
Abstract
Lysosomal diseases cover a wide spectrum of inborn metabolic disorders and are characterized by a remarkable variability in pathophysiology and clinical presentation. This first chapter presents an overview of the natural history and biochemical characteristics of lysosomal storage disorders and some of the developments in therapy that have been made in the past decades and a brief look at potential future therapies.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others

Keywords
These keywords were added by machine and not by the authors. This process is experimental and the keywords may be updated as the learning algorithm improves.
1.1 Introduction
Lysosomal storage diseases (LSDs) comprise a group of more than 50 distinct inherited metabolic diseases, each of which is caused by a specific deficient function of a lysosomal enzyme or transporter or by defects in lysosomal biogenesis or vesicular trafficking.
Since the discovery of the lysosome as a cellular organelle in 1955, enormous advances have been made in the understanding of the complex lysosomal biology. This knowledge led to the first pioneering studies on treatment of LSDs, by Hobbs and colleagues using bone-marrow transplantation in a patient with Hurler’s disease [1] and by Brady and co-workers on intravenous enzyme supplementation in patients with non-neuronopathic Gaucher disease [2].
In this chapter a concise overview of the history of lysosomal storage diseases, lysosomal biology, lysosomal storage diseases and current and future therapies is presented.
1.2 History
In 1955, the Belgium biochemist Christian de Duve described a new intracellular compartment which he later called the “lysosome” (Greek: lysis = to split; somos = body) [3, 4]. De Duve was awarded the Nobel Prize in Physiology and Medicine for his work in 1974.
The first description of a disease that was recognized as an LSD only much later is Gaucher disease. Philippe Gaucher was a French medical student who, in 1882, reported a patient with an enlarged spleen [5]. Microscopic studies revealed abnormal cells which were later called “Gaucher cells”. In 1924, a German medical doctor succeeded in isolating a special fatty substance from the spleen of patients with this disorder, which was already named “Gaucher disease”. Ten years later additional studies showed that this fatty substance was a glucocerebroside, a component of the plasma membrane of the red and white blood cells.
A significant number of other diseases of which it later became clear that they were all to be classified as LSDs, were already described at the end of the nineteenth and the beginning of the twentieth century. For instance, Fabry disease was reported for the first time independently by two dermatologists, Johannes Fabry from Germany and William Anderson from the UK [6, 7]. The British army doctor C.A. Hunter reported the first cases of what later became known as Hunter disease (mucopolysaccharidosis type II) in 1917 [8] and in 1919 the German paediatrician Gertrud Hurler reported the first cases of patients with the disorder later known as Hurler’s disease (mucopolysaccharidosis type I) [9].
The mechanism of lysosomal storage diseases was first discovered in 1963 in Pompe disease. This disease was reported for the first time by the Dutch pathologist J.C. Pompe in 1932 in a 7-month-old infant who had died from cardiac hypertrophy [10]. Dr Pompe discovered that this disease involved accumulation of large quantities of glycogen in almost all studied tissues. He called the disease “cardiomegalia glycogenica diffusa”. Gerty Cori, who, together with her husband Carl Ferdinand Cori, had received the Nobel Prize in Physiology or Medicine in 1947 for their work on glycogen degradation, recognized in 1954 that Pompe disease involved abnormal glycogen degradation and classified Pompe disease as “glycogen storage disease type 2” [11]. It was the Belgian biochemist Henri-Géry Hers, a former colleague of de Duve, who finally discovered that the glycogen storage in patients with Pompe disease was caused by a deficiency of the lysosomal enzyme acid-alpha-glucosidase [12]. His discovery of an inherited deficiency of a lysosomal enzyme as the cause of a disease thus marks the development of the concept of LSDs and the start of many new discoveries relating clinical diseases to dysfunction of the lysosomal machinery.
1.3 Lysosomal Biology
The lysosome is a membrane enclosed organelle, containing many unique hydrolytic enzymes, including proteases, nucleases, glycosidases, phosphatases, sulphatases, lipases and phospholipases. In addition, numerous lysosomal transmembrane proteins are recognized and involved in the complex lysosomal biology. Together they transport a large number of biological molecules and hydrolases degrade proteins, nucleic acids, phospholipids and oligosaccharides. These macromolecules can be derived from intracellular material, such as “worn out” organelles or from extracellular material transported to the lysosome after endocytosis or phagocytosis. The lysosomal enzymes are optimally active in an acidic environment with a pH of about 4.5–5. The low pH optimum of the hydrolases compared to the average cytosolic pH of 7.2 requires a gradient over the unique lysosomal membrane. This gradient is maintained by an ATP-driven H+-pump, pumping H+ ions into the lysosome. While this V-type ATPase pump uses the energy derived from converting ATP to ADP, the H+ gradient thus created also serves as a source of energy for transport of small metabolites across the membrane.
Both the encapsulating membrane and the pH gradient protect the cell from the multiple degradation effects of lysosomal enzymes. Naturally, the digestive enzymes themselves also need to be protected from digestion. For the membrane bound proteins, like transporters, this is realized through an unusual high glycosylation. These sugars form a protective layer keeping the lysosomal proteases from the proteins [13].
The substrates for the lysosomal machinery can reach the lysosome in three ways. Through endocytosis, the cell takes up macromolecules from the extracellular fluid. This way, early endosomes are formed where a selection takes place in the products that should be broken down and those that can be recycled without a full degradation. By sorting the macromolecules and receiving newly formed hydrolases from the Golgi apparatus a late endosome is formed, where hydrolytic digestion begins. Through fusion with existing lysosomes, the pH drops and digestion is facilitated more readily. Once the slowly digestible residues are left over, the organelle most resembles the classic lysosome; compact and dense.
A second pathway for degradation is called autophagy. In this process the cell forms a double membrane around an obsolete intracellular structure or organelle. Once a closed sack is formed, it fuses with a lysosome and digestion starts. In a state of low extracellular nutrient supply such as starvation, autophagy can provide the cell with metabolites needed for survival.
Some specialized cells have the ability to form phagosomes. This way, a cell is able to take up large particles and microorganisms. The processing of the phagosomal contents is similar to that of the autophagosome [14].
The lysosomal hydrolases and membrane proteins are composed and folded in the endoplasmatic reticulum. They are subsequently transported to late endosomes through the Golgi apparatus and especially the trans-Golgi-network (TGN). In the TGN the proteins are selected for their specific destination. The lysosomal hydrolases are recognized because they carry specific mannose-6-phosphate (M6P) markers, which are added in the Golgi network to the N-linked oligosaccharides of these enzymes. In the TGN, transmembrane M6P-receptors recognize and bind the flagged enzymes. While binding takes place at a pH of around 6.7, the receptor releases the oligosaccharide at a lower pH of around 6. On average, this is the pH in late endosomes. This leads to a gradual release of the enzymes as the environment gets more acidic in the process of maturation into a lysosome. After this dissociation, the M6P receptors are returned to the TGN, where they enter the cycle once more [15].
Once the lysosome has degraded its contents, transmembrane transporters facilitate relocation of metabolites to the cytosol, and other waste products leave the cell through exocytosis.
Lysosomes and endosomes have several other important functions besides processing cellular waste material. Among these functions are antigen presentation, signal transduction, cell division and neurotransmission. This wide range of tasks might aid our understanding why storage of an indigestible waste product might lead to cell and organ dysfunction in lysosomal storage diseases.
In lysosomal disease it can be either a soluble hydrolytic enzyme that is deficient or any of many regulatory and transport enzymes. The classic hypothesis of cytotoxicity stated that accumulation would eventually overwhelm the cell, impair its normal functions and eventually lead to cell death [16]. Ongoing research in cellular processes learns that the mechanism leading from the relatively simple single enzyme defect with accumulation of material to clinical disease is much more complex. It is now clear that many secondary effects contribute to cell and organ damage. Some of these effects involve ER stress, impaired pH regulation, insufficient M6P recycling, disturbed Ca2+ homeostasis, impaired signal transduction and reduced ability of autophagy [17–19].
1.4 Clinical Aspects of Lysosomal Storage Diseases
The lysosomal storage diseases (LSDs) comprise a quantitatively important group within the field of inherited metabolic disorders. The diseases of this heterogeneous group might be classified by the storage products or by the affected cellular mechanism. An extensive overview of lysosomal storage disorders and inherited defects in lysosome-like organelles is presented in Table 1.1.
LSDs are considered rare genetic disorders, but taken together they have an incidence of more than 1 in 8000 births [18]. Incidence of any LSD also highly relies on the ethnicity of the population studied. For example Gaucher disease has an incidence of 1:450 to 1:1,000 is Ashkenazi Jewish families, compared to a general incidence of 1:60,000 [20]. For mucopolysaccharidosis type I (MPS I) the incidence in the Republic of Ireland is around 1:26,000 births. Within this cohort, the incidence among the Irish Traveller Community is even much higher, namely 1:371 [21]. In other countries the incidence of MPS I can be significantly lower, ranging from 1.19:100,000 in the Netherlands [22] to 0.11:100,000 in Taiwan [23].
In general, the characteristic signs and symptoms of any LSD reflect the cell type that is the principal site of substrate deposit. Which cells are affected mainly depends on the availability of substrate for the deficient enzyme. For example, in Pompe disease α-glucosidase is deficient, which is an enzyme involved in glycogen breakdown. Consequently the skeletal muscles, the heart and the liver are the most affected organs in Pompe disease, as these are the most active organs in glycogen metabolism. But as lysosomal proteins are distributed ubiquitously, ultimately any cell type may be involved [24].
Most of the LSDs display a remarkable clinical heterogeneity with a spectrum ranging from severe disease to an attenuated phenotype. In many LSDs, an underlying genetic defect has been identified and although no perfect concordance in the relationship between phenotype and genotype has been found, null-mutations have been associated with more severe disease and young age of onset. Phenotypes are often biochemically indistinguishable. Prognosis is thus mainly based on clinical presentation.
Different from most inborn errors of metabolism, the LSDs do not present with “intoxication type” symptoms with acute decompensation with altered mental status, but have a gradual onset of symptoms and an invariably progressive course. With the development of disease modifying treatments options, some previously severe and fatal LDSs have now turned into more chronic conditions, with survival well into adulthood, displaying a new phenotype of the disease [25].
As much as two-thirds of the LSDs may present with neurological disease. This can range from central nervous system involvement leading to developmental delay and behavioural problems to peripheral neuropathy [26]. Also organomegaly, corneal disease, skeletal involvement and connective tissue disease are often seen.
1.5 Current Therapeutic Options in Lysosomal Storage Diseases
During the last decades, much progress has been made in the development of therapies for lysosomal storage diseases (LSDs). For some disorders, disease modifying treatment is now available. As none of the disease can yet be completely cured, there is still a large role for supportive care. Main goals in the treatment of LSDs are to ameliorate symptoms and prevent complications.
1.5.1 Supportive Care
Regardless of the type of LSD, as long as there is no curative therapy available, supportive care is and will be the corner stone of treatment. Supportive care should be coordinated by a physician with experience in lysosomal storage disorders. Depending on the expected complications, other specialists such as pulmonologists, cardiologists, otorhinolaryngologists, anaesthesiologists, orthopaedic surgeons, physiotherapists and neurosurgeons should be involved in the management and prevention of symptoms and complications. Besides providing “standard care” every specialist thus involved should be aware of the typical problems that may occur in patients with a lysosomal storage disease. These issues can be life threatening complications, such as cervical instability and cardiac failure, or may primarily affect quality of life, such as immobility, loss of vision and hearing impairment. Arising from a completely unique pathophysiology, these complications require special and multidisciplinary attention [27, 28].
In many LSDs, surgical interventions are frequently needed. Surgical interventions may include carpal tunnel release, ventriculoperitoneal shunting, heart valve replacement or orthopaedic interventions [29]. It is advised that surgical procedures take place in specialized centres. Especially general anaesthesia might be challenging as some of the lysosomal storage diseases such as mucopolysaccharidosis may present with a distorted anatomy due to clinical features as an enlarged tongue, a short stiff neck and odontoid dysplasia [30, 31].
Aside from somatic care, patients and families with lysosomal storage disorders in general require psychosocial support. The decrease in health related quality of life is not only associated with disease severity, but also with the inability to maintain friends or a hobby [32]. Educational programmes aimed to improve knowledge and promote self support in patients and their care takers may substantially improve quality of life.
1.5.2 Disease Modifying Treatment Options
1.5.2.1 Hematopoietic Stem Cell Transplantation
Disease modifying therapies have been available for lysosomal storage diseases only since the early 1980s, after Hobbs and colleagues performed the first bone marrow transplantation in a patient with Hurler disease. This was successful, with engrafted survival as well as decrease in glycosaminoglycans excretion, reduction in liver size and an arrest in the deterioration of cognitive development [1]. After this encouraging result, bone marrow transplantations have been performed in many other LSDs. It was the discovery of the mechanism of “cross-correction” that led to the hypothesis that this procedure may benefit patients with a lysosomal storage disease.
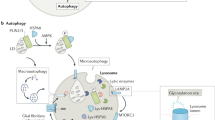
In 1968 Elisabeth Neufeld and her group reported that the biochemical defects in co-cultured skin fibroblasts of Hunter and Hurler patients were corrected [33]. In addition, they showed that co-culturing with control fibroblasts also resulted in correction of the biochemical defect. Later studies demonstrated that this process of cross-correction was based on the phenomenon that lysosomal enzymes can be excreted and imported by cultured cells. Most of the lysosomal enzymes are targeted for their lysosomal destination by a residue which is recognized by a mannose-6-phosphate receptor (M6P receptor). For enzymes that are not transported to the lysosome, but is instead secreted from the cell, a surface M6P receptor facilitates their re-uptake. These enzymes can subsequently reach the lysosome through the endocytic pathway (see Fig. 1.1).

Schematic overview of the lysosome and related intracellular processes. 1: Exocytosis of proteins from the Golgi network. 2: Mannose-6-Phosphate (M6P) receptors return to the cell membrane. 3: M6P receptors facilitate uptake of labelled hydrolytic enzymes. 4: M6P receptors regulate transport of newly formed hydrolytic enzymes from the trans-Golgi-network (TGN) to the endosomal/lysosomal system. M6P receptors can be recycled from the LE to the TGN. 5: Receptor mediated endocytosis. 6: Non-receptor mediated endocytosis. 7: Autophagy. 8: Exocytosis. AV autophagosome, CG cis-Golgi-network, TG trans-Golgi-network, ER endoplasmatic reticulum, CCV clathrin coated vesicle, EE early endosome, LE late endosome, LYS lysosome, CCP clathrin coated pit
Cross-correction uses this physiologic process and the essential fact that secreted enzymes are not just re-captured by the excreting cell, but might also be captured by any neighbouring cell with a surface M6P receptor.
As it appears that in most of the LSDs an enzyme activity of only 10–20 % of normal may already substantially improve the clinical outcome, cross-correction is an important mechanism in developing therapeutic strategies [34].
So, through performing bone marrow transplantations, enzyme producing and secreting cells become available in patients with a lysosomal storage disease. Due to its size, secreted enzymes are not able to pass the blood barrier, but surprisingly neurodegeneration is halted in transplanted patients with some types of LSDs. In addition, a decrease in white matter lesions on brain MRI has been reported [35]. This is attributed to the fact that microglia cells in the brain are derived from bone marrow precursors. After a haematopoietic stem cell transplantation (HSCT), the deficient microglia cells are gradually replaced by enzyme secreting cells, providing the surrounding neuronal cells with the deficient enzyme. Partial or late onset neurological benefits can be due to the slow rate of replacement for microglia cells. Unfortunately, neurological damage already present before HSCT is often irreversible. As a consequence, early—preferably presymptomatic—diagnosis and early initiation of treatment is of utmost importance to improve the outcome of HSCT.
As the brain is populated with microglia cells, other tissue macrophages also infiltrate organs throughout the body. These haematopoietic precursors migrate to the liver to form enzyme producing Kupffer cells and in the lungs they replace deficient alveolar macrophages. Due to their wide tissue distribution they increase the amount of available enzyme [36].
Because of the risks of morbidity and mortality associated with HSCT, the procedure was initially indicated only for patients with the more severe phenotypes of the diseases. Currently most experience is in patients with severe MPS I, also known as Hurler phenotype. In 2005 the European Group for Blood and Marrow Transplant (EBMT) developed transplantation guidelines for these patients.
Based on the success of HSCT, this was subsequently tried in many other LSDs with often disappointing results. However, recent advances in treatment regimen and the donor source may chance these policies in the near future [36].
1.5.2.2 Enzyme Replacement Therapy
The ability of cells to reuptake lysosomal hydrolases is not just the mechanism on which haematopoietic stem cell transplantation relies; it is also the basis of enzyme supplementation or replacement therapy (ERT). With ERT the enzyme is not produced endogenously, but infused at regular intervals as a recombinant enzyme.
As early as 1973, the first attempts to use enzyme replacement therapy (ERT) in LSDs were made by intravenous injection of urine-derived hexosaminidase A in a patient with Sandhoff disease. Biochemically this resulted in a significant reduction of the accumulated globoside in the circulation, but no clinical improvement was noted [37].
As an alternative source for enzyme, the human placenta was later investigated as option for ERT, first for Fabry disease, later for Gaucher disease [2, 38]. Glucocerebrosidase isolated from human placentae was made available in sufficient quantities and the first clinical trials were initiated. In order to target the exogenous enzyme to macrophages (the most affected cells in Gaucher disease) the glycosylation signal on the enzyme was modified to contain mannose-terminal oligosaccharide side chains. In the early 1990s the first patients with non-neuronopathic Gaucher disease were treated with this modified glucocerebrosidase and an excellent clinical response was observed with a decrease in spleen and liver size and resolution of anaemia and thrombocytopenia and improvement of skeletal disease [39]. In 1991 this first clinically effective ERT was approved by the regulatory authorities in the USA and later in many other countries. Later, Chinese hamster ovary cells were genetically modified to produce glucocerebrosidase which is subsequently processed to express a mannose terminated oligosaccharide residue. This enzyme, imiglucerase, was approved in 1994 for the use in patients with Gaucher disease. At present, over 4,000 patients are treated with this product.
The remarkable success of ERT in Gaucher disease led to increased attention for new therapeutic strategies in lysosomal storage disease in general and ERT in particular. This resulted in the development more recombinant enzymes, as shown in Table 1.2.
It is important to realize that where in Gaucher disease the recombinant enzyme is effectively targeted for only one cell type, this is insufficient in other LSDs as they encompass many more cell types. In addition, it is increasingly recognized that antibody formation may be a great challenge in some of the enzyme replacement therapies, as this might lessen the available enzyme and its effects. Despite these challenges, ERT has proven to significantly reduce some of the somatic signs and symptoms in LSDs and improve the quality of life of the patients.
1.5.2.3 Substrate Reduction Therapy
A different approach to the treatment of LSDs is substrate reduction therapy. By partially inhibiting the synthesis of the substrate for the deficient enzyme, the influx into the lysosome and subsequent accumulation may be reduced. Any residual activity of the deficient enzyme might be enough for the lysosome to cope with the amount of macromolecules still synthesized. At present, N-butyldeoxynojirimycin (miglustat) is the only registered substrate reduction therapy in LSDs. This imino-sugar inhibits the synthesis of glucosphingolipids and is registered for Gaucher disease and Niemann Pick type C. Clinical trials are being performed to confirm its beneficial effects and safety in other LSDs [40, 41].
1.6 Future Therapies
As more knowledge on the pathophysiology processes involved in lysosomal storage diseases (LSDs) is gained, new treatment options are considered and studied. Although a number of successful therapies have been introduced during the last decades, many LSDs remain without a proper disease modifying option. Also, there are limitations to the existing therapies. Often, the central nervous system involvement is not or only partially treated, resulting in ongoing brain disease.
New therapeutic options are therefore still very much needed. Several promising possibilities will be discussed in some detail.
1.6.1 Gene Therapy
Lysosomal storage diseases appear to be ideal candidates for gene therapy. Firstly, most LSDs are caused by a single genetic defect. Secondly, the involved genes are not subject to a complex regulatory system. Various experiments with both microencapsulation and viral vector delivery systems have been performed. Although the in vitro and non-human in vivo results are often promising, clinical experience is still limited.
In gene therapy, the aim is to restore enzyme availability in one of two ways. The first technique allows genes to be transfected into the subject’s cells by viral vectors such as adeno-associated or lentiviruses. Once the gene is expressed, these cells will produce the specific enzyme which will not only provide for their own lysosomal degradation of the storage product, but through cross-correction will also treat cells in their vicinity.
Another form of gene therapy consists of the so called microencapsulation technique. In this situation, genetically modified cell are encapsulated in a semipermeable membrane and then brought into the host. The membrane allows for exchange of molecules such as nutrients and metabolites between the cell and its environment, while preventing the access of the immune system. In microencapsulation cross-correction is again the mechanism with which the surrounding cells receive their deficient enzyme [42, 43].
1.6.2 Small Molecules Therapy
Enzyme deficiency is the result of a misbalance between synthesis, degradation and functionality of the enzyme. Misfolding, for instance, leads to premature degradation of enzymes. Small molecule therapies aim to restore the balance.
In chaperone therapy, enzyme stabilization is facilitated by administering a low dose of hydrolase inhibitory molecules. The otherwise degraded enzyme might in this way bypass the cell’s quality control mechanism and still perform its function in the lysosome. No human in vivo trials have been published to date [43, 44].
Substrate reduction therapy (SRT) is a form of small molecule therapy. The clinical success of miglustat use in Gaucher and Niemann Pick type C disease has prompted more research. Both miglustat’s efficacy in other LSDs and alternative substrate reducing agents are currently under investigation. At present, phase 2 clinical trials with eliglustat are performed, which might prove a more potent form of SRT in Gaucher disease.
1.6.3 Alternative Uses of Known Therapies
In addition to the search for new therapies, new utilities for known therapies are studied. For example, combining therapies in order to target different aspects of disease at the same time might improve clinical outcome in LSDs. Both additional and synergistic effects have been noted in murine models [45]. No clinical data is available at this point. The combined therapies might be LSD-specific such as ERT and SRT for Fabry disease, but also commonly used drugs as non-steroidal anti-inflammatory drugs [43].
As well as combining two or more therapies, an alternative route of administration might also improve efficacy of known drugs. For instance, direct intra-thecal injection allows for bypassing the blood–brain barrier. Intrathecal enzyme injection has been tried in a patient with MPS I and MPS VI, with some clinical improvement [46] and is currently studied in MPS II and MPS III patients. Another way of combining two therapeutic strategies may consist of genetic modification of autologous bone marrow. This method was used by Aubourg and colleagues [47]. They performed an autologous HSCT in two patients with the peroxisomal inborn error X-linked adrenoleukodystrophy. Before re-infusion the patients CD34+ cells were genetically corrected ex vivo to produce the deficient enzyme. Thus genetic modification and transplantation techniques may decrease the risks and side effects of these techniques, and improve efficacy.
References
Hobbs, J.R., et al.: Reversal of clinical features of Hurler’s disease and biochemical improvement after treatment by bone-marrow transplantation. Lancet 2, 709–712 (1981)
Brady, R.O., Pentchev, P.G., Gal, A.E., Hibbert, S.R., Dekaban, A.S.: Replacement therapy for inherited enzyme deficiency. Use of purified glucocerebrosidase in Gaucher’s disease. N. Engl. J. Med. 291, 989–993 (1974)
De Duve, C.: From cytases to lysosomes. Fed. Proc. 23, 1045–1049 (1964)
De Duve, C.: Exploring cells with a centrifuge. Science 189, 186–194 (1975)
Gaucher, P.C.E.: De l’epithélioma primitive de la rate, hypertrophie idiopathique de la rate sans leucémie. PhD thesis, Faculté de Médicine, Paris (1882)
Fabry, J.: Ein Beitrag zur Kenntnis der Purpura Haemorrhagica nodularis (Purpura papulosa haemorrhagica Hebrae). Arch. Dermatol. Res. 43, 187–200 (1898)
Anderson, W.: A case of ‘angiokeratoma’. Br. J. Dermatol. 10, 113–117 (1898)
Hunter, C.: A rare disease in two brothers. Proc. R. Soc. Med. 10, 104–116 (1917)
Hurler, G.: Ûber einem Typ multipler Abartungen, vorwiegend am Skelettsystem. Z. Kinderheilkd. 24, 220–234 (1919)
Pompe, J.C.: Over idiopatische hypertrofie van het hart. Ned. Tijdschr. Geneeskd. 76, 304–311 (1932)
CORI, G.T.: Glycogen structure and enzyme deficiencies in glycogen storage disease. Harvey Lect. 48, 145–171 (1952)
HERS, H.G.: Alpha-Glucosidase deficiency in generalized glycogenstorage disease (Pompe’s disease). Biochem. J. 86, 11–16 (1963)
Alberts, B., et al.: Intracellular compartments and protein sorting. In: Alberts, B. (ed.) Molecular Biology of the Cell. Garland Science, New York (2007)
Doherty, G.J., McMahon, H.T.: Mechanisms of endocytosis. Annu. Rev. Biochem. 78, 857–902 (2009)
Storch, S., Braulke, T.: Transport of lysosomal enzymes. In: Saftig, P. (ed.) Lysosomes, Georgetown: Landes Bioscience: Springer Science+Business Media, New York, pp. 17–22 (2005)
Desnick, R.J., Thorpe, S.R., Fiddler, M.B.: Toward enzyme therapy for lysosomal storage diseases. Physiol. Rev. 56, 57–99 (1976)
Walkley, S.U.: Pathogenic cascades in lysosomal disease—why so complex? J. Inherit. Metab. Dis. 32, 181–189 (2009)
Schultz, M.L., Tecedor, L., Chang, M., Davidson, B.L.: Clarifying lysosomal storage diseases. Trends Neurosci. 34, 401–410 (2011)
Parkinson-Lawrence, E.J., et al.: Lysosomal storage disease: revealing lysosomal function and physiology. Physiology (Bethesda) 25, 102–115 (2010)
Weinstein, L.B.: Selected genetic disorders affecting Ashkenazi Jewish families. Fam. Community Health 30, 50–62 (2007)
Murphy, A.M., Lambert, D., Treacy, E.P., O’Meara, A., Lynch, S.A.: Incidence and prevalence of mucopolysaccharidosis type 1 in the Irish republic. Arch. Dis. Child. 94, 52–54 (2009)
Poorthuis, B.J., et al.: The frequency of lysosomal storage diseases in The Netherlands. Hum. Genet. 105, 151–156 (1999)
Lin, H.Y., et al.: Incidence of the mucopolysaccharidoses in Taiwan, 1984–2004. Am. J. Med. Genet. A 149A, 960–964 (2009)
van der Ploeg, A.T., Reuser, A.J.: Pompe’s disease. Lancet 372, 1342–1353 (2008)
Pastores, G.M.: Clinical perspectives. In: Lysosomal storage disorders: principles and practice Pastores, Hackensack NJ: World Scientific, pp. 23–46 (2010)
Bellettato, C.M., Scarpa, M.: Pathophysiology of neuropathic lysosomal storage disorders. J. Inherit. Metab. Dis. 33, 347–362 (2010)
Muenzer, J., Wraith, J.E., Clarke, L.A.: Mucopolysaccharidosis I: management and treatment guidelines. Pediatrics 123, 19–29 (2009)
Muenzer, J., et al.: Multidisciplinary management of Hunter syndrome. Pediatrics 124, e1228–e1239 (2009)
Arn, P., Wraith, J.E., Underhill, L.: Characterization of surgical procedures in patients with mucopolysaccharidosis type I: findings from the MPS I Registry. J. Pediatr. 154, 859–864 (2009)
Walker, R.W., Darowski, M., Morris, P., Wraith, J.E.: Anaesthesia and mucopolysaccharidoses. A review of airway problems in children. Anaesthesia 49, 1078–1084 (1994)
Moores, C., Rogers, J.G., McKenzie, I.M., Brown, T.C.: Anaesthesia for children with mucopolysaccharidoses. Anaesth. Intensive Care 24, 459–463 (1996)
Hatzmann, J., et al.: Predicting health-related quality of life of parents of children with inherited metabolic diseases. Acta Paediatr. 98, 1205–1210 (2009)
Fratantoni, J.C., Hall, C.W., Neufeld, E.F.: Hurler and Hunter syndromes: mutual correction of the defect in cultured fibroblasts. Science 162, 570–572 (1968)
Sands, M.S., Davidson, B.L.: Gene therapy for lysosomal storage diseases. Mol. Ther. 13, 839–849 (2006)
Lucke, T., et al.: Developmental outcome in five children with Hurler syndrome after stem cell transplantation: a pilot study. Dev. Med. Child Neurol. 49, 693–696 (2007)
Boelens, J.J., Prasad, V.K., Tolar, J., Wynn, R.F., Peters, C.: Current international perspectives on hematopoietic stem cell transplantation for inherited metabolic disorders. Pediatr. Clin. North Am. 57, 123–145 (2010)
Johnson, W.G., et al.: Intravenous injection of purified hexosaminidase A into a patient with Tay-Sachs disease. Birth Defects Orig. Artic. Ser. 9, 120–124 (1973)
Brady, R.O., et al.: Replacement therapy for inherited enzyme deficiency. Use of purified ceramidetrihexosidase in Fabry’s disease. N. Engl. J. Med. 289, 9–14 (1973)
Barton, N.W., et al.: Replacement therapy for inherited enzyme deficiency—macrophage-targeted glucocerebrosidase for Gaucher’s disease. N. Engl. J. Med. 324, 1464–1470 (1991)
Patterson, M.C., Vecchio, D., Prady, H., Abel, L., Wraith, J.E.: Miglustat for treatment of Niemann-Pick C disease: a randomised controlled study. Lancet Neurol. 6, 765–772 (2007)
Cox, T., et al.: Novel oral treatment of Gaucher’s disease with N-butyldeoxynojirimycin (OGT 918) to decrease substrate biosynthesis. Lancet 355, 1481–1485 (2000)
Seregin, S.S., Amalfitano, A.: Gene therapy for lysosomal storage diseases: progress, challenges and future prospects. Curr. Pharm. Des. 17, 2558–2574 (2011)
Beck, M.: Therapy for lysosomal storage disorders. IUBMB Life 62, 33–40 (2010)
Platt, F.M., Lachmann, R.H.: Treating lysosomal storage disorders: current practice and future prospects. Biochim. Biophys. Acta 1793, 737–745 (2009)
Hawkins-Salsbury, J.A., Reddy, A.S., Sands, M.S.: Combination therapies for lysosomal storage disease: is the whole greater than the sum of its parts? Hum. Mol. Genet. 20, R54–R60 (2011)
Lachmann, R.H.: Enzyme replacement therapy for lysosomal storage diseases. Curr. Opin. Pediatr. 23, 588–593 (2011)
Cartier, N., et al.: Hematopoietic stem cell gene therapy with a lentiviral vector in X-linked adrenoleukodystrophy. Science 326, 818–823 (2009)
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2013 Springer Science+Business Media New York
About this chapter
Cite this chapter
Langereis, E.J., Wijburg, F.A. (2013). Lysosomal Diseases and Therapeutic Options: An Overview. In: Boelens, J., Wynn, R. (eds) Stem Cell Therapy in Lysosomal Storage Diseases. Stem Cell Biology and Regenerative Medicine. Humana Press, New York, NY. https://doi.org/10.1007/978-1-4614-8357-1_1
Download citation
DOI: https://doi.org/10.1007/978-1-4614-8357-1_1
Published:
Publisher Name: Humana Press, New York, NY
Print ISBN: 978-1-4614-8356-4
Online ISBN: 978-1-4614-8357-1
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)