperiosteal hemorrhage. The high signal intensity of the affected bones on fat-suppressed T1-WI images, which may represent the sequestered RBCs in the infarcted marrow [13, 78], can be very helpful in the differentiation of bone infarcts from osteomyelitis (Fig. 25).


A 15-year-old boy with SCD, presenting with headache. (a) Coronal T2-WI image shows high signal intensity in the skull consistent with bone marrow edema (arrows), as well as mixed abnormal signal intensity in the subgaleal fluid collection consistent with hemorrhage (open arrow). (b) Axial DWI (b = 1,000) shows an area of increased signal in the skull, compatible with bone infarct (arrows)


A 17-year-old man presenting with headache. Sagittal T1-WI image shows high signal intensity of the bone marrow in the parietal bone consistent with bone infarct with hemorrhage (arrow) (Adapted with permission from Lippincott Williams and Wilkins/Wolters Kluwer Health: Journal of Computer Assisted Tomography [13], copyright (2013))
6.1.3 Osteomyelitis
Osteomyelitis is less common than bone infarcts and typically affects long bones. However, osteomyelitis may also occur in craniofacial bones, particularly the mandible, because of its relatively poor blood supply [13, 68] (Fig. 26). Differentiation between infarct and osteomyelitis in early presentation with imaging alone can be quite challenging. The typical MR imaging findings, including bone marrow edema, heterogeneous bone marrow enhancement, and soft tissue swelling, can be seen in osteomyelitis as well as bone infarcts. In the presence of fluid collections, MR susceptibility artifact is suggestive of blood products from subperiosteal hematoma, which is more commonly seen with bone infarcts. Infectious fluid collections may exhibit restricted diffusion [13].


A 25-year-old man presenting with left jaw swelling and pain. Coronal contrast-enhanced CT shows peripherally enhancing low density fluid collections medial and lateral to the left mandibular ramus (arrows), consistent with subperiosteal abscess formation
6.2 Temporal Bone
Sensorineural hearing loss (SNHL) is a well-recognized inner ear complication of SCD [68, 79]. The most accepted pathogenesis of SNHL is the recurrent vaso-occlusion of the labyrinthine blood vessels, either in the distribution of the anteroinferior cerebellar artery or a branch of the basilar artery, which can result in labyrinthine hemorrhage (LH) and labyrinthitis ossificans (LO) [79]. In the past, LH was considered relatively rare in patients of SCD with sudden-onset SNHL and vertigo, but it was recently shown that abnormalities can be detected by MR imaging in approximately one-third of SCD patients with inner ear symptoms, with males preferentially affected [79].
High signal intensity on non-contrast high-resolution T1-WI images is a characteristic finding for LH. Contrast-enhanced T1-WI images can exclude tumors such as schwannomas and hemangiomas that would show intense and localized enhancement, and fat-suppressed MR imaging or high-resolution temporal bone CT may be used to exclude lipoma and fat-containing tumors (Fig. 27).


A 14-year-old man presenting with sudden onset of right-sided hearing loss. Axial T1-WI image shows high signal intensity in the right cochlea (arrow) compared to left, suggesting labyrinthine hemorrhage
The characteristic imaging finding of LO is the high density of the membranous labyrinth on CT. On high-resolution T2-WI images, the normal T2 high signal from the fluid in the membranous labyrinth is lost. LO is the end stage of labyrinthitis, characterized pathologically by proliferation of fibroblasts and finally osteoblasts. In patients with SCD presenting with inner ear complaints, dedicated temporal bone imaging should be performed, preferably by MR imaging (Fig. 28).


A 12-year-old boy presenting with hearing loss. (a) Axial high-resolution T2-WI image (DRIVE sequence) shows signal loss of the right cochlea (asterisk). (b) Axial CT shows ossification of the cochlea corresponding to signal loss on MRI (a), consistent with labyrinthitis ossificans (arrow)
7 Advanced Imaging
7.1 Diffusion Tensor Imaging (DTI)
Diffusion tensor imaging (DTI), which measures the directional tendencies of Brownian water molecule motion in biologic tissues, has a potential to detect and quantify microstructural brain changes earlier than conventional MRI [80]. This advanced MRI technique can show the orientation and integrity of white matter fibers in vivo. The apparent diffusion coefficient (ADC), which is comparable to mean diffusivity as a measure of the degree of restriction to water diffusion, and fractional anisotropy (FA), a measure of the preponderant directionality of water diffusion, are two of the most frequently used DTI metrics for measuring microstructural tissue damage in patients with brain disease [80]. DTI directionality information is used for fiber tractography, which is the only way to obtain 3D fiber architecture of white matter tracts in vivo [80].
Using region of interest (ROI) based analysis in a study comparing SCD patients with controls, FA values in the prefrontal segment of the corpus callosum (which covers the first sixth of the corpus callosum), upper frontal white matter area, centrum semiovale, anterior periventricular white matter area, posterior periventricular white matter area, and brain stem were significantly lower in SCD patients than controls. ADC values were significantly higher in SCD patients compared to controls in the prefrontal segment of the corpus callosum and sensory segment of the corpus callosum (which covers the posterior one-third but not including the posterior one-fourth), caudate nucleus, thalamus, and pons [81]. The increase in ADC along the fibers may indicate extracellular water content increase secondary to axonal loss and fiber density reduction, both of which could be attributed to chronic ischemia. One study found a significant reduction in DTI fiber counts in the corpus callosum of SCD patients compared to controls. A significant reduction in DTI fiber counts was also observed in the corticospinal tracts bilaterally. These results can be explained by axonal damage due to vasculopathy [81].
7.2 Perfusion Imaging
Perfusion imaging can be achieved with contrast-enhanced CT, contrast-enhanced MRI, non-contrast MRI with arterial spin labeling (ASL) technique, and positron emission tomography (PET) imaging [82]. Perfusion imaging does not affect treatment decisions in SCD patients. However, there is value in studying brain perfusion in SCD to better understand the pathophysiology of the disease. Perfusion imaging allows the calculation of relative cerebral blood flow (CBF) and blood volume in the brain. Because of the very high radiation dose compared to conventional head CT, CT perfusion and PET imaging should be cautiously used in children [82]. Perfusion MRI techniques commonly used include dynamic susceptibility contrast (DSC) MRI and ASL. Compared with ASL, DSC MRI perfusion provides higher spatial resolution, requires shorter scanning time, and can measure other hemodynamic parameters such as cerebral blood volume (CBV) and mean transit time (MTT) simultaneously [83]. ASL using either continuous or pulsed ASL sequence is a noninvasive MR perfusion imaging technique that uses arterial blood water protons as an endogenous tracer of perfusion [84–86] (Fig. 29). Perfusion imaging studies can show changes of cerebral blood flow immediately after the event [87]. ASL studies of whole brain CBF in SCD patient compared to controls have been controversial [84–86]. ASL has been used to correlate CBF with both full-scale and performance IQ [85]. Using ASL, studies have identified right-left CBF asymmetries that could be early indicators of subclinical pathological changes in microvasculature and/or hemodynamics in SCD patients. The described perfusion asymmetries, however, could not always be associated with the presence of ischemic lesions [84–86].


A 10-year-old boy with SCD presenting with acute visual symptoms. (a) Axial DWI (b = 1,000) shows mildly increased signal in the bilateral parietal lobes. (b) Arterial spin labeling perfusion image shows decreased blood flow in the bilateral parieto-occipital lobes (arrows). (c) Axial DWI (b = 1,000) 2 days after the initial MRI shows discrete areas of diffusion restriction in the bilateral parietal lobes (arrows) and in the left frontal periventricular watershed distribution (open arrows)
7.3 MR Spectroscopy (MRS)
MR spectroscopy (MRS) is an advanced MR imaging method for quantitative and qualitative assessment of the metabolic state of brain tissue. Reports of MRS in SCD are limited. In 1992, one study showed that normally appearing brain tissues on conventional MRI did not show abnormal MRS findings [88], meaning there was no additional increase in sensitivity in disease detection with MRS. The same group reported a reversible temporary decrease of N-acetylaspartate (NAA), which is considered a marker of neuronal tissue, at the time of a stroke episode [89]. Another study showed that NAA was increased in the basal ganglia and generally in the brain of SCD patients [90]. These results suggest that brain NAA appears not to be a reliable marker of viable neurons in SCD patients at this moment.
7.4 Quantitative MRI (qMRI)
Remarkable advances in MR imaging have led to the development of a variety of quantitative MR imaging (qMRI) techniques, including diffusion-weighted imaging (DWI), diffusion tensor imaging (DTI), relaxometry, magnetization transfer imaging, perfusion imaging, MR spectroscopy, and volumetry [91].
qMRI studies of T1 spin–lattice relaxation times have shown reduced gray matter relaxivity in pediatric patients with SCD compared to healthy controls [92, 93], especially in the caudate nucleus and in cortical gray matter. In addition, volumetric deficits of gray matter in patients with SCD were reported in the absence of volumetric deficits in the white matter [94]. These observations support the idea that gray matter is selectively vulnerable to injury in pediatric SCD patients. In the future, the use of qMRI techniques, such as T1 values and volumetry, may serve as early markers of brain damage in young SCD patients [92–94].
qMRI studies examined with mixed TSE pulse sequence showed T1 lengthening, T2 and secular-T2 shortening, and an increase in bone marrow volume in patients with SCD [75, 76]. The T1 lengthening supports the fact that there is less yellow marrow volume and more red marrow volume in patients with SCD, which suggests the failure of red-to-yellow marrow conversion. T2 and secular-T2 shortening also supports the failure of red-to-yellow marrow conversion. Increased iron deposition as a result of hemolysis in subjects with severe disease and repeated blood transfusions can account for the shortening of T2 and secular-T2 times. By using the specific changes seen in T1, T2, and secular-T2 relaxation times, qMRI can be a useful tool in the assessment and monitoring of disease severity in patients with SCD [75, 76]. In addition, qMRI has been used to evaluate the extracranial structures, such as salivary glands [95] and lacrimal glands [96].
7.5 Nuclear Medicine
Brain perfusion single-photon emission computed tomography (SPECT) is a functional neuroimaging technique that allows for noninvasive evaluation of the physiological and pathophysiological status of the brain. SPECT imaging may demonstrate cerebral perfusion deficits in SCD patients with normal brain MR images. One study found that neurologically intact adult SCD patients exhibit brain perfusion decreases in areas such as the basal ganglia, thalamus, watershed areas of the anterior circulation, and cerebellar and occipital cortex [97]. These findings may contribute to the understanding of the mechanisms underlying the evolution of cerebral vasculopathy in SCD patients. Calculation of cerebrovascular reserve with acetazolamide (Diamox)-induced cerebrovascular vasodilatation in conjunction with SPECT imaging has a role in identifying SCD patients who are at a high risk for stroke [98].
S ince the late 1990s, perfusion and metabolic imaging techniques, such as positron emission tomography (PET), have been increasingly used to investigate the microvascular blood flow in patients with SCD. One study showed that PET may detect abnormalities missed by structural MRI in patients with SCD and a history of stroke [99]. Another study found that metabolic compromise in SCD patients exceeded the corresponding anatomic abnormality demonstrated on MRI [100].
8 Management
8.1 Prevention of Primary Stroke
Early screening of SCD patients with TCD ultrasound is the gold standard for assessing stroke risk [25]. In the absence of early TCD screening, stroke occurs in 7.4 % and 11 % of patients with SCD by 14 and 20 years of age, respectively, [16, 101] and by age 30 and 45 years, 15 % and 24 % will experience overt stroke, respectively [16, 102]. In a newborn cohort, an early TCD screening and transfusion program employed in patients at risk decreased the overt stroke risk to 1.9 % by 18 years of age [103]. It has been recommended to obtain serial TCD and brain MRI/MRA studies to monitor the effectiveness of transfusion in children with abnormal TCD velocities [104] (Fig. 30).


An 8-year-old boy with SCD presenting with annual follow-up with transcranial Doppler screening. Peak systolic velocity (PSV) in the left MCA was 258.7 cm/s (<200, normal; 200–249, conditional; ≥250 abnormal). Follow-up MRA showed left MCA stenosis (not shown)
The time-averaged mean of the maximal velocity (TAMMV) in large intracranial arteries is a powerful, noninvasive tool that can be utilized to classify an individual patient’s risk of stroke [25]. According to the TCD criteria for SCD, TAMMV < 170 cm/s in any intracranial artery is considered normal, and TAMMV of 170–199 cm/s is conditional. TAMMV > 200 cm/s is considered abnormal and indicates the need for transfusion. TAMMV > 200 cm/s is associated with a 40 % risk of stroke within the next 3 years [105]. The Stroke Prevention Trial in Sickle Cell Anemia (STOP) documented that chronic transfusion therapy in patients with a mean blood flow velocity of >200 cm/s with TCD prevented the initial occurrence of stroke, decreasing the annual incidence of stroke from 10 % to <1 % [25]. TCD is repeated every 12 months after a normal scan and every 3 months with a conditional scan. Although TAMMV is the gold standard TCD measurement in the setting of SCD, in clinical practice, peak systolic velocity (PSV) measurements are more commonly used in vascular ultrasound practice and have been shown to be highly correlated with TAMMV in the prediction of future stroke [106] (Fig. 30). However, a recent abstract has demonstrated that PSV alone may overestimate the risk of stroke in children, demonstrating the importance of TAMMV as the primary initial measurement for stroke-risk stratification [107].
8.2 Prevention of Secondary Stroke
SCIs that may be found in approximately 20–37 % of children with SCD are associated with increased risk of subsequent overt stroke [42, 44, 53]. Transfusion therapy has been shown to reduce overt strokes in patients with SCIs. Despite transfusion treatment, there remains a 20 % secondary stroke rate [108] and a 25 % likelihood of additional SCIs [51]. Hydroxyurea (HU) has also been shown to reduce TCD velocities and has shown some benefit in reducing the risk of secondary stroke [109]. A randomized clinical trial, Stroke With Transfusions Changing to Hydroxyurea (SWiTCH), evaluated HU with phlebotomy as an alternative to the ongoing transfusion therapy, but the study was closed early after an interim analysis revealed equivalent liver iron between the two groups, failing to meet the primary end point [110]. MRI can monitor the progression and extent of the ischemic change, and MRA can monitor the stenosis and/or occlusion of the intra- and extracranial arteries (Fig. 14).
9 Summary
Sickle cell disease (SCD) is one of the most important hereditary hematologic disorders which may result in severe neurovascular complications due to vaso-occlusion. Radiological evaluation is extremely important in daily practice, such as TCD in stroke-risk screening with velocity measurements of the cerebral arteries, CT in detecting hemorrhagic strokes, MRI in diagnosing acute infarct, white matter changes and cerebral vasculopathy, and catheter angiogram in evaluating precise vascular features and in treatment planning. Radiologic imaging features may also support the current understanding of the pathophysiology underlying neurovascular complications of SCD. Advanced MR imaging and nuclear imaging have the potential to provide additional findings that may improve the management of the neurovascular complications of SCD, but further investigations are still necessary. Finally, radiologists and clinicians should be familiar with the pathophysiology and imaging findings of the neurovascular complications of SCD as well as various complications in the craniofacial region and spine, which may be crucial for timely diagnosis and appropriate treatment.
References
Rees DC, Williams TN, Gladwin MT (2010) Sickle-cell disease. Lancet 376:2018–2031
Steinberg MH (2008) Sickle cell anemia, the first molecular disease: overview of molecular etiology, pathophysiology, and therapeutic approaches. Sci World J 8:1295–1324
Steen RG, Hankins GM, Xiong X, Wang WC, Beil K, Langston JW, Helton KJ (2003) Prospective brain imaging evaluation of children with sickle cell trait: initial observations. Radiology 228:208–215
Kanter J, Kruse-Jarres R (2013) Management of sickle cell disease from childhood through adulthood. Blood Rev 27:279–287
Connes P, Verlhac S, Bernaudin F (2013) Advances in understanding the pathogenesis of cerebrovascular vasculopathy in sickle cell anaemia. Br J Haematol 161:484–498
Lonergan GJ, Cline DB, Abbondanzo SL (2001) Sickle cell anemia. Radiographics 21:971–994
Moran CJ, Siegel MJ, DeBaun MR (1998) Sickle cell disease: imaging of cerebrovascular complications. Radiology 206:311–321
Moritani T, Numaguchi Y, Lemer NB, Rozans MK, Robinson AE, Hiwatashi A, Westesson PL (2004) Sickle cell cerebrovascular disease: usual and unusual findings on MR imaging and MR angiography. Clin Imaging 28:173–186
Glushakova S, Balaban A, McQueen PG, Coutinho R, Miller JL, Nossal R, Fairhurst RM, Zimmerberg J (2014) Hemoglobinopathic erythrocytes affect the intraerythrocytic multiplication of Plasmodium falciparum in vitro. J Infect Dis 210:1100–1109
Halasa NB, Shankar SM, Talbot TR, Arbogast PG, Mitchel EF, Wang WC, Schaffner W, Craig AS, Griffin MR (2007) Incidence of invasive pneumococcal disease among individuals with sickle cell disease before and after the introduction of the pneumococcal conjugate vaccine. Clin Infect Dis 44:1428–1433
Quinn CT, Rogers ZR, McCavit TL, Buchanan GR (2010) Improved survival of children and adolescents with sickle cell disease. Blood 115:3447–3452
Platt OS, Brambilla DJ, Rosse WF, Milner PF, Castro O, Steinberg MH, Klug PP (1994) Mortality in sickle cell disease. Life expectancy and risk factors for early death. N Engl J Med 330:1639–1644
Watanabe M, Saito N, Nadgir RN, Liao JH, Flower EN, Steinberg MH, Sakai O (2013) Craniofacial bone infarcts in sickle cell disease: clinical and radiological manifestations. J Comput Assist Tomogr 37:91–97
Gladwin MT, Vichinsky E (2008) Pulmonary complications of sickle cell disease. N Engl J Med 359:2254–2265
Ejindu VC, Hine AL, Mashayekhi M, Shorvon PJ, Misra RR (2007) Musculoskeletal manifestations of sickle cell disease. Radiographics 27:1005–1021
Ohene-Frempong K, Weiner SJ, Sleeper LA, Miller ST, Embury S, Moohr JW, Wethers DL, Pegelow CH, Gill FM (1998) Cerebrovascular accidents in sickle cell disease: rates and risk factors. Blood 91:288–294
Pegelow CH, Macklin EA, Moser FG, Wang WC, Bello JA, Miller ST, Vichinsky EP, DeBaun MR, Guarini L, Zimmerman RA, Younkin DP, Gallagher DM, Kinney TR (2002) Longitudinal changes in brain magnetic resonance imaging findings in children with sickle cell disease. Blood 99:3014–3018
Miller ST, Macklin EA, Pegelow CH, Kinney TR, Sleeper LA, Bello JA, DeWitt LD, Gallagher DM, Guarini L, Moser FG, Ohene-Frempong K, Sanchez N, Vichinsky EP, Wang WC, Wethers DL, Younkin DP, Zimmerman RA, DeBaun MR, Cooperative Study of Sickle Cell D (2001) Silent infarction as a risk factor for overt stroke in children with sickle cell anemia: a report from the Cooperative Study of Sickle Cell Disease. J Pediatr 139:385–390
Sidani CA, Ballourah W, El Dassouki M, Muwakkit S, Dabbous I, Dahoui H, Al-Kutoubi A, Abboud MR (2008) Venous sinus thrombosis leading to stroke in a patient with sickle cell disease on hydroxyurea and high hemoglobin levels: treatment with thrombolysis. Am J Hematol 83:818–820
Vichinsky EP, Neumayr LD, Gold JI, Weiner MW, Rule RR, Truran D, Kasten J, Eggleston B, Kesler K, McMahon L, Orringer EP, Harrington T, Kalinyak K, De Castro LM, Kutlar A, Rutherford CJ, Johnson C, Bessman JD, Jordan LB, Armstrong FD, Neuropsychological Dysfunction, Neuroimaging Adult Sickle Cell Anemia Study Group (2010) Neuropsychological dysfunction and neuroimaging abnormalities in neurologically intact adults with sickle cell anemia. JAMA 303:1823–1831
Mackin RS, Insel P, Truran D, Vichinsky EP, Neumayr LD, Armstrong FD, Gold JI, Kesler K, Brewer J, Weiner MW, Neuropsychological Dysfunction, Neuroimaging Adult Sickle Cell Anemia Study Group (2014) Neuroimaging abnormalities in adults with sickle cell anemia: associations with cognition. Neurology 82:835–841
Charache S, Terrin ML, Moore RD, Dover GJ, Barton FB, Eckert SV, McMahon RP, Bonds DR (1995) Effect of hydroxyurea on the frequency of painful crises in sickle cell anemia. Investigators of the Multicenter Study of Hydroxyurea in Sickle Cell Anemia. N Engl J Med 332:1317–1322
DeBaun MR, Gordon M, McKinstry RC, Noetzel MJ, White DA, Sarnaik SA, Meier ER, Howard TH, Majumdar S, Inusa BP, Telfer PT, Kirby-Allen M, McCavit TL, Kamdem A, Airewele G, Woods GM, Berman B, Panepinto JA, Fuh BR, Kwiatkowski JL, King AA, Fixler JM, Rhodes MM, Thompson AA, Heiny ME, Redding-Lallinger RC, Kirkham FJ, Dixon N, Gonzalez CE, Kalinyak KA, Quinn CT, Strouse JJ, Miller JP, Lehmann H, Kraut MA, Ball WS Jr, Hirtz D, Casella JF (2014) Controlled trial of transfusions for silent cerebral infarcts in sickle cell anemia. N Engl J Med 371:699–710
Hulbert ML, Scothorn DJ, Panepinto JA, Scott JP, Buchanan GR, Sarnaik S, Fallon R, Chu JY, Wang W, Casella JF, Resar L, Berman B, Adamkiewicz T, Hsu LL, Smith-Whitley K, Mahoney D, Woods G, Watanabe M, DeBaun MR (2006) Exchange blood transfusion compared with simple transfusion for first overt stroke is associated with a lower risk of subsequent stroke: a retrospective cohort study of 137 children with sickle cell anemia. J Pediatr 149:710–712
Adams RJ, McKie VC, Hsu L, Files B, Vichinsky E, Pegelow C, Abboud M, Gallagher D, Kutlar A, Nichols FT, Bonds DR, Brambilla D (1998) Prevention of a first stroke by transfusions in children with sickle cell anemia and abnormal results on transcranial Doppler ultrasonography. N Engl J Med 339:5–11
Peinemann F, Bartel C, Grouven U (2013) First-line allogeneic hematopoietic stem cell transplantation of HLA-matched sibling donors compared with first-line ciclosporin and/or antithymocyte or antilymphocyte globulin for acquired severe aplastic anemia. Cochrane Database Syst Rev 7, CD006407
Webb J, Kwiatkowski JL (2013) Stroke in patients with sickle cell disease. Expert Rev Hematol 6:301–316
Arkuszewski M, Melhem ER, Krejza J (2010) Neuroimaging in assessment of risk of stroke in children with sickle cell disease. Adv Med Sci 55:115–129
Smajlovic D, Sinanovic O (2004) Sensitivity of the neuroimaging techniques in ischemic stroke. Med Arh 58:282–284
Paonessa A, Limbucci N, Tozzi E, Splendiani A, Gallucci M (2010) Radiological strategy in acute stroke in children. Eur J Radiol 74:77–85
Roach ES, Golomb MR, Adams R, Biller J, Daniels S, Deveber G, Ferriero D, Jones BV, Kirkham FJ, Scott RM, Smith ER, American Heart Association Stroke Council, Council on Cardiovascular Disease in the Young (2008) Management of stroke in infants and children: a scientific statement from a Special Writing Group of the American Heart Association Stroke Council and the Council on Cardiovascular Disease in the Young. Stroke 39:2644–2691
Gonzalez RG, Schaefer PW, Buonanno FS, Schwamm LH, Budzik RF, Rordorf G, Wang B, Sorensen AG, Koroshetz WJ (1999) Diffusion-weighted MR imaging: diagnostic accuracy in patients imaged within 6 hours of stroke symptom onset. Radiology 210:155–162
Rydberg JN, Hammond CA, Grimm RC, Erickson BJ, Jack CR Jr, Huston J 3rd, Riederer SJ (1994) Initial clinical experience in MR imaging of the brain with a fast fluid-attenuated inversion-recovery pulse sequence. Radiology 193:173–180
Cheng AL, Batool S, McCreary CR, Lauzon ML, Frayne R, Goyal M, Smith EE (2013) Susceptibility-weighted imaging is more reliable than T2*-weighted gradient-recalled echo MRI for detecting microbleeds. Stroke 44:2782–2786
Strouse JJ, Lanzkron S, Urrutia V (2011) The epidemiology, evaluation and treatment of stroke in adults with sickle cell disease. Expert Rev Hematol 4:597–606
Frietsch T, Ewen I, Waschke KF (2001) Anaesthetic care for sickle cell disease. Eur J Anaesthesiol 18:137–150
Maeda M, Tsuchida C (1999) “Ivy sign” on fluid-attenuated inversion-recovery images in childhood moyamoya disease. AJNR Am J Neuroradiol 20:1836–1838
Brandao RA, de Carvalho GT, Reis BL, Bahia E, de Souza AA (2009) Intracranial aneurysms in sickle cell patients: report of 2 cases and review of the literature. Surg Neurol 72:296–299 (discussion 299)
Santhosh K, Kesavadas C, Thomas B, Gupta AK, Thamburaj K, Kapilamoorthy TR (2009) Susceptibility weighted imaging: a new tool in magnetic resonance imaging of stroke. Clin Radiol 64:74–83
Lu L, Zhang LJ, Poon CS, Wu SY, Zhou CS, Luo S, Wang M, Lu GM (2012) Digital subtraction CT angiography for detection of intracranial aneurysms: comparison with three-dimensional digital subtraction angiography. Radiology 262:605–612
Sailer AM, Wagemans BA, Nelemans PJ, de Graaf R, van Zwam WH (2014) Diagnosing intracranial aneurysms with MR angiography. systematic review and meta-analysis. Stroke 45:119–126
Kwiatkowski JL, Zimmerman RA, Pollock AN, Seto W, Smith-Whitley K, Shults J, Blackwood-Chirchir A, Ohene-Frempong K (2009) Silent infarcts in young children with sickle cell disease. Br J Haematol 146:300–305
Pegelow CH, Wang W, Granger S, Hsu LL, Vichinsky E, Moser FG, Bello J, Zimmerman RA, Adams RJ, Brambilla D, Trial S (2001) Silent infarcts in children with sickle cell anemia and abnormal cerebral artery velocity. Arch Neurol 58:2017–2021
Dowling MM, Quinn CT, Rogers ZR, Buchanan GR (2010) Acute silent cerebral infarction in children with sickle cell anemia. Pediatr Blood Cancer 54:461–464
Kinney TR, Sleeper LA, Wang WC, Zimmerman RA, Pegelow CH, Ohene-Frempong K, Wethers DL, Bello JA, Vichinsky EP, Moser FG, Gallagher DM, DeBaun MR, Platt OS, Miller ST (1999) Silent cerebral infarcts in sickle cell anemia: a risk factor analysis. The Cooperative Study of Sickle Cell Disease. Pediatrics 103:640–645
Wang WC, Gallagher DM, Pegelow CH, Wright EC, Vichinsky EP, Abboud MR, Moser FG, Adams RJ (2000) Multicenter comparison of magnetic resonance imaging and transcranial Doppler ultrasonography in the evaluation of the central nervous system in children with sickle cell disease. J Pediatr Hematol Oncol 22:335–339
Arkuszewski M, Krejza J, Chen R, Ichord R, Kwiatkowski JL, Bilello M, Zimmerman R, Ohene-Frempong K, Melhem ER (2014) Sickle cell anemia: Intracranial stenosis and silent cerebral infarcts in children with low risk of stroke. Adv Med Sci 59:108–113
Schatz J, White DA, Moinuddin A, Armstrong M, DeBaun MR (2002) Lesion burden and cognitive morbidity in children with sickle cell disease. J Child Neurol 17:891–895
DeBaun MR, Armstrong FD, McKinstry RC, Ware RE, Vichinsky E, Kirkham FJ (2012) Silent cerebral infarcts: a review on a prevalent and progressive cause of neurologic injury in sickle cell anemia. Blood 119:4587–4596
Steen RG, Miles MA, Helton KJ, Strawn S, Wang W, Xiong X, Mulhern RK (2003) Cognitive impairment in children with hemoglobin SS sickle cell disease: relationship to MR imaging findings and hematocrit. AJNR Am J Neuroradiol 24:382–389
Hulbert ML, McKinstry RC, Lacey JL, Moran CJ, Panepinto JA, Thompson AA, Sarnaik SA, Woods GM, Casella JF, Inusa B, Howard J, Kirkham FJ, Anie KA, Mullin JE, Ichord R, Noetzel M, Yan Y, Rodeghier M, Debaun MR (2011) Silent cerebral infarcts occur despite regular blood transfusion therapy after first strokes in children with sickle cell disease. Blood 117:772–779
Casella JF, King AA, Barton B, White DA, Noetzel MJ, Ichord RN, Terrill C, Hirtz D, McKinstry RC, Strouse JJ, Howard TH, Coates TD, Minniti CP, Campbell AD, Vendt BA, Lehmann H, Debaun MR (2010) Design of the silent cerebral infarct transfusion (SIT) trial. Pediatr Hematol Oncol 27:69–89
Steen RG, Emudianughe T, Hankins GM, Wynn LW, Wang WC, Xiong X, Helton KJ (2003) Brain imaging findings in pediatric patients with sickle cell disease. Radiology 228:216–225
Gibbs WN, Opatowsky MJ, Burton EC (2012) AIRP best cases in radiologic-pathologic correlation: cerebral fat embolism syndrome in sickle cell beta-thalassemia. Radiographics 32:1301–1306
Mossa-Basha M, Izbudak I, Gurda GT, Aygun N (2012) Cerebral fat embolism syndrome in sickle cell anaemia/beta-thalassemia: importance of susceptibility-weighted MRI. Clin Radiol 67:1023–1026
Currie S, Raghavan A, Batty R, Connolly DJ, Griffiths PD (2011) Childhood moyamoya disease and moyamoya syndrome: a pictorial review. Pediatr Neurol 44:401–413
Amlie-Lefond C, Bernard TJ, Sebire G, Friedman NR, Heyer GL, Lerner NB, DeVeber G, Fullerton HJ, International Pediatric Stroke Study Group (2009) Predictors of cerebral arteriopathy in children with arterial ischemic stroke: results of the International Pediatric Stroke Study. Circulation 119:1417–1423
Dobson SR, Holden KR, Nietert PJ, Cure JK, Laver JH, Disco D, Abboud MR (2002) Moyamoya syndrome in childhood sickle cell disease: a predictive factor for recurrent cerebrovascular events. Blood 99:3144–3150
Lin N, Baird L, Koss M, Kopecky KE, Gone E, Ullrich NJ, Scott RM, Smith ER (2011) Discovery of asymptomatic moyamoya arteriopathy in pediatric syndromic populations: radiographic and clinical progression. Neurosurg Focus 31:E6
Arias EJ, Derdeyn CP, Dacey RG Jr, Zipfel GJ (2014) Advances and surgical considerations in the treatment of moyamoya disease. Neurosurgery 74:S116–S125
Adelson PD, Scott RM (1995) Pial synangiosis for moyamoya syndrome in children. Pediatr Neurosurg 23:26–33
Robertson RL, Burrows PE, Barnes PD, Robson CD, Poussaint TY, Scott RM (1997) Angiographic changes after pial synangiosis in childhood moyamoya disease. AJNR Am J Neuroradiol 18:837–845
Kennedy BC, McDowell MM, Yang PH, Wilson CM, Li S, Hankinson TC, Feldstein NA, Anderson RC (2014) Pial synangiosis for moyamoya syndrome in children with sickle cell anemia: a comprehensive review of reported cases. Neurosurg Focus 36:E12
Calviere L, Viguier A, Guidolin B, Tall P, Larrue V (2007) Cervical artery stenoses in sickle cell disease. Eur Neurol 58:120–121
Deane CR, Goss D, Bartram J, Pohl KR, Height SE, Sibtain N, Jarosz J, Thein SL, Rees DC (2010) Extracranial internal carotid arterial disease in children with sickle cell anemia. Haematologica 95:1287–1292
Telfer PT, Evanson J, Butler P, Hemmaway C, Abdulla C, Gadong N, Whitmarsh S, Kaya B, Kirkham FJ (2011) Cervical carotid artery disease in sickle cell anemia: clinical and radiological features. Blood 118:6192–6199
Verlhac S, Balandra S, Cussenot I, Kasbi F, Vasile M, Kheniche A, Elmaleh-Berges M, Ithier G, Benkerrou M, Bernaudin F, Sebag G (2014) Extracranial carotid arteriopathy in stroke-free children with sickle cell anemia: detection by submandibular Doppler sonography. Pediatr Radiol 44:587–596
Saito N, Nadgir RN, Flower EN, Sakai O (2010) Clinical and radiologic manifestations of sickle cell disease in the head and neck. Radiographics 30:1021–1034
Steen RG, Reddick WE, Glass JO, Wang WC (1998) Evidence of cranial artery ectasia in sickle cell disease patients with ectasia of the basilar artery. J Stroke Cerebrovasc Dis 7:330–338
Frye RE (2009) Reversible posterior leukoencephalopathy syndrome in sickle-cell anemia. Pediatr Neurol 40:298–301
Geevasinga N, Cole C, Herkes GK, Barnett Y, Lin J, Needham M (2014) Sickle cell disease and posterior reversible leukoencephalopathy. J Clin Neurosci 21:1329–1332
Henderson JN, Noetzel MJ, McKinstry RC, White DA, Armstrong M, DeBaun MR (2003) Reversible posterior leukoencephalopathy syndrome and silent cerebral infarcts are associated with severe acute chest syndrome in children with sickle cell disease. Blood 101:415–419
Khademian Z, Speller-Brown B, Nouraie SM, Minniti CP (2009) Reversible posterior leuko-encephalopathy in children with sickle cell disease. Pediatr Blood Cancer 52:373–375
Kolovou V, Zampakis P, Ginopoulou A, Varvarigou A, Kaleyias J (2013) Reversible posterior leukoencephalopathy syndrome after blood transfusion in a pediatric patient with sickle cell disease. Pediatr Neurol 49:213–217
Elias EJ, Liao JH, Jara H, Watanabe M, Nadgir RN, Sakai Y, Erbay K, Saito N, Ozonoff A, Steinberg MH, Sakai O (2013) Quantitative MRI analysis of craniofacial bone marrow in patients with sickle cell disease. AJNR Am J Neuroradiol 34:622–627
Liao JH, Jara H, Nadgir R, Elias E, Nowrouzi N, Saito N, Steinberg MH, Sakai O (2013) qMRI relaxometry of mandibular bone marrow: a monomodal distribution in sickle cell disease. J Magn Reson Imaging 37:1182–1188
Marlow TJ, Brunson CY, Jackson S, Schabel SI (1998) “Tower vertebra”: a new observation in sickle cell disease. Skeletal Radiol 27:195–198
Jain R, Sawhney S, Rizvi SG (2008) Acute bone crises in sickle cell disease: the T1 fat-saturated sequence in differentiation of acute bone infarcts from acute osteomyelitis. Clin Radiol 63:59–70
Saito N, Watanabe M, Liao J, Flower EN, Nadgir RN, Steinberg MH, Sakai O (2011) Clinical and radiologic findings of inner ear involvement in sickle cell disease. AJNR AmJ Neuroradiol 32:2160–2164
Le Bihan D, Mangin JF, Poupon C, Clark CA, Pappata S, Molko N, Chabriat H (2001) Diffusion tensor imaging: concepts and applications. J Magn Reson Imaging 13:534–546
Balci A, Karazincir S, Beyoglu Y, Cingiz C, Davran R, Gali E, Okuyucu E, Egilmez E (2012) Quantitative brain diffusion-tensor MRI findings in patients with sickle cell disease. AJR Am J Roentgenol 198:1167–1174
Behpour AM, Shah PS, Mikulis DJ, Kassner A (2013) Cerebral blood flow abnormalities in children with sickle cell disease: a systematic review. Pediatr Neurol 48:188–199
Kirkham FJ, Calamante F, Bynevelt M, Gadian DG, Evans JP, Cox TC, Connelly A (2001) Perfusion magnetic resonance abnormalities in patients with sickle cell disease. Ann Neurol 49:477–485
Oguz KK, Golay X, Pizzini FB, Freer CA, Winrow N, Ichord R, Casella JF, van Zijl PC, Melhem ER (2003) Sickle cell disease: continuous arterial spin-labeling perfusion MR imaging in children. Radiology 227:567–574
Strouse JJ, Cox CS, Melhem ER, Lu H, Kraut MA, Razumovsky A, Yohay K, van Zijl PC, Casella JF (2006) Inverse correlation between cerebral blood flow measured by continuous arterial spin-labeling (CASL) MRI and neurocognitive function in children with sickle cell anemia (SCA). Blood 108:379–381
van den Tweel XW, Nederveen AJ, Majoie CB, van der Lee JH, Wagener-Schimmel L, van Walderveen MA, Poll The BT, Nederkoorn PJ, Heijboer H, Fijnvandraat K (2009) Cerebral blood flow measurement in children with sickle cell disease using continuous arterial spin labeling at 3.0-Tesla MRI. Stroke 40:795–800
Sanelli PC, Sykes JB, Ford AL, Lee JM, Vo KD, Hallam DK (2013) Imaging and treatment of patients with acute stroke: an evidence-based review. AJNR Am J Neuroradiol 35:1045–1051
Wang Z, Bogdan AR, Zimmerman RA, Gusnard DA, Leigh JS, Ohene-Frempong K (1992) Investigation of stroke in sickle cell disease by 1H nuclear magnetic resonance spectroscopy. Neuroradiology 35:57–65
Wang Z, Zimmerman RA, Sauter R (1996) Proton MR spectroscopy of the brain: clinically useful information obtained in assessing CNS diseases in children. AJR Am J Roentgenol 167:191–199
Steen RG, Ogg RJ (2005) Abnormally high levels of brain N-acetylaspartate in children with sickle cell disease. AJNR Am J Neuroradiol 26:463–468
Watanabe M, Liao JH, Jara H, Sakai O (2013) Multispectral quantitative MR imaging of the human brain: lifetime age-related effects. Radiographics 33:1305–1319
Steen RG, Langston JW, Ogg RJ, Xiong X, Ye Z, Wang WC (1999) Diffuse T1 reduction in gray matter of sickle cell disease patients: evidence of selective vulnerability to damage? Magn Reson Imaging 17:503–515
Steen RG, Schroeder J (2003) Age-related changes in the pediatric brain: proton T1 in healthy children and in children with sickle cell disease. Magn Reson Imaging 21:9–15
Steen RG, Emudianughe T, Hunte M, Glass J, Wu S, Xiong X, Reddick WE (2005) Brain volume in pediatric patients with sickle cell disease: evidence of volumetric growth delay? AJNR Am J Neuroradiol 26:455–462
Liao J, Saito N, Ozonoff A, Jara H, Steinberg M, Sakai O (2012) Quantitative MRI analysis of salivary glands in sickle cell disease. Dentomaxillofac Radiol 41:630–636
Buch K, Watanabe M, Elias EJ, Liao JH, Jara H, Nadgir RN, Saito N, Steinberg MH, Sakai O (2014) Quantitative magnetic resonance imaging analysis of the Lacrimal gland in sickle cell disease. J Comput Assist Tomogr 38:674–680
Deus-Silva L, Bonilha L, Damasceno BP, Costa AL, Yasuda CL, Costa FF, Santos AO, Etchebehere EC, Oquendo-Nogueira R, Fockink R, de Freitas CF, Camargo EE, Li LM, Cendes F, Saad ST (2013) Brain perfusion impairment in neurologically asymptomatic adult patients with sickle-cell disease shown by Voxel-based analysis of SPECT images. Front Neurol 4:207
Kedar A, Drane WE, Shaeffer D, Nicole M, Adams C (2006) Measurement of cerebrovascular flow reserve in pediatric patients with sickle cell disease. Pediatr Blood Cancer 46:234–238
Powars DR, Conti PS, Wong WY, Groncy P, Hyman C, Smith E, Ewing N, Keenan RN, Zee CS, Harold Y, Hiti AL, Teng EL, Chan LS (1999) Cerebral vasculopathy in sickle cell anemia: diagnostic contribution of positron emission tomography. Blood 93:71–79
Reed W, Jagust W, Al-Mateen M, Vichinsky E (1999) Role of positron emission tomography in determining the extent of CNS ischemia in patients with sickle cell disease. Am J Hematol 60:268–272
Quinn CT, Rogers ZR, Buchanan GR (2004) Survival of children with sickle cell disease. Blood 103:4023–4027
Switzer JA, Hess DC, Nichols FT, Adams RJ (2006) Pathophysiology and treatment of stroke in sickle-cell disease: present and future. Lancet Neurol 5:501–512
Bernaudin F, Verlhac S, Arnaud C, Kamdem A, Chevret S, Hau I, Coic L, Leveille E, Lemarchand E, Lesprit E, Abadie I, Medejel N, Madhi F, Lemerle S, Biscardi S, Bardakdjian J, Galacteros F, Torres M, Kuentz M, Ferry C, Socie G, Reinert P, Delacourt C (2011) Impact of early transcranial Doppler screening and intensive therapy on cerebral vasculopathy outcome in a newborn sickle cell anemia cohort. Blood 117:1130–1140, quiz 436
Sheehan VA, Hansbury EN, Smeltzer MP, Fortner G, McCarville MB, Aygun B (2013) Transcranial Doppler velocity and brain MRI/MRA changes in children with sickle cell anemia on chronic transfusions to prevent primary stroke. Pediatr Blood Cancer 60:1499–1502
Adams RJ, Nichols FT, Figueroa R, McKie V, Lott T (1992) Transcranial Doppler correlation with cerebral angiography in sickle cell disease. Stroke 23:1073–1077
Jones A, Granger S, Brambilla D, Gallagher D, Vichinsky E, Woods G, Berman B, Roach S, Nichols F, Adams RJ (2005) Can peak systolic velocities be used for prediction of stroke in sickle cell anemia? Pediatr Radiol 35:66–72
Ishola T, Quinn CT (2013) Transcranial Doppler peak systolic velocities overestimate the risk of stroke in sickle cell anemia. Blood 122:2240
Scothorn DJ, Price C, Schwartz D, Terrill C, Buchanan GR, Shurney W, Sarniak I, Fallon R, Chu JY, Pegelow CH, Wang W, Casella JF, Resar LS, Berman B, Adamkiewicz T, Hsu LL, Ohene-Frempong K, Smith-Whitley K, Mahoney D, Scott JP, Woods GM, Watanabe M, Debaun MR (2002) Risk of recurrent stroke in children with sickle cell disease receiving blood transfusion therapy for at least five years after initial stroke. J Pediatr 140:348–354
Madden NA, Jones GL, Kalpatthi R, Woods G (2014) Practice patterns of stroke screening and hydroxyurea use in children with sickle cell disease: a survey of health care providers. J Pediatr Hematol Oncol 36:e382–e386
Ware RE, Helms RW, Investigators SW (2012) Stroke with transfusions changing to Hydroxyurea (SWiTCH). Blood 119:3925–3932
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2016 Springer Science+Business Media New York
About this entry
Cite this entry
Fujita, A. et al. (2016). Sickle Cell Disease and Stroke. In: Saba, L., Raz, E. (eds) Neurovascular Imaging. Springer, New York, NY. https://doi.org/10.1007/978-1-4614-9029-6_11
Download citation
DOI: https://doi.org/10.1007/978-1-4614-9029-6_11
Published:
Publisher Name: Springer, New York, NY
Print ISBN: 978-1-4614-9028-9
Online ISBN: 978-1-4614-9029-6
eBook Packages: MedicineReference Module Medicine