
Abstract
Waldenström’s macroglobulinemia (WM) is a B-cell lymphoproliferative disorder defined by a lymphoplasmacytic infiltration in the bone marrow or lymphatic tissue and a monoclonal immunoglobulin M (IgM) protein in the serum [1, 2]. The infiltration of the bone marrow and extramedullary sites by malignant B-cells, as well as elevated IgM levels, accounts for the symptoms associated with this disease. This may result in patients developing constitutional symptoms, pancytopenia, organomegaly, neuropathy, symptoms associated with immunoglobulin deposition, or hyperviscosity [3, 4]. There is significant heterogeneity, however, in the symptoms with which patients present. While some patients present with the symptoms listed above, many are asymptomatic at the time of diagnosis.
Waldenström’s macroglobulinemia remains incurable with current therapy with a median survival for symptomatic patients of approximately 8 years [5]. However, because many patients are diagnosed with this disease at an advanced age, approximately half of the patients die from causes unrelated to Waldenström’s macroglobulinemia. Due to the incurable nature of the disease, the heterogeneity of clinical presentation, as well as the comorbid conditions and competing causes of death, the decision to treat patients and the choice of treatment can be rather complex. A number of consensus meetings have listed reasonable treatment options [6–8], but the treating physician is still faced with a difficult treatment decision in a patient with an uncommon disease.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others


Keywords
- Chronic Lymphocytic Leukemia
- Mantle Cell Lymphoma
- Marginal Zone Lymphoma
- Immunoglobulin Deposition
- Lymphoplasmacytic Lymphoma
These keywords were added by machine and not by the authors. This process is experimental and the keywords may be updated as the learning algorithm improves.
Introduction
Waldenström’s macroglobulinemia (WM) is a B-cell lymphoproliferative disorder defined by a lymphoplasmacytic infiltration in the bone marrow or lymphatic tissue and a monoclonal immunoglobulin M (IgM) protein in the serum [1, 2]. The infiltration of the bone marrow and extramedullary sites by malignant B-cells, as well as elevated IgM levels, accounts for the symptoms associated with this disease. This may result in patients developing constitutional symptoms, pancytopenia, organomegaly, neuropathy, symptoms associated with immunoglobulin deposition, or hyperviscosity [3, 4]. There is significant heterogeneity, however, in the symptoms with which patients present. While some patients present with the symptoms listed above, many are asymptomatic at the time of diagnosis.
Waldenström’s macroglobulinemia remains incurable with current therapy with a median survival for symptomatic patients of approximately 8 years [5]. However, because many patients are diagnosed with this disease at an advanced age, approximately half of the patients die from causes unrelated to Waldenström’s macroglobulinemia. Due to the incurable nature of the disease, the heterogeneity of clinical presentation, as well as the comorbid conditions and competing causes of death, the decision to treat patients and the choice of treatment can be rather complex. A number of consensus meetings have listed reasonable treatment options [6–8], but the treating physician is still faced with a difficult treatment decision in a patient with an uncommon disease.
Epidemiology
The overall incidence of Waldenström’s macroglobulinemia is approximately 5 per million persons per year accounting for approximately 1–2 % of hematological cancers [9, 10]. The incidence of this disease is highest among Caucasians and is rare in other population groups [11]. The median age at diagnosis varies between 63 and 68 years, and the majority of new patients (55–70 %) are male [3].
Patients with monoclonal gammopathy of undetermined significance (MGUS) are at increased risk for progression to Waldenström’s macroglobulinemia [12]. In a population-based study of 1,384 individuals with MGUS, researchers showed an increased risk factor of 46 for developing Waldenström’s macroglobulinemia [12]. The rate of progression from IgM MGUS to Waldenström’s macroglobulinemia was further noted to be 1.5–2 % a year [13–15].
While the development of Waldenström’s macroglobulinemia is thought to be sporadic, there are a few studies demonstrating familial linkage and predisposition to the disease [16–18]. Both familial clustering of Waldenström’s macroglobulinemia and a notable increase (~10-fold) in the frequency of IgM MGUS in first-degree relatives of Waldenström’s patients are suggestive of familial risk [17]. Under the assumption that Waldenström’s macroglobulinemia and IgM MGUS share common susceptibility genes, strong linkages involving chromosomes 1q, 3q, and 4q have been identified [13]. Additionally, several studies have indicated a causal relationship between MGUS/Waldenström’s macroglobulinemia and chronic antigenic stimulation [18–21]. Recently, it was shown that 11 % of patients with IgM MGUS/Waldenström’s macroglobulinemia reacted with paratarg-7 (P-7), a protein of unknown function [22]. Analyses of relatives of patients with IgM MGUS/Waldenström’s macroglobulinemia with an anti-P-7-paraprotein showed that the hyperphosphorylated state of this protein (pP7) is inherited as a dominant trait, and carriers of pP7 have more than a sixfold increased risk of developing IgM MGUS/Waldenström’s macroglobulinemia (p = 0.001) [22]. Thus, pP-7 is the first biological entity that provides a plausible explanation for the familial clustering of cases of IgM MGUS/Waldenström’s macroglobulinemia.
Diagnosis
Attempts to better define Waldenström’s macroglobulinemia have been made in recent years by the World Health Organization (WHO) Lymphoma Classification [23], the consensus group formed at the Second International Workshop on Waldenström’s Macroglobulinemia [1], and the Mayo Clinic [24]. However, the respective definitions of the diagnostic criteria for Waldenström’s macroglobulinemia by these groups are not identical. All groups recognize Waldenström’s macroglobulinemia as a lymphoplasmacytic lymphoma associated with an IgM monoclonal protein in the serum. The WHO definition includes lymphomas other than lymphoplasmacytic lymphoma and also allows the monoclonal protein to be IgG or IgA. In contrast, the Second International Workshop on Waldenström’s Macroglobulinemia restricts the diagnosis exclusively to cases with lymphoplasmacytic lymphoma and an IgM monoclonal protein. The Second International Workshop on Waldenström’s Macroglobulinemia also eliminated the requirement for either a minimum amount of bone marrow involvement or a threshold concentration of IgM in the serum to fulfill the diagnosis, allowing for any detectable amount of either. In contrast, Mayo Clinic criteria require at least 10 % marrow involvement by lymphoplasmacytic lymphoma in asymptomatic patients. Furthermore, in regard to pathologic features, the WHO criteria focus predominantly on nodal involvement, whereas studies at Mayo Clinic indicate that most cases of Waldenström’s macroglobulinemia are bone marrow-based.
Lymphoplasmacytic lymphoma involving either the bone marrow or the extramedullary sites typically exhibits a cytologic spectrum ranging from small lymphocytes with clumped chromatin, inconspicuous nucleoli, and sparse cytoplasm to well-formed plasma cells [1, 25]. Frequently present are “plasmacytoid lymphocytes,” which have cytologic features of both lymphocytes and plasma cells, although the cytologic composition and the degree of plasmacytic differentiation vary from case to case. Nodal involvement is typically characterized by paracortical and hilar infiltration with frequent sparing of the subscapular and marginal sinuses. The bone marrow usually has some combination of nodular, paratrabecular, and interstitial infiltration; in approximately one-half of cases, plasma cells that contain Dutcher bodies are present.
The broad cytologic spectrum of the malignant cells composing Waldenström’s macroglobulinemia tumors is reflected in their immunophenotypic attributes. A monotypic lymphocytic component is almost always detected, typically with high levels of surface CD19, CD20, and immunoglobulin light chain expression [25]. The lymphoid component typically lacks CD10. In approximately 40 % of cases, the lymphocytes show some degree of CD5 expression; however, these cases usually do not express this antigen as strongly as tumor cells derived from patients with chronic lymphocytic leukemia (CLL)/small lymphocytic lymphoma or mantle cell lymphoma. By comparison, the plasmacytic component expresses the same immunoglobulin light chain as the lymphocytic component, is positive for CD138 (particularly when assessed by immunohistochemistry), and shows diminished expression of B-cell-associated antigens such as CD19, CD20, and PAX5. Typically, the lymphoplasmacytic lymphoma cells are positive for surface IgM, but on the basis of the WHO criteria, they may express any immunoglobulin isotype. In cases with isotype switch, the phenotype of the plasma cells closely resembles that of myelomatous plasma cells with strong CD38 and CD138 co-expression and complete lack of CD19. Waldenström’s macroglobulinemia tumor cells have also been shown to be CD25+, CD27+, CD75−, FMC7+, Bcl2+, and Bcl6−.
Conventional cytogenetic analyses initially determined the deletion of 6q to be the most common recurrent chromosomal abnormality in Waldenström’s macroglobulinemia, identified in approximately 50 % of patients [26]. Schop et al. observed 23 % of patients with an abnormal karyotype to have a 6q deletion, while FISH analysis confirmed deletion of 6q in 42 % of patients [27]. Further studies to assess minimal areas of deletion used multiple FISH probes on the 6q arm, and a minimal deleted region at 6q23–24.3 was suggested [28]. Although the deletion of 6q is present in around 50 % of WM patients, its presence cannot be used for diagnosis as it is widely observed in several B-cell malignancies, such as marginal zone lymphomas, multiple myeloma, and chronic lymphocytic leukemias [29–32].
Preliminary data obtained from whole genome sequencing of 30 Waldenström’s macroglobulinemia cases have recently been reported [33]. Strikingly, a mutation in MYD88 leading to a leucine to proline substitution in codon 265 (L265P) was found in 90 % of the cases (46/51). The MYD88 mutation provides a potential biomarker for differentiating Waldenström’s macroglobulinemia from other related entities such as marginal zone lymphoma, where MYD88 L265P was detected in less than 10 % of cases. Furthermore, the low prevalence of MYD88 mutations in IgM MGUS suggests either that the abnormality is associated with disease progression or that there is more than one type of IgM MGUS, with only some types progressing to Waldenström’s macroglobulinemia.
Gene expression profile (GEP) analysis of Waldenström’s macroglobulinemia also provides valuable information regarding the transcriptional signature of the disease. Data gathered from two independent studies highlight the similarities and differences in GEP between Waldenström’s macroglobulinemia, CLL, multiple myeloma, normal B-cells, and normal plasma cells [34, 35]. These studies specifically highlight similarities between GEP in malignant Waldenström’s macroglobulinemia cells and CLL. When analyzed in unsupervised clusters, Waldenström’s macroglobulinemia clustered far more with CLL expressions than with multiple myeloma [34]. Both Waldenström’s macroglobulinemia and CLL have strong B-cell signatures, characterized by the common marker CD20, and are defined by low proliferation rates and a lack of IgH translocations [35]. The GEP of both Waldenström’s macroglobulinemia and CLL shared similar profiles, particularly with regard to cell markers and IL-10 [34, 35].
One of the most significant findings in both studies was the high level of IL-6 transcript expression in Waldenström’s macroglobulinemia compared to multiple myeloma, CLL, and normal B-cells [34, 35]. IL-6 is a potent inflammatory cytokine that stimulates both local and systemic activating physiological functions in a multitude of cells [36]. Locally, it acts to increase lymphocyte activity, including antibody production. Additionally, IL-6 plays a key role by activating the MAPK pathway. While there were no specific mutations found in MAPK, its activity was notably increased, likely correlating with the upregulation of IL-6 [34]. The increase in IL-6 expression in Waldenström’s macroglobulinemia cells, more so than in normal B-cells, is suggestive of its autocrine activity.
Interestingly, IL-6 binds to the tyrosine kinase receptor Janus kinases (JAK) 1 and 2, which activate the downstream transcription factor Stat3, leading to increases in gene transcription, IgM production, and the activation of other signaling pathways [37]. Recently, a functional relationship between IL-6, Rantes (CCL5), and IgM secretion was observed and appears to be mediated through the JAK/STAT and PI3K pathways [38]. For the moment, the specific mechanisms of hyperimmunoglobulin secretion in Waldenström’s macroglobulinemia are still not known. GEP data combined with studies of the JAK/STAT pathway could be useful in future investigations into the pathogenic role of IL-6 and the JAK/STAT pathway in Waldenström’s macroglobulinemia.
Clinical Presentation
The infiltration of the bone marrow with malignant cells and the high levels of serum IgM protein circulating in patients with Waldenström’s macroglobulinemia are responsible for the majority of the morbidity associated with this malignancy. While some patients with Waldenström’s macroglobulinemia have no symptoms at diagnosis, others present with anemia, bleeding, or neurological complaints [39]. Additionally, as IgM protein is capable of forming large pentameric molecules in the circulation, many patients present with symptoms associated with immunoglobulin deposition and hyperviscosity syndrome [3]. Symptoms due to hyperviscosity syndrome have been reported in around 30 % of Waldenström’s macroglobulinemia patients and include skin and mucosal bleeding, retinopathy and visual disturbances, and cold sensitivity [39, 40].
Due to a shortage of effective therapies and a wide variability in clinical presentation and comorbidities, the process involved in deciding when and how to treat patients diagnosed with Waldenström’s macroglobulinemia can be a challenging one. However, before treatment can even be considered, an appropriate differential diagnosis between Waldenström’s macroglobulinemia, IgM MGUS, and smoldering Waldenström’s macroglobulinemia must be made as the appropriate treatment strategy may vary depending on the diagnosis. To this end, Mayo Clinic has created diagnostic criteria to differentiate between these IgM gammopathies based on the extent of bone marrow involvement and the presence or absence of symptomatic disease (see Table 24.1) [24].
Prognostic Factors
Following a diagnosis of Waldenström’s macroglobulinemia, the next step is to determine how best to manage the disease using a risk-adapted approach. Criteria commonly used for risk stratification are shown in Table 24.2. A multicenter collaborative project known as the International Prognostic Staging System for Waldenstrom’s Macroglobulinemia (IPSSWM) has incorporated five adverse prognostic factors to define three different risk groups for patients with Waldenström’s macroglobulinemia [3]. These factors include age >65 years, hemoglobin <11.5 g/dL, platelet count <100,000/μL, β2-microglobulin >3 mg/L, and monoclonal IgM protein >7 g/dL. Patients with 0–1, 2, or >2 of these factors are considered to be at low-, intermediate-, or high-risk, respectively, with corresponding 5-year survival rates of 87, 68, and 37 % [41]. While not currently used to determine the most appropriate treatment regimen, understanding a patient’s level of risk may be taken into account in deciding if and when treatment is necessary. Conversely, many asymptomatic patients may not require any therapy at all. For example, in a study by Garcia-Sanz et al., 50 % of patients who were asymptomatic at diagnosis did not require therapy for almost 3 years [39]. Similarly, one in ten patients who were managed with a watch-and-wait approach did not require therapy for 10 years. Taken together, these data underscore the need to carefully consider a patient’s prognostic risk prior to the initiation of any treatment to limit therapy to only those patients in whom it is required.
Indications for Treatment
To better determine which patients with Waldenström’s macroglobulinemia should receive treatment, a consensus panel at the Second International Workshop on Waldenström’s Macroglobulinemia agreed that therapy should be initiated in patients with a defined set of clinical findings and/or laboratory values [42]. Specifically, treatment was deemed appropriate in patients presenting with any of the following: constitutional symptoms including fever, night sweats, or weight loss; lymphadenopathy or splenomegaly; hemoglobin <10 g/dL or a platelet count lower than 100 × 109/L due to bone marrow infiltration; complications of the disease including symptomatic sensorimotor peripheral neuropathy, systemic amyloidosis, renal insufficiency, or symptomatic cryoglobulinemia. It was also recommended that patients with IgM MGUS and smoldering (asymptomatic) Waldenström’s macroglobulinemia with preserved hematologic function should be managed with a watch-and-wait approach. Additionally, all patients should be evaluated for symptoms of hyperviscosity syndrome (rarely observed with IgM levels <4 g/dL) such as visual deterioration, neurological symptoms, or unexplained bleeding, and should undergo plasmapheresis if necessary prior to receiving chemotherapy or immunotherapy [43].
Initial Therapy
Initial therapy for previously untreated patients with symptomatic Waldenström’s macroglobulinemia may involve various chemotherapeutic combinations with or without the addition of the CD20+-targeted antibody rituximab [44]. Rituximab is also used successfully as a single agent as first-line treatment in low-risk patients with symptomatic Waldenström’s macroglobulinemia. Treatment regimens containing nucleoside analogs (NA) such as fludarabine, with combinations including fludarabine/cyclophosphamide/rituximab (FCR) and fludarabine/rituximab (FR), have demonstrated good efficacy in symptomatic Waldenström’s macroglobulinemia patients. In a multicenter prospective study of 43 previously untreated patients with symptomatic disease, the FCR regimen was associated with an overall response rate of 79 %, including 11.6 % complete remission and 20.9 % very good partial remissions [45]. However, significant myelosuppression may limit the utility of this combination, as grade 3–4 neutropenia was reported in 45 % of courses and was the main reason for treatment discontinuation. Similarly, a separate study examined the combination of six cycles of fludarabine and eight infusions of rituximab (FR) [7]. Of the 43 patients enrolled, complete responses were achieved in two patients, with 81 % of patients achieving either a very good partial response or partial response. Neutropenia, thrombocytopenia, and pneumonia of grade 3 or higher were reported in 63 % of patients receiving the FR combination.
While NA-based therapies have demonstrated activity in the treatment of Waldenström’s macroglobulinemia, an increased incidence of transformation to non-Hodgkin’s lymphoma and the development of myelodysplasia have been associated with the use of these agents. A recent study followed 439 patients with Waldenström’s macroglobulinemia, of which 193 were previously treated with NA, 136 were treated without an NA, and 110 of whom had follow-up without treatment, for a median of 5 years [46, 47]. Overall, 5 % of patients transformed and 2 % developed myelodysplasia among the NA-treated cohort whereas only one patient transformed within the other groups. These data suggest that while NA-based therapeutic regimens are effective, the additional long-term risks associated with these therapies must be taken into account by clinicians when deciding upon an initial treatment strategy for patients with Waldenström’s macroglobulinemia.
Initially considered to be the standard of care, alkylating agents have also been used successfully in the treatment of Waldenström’s macroglobulinemia. Over time, combinations of alkylating agents, including chlorambucil and cyclophosphamide, with vinca alkaloids, nucleoside analogs, and/or anthracyclines have been studied and deemed effective [48–51]. The addition of rituximab to alkylating agent-based combinations has further increased patient response rates. In a prospective, randomized trial including 34 Waldenström’s macroglobulinemia patients treated with R-CHOP and 30 patients treated with CHOP but no rituximab, a significantly higher overall response rate was achieved in the patient group receiving chemoimmunotherapy (94 % vs. 67 %, p = 0.0085) as compared to chemotherapy alone, with no major differences noted in toxicity [52]. Furthermore, patients in the R-CHOP group experienced a significantly longer time to treatment failure (median of 63 months) as compared to patients in the CHOP arm (22 months p = 0.0033).
Significant activity coinciding with improved toxicity profiles has been achieved in Waldenström’s macroglobulinemia with other alkylating agents administered in combination with rituximab, suggesting that such regimens may be preferable as initial therapy for this disease [43]. For example, a regimen including dexamethasone, rituximab, and cyclophosphamide (DRC) yielded an overall response rate of 83 % in previously untreated Waldenström’s macroglobulinemia patients, of which 7 % were complete responders [53]. Furthermore, only 9 % of patients experienced grade 3 or 4 neutropenia. Alkylating agents combined with rituximab are also useful in treating relapsed or refractory patients. Treon et al. reported an overall response rate of 83.3 % in 30 such WM patients treated with bendamustine in combination with rituximab (BR) [54]. While the therapy was well tolerated, there was an increased incidence of myelosuppression in patients who had previously been treated with nucleoside analogs, as has been reported previously [47]. Further support for the use of BR as initial therapy comes from a comparison with R-CHOP in 41 Waldenström’s macroglobulinemia patients [55]. When compared with R-CHOP, treatment with BR resulted in fewer relapses, was better tolerated, and was associated with a longer progression-free survival, despite identical response rates for both regimens.
Rapid and durable patient responses have also been achieved with the proteasome inhibitor bortezomib when used in combination with rituximab in Waldenström’s macroglobulinemia. When bortezomib, dexamethasone, and rituximab (BDR) were administered to 23 previously untreated, but symptomatic Waldenström’s macroglobulinemia patients, overall response rates neared 96 % with responses occurring at a median of 1.4 months [56]. However, a high incidence of peripheral neuropathy led to the discontinuation of bortezomib in 61 % of patients. A separate study by Ghobrial et al. reported overall response rates of 88 % when bortezomib and rituximab were administered concurrently in patients with symptomatic Waldenström’s macroglobulinemia [57]. However, in this study, no grade 3 or 4 neuropathies were documented, with the most significant adverse event being neutropenia, which occurred in 12 % of patients.
The therapeutic benefit of adding rituximab to chemotherapeutic regimens for the treatment of Waldenström’s macroglobulinemia has been well documented. However, when used as a single agent, rituximab has been associated with response rates ranging from 29 to 65 %, making single agent rituximab a viable option in the treatment of Waldenström’s macroglobulinemia, specifically in low-risk patients with symptomatic disease and modest hematologic compromise and in patients with IgM-related neuropathy requiring treatment [43]. In a study of 69 symptomatic patients, 35 of whom had received treatment previously, overall response rates of 52 % were reported following administration of rituximab as a single agent [58]. Yet, when using rituximab as a single agent, clinicians must be made aware of the paradoxical rituximab-associated increase in IgM protein levels occurring in some patients, known as the rituximab “flare” [43, 59]. While IgM levels may remain elevated out to 4 months following treatment with rituximab, this does not necessarily indicate a treatment failure, although additional plasmapheresis may be necessary to alleviate symptoms of hyperviscosity.
Based on the array of agents that are clinically active in this disease, a risk-adapted approach to the management of Waldenström’s macroglobulinemia is recommended. Three groups of patients can be identified [43]. Patients with IgM MGUS or smoldering (asymptomatic) Waldenström’s macroglobulinemia and preserved hematological function constitute a low-risk group. Symptomatic Waldenström’s macroglobulinemia patients with modest hematological compromise, IgM-related neuropathy, or hemolytic anemia have an intermediate risk of disease progression and subsequent morbidity or mortality. Waldenström’s macroglobulinemia patients who have significant constitutional symptoms, profound hematological compromise, bulky disease, or hyperviscosity have a high-risk of disease progression and early mortality.
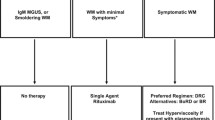
Utilizing the risk groups outlined above, we recommend the following: (1) Patients with IgM MGUS or smoldering (asymptomatic) Waldenström’s macroglobulinemia and preserved hematological function should be observed without initial pharmacotherapy. (2) Symptomatic Waldenström’s macroglobulinemia patients with modest hematological compromise, IgM-related neuropathy requiring treatment, or hemolytic anemia unresponsive to corticosteroids should receive standard doses of rituximab alone without maintenance therapy. (3) Waldenström’s macroglobulinemia patients who have significant constitutional symptoms, profound hematological compromise, bulky disease, or hyperviscosity should be treated with the DRC regimen (dexamethasone, rituximab, cyclophosphamide). Any patient with symptoms of hyperviscosity should first be started on plasmapheresis (see mSMART algorithm in Fig. 24.1) [43].

Mayo Clinic (Mayo stratification of macroglobulinemia and risk-adapted therapy [mSMART]) consensus for management of newly diagnosed Waldenström’s macroglobulinemia (WM) [43]. MGUS = monoclonal gammopathy of undetermined significance. SI conversion factor: To convert hemoglobin values to g/L, multiply by 10
Management of Relapsed Disease
Despite the high overall response rates associated with the aforementioned treatment regimens and the introduction of new therapeutic agents in the past few decades, studies have not demonstrated a significant improvement in the outcome of patients with Waldenström’s macroglobulinemia treated over the last 25 years [60]. These data underscore the need for more effective agents to further improve patient survival, especially in those who have failed previous treatment regimens. Fortunately, new therapies and treatment combinations are currently in clinical testing in patients with refractory and relapsed disease. For example, drugs classified as immunomodulators (IMiDs), including thalidomide and lenalidomide, have been studied in Waldenström’s macroglobulinemia in combination with rituximab as these agents enhance rituximab-mediated, antibody-dependent, cell-mediated cytotoxicity [61]. However, despite relatively high overall response rates, the use of both thalidomide and lenalidomide has been associated with substantial toxicity [62]. In the case of lenalidomide and rituximab, the clinical trial was closed early due to reports of significant anemia, which occurred in 13 of 16 enrolled patients [63]. Thus, while these agents have demonstrated significant activity and durable responses, further studies are necessary to identify the optimal dose of drug required to achieve maximal activity with minimal toxicity.
The mammalian target of rapamycin (mTOR) inhibitor everolimus has also been studied in Waldenström’s macroglobulinemia, due to the known role of the PI3K/Akt/mTOR signal transduction pathway as a driver of tumor survival in various hematologic malignancies, including Waldenström’s macroglobulinemia [64]. When used as a single agent in 50 patients with symptomatic, relapsed, or refractory Waldenström’s macroglobulinemia, overall response rates reached 70 % with a 12-month progression-free survival of 62 % [65]. However, significant toxicities occurred with the use of everolimus, with 56 % of patients developing grade 3 or higher toxicities requiring dose reductions in 52 % of patients. Yet while bearing in mind its toxicity profile, single agent everolimus appears to be a potential new therapeutic option for the treatment of Waldenström’s macroglobulinemia.
As preclinical studies indicated activity of the nonselective histone deacetylase inhibitor panobinostat in Waldenström’s macroglobulinemia cell lines, this agent has also been studied in a phase II trial of 27 patients with refractory or relapsed/refractory disease [66]. Panobinostat was observed to be an active therapeutic agent in this patient population with an overall response rate of 60 %. Due to a high incidence of hematological toxicities, the initial protocol required modifications to decrease the panobinostat dose from 30 mg 3 times per week to 25 mg 3 times per week; the lower dosing schedule was better tolerated.
In addition to chemotherapeutics, novel immunotherapies targeting CD20 are currently in development in an effort to try and improve upon the response rates achieved with single agent rituximab while limiting the rituximab “flare” in IgM. One such monoclonal antibody is ofatumumab (OFA), which targets an epitope encompassing both the large and small extracellular loops of CD20, whereas rituximab targets only the large loop alone [67]. OFA has been studied as a single agent in a cohort of 37 patients with Waldenström’s macroglobulinemia, 28 of whom had received a median of three prior therapies [68]. An overall response rate of 59 % was reported along with a lower incidence of IgM “flare” as compared to rituximab. The toxicity profile, which included the development of infection in 15 patients, was deemed to be acceptable, making OFA another potential therapeutic option in Waldenström’s macroglobulinemia, especially in patients with refractory disease.
Lastly, stem cell transplantation is another potential option for the treatment of patients with advanced Waldenström’s macroglobulinemia. Autologous stem cell transplants are relatively well tolerated and long-lasting complete responses have been reported [43]. In a retrospective analysis of 158 young, but heavily pretreated, patients with Waldenström’s macroglobulinemia who underwent autologous stem cell transplantation (ASCT), nearly half of the patients remained in remission at 5 years, with a non-relapse mortality rate of only 3.8 %. Five-year progression-free survival and overall survival rates were 40 % and 68.5 %, respectively [69]. While additional prospective studies are warranted, these initial data suggest that ASCT may have a place in the treatment of Waldenström’s macroglobulinemia, especially in younger, heavily pretreated, or relapsed patients. A similar retrospective study has also been performed to assess the role of allogeneic stem cell transplantation (alloSCT) in the treatment of Waldenström’s macroglobulinemia. Kyriakou et al. assessed 86 patients with Waldenström’s macroglobulinemia who received an allograft after either myeloablative (MAC) or reduced-intensity conditioning (RIC) regimens [69]. However, both the MAC and RIC regimens were associated with significantly higher risks of non-relapse mortality at 3 years (33 % and 23 %, respectively) as compared with ASCT, and therefore alloSCT is not considered an appropriate therapeutic option for patients with Waldenström’s macroglobulinemia outside of a clinical trial.
As there is currently no standard approach to the management of patients with relapsed Waldenström’s macroglobulinemia, our approach (Fig. 24.2) is to consider all patients for participation in a clinical trial either as definitive therapy for their disease or as preparative therapy prior to transplant [43]. For patients who are ineligible or unwilling to go on a clinical trial, the choice of therapy is determined by their response to frontline treatment. Because responses to initial therapies are often delayed and can occur 12 months or more after initiating treatment, we recommend using a 2-year cutoff to determine treatment. For patients with a durable response that lasted >2 years, the original therapy can be repeated. For patients who have an inadequate response to initial therapy or a response lasting <2 years, an alternative agent or combination should be used. An autologous stem cell transplant should be considered in all eligible patients with relapsed disease.

Mayo Clinic (Mayo stratification of macroglobulinemia and risk-adapted therapy [mSMART]) consensus for management of relapsed Waldenström’s macroglobulinemia [43]
Summary
Waldenström’s macroglobulinemia is a rare disease, and practicing hematologists and oncologists may infrequently treat these patients. Patients may present with a spectrum of clinical findings, and many patients do not require treatment initially. When patients do require therapy, it is important to select therapies that do not negatively impact future treatment options. To provide a simple risk-adapted approach to managing patients with Waldenström’s macroglobulinemia, we have outlined a rational approach to this disease [43]. These recommendations are regularly modified as new data become available and the most current guidelines are available at www.mSMART.org.
References
Owen RG, Treon SP, Al-Katib A, et al. Clinicopathological definition of Waldenstrom’s macroglobulinemia: consensus panel recommendations from the Second International Workshop on Waldenstrom’s macroglobulinemia. Semin Oncol. 2003;30:110–5.
Dimopoulos MA, Kyle RA, Anagnostopoulos A, Treon SP. Diagnosis and management of Waldenstrom’s macroglobulinemia. J Clin Oncol. 2005;23:1564–77.
Dimopoulos MA, Panayiotidis P, Moulopoulos LA, Sfikakis P, Dalakas M. Waldenstrom’s macroglobulinemia: clinical features, complications, and management. J Clin Oncol. 2000;18:214–26.
Vijay A, Gertz MA. Waldenstrom macroglobulinemia. Blood. 2007;109:5096–103.
Kastritis E, Kyrtsonis MC, Hatjiharissi E, et al. No significant improvement in the outcome of patients with Waldenström’s macroglobulinemia treated over the last 25 years. Am J Hematol. 2011;86(6):479–83.
Gertz MA, Anagnostopoulos A, Anderson K, et al. Treatment recommendations in Waldenstrom’s macroglobulinemia: consensus panel recommendations from the second international workshop on Waldenstrom’s macroglobulinemia. Semin Oncol. 2003;30:121–6.
Treon SP, Gertz MA, Dimopoulos M, et al. Update on treatment recommendations from the third international workshop on Waldenstrom’s macroglobulinemia. Blood. 2006;107:3442–6.
Dimopoulos MA, Gertz MA, Kastritis E, et al. Update on treatment recommendations from the fourth international workshop on Waldenstrom’s macroglobulinemia. J Clin Oncol. 2009;27:120–6.
Herrinton LJ, Weiss NS. Incidence of Waldenstrom’s macroglobulinemia. Blood. 1993;82:3148–50.
Groves FD, Travis LB, Devesa SS, Ries LA, Fraumeni Jr JF. Waldenstrom’s macroglobulinemia: incidence patterns in the United States, 1988–1994. Cancer. 1998;82:1078–81.
Benjamin M, Reddy S, Brawley OW. Myeloma and race: a review of the literature. Cancer Metastasis Rev. 2003;22:87–93.
Kyle RA, Therneau TM, Rajkumar SV, et al. A long-term study of prognosis in monoclonal gammopathy of undetermined significance. N Engl J Med. 2002;346(8):564–9.
McMaster ML, Goldin LR, Bai Y, et al. Genome wide linkage screen for Waldenstrom macroglobulinemia susceptibility loci in high-risk families. Am J Hum Genet. 2006;79(4):695–701.
Kyle RA, Therneau TM, Rajkumar SV, et al. Long-term follow-up of IgM monoclonal gammopathy of undetermined significance. Blood. 2003;102(10):3759–64.
Kyle RA, Therneau TM, Rajkumar SV, et al. Long-term follow-up of IgM monoclonal gammopathy of undetermined significance. Semin Oncol. 2003;30(2): 169–71.
Treon SP, Hunter ZR, Aggarwal A, et al. Characterization of familial Waldenstrom’s macroglobulinemia. Ann Oncol. 2006;17(3):488–94.
McMaster ML. Familial Waldenstrom’s macroglobulinemia. Semin Oncol. 2003;30(2):146–52.
Royer RH, Koshiol J, Giambarresi TR, et al. Differential characteristics of Waldenstrom macroglobulinemia according to patterns of familial aggregation. Blood. 2010;115(22):4464–71.
Aoki H, Takishita M, Kosaka M, Saito S. Frequent somatic mutations in D and/or JH segments of Ig gene in Waldenstrom’s macroglobulinemia and chronic lymphocytic leukemia (CLL) with Richter’s syndrome but not in common CLL. Blood. 1995;85(7):1913–9.
Wagner SD, Martinelli V, Luzzatto L. Similar patterns of V kappa gene usage but different degrees of somatic mutation in hairy cell leukemia, prolymphocytic leukemia, Waldenstrom’s macroglobulinemia, and myeloma. Blood. 1994;83(12):3647–53.
Martin-Jimenez P, Garcia-Sanz R, Balanzategui A, et al. Molecular characterization of heavy chain immunoglobulin gene rearrangements in Waldenstrom’s macroglobulinemia and IgM monoclonal gammopathy of undetermined significance. Haematologica. 2007;92(5):635–42.
Grass S, Preuss KD, Wikowicz A, et al. Hyperphosphorylated paratarg-7: a new molecularly defined risk factor for monoclonal gammopathy of undetermined significance of the IgM type and Waldenstrom macroglobulinemia. Blood. 2011; 117(10):2918–23.
Swerdlow SH, Campo E, Harris NL, et al. WHO classification of tumours of haematopoietic and lymphoid tissues, vol. 2. 4th ed. Geneva, Switzerland: International Agency for Research on Cancer (IARC); 2008. p. 441.
Kyle RA, Rajkumar SV. Criteria for diagnosis, staging, risk stratification and response assessment of multiple myeloma. Leukemia. 2009;23:3–9.
Morice WG, Chen D, Kurtin PJ, Hanson CA, McPhail ED. Novel immunophenotypic features of marrow lymphoplasmacytic lymphoma and correlation with Waldenstrom’s macroglobulinemia. Mod Pathol. 2009;22:807–16.
Mansoor A, Medeiros LJ, Weber DM, et al. Cytogenetic findings in lymphoplasmacytic lymphoma/Waldenstrom macroglobulinemia. Chromosomal abnormalities are associated with the polymorphous subtype and an aggressive clinical course. Am J Clin Pathol. 2001;116(4):543–9.
Schop RF, Kuehl WM, Van Wier SA, et al. Waldenstrom macroglobulinemia neoplastic cells lack immunoglobulin heavy chain locus translocations but have frequent 6q deletions. Blood. 2002;100(8):2996–3001.
Schop RF, Van Wier SA, Xu R, et al. 6q deletion discriminates Waldenstrom macroglobulinemia from IgM monoclonal gammopathy of undetermined significance. Cancer Genet Cytogenet. 2006;169(2): 150–3.
Braggio E, Dogan A, Keats JJ, et al. Genomic analysis of marginal zone and lymphoplasmacytic lymphomas identified common and disease-specific abnormalities. Mod Pathol. 2012;25(5):651–60.
Ferreira BI, Garcia JF, Suela J, et al. Comparative genome profiling across subtypes of lowgrade B-cell lymphoma identifies type-specific and common aberrations that target genes with a role in B-cell neoplasia. Haematologica. 2008;93(5):670–9.
Rinaldi A, Mian M, Chigrinova E, et al. Genome-wide DNA profiling of marginal zone lymphomas identifies subtype-specific lesions with an impact on the clinical outcome. Blood. 2011;117(5):1595–604.
Dohner H, Stilgenbauer S, Benner A, et al. Genomic aberrations and survival in chronic lymphocytic leukemia. N Engl J Med. 2000;343(26):1910–6.
Treon SP, Xu L, Yang G, et al. MYD88 L265P somatic mutation in Waldenström’s macroglobulinemia. N Engl J Med. 2012;367(9):826–33.
Chng WJ, Schop RF, Price-Troska T, et al. Gene expression profiling of Waldenstrom macroglobulinemia reveals a phenotype more similar to chronic lymphocytic leukemia than multiple myeloma. Blood. 2006;108(8):2755–63.
Gutierrez NC, Ocio EM, de Las Rivas J, et al. Gene expression profiling of B lymphocytes and plasma cells from Waldenstrom’s macroglobulinemia: comparison with expression patterns of the same cell counterparts from chronic lymphocytic leukemia, multiple myeloma and normal individuals. Leukemia. 2007;21(3):541–9.
Hodge DR, Hurt EM, Farrar WL. The role of IL-6 and STAT3 in inflammation and cancer. Eur J Cancer. 2005;41(16):2502–12.
Hodge LS, Ansell SM. Jak/stat pathway in Waldenstrom’s macroglobulinemia. Clin Lymphoma Myeloma Leuk. 2011;11(1):112–4.
Elsawa SF, Novak AJ, Ziesmer SC, et al. Comprehensive analysis of tumor microenvironment cytokines in waldenstrom macroglobulinemia identifies CCL5 as a novel modulator of IL-6 activity. Blood. 2011;118(20):5540–9.
Garcia-Sanz R, Montoto S, Torrequebrada A, et al. Waldenstrom macroglobulinemia: presenting features and outcome in a series with 217 cases. Br J Haematol. 2001;115:575–82.
Stone MJ, Pascual V. Pathophysiology of Waldenstrom’s macroglobulinemia. Haematologica. 2010;95:359–64.
Morel P, Duhamel A, Gobbi P, et al. International prognostic scoring system for Waldenstrom macroglobulinemia. Blood. 2009;113:4163–70.
Kyle RA, Treon SP, Alexanian R, et al. Prognostic markers and criteria to initiate therapy in Waldenstrom’s macroglobulinemia: consensus panel recommendations from the second international workshop on Waldenstrom’s macroglobulinemia. Semin Oncol. 2003;30:116–20.
Ansell SM, Kyle RA, Reeder CB, et al. Diagnosis and management of Waldenstrom macroglobulinemia: mayo stratification of macroglobulinemia and risk-adapted therapy (mSMART) guidelines. Mayo Clin Proc. 2010;85:824–33.
Tedeschi A, Benevolo G, Varettoni M, et al. Fludarabine plus cyclophosphamide and rituximab in Waldenstrom macroglobulinemia: an effective but myelosuppressive regimen to be offered to patients with advanced disease. Cancer. 2012;118:434–43.
Treon SP, Branagan AR, Ioakimidis L, et al. Long-term outcomes to fludarabine and rituximab in Waldenstrom macroglobulinemia. Blood. 2009;113: 3673–8.
Leleu X, Soumerai J, Roccaro A, et al. Increased incidence of transformation and myelodysplasia/acute leukemia in patients with Waldenstrom macroglobulinemia treated with nucleoside analogs. J Clin Oncol. 2009;27:250–5.
Annibali O, Petrucci MT, Martini V, et al. Treatment of 72 newly diagnosed Waldenstrom macroglobulinemia cases with oral melphalan, cyclophosphamide, and prednisone: results and cost analysis. Cancer. 2005;103:582–7.
Petrucci MT, Avvisati G, Tribalto M, Giovangrossi P, Mandelli F. Waldenstrom’s Macroglobulinaemia: results of a combined oral treatment in 34 newly diagnosed patients. J Intern Med. 1989;226:443–7.
Leblond V, Levy V, Maloisel F, et al. Multicenter, randomized comparative trial of fludarabine and the combination of cyclophosphamide-doxorubicin-prednisone in 92 patients with Waldenstrom macroglobulinemia in first relapse or with primary refractory disease. Blood. 2001;98:2640–4.
Tamburini J, Levy V, Chaleteix C, Fermand JP, Delmer A, Stalniewicz L, et al. Fludarabine plus cyclophosphamide in Waldenstrom’s macroglobulinemia: results in 49 patients. Leukemia. 2005;19: 1831–4.
Buske C, Hoster E, Dreyling M, et al. The addition of rituximab to front-line therapy with CHOP (R-CHOP) results in a higher response rate and longer time to treatment failure in patients with lymphoplasmacytic lymphoma: results of a randomized trial of the German Low-Grade Lymphoma Study Group (GLSG). Leukemia. 2009;23:153–61.
Dimopoulos MA, Anagnostopoulos A, Kyrtsonis MC, et al. Primary treatment of Waldenstrom macroglobulinemia with dexamethasone, rituximab, and cyclophosphamide. J Clin Oncol. 2007;25:3344–9.
Treon SP, Hanzis C, Tripsas C, et al. Bendamustine therapy in patients with relapsed or refractory Waldenstrom’s macroglobulinemia. Clin Lymphoma Myeloma Leuk. 2011;11:133–5.
Rummel MJ, Niederle N, von Grunhagen U, et al. Bendamustine plus rituximab versus CHOP plus rituximab as first-line treatment in patients with indolent lymphomas and Waldenstrom’s macroglobulinemia. In: Sixth International Workshop on Waldenstrom’s Macroglobulinemia. Venice, Italy 2010.
Treon SP, Ioakimidis L, Soumerai JD, et al. Primary therapy of Waldenstrom macroglobulinemia with bortezomib, dexamethasone, and rituximab: WMCTG clinical trial 05–180. J Clin Oncol. 2009;27:3830–5.
Ghobrial IM, Xie W, Padmanabhan S, et al. Phase II trial of weekly bortezomib in combination with rituximab in untreated patients with Waldenstrom macroglobulinemia. Am J Hematol. 2010;85:670–4.
Gertz MA, Rue M, Blood E, Kaminer LS, Vesole DH, Greipp PR. Multicenter phase 2 trial of rituximab for Waldenstrom macroglobulinemia (WM): an Eastern Cooperative Oncology Group Study (E3A98). Leuk Lymphoma. 2004;45:2047–55.
Ghobrial IM, Fonseca R, Greipp PR, et al. Initial immunoglobulin M ‘flare’ after rituximab therapy in patients diagnosed with Waldenstrom macroglobulinemia. Cancer. 2004;101:2593–8.
Kastritis E, Kyrtsonis M-C, Hatjiharissi E, et al. No significant improvement in the outcome of patients with Waldenström’s macroglobulinemia treated over the last 25 years. Am J Hematol. 2011;86:479–83.
Davies FE, Raje N, Hideshima T, et al. Thalidomide and immunomodulatory derivatives augment natural killer cell cytotoxicity in multiple myeloma. Blood. 2001;98:210–6.
Treon SP, Soumerai JD, Branagan AR, et al. Thalidomide and rituximab in Waldenstrom macroglobulinemia. Blood. 2008;112:4452–7.
Treon SP, Soumerai JD, Branagan AR, et al. Lenalidomide and rituximab in Waldenstrom’s macroglobulinemia. Clin Cancer Res. 2009;15:355–60.
Leleu X, Jia X, Runnels J, et al. The Akt pathway regulates survival and homing in Waldenstrom macroglobulinemia. Blood. 2007;110:4417–26.
Ghobrial IM, Gertz M, Laplant B, et al. Phase II trial of the oral mammalian target of rapamycin inhibitor everolimus in relapsed or refractory Waldenstrom macroglobulinemia. J Clin Oncol. 2010;28:1408–14.
Ghobrial IM, Poon T, Rourke M, et al. Phase II trial of single agent pabinostat (LBH589) in relapsed or relapsed/refractory Waldenstrom macroglobulinemia. San Diego, CA: American Society of Hematology; 2010.
Cheson BD. Ofatumumab, a novel anti-CD20 monoclonal antibody for the treatment of B-cell malignancies. J Clin Oncol. 2010;28:3525–30.
Furman RR, Eradat H, DiRienzo CG, et al. A phase II trial of ofatumumab in subjects with Waldenstroms macroglobulinemia. San Diego, CA: American Society for Hematology; 2011.
Kyriakou C, Canals C, Sibon D, et al. High-dose therapy and autologous stem-cell transplantation in Waldenstrom macroglobulinemia: the Lymphoma Working Party of the European Group for Blood and Marrow Transplantation. J Clin Oncol. 2010;28: 2227–32.
Kyriakou C, Canals C, Cornelissen JJ, et al. Allogeneic stem-cell transplantation in patients with Waldenstrom macroglobulinemia: report from the Lymphoma Working Party of the European Group for Blood and Marrow Transplantation. J Clin Oncol. 2010;28: 4926–34.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2014 Mayo Foundation for Medical Education and Research
About this chapter
Cite this chapter
Ansell, S.M., Hodge, L.S., Hayman, S.R. (2014). Waldenström’s Macroglobulinemia. In: Gertz, M., Rajkumar, S. (eds) Multiple Myeloma. Springer, New York, NY. https://doi.org/10.1007/978-1-4614-8520-9_24
Download citation
DOI: https://doi.org/10.1007/978-1-4614-8520-9_24
Published:
Publisher Name: Springer, New York, NY
Print ISBN: 978-1-4614-8519-3
Online ISBN: 978-1-4614-8520-9
eBook Packages: MedicineMedicine (R0)