
Abstract
B-cell chronic lymphocytic leukemia (CLL) is the most frequent human leukemia and it occurs in two forms, indolent and aggressive. Although clinical features and genetic abnormalities in CLL are well documented, molecular details underlying the disease are still under investigation.
MicroRNAs are small noncoding RNAs involved in a variety of cellular processes and expressed in a tissue-specific manner. MicroRNAs have the ability to regulate gene expression. In physiological conditions, microRNAs act as gene expression controllers by targeting the mRNA or inhibiting its translation. Their deregulation can lead to an alteration of the expression level of many genes which can induce the development or promote the progression of tumors.
In CLL, microRNAs can function as oncogenes, tumor suppressor genes, and/or can be used as markers for disease onset/progression. For example, in indolent CLL, 13q14 deletions targeting miR-15/16 initiate the disease, while in aggressive CLL miR-181 targets the critical TCL1 oncogene and can also be used as a progression marker.
Here we discuss the foremost findings about the role of microRNAs in CLL pathogenesis, and how this knowledge can be used to identify new approaches to treat CLL.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others


Keywords
CLL: Characteristics and Outcomes
Chronic lymphocytic leukemia (CLL) is the most common human leukemia, accounting for ~30 % of all cases of adult leukemia. In the United States, almost 15,000 new cases are diagnosed each year [42]. CLL is mostly a disease of elderly people, and the incidence increases linearly with each decade [12]. This disease occurs in two forms, aggressive and indolent, both characterized by the progressive accumulation of functionally incompetent B-lymphocytes expressing CD5 antigen on their surface [12]. More than 90 % of the leukemic cells are nondividing and are at the G0/G1 phase of the cell cycle [12]. However, several reports showed that high lymphocyte count in CLL patients is also triggered by the presence of proliferating cells from the bone morrow, spleen, or lymph nodes [22, 52, 79]. CLL cells are also quite resistant to apoptosis [12].
The clinical course of CLL is highly variable, but several prognostic markers have been identified so far to facilitate the clinical management of CLL patients, such as the mutational status of the immunoglobulin heavy-chain variable-region gene (IgH VH), the expression levels of the 70 kD zeta-associated protein (ZAP-70), and the presence of different chromosomal alterations [58, 70]. CLLs with unmutated IgH VH gene and high expression of the ZAP-70 usually have an aggressive course, whereas patients with mutated VH clones and low ZAP-70 expression have an indolent course [23]. Genomic alterations in CLL are also important independent predictors of disease progression and survival [29]; however, the molecular basis of these associations was largely unknown until recently. Genomic aberrations are detected by fluorescence in situ hybridization (FISH) in over 80 % of CLL cases and include 13q, 11q, 17p, and 6q deletions and trisomy 12 [29]. The incidence of these genetic abnormalities is ~50 % for deletion of 13q14, ~10 % for deletion of 11q23, ~15 % for trisomy 12, 7–10 % for deletion of 17p, and 2–5 % for deletion of 6q [56, 81]. Prognosis is worst in patients with 17p deletion, followed by 11q deletion, trisomy 12, and normal karyotype (negative FISH panel), while patients with deletion of 13q as the only abnormality have the best prognosis [56, 95]. Cytogenetic abnormalities can be used to identify subsets of patients with different clinical course, time to progression, and survival rates. According to recent studies, three risk groups can be differentiated: (1) low-risk: patients with a normal karyotype or isolated 13q deletion; (2) intermediate-risk: subjects with 11q deletion, trisomy 12, or 6q deletion; and (3) high-risk: patients with 17p deletion or a complex karyotype [54]. Approximately one third of patients never require treatment; in another third the initial indolent phase is followed by progression of the disease, and the remaining third has aggressive disease at the onset and needs immediate treatment [27]. Because several CLL cases show discordant prognostic factors, the identification of new parameters able to relate disease activity and clinical outcome is essential for patient management.
Signatures of MicroRNAs in CLLs
The miRNAs are a large family of highly conserved noncoding genes thought to be involved in temporal and tissue-specific gene regulation [3]. miRNAs represent an evolving class of gene products with generally unknown function and are usually excised from 70- to 80-nt stem-loop RNA precursor structures. Derived from transcripts transcribed by RNA polymerase II [14], microRNAs are made via a two-step processing mechanism from a primary transcript (pri-miRNA) through an intermediate 60–90 nucleotide stem-loop structure (pre-mRNA) to the final mature microRNA. Dicer and Argonaute family members are required for the miRNA precursor processing reaction [2]. In mammals, single-stranded microRNA binds specific messenger RNA (mRNA) through sequences that are significantly, though not completely, complementary to the target mRNA, mainly to the 3′ untranslated region (3′ UTR) [2]. By a mechanism that is not fully characterized, the bound mRNA remains untranslated, resulting in reduced levels of the corresponding protein; alternatively, the bound mRNA can be degraded, resulting in reduced levels of both the corresponding transcript and consequently the protein. It was estimated that there could be from 300 to 1,000 microRNA genes in the mammalian genome (~1–3 % of known genes are represented by microRNAs). The function of most microRNAs is not known. However, recent reports revealed functions of several microRNAs: hematopoietic B-cell lineage fate (miR-181), B-cell survival (miR-15a and miR-16-1), cell proliferation control (miR-125b and let-7), brain patterning (miR-430), pancreatic cell insulin secretion (miR-375), and adipocyte development (miR-375), reviewed in [38]. Recently, several microRNAs were also linked to several types of cancer [6] and DNA methylation [32]. Moreover, microRNAs can modulate gene expression in a tissue-specific manner and are able to bind target mRNAs, either inhibiting their translation or promoting their degradation [41].
Since the first association between microRNAs and cancer has been demonstrated by Calin et al. [16], it was clear that these genes could play a role in the clinical management of cancer patients. Numerous reports further confirmed that microRNAs are differentially expressed in cancers, thus suggesting that their deregulation could play tumor suppressor or oncogenic roles in cancer pathogenesis [90].
MicroRNA expression profiles revealed several remarkable outcomes that could be applied to the clinic. MicroRNA profiles can be used to distinguish normal B-cells from malignant CLL cells and, more importantly, they are associated with prognosis, progression, and drug resistance in CLL [33]. In particular, a signature profile was reported, describing 13 microRNAs that differentiate aggressive and indolent CLLs [18]. Another report showed that the expression profile of 32 microRNAs is able to discriminate between cytogenetic subgroups [88]. For instance, patients with high levels of miR-21 had a higher risk of death compared to patients with low expression levels [74]. Likewise, high expression of miR-155 was reported in the aggressive form of CLL [19]. Intriguingly, we recently found that expression levels of miR-181b can not only distinguish between indolent and aggressive cohorts of patients but also predict time to treatment, acting as a biomarker of the disease progression. We studied serial time points derived from the same patients and found that expression of miR-181b decreases along with the severity of the disease. These new findings highlight the importance of miR-181b in clinics, suggesting that expression levels of microRNAs can be used not only to classify patients according to the gravity of the pathology but also for tracking the disease course [89]. Moreover, microRNA signature can be also used to predict refractoriness to fludarabine treatment in CLL [33]. To clarify if microRNAs are directly involved in the development of fludarabine resistance, Ferracin et al. analyzed the expression of microRNAs before and after fludarabine therapy in patients classified as responder or refractory and identified a microRNA signature able to distinguish between these two classes. Expression levels of several microRNAs were also able to predict fludarabine resistance in an independent test cohort. Among these microRNAs, miR-148a, miR-222, and miR-21 exhibited a significantly higher expression in nonresponders either before or after treatment. Recently, Zenz et al. found that fludarabine refractory CLLs are frequently characterized by lower levels of miR-34a [96], and low expression of miR-34a was associated with fludarabine resistance even in the absence of p53 aberrations [96].
To conclude, microRNA expression levels can distinguish normal B-cells from CLL, discriminate between indolent and aggressive CLL forms, indicate the progression of the disease, and separate responder and refractory cohorts of patients. These findings provide new roles for microRNAs as markers for CLL development/sensitivity to treatment [33] and potential predictors of time to treatment [89].
Role of MicroRNAs in CLL
Besides using microRNAs’ expression levels as tools to discriminate different CLL forms or to keep track of disease progression, researchers have recently focused on the molecular impact of microRNA deregulation in CLL. Interestingly, the miR-15/16 cluster, miR-29, miR-181 family members, and miRs-34b/c were found as the most deregulated microRNAs in CLL. The same microRNAs were found to regulate gene expression patterns, helping to clarify molecular steps that lead to the onset of the disease or drive its progression.
MicroRNA 15a/16-1. In CLL, deletion at chromosome 13q14.3 is the most frequent genomic aberration (about 50 % of cases) and is associated with the longest treatment-free interval [29]. The first attempts to identify tumor suppressor genes at the 13q14 locus by using positional cloning and sequencing of a region of more than 1 Mb failed [13, 53]; moreover, none of the known genes in this region were found to be down-regulated in CLL by deletions or mutations [13, 51, 53, 73]. In 2001 we generated somatic cell hybrids using mouse and CLL cells carrying 13q14 deletion and translocations, and we identified a 30-kb region of deletion between exon 2 and exon 5 of the LEU2 gene [16, 62]. Interestingly, the translocation breakpoint was mapped to the same region [16, 62]. Since LEU2 had previously been sequenced and excluded as a candidate tumor suppressor gene in 13q14 [13, 51, 53, 92], we continued to investigate that region and finally discovered a cluster of two noncoding microRNA genes, miR-15a and miR-16-1, located exactly within the deleted region and near the translocation breakpoint [16]. Accordingly, the miR-15a/16-1 cluster was found deleted or its expression down-regulated in ~66 % of CLL cases [16, 80]. In contrast, expression levels of the other genes in the region (DLEU1, DLEU2, and RFP5) were not affected by the 13q14 deletions [13, 53, 62].
The first genetic manipulation in mice that confirmed the importance of miR-15a/16-1 deletion in CLL was carried out by Dr. Dalla-Favera and colleagues [45]. These authors designed a model with conditional alleles that resembled either the loss of the minimal deleted region (Mdr), already characterized in human CLL and entirely spans the DLEU2 gene [53], or the specific miR-15a/16-1 cluster deletion, without altering Dleu2 expression [45]. Both Mdr and miR-15a/16-1 knockout strains at 1 year of age presented approximately 50 % of CD5+ B220+ B-cells among mononuclear cells in the peritoneum vs. 15 % in control animals. In total, mice with CLL were 27 % of Mdr KO and 21 % of miR-15a/16-1 KO, while some type of clonal B-cell proliferation affected 42 % of Mdr KO and 26 % of miR-15a/16-1 KO mice between 15 and 18 months of age. Mdr KO animals lived less than WT siblings and eventually succumbed to leukemias, while the differential survival between miR-15a/16-1 and their WT littermates was not statistically significant, providing evidence that the latter were affected by a phenotype milder than the former. Because of the more aggressive disease shown by Mdr KO mice, it is likely that other elements included in the Mdr locus, like the DLEU2 gene itself, may participate to CLL tumor suppression [45]. Mechanisms leading to B-cell proliferations were investigated with different approaches. MiR-15a/16-1 KO B-cells were shown to begin DNA synthesis earlier than WT B-cells [45]. The authors also analyzed levels of phosphorylated retinoblastoma (pRb) protein, an indicator of entry into the cell cycle, in mitogen-stimulated B-cells isolated from miR-15a/16-1 KO or Mdr KO and WT animals. PRb was produced in both KO B-cells at earlier time points than in WT B-cells. Individual contributions of miR-15a/16-1 cluster vs. DLEU2 gene to the lympho-proliferation were dissected, generating an inducible system where these two genetic elements underwent separate in vitro re-expression in a human cell line derived from a 13q14 KO CLL. These findings demonstrated that impaired proliferation occurred in miR-15a/16-1 expressing cells, with a higher fraction of cells in G0/G1 phase, but not in those expressing Dleu2, thus suggesting a possible control of the inhibition of G0/G1 phase transition by miR-15a/16-1 [45].
The importance of the miR-15a/16-1 cluster in CLL was confirmed in a study of CLL development in New Zealand black (NZB) mice, the only mouse strain that naturally develops CLL [72]. In NZB mice, CLL arises late in life, with an autoimmune phenotype and B-cell hyper-proliferation followed by slow progression to late-onset CLL [71, 93]. Older NZB animals show a clonal expansion of the subpopulation of B-1 B-cells similar to that found in human CLL [71, 93]. Linkage analysis has found that the mouse genomic region homologous to 13q14 is one of the loci associated with CLL development. Subsequent DNA sequencing resulted in the identification of a point mutation in miR-15a/16-1 precursor causing a decrease of miR-16-1 expression in NZB lymphoid tissues, accompanied by elevated levels of Bcl-2 [72]. Accordingly, lymphoid tissues from NZB mice were analyzed for the levels of mature miR-16-1 and showed reduced expression of this microRNA. Finally, delivery of exogenous miR-16-1 to a NZB malignant cell line led to cell cycle alterations such as decrease in S phase cells and G1 arrest [72]. Other strains of mice, including the NZW strain, the closest relative of NZB, did not show the mutation in miR-15a/16-1 precursor.
B-cell lymphoma 2 (BCL2) is a central player in the genetic program of eukaryotic cells, promoting survival by inhibiting cell death [26]. Over-expression of Bcl2 protein has been reported in many types of human cancers, including leukemias, lymphomas, and carcinomas [76]. In follicular lymphomas and in a fraction of diffuse large B-cell lymphomas, BCL2 is activated due to the translocation t(14,18)(q32;q21), which places the BCL2 gene under the control of Ig heavy-chain enhancers, resulting in the over-expression of the gene [83, 84]. In CLL, malignant B-cells over-express Bcl2 [44]; however, with the exception of less than 5 % of cases, in which the BCL2 gene is juxtaposed to Ig loci [1], no mechanism has been discovered to explain BCL2 up-regulation in CLL. MiR-15a and miR-16-1 expression is inversely correlated to Bcl2 expression in CLL and these microRNAs negatively regulate BCL2 at the posttranscriptional level [25]. Since BCL2 is a predicted target of both miR-15a and miR-16-1, the down-regulation of these microRNAs in a leukemic cell line resulted in an increase of Bcl2 expression with consequent inhibition of apoptosis [25]. Interestingly, miR-15a/16-1 expression also resulted in growth inhibition of tumor engraftment of leukemic cells in nude mice, confirming the tumor suppression properties of these microRNAs [15]. In summary, Bcl2 over-expression driven by down-regulation of miR-15a and miR-16-1 seems to be a regulatory mechanism involved in the pathogenesis of a large part of human CLL. These studies determined that the miR-15a/16-1 cluster functions as a tumor suppressor in CLL by inhibiting Bcl2, and deletions at 13q14 represent an initializing step in CLL development [25]. In this respect miR-15a/16-1 have promise to be used as a drug for CLL.
Since the indolent form of the disease is often characterized by 13q14 deletion, it is likely that up-regulation of BCL2 plays a major role in this subset of CLLs. Evidence for this hypothesis came from Dr. Reed and colleagues, who used two previously described mouse models, one with Bcl2 over-expression in the lymphoid system [43] and the second with up-regulation of a specific isoform of TRAF2 (tumor necrosis factor (TNF) receptor-associated factor 2) in B- and T-cells [47]. TRAF2 can bind to the TNF receptor family and mediate the activation of NF-kB by TNF proteins [24]; TNF-mediated signaling increased lymphocyte proliferation and survival [36].
TRAF2 transgenic mice failed to develop a frank leukemia, but showed an increased number of B-cells accompanied by lympho-adenopathy and splenomegaly [47]. BCL2 transgenic animals, which were designed with a construct mimicking the t(14;18) translocation, juxtaposing BCL2 gene with the immunoglobulin heavy-chain locus at 14q32 as reported in human follicular lymphomas, did not develop malignancies either, presenting only prolonged in vitro B-cell survival and in vivo polyclonal B-cell expansions [43].
TRAF2DN-BCL2 double transgenic mice, on the other hand, displayed severe splenomegaly, and most animals were affected by a CLL-like disease with high B-cell blood count [94]. While single transgenics showed a normal lifespan, the double ones survived only between 6 and 14 months. Because of their complex features, it was not clear whether TRAF2DN-BCL2 transgenics were a model of indolent or aggressive CLL [67].
Based on these findings, 13q14 deletions could induce CLL development by a molecular mechanism resembling the oncogenic events in TRAF2DN/BCL2 transgenics[60]. In fact, in addition to miR-15a/16-1, the 13q14 region deleted in indolent CLL contains the DLEU7 gene, located telomeric to miR-15a/16-1, [59]. Our report showed that DLEU7 is a cooperating tumor suppressor along with miR-15a/16-1, and we recently confirmed that DLEU7 deletions result in the induction of TNF signaling through TRAFs, while miR-15a/16-1 deletions cause a constitutive increase of Bcl2 expression.
DLEU7 was previously identified as a candidate tumor suppressor gene at 13q14 [37]. Recently, Ouillette et al., by using microarray technology, have displayed that the minimal deleted region at 13q14 in CLL contains DLEU7 gene [59]. Since DLEU7 is the only protein coding gene located within the reported minimal deleted region at 13q14, we investigated whether DLEU7 can cooperate with miR-15a/16-1 [60]. Sequencing of DLEU7 coding exons failed to find mutations in CLL samples, although a previous study reported hyper-methylation of DLEU7 promoter, with consequent silencing of this gene in 61 % of CLL cases [37]. Real time RT-PCR experiments confirmed that expression of DLEU7 in CLL samples is decreased when compared to normal CD19+ B-cells. MiR-15a/16-1 were also found down-regulated in the same CLL samples [60].
Since recent studies confirmed a significant role for the NF-kB pathway in the pathogenesis of CLL [67], we examined whether Dleu7 might function as an inhibitor of NF-kB. In the inactive state, NF-kB proteins are bound to IκB proteins in the cytoplasm; after stimulation, IκB is degraded and NF-kB translocates the nucleus [11, 21, 34]. Induction of NF-kB can be driven by a variety of stimuli, including exposure to members of the TNF superfamily, chemotherapy, and ionizing radiation [7, 85, 91]. Activation of NF-kB prevents B-cells from undergoing apoptosis and regulates growth and differentiation [7, 85, 91]. In B-cells, it has been shown that transgenic expression of the TNF ligand APRIL resulted in an expansion of B220+ CD5+ cells [68]. APRIL binds BCMA (B-cell maturation antigen) and TACI [36], which stimulate the NF-kB pathway, thus suggesting that NF-kB activation through TACI and BCMA is important in the pathogenesis of CLL [60]. Moreover, nuclear factor of activated T-cells (NFAT) can also be activated by TACI and BCMA [48]; NFAT was previously reported as a hallmark of unstimulated CLL cells [8, 78].
Since DLEU7 is located within the 13q14-deleted region and NF-kB/NFAT activation can be critical in CLL pathogenesis, we studied whether Dleu7 expression has an effect on NF-kB and NFAT activation by TACI and BCMA. Our experiments showed that Dleu7 expression inhibits NF-kB activation by BCMA over fivefold, while activation by TACI was inhibited over fourfold [60]. Also, Dleu7 expression can inhibit NFAT activation by TACI and BCMA approximately eightfold. Thus, we concluded that Dleu7 functions as NFAT and NF-kB inhibitor [60].
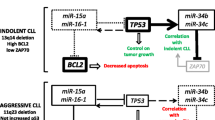
In conclusion, miR-15a/16-1 deletion is an initializing step in CLL development, eliciting control on Bcl2 expression level and cooperating with DLEU7 in promoting the activation of NF-kB and NFAT via TACI and BCMA. Moreover, we also recently discovered a miR-15a/16-1-TP53 feedback circuitry, in which p53 directly transactivates miR-15a/16-1 promoter, while miR-15a/16-1 cluster targets TP53 expression [31].
MicroRNA 34b/c. It is currently unknown how the 11q, 17p, and 13q deletions contribute to CLL pathogenesis and progression [29]. However, it has been proved that the loss of the long arm of chromosome 11 includes the region where the miR-34b/c cluster is located [5], while deletion of 17p leads to abrogation of the p53 tumor suppressor [50] and 13q deletion involves miR15a/16-1 down-regulation. To establish the possible existence of molecular interactions between these chromosomal alterations, we investigated if the miR-15a/16-1 cluster, tumor protein p53, and miR-34b/c cluster are connected in a molecular pathway that could explain the prognostic implications (aggressive vs. indolent form) of 11q, 17p, and 13q deletions in CLL [31].
Several TP53 binding sites were found upstream of the miR-15a/16-1 on chromosome 13 and of the miR-34b/c on chromosome 11. Chromatin immunoprecipitation analysis revealed that TP53 directly binds to its predicted binding sites on both chromosomes 13 and 11. Thus, TP53 can induce the expression of both these microRNAs [31]. On the other hand, miR-15a/16-1 target TP53, while a binding site for the miR-34 family was predicted in ZAP-70 mRNA [31]. These interactions could lead to different outcomes via feedback circuits involving protein coding genes and microRNAs [31]. In this model, TP53 (on chromosome 17p) represents the molecular connection between miR-15a/16-1 (on chromosome 13q) and miR-34b/c (on chromosome 11q) [31].
In 13q-deleted patients, the loss of miR-15a/16-1 expression shifts the balance not only toward higher levels of the anti-apoptotic protein Bcl2 [15, 25] but also toward higher levels of the tumor suppressor protein p53. Consequently, in 13q patients, while the number of apoptotic cells may decrease because of the increased levels of Bcl2, the p53 tumor suppressor pathway remains intact, thus keeping the increase in tumor burden relatively low. This finding could explain how 13q deletions are associated with the indolent form of CLL. Moreover, increased p53 levels in patients with 13q deletions are associated with transactivation of miR-34b/c and with reduced levels of ZAP-70 [70], and further supporting the indolent course of CLLs carrying 13q deletions.
CLL patients with 11q deletion, instead, express significantly lower levels of miR-34b/c and significantly higher levels of ZAP-70, both at mRNA and protein levels. These patients show poorer overall survival than patients with normal cytogenetic profiles and lower levels of ZAP-70. In these patients, TP53 is not upregulated because miR-15a/16-1 are not deleted, and this condition is associated with lower control on apoptosis [31].
In conclusion, we demonstrated that a microRNA/TP53 feedback circuitry is associated with the pathogenesis of CLL. These results also showed that restoring expression of miR-15a/16-1 indirectly affects expression of the miR-34 family by modulating levels of TP53 expression. Moreover, the miR-34 family is a downstream target of p53, and its over-expression can cause p53-like effects on apoptosis or cell cycle arrest [31].
MicroRNA 29. In both indolent and aggressive CLLs, miR-29 is over-expressed compared to normal B-cells, but its role in development/progression of CLLs is still unclear. In addition, expression levels of miR-29 are higher in indolent than in aggressive CLLs [17, 66, 77]. These results prompted us to evaluate the role of this microRNA in CLL. The up-regulation of miR-29 in indolent CLL compared to normal B-cells implies an oncogenic function for this microRNA, initiating or at least significantly contributing to the pathogenesis of CLL [17, 66, 77]. On the other hand, we showed that expression levels of TCL1 and miR-29 are inversely correlated, and that miR-29 targets TCL1 expression [66], thus suggesting a possible tumor suppressor function for miR-29 in aggressive CLL. Furthermore, a microRNA signature was published with 13 microRNAs that differentiate aggressive and indolent CLLs [18]. Intriguingly, of the four down-regulated microRNAs in aggressive CLL, three are different isoforms of miR-29 (miR-29a-2, miR-29b-2, and miR-29c) [18], strongly suggesting that deregulation of miR-29 can play a role in the pathogenesis of aggressive CLLs. In addition, expression of members of the miR-29 family could discriminate between CLL samples with good and bad prognosis [17].
In order to study the role of miR-29 in B-cell leukemias, we designed a transgenic mouse characterized by over-expression of miR-29 in B-cells. In splenocytes from these transgenics we reported an increase in CD5+ CD19+ IgM+ B-cell populations, a hallmark of CLL [77]. Eighty-five percent of miR-29 animals showed a marked growth of CD5+ B-cells that, between 12 and 14 months of age, represented up to 50 % of total B-cells. Only 20 % of the transgenics died because of leukemia between 24 and 26 months of age. These data led us to conclude that miR-29 mice mimicked the indolent form of CLL. In fact, the percentage of leukemic cells increased with age, from 20 % of all B-cells in mice below 15 months of age to more than 65 % in mice above 20 months of age, indicating a gradual progression of indolent CLL [77]. Using BrdU incorporation experiments to measure the proliferative capacity of leukemic cells, we confirmed a significantly increased proliferation in miR-29 transgenic B-cells compared to wild type CD19+ cells, where no proliferation was found. Thus, miR-29 over-expression seems to play a role in promoting B-cell proliferation. Furthermore, since immune incompetence and progressive hypogammaglobulinemia are typical features of human CLL, immune response to SRBC antigen and serum levels of immunoglobulins were analyzed in miR-29 mice and their wild type littermates. Both parameters were drastically decreased in transgenic animals, confirming that miR-29 transgenics mimic the indolent course of human CLL [77].
In aggressive CLLs, the down-regulation of miR-29 appears to be involved in Tcl1 over-expression, along with miR-181 [66]. Activation of the TCL1 oncogene is a central initiating event in the pathogenesis of aggressive CLL. TCL1 (T cell leukemia/lymphoma 1) was originally identified as a target of translocations and inversions at 14q32.1 in T-cell prolymphocytic leukemias (T-PLL) [87]. High Tcl1 expression in human CLL correlates with aggressive phenotype [40]. Tcl1 functions as a promoter of the PI3K–Akt(PKB) oncogenic pathway [46, 64], activating Akt, driving its nuclear translocation and leading to an increased proliferation, inhibition of apoptosis, and transformation [64]. At the same time, Tcl1 activates NF-kB, inhibits AP-1 [65], and restrains DNMT3a [61], which is involved in epigenetic deregulation of gene expression. This leads to defects in cell death, increased survival, and CLL pathogenesis.
Recently we investigated whether TCL1 expression in CLL is regulated by microRNAs [66]. MiR-29b and miR-181b are down-regulated in aggressive CLLs with 11q deletions and are predicted to target Tcl1 [66]. Interestingly, miR-181 is differentially expressed in B-cells, and TCL1 is mostly a B-cell-specific gene [69], thus suggesting that Tcl1 might be a target of miR-181 not only in CLL cells but also in normal B-lymphocytes. We therefore proceeded to verify if these microRNAs really target Tcl1 expression. Our experiments revealed that co-expression of Tcl1 with miR-29 and miR-181 significantly decreased Tcl1 expression [66], and we consequently concluded that miR-29b and miR-181b target TCL1 expression on mRNA and protein levels [66]. Concordantly, we found an inverse correlation between miR-29b and miR-181b expression and Tcl1 protein expression in CLL samples, which further supports the idea that Tcl1 expression in CLL is, at least in part, regulated by miR-29 and miR-181 [66].
Since TCL1 expression is regulated by microRNAs, like miR-29 and miR-181, that target the 3′ UTR region of the gene, we generated transgenic mice of Eμ-TCL1 Full Length (Eμ-TCL1 FL), including both the 3′ and 5′ UTRs of TCL1 under a B-cell-specific promoter [30]. These animals showed the development of a CLL-like leukemia between 16 and 20 months of age and a population of CD5+ CD23+ B-cells accumulated in spleens and lymph nodes of these mice. Immunological abnormalities like hypoimmunoglobulinemia, impaired immune response, and abnormal levels of cytokines were also found in Eμ-TCL1 FL animals and were similar to those observed in human CLL [30]. In conclusion, both classical Eμ-TCL1 and Eμ-TCL1 FL transgenic mouse models of CLL displayed important biological similarities with their human counterpart that went beyond the simple resemblance between the two leukemias. Our study demonstrated that TCL1 up-regulation in mouse B-cells results in aggressive CLL [9].
In conclusion, the current idea of the role of miR-29 in CLL is associated with its effect on Tcl1 expression levels in both indolent and aggressive forms. Since TCL1 is generally not expressed in indolent CLL [66], it likely does not play an important function in indolent CLL, and its down-regulation due to miR-29 over-expression does not slow indolent CLL development. Up-regulation of miR-29 expression is not sufficient to cause aggressive CLL; on the other hand, up-regulation of Tcl1 is absolutely required for the initiation of the aggressive form of CLL. Down-regulation of miR-29 expression in aggressive CLL (compared to the indolent form) contributes to up-regulation of Tcl1 and development of aggressive CLL [63].
Effects of Polymorphisms and Epigenetic Regulation on microRNAs Expression
The complexity of the pathways involving microRNAs in CLL development/progression was found to extend beyond their ability to directly regulate gene expression. MicroRNA expression can respond to the presence of single nucleotide polymorphisms (SNPs) and can also be altered by transactivator factors [4]. Moreover, deregulation of epigenetic processes can modify microRNA expression, leading to a diverse progression of the disease and a different prognosis [75].
A good example of SNPs being involved in altered microRNA expression is offered by miR-34a [4]. MiR-34a has been implicated in the CLL response to DNA damage through a p53-mediated induction [28, 55, 96]. TP53 protein transactivates miR-34a on chromosome 1p36, inducing tumor suppressor effects, enhancing apoptosis and cycle arrest [10, 20, 39, 82]. The presence of a SNP 309 in the intronic region of the promoter of ubiquitin ligase MDM2 leads to increased expression of MDM2, which binds p53 [4]. In patients with intact p53, it has been reported that the presence of this SNP inhibits p53 transactivation effects on miR-34a and can induce down-regulation of miR34a [4]. In many types of cancer this SNP has been associated with accelerated tumor formation and poor prognosis [35, 49, 57]. Asslaber et al. have shown that the GG-genotype of MDM2 SNP 309 is associated with reduced overall survival and treatment-free survival in CLL. CLL cells of patients with the GG-genotype had a significantly lower mean expression of miR-34a as compared with the TT-genotype, suggesting attenuation of the p53 pathway by the SNP 309. MiR-34a levels in cells with the heterozygous GT-genotype were found between those with the GG- and the TT-genotype. Thus, the presence of this SNP restrains p53 activity on miR-34a expression in CLL patients without p53 deletion/mutation [4].
MicroRNAs can be also involved in epigenetic gene regulation with positive and negative feedback circuits [75]. The histone deacetylases (HDACs) are chromatin-modulating enzymes that catalyze the removal of acetyl groups on specific lysines around gene promoters [86]. Moreover, they can trigger the demethylation of lysine 4 on histones (H3K4me2/3), thus promoting chromatin compaction and leading to epigenetic gene silencing [86]. Recent data established that HDACs can also silence microRNAs. In particular, it has been observed that miR-15a/16-1 are silenced by epigenetic mechanisms in 30–35 % of CLL samples, therefore cooperating with 13q14 deletion to account for the low expression levels of these microRNAs in CLL [75]. Indeed, it has been found that HDAC1–3 are over-expressed in CLL but not in normal lymphocytes, hence identifying an independent mechanism for the silencing of miR-15a/16-1 [75].
In samples with monoallelic 13q14 deletion it has been observed that the HDACs repressed miR-15a/16-1 expression on the residual allele, providing an example of functional cooperation between a genetic and an epigenetic mechanism to achieve gene repression. Induction of miR-15a/16-1 in response to HDAC inhibition is associated with activation of cell death. Future prospective trials should evaluate the specific impact of epigenetic silencing of miR-15a/16-1 on disease behavior and progression that could represent a new therapeutic strategy to antagonize an important survival mechanism in cells. CLL patients who exhibit such epigenetic silencing may represent a group that will possibly benefit from HDAC inhibitor-based therapy [75].
Conclusions
CLL is a heterogeneous disease. Karyotypic aberrations are strongly prognostic of survival, as well as IgH VH mutational status and ZAP-70 expression. Lately, microRNA expression has been considered as a new important tool in the management of the disease. In the order of highest to lowest risk, the genomic categories so far identified are: 17p deletion, 11q deletion, trisomy 12, normal FISH, and 13q deletion. Patients with 17p deletion respond poorly to treatment while patients with 11q deletion CLL show a better response to treatment, even if it progresses early. Moreover, unmutated IgH VH/ZAP-70-positive patients have increased rates of progression and reduced remission durations.
MicroRNAs are differentially expressed in cancers, and their deregulation could play tumor suppressor or oncogenic roles in cancer pathogenesis. MicroRNA expression profiles have been found to be useful tools to distinguish normal B-cells from malignant CLL cells and can be correlated with prognosis, progression, and drug resistance of CLL. MicroRNAs modify gene expression, and their deregulation involves downstream effects on cell cycle and proliferation. Deletion of miR-15a/16-1 has been correlated to Bcl2 up-regulation in indolent CLL, while down-regulation of miR-29 and miR-181 has been correlated to Tcl1 up-regulation in aggressive CLL. On the other hand, over-expression of miR-29 in B-cells results in development of indolent CLL. MiR-34 family members are involved in a finely regulated feedback circuitry with p53 and miR-15a/16-1 in 13q-deleted CLL, thus suggesting that the interplay between microRNAs and genes is bidirectional.
Deregulation of microRNAs can be a consequence of chromosomal alteration, epigenetic modulation, or interaction with other genes. In fact, microRNAs can be epigenetically silenced, suggesting a new cooperating system of abnormal regulation of these molecules. The study of these mechanisms can clarify the role of microRNAs in the development and progression of CLL and allow the identification of new targets for therapy.
References
Adachi M, Tefferi A, Greipp PR, et al. Preferential linkage of bcl-2 to immunoglobulin light chain gene in chronic lymphocytic leukemia. J Exp Med. 1990;171(2):559–64.
Ambros V. MicroRNA pathways in flies and worms: growth, death, fat, stress, and timing. Cell. 2003;113(6):673–6.
Ambros V. The functions of animal microRNAs. Nature. 2004;431(7006):350–5.
Asslaber D, Pinon JD, Seyfried I, et al. MicroRNA-34a expression correlates with MDM2 SNP309 polymorphism and treatment-free survival in chronic lymphocytic leukemia. Blood. 2010;115(21):4191–7.
Auer RL, Riaz S, Cotter FE. The 13q and 11q B-cell chronic lymphocytic leukaemia-associated regions derive from a common ancestral region in the zebrafish. Br J Haematol. 2007;137(5):443–53.
Bartel DP. MicroRNAs: genomics, biogenesis, mechanism, and function. Cell. 2004;116(2):281–97.
Beg AA, Baltimore D. An essential role for NF-kappaB in preventing TNF-alpha-induced cell death. Science. 1996;274(5288):782–4.
Berland R, Wortis HH. An NFAT-dependent enhancer is necessary for anti-IgM-mediated induction of murine CD5 expression in primary splenic B cells. J Immunol. 1998;161(1):277–85.
Bichi R, Shinton SA, Martin ES, et al. Human chronic lymphocytic leukemia modeled in mouse by targeted TCL1 expression. Proc Natl Acad Sci U S A. 2002;99(10):6955–60.
Bommer GT, Gerin I, Feng Y, et al. p53-Mediated activation of miRNA34 candidate tumor-suppressor genes. Curr Biol. 2007;17(15):1298–307.
Brockman JA, Scherer DC, McKinsey TA, et al. Coupling of a signal response domain in I kappa B alpha to multiple pathways for NF-kappa B activation. Mol Cell Biol. 1995;15(5):2809–18.
Bullrich F, Croce CM. Molecular biology of chronic lymphocytic leukemia. In: Cheson B, editor. Chronic lymphocytic leukemias, 2nd ed, revised and expanded. New York: Marcel Dekker; 2001. p. 9–32.
Bullrich F, Fujii H, Calin G, et al. Characterization of the 13q14 tumor suppressor locus in CLL: identification of ALT1, an alternative splice variant of the LEU2 gene. Cancer Res. 2001;61(18):6640–8.
Cai X, Hagedorn CH, Cullen BR. Human micrornas are processed from capped, polyadenylated transcripts that can also function as mrnas. RNA. 2004;10:1957–66.
Calin GA, Cimmino A, Fabbri M, et al. MiR-15a and miR-16-1 cluster functions in human leukemia. Proc Natl Acad Sci U S A. 2008;105(13):5166–71.
Calin GA, Dumitru CD, Shimizu M, et al. Frequent deletions and down-regulation of micro- RNA genes miR15 and miR16 at 13q14 in chronic lymphocytic leukemia. Proc Natl Acad Sci U S A. 2002;99(24):15524–9.
Calin GA, Ferracin M, Cimmino A, et al. A microRNA signature associated with prognosis and progression in chronic lymphocytic leukemia. N Engl J Med. 2005;353(17):1793–801.
Calin GA, Liu CG, Sevignani C, et al. MicroRNA profiling reveals distinct signatures in B cell chronic lymphocytic leukemias. Proc Natl Acad Sci U S A. 2004;101(32):11755–60.
Calin GA, Pekarsky Y, Croce CM. The role of microRNA and other non-coding RNA in the pathogenesis of chronic lymphocytic leukemia. Best Pract Res Clin Haematol. 2007;20(3):425–37.
Chang TC, Wentzel EA, Kent OA, et al. Transactivation of miR-34a by p53 broadly influences gene expression and promotes apoptosis. Mol Cell. 2007;26(5):745–52.
Chen Z, Hagler J, Palombella VJ, et al. Signal-induced site-specific phosphorylation targets I kappa B alpha to the ubiquitin-proteasome pathway. Genes Dev. 1995;9(13):1586–97.
Chiorazzi N. Cell proliferation and death: forgotten features of chronic lymphocytic leukemia B cells. Best Pract Res Clin Haematol. 2007;20(3):399–413.
Chiorazzi N, Rai KR, Ferrarini M. Chronic lymphocytic leukemia. N Engl J Med. 2005;352(8):804–15.
Chung JY, Park YC, Ye H, et al. All TRAFs are not created equal: common and distinct molecular mechanisms of TRAF-mediated signal transduction. J Cell Sci. 2002;115(Pt 4):679–88.
Cimmino A, Calin GA, Fabbri M, et al. miR-15 and miR-16 induce apoptosis by targeting BCL2. Proc Natl Acad Sci U S A. 2005;102(39):13944–9.
Cory S, Adams JM. The Bcl2 family: regulators of the cellular life-or-death switch. Nat Rev Cancer. 2002;2(9):647–56.
Dighiero G, Binet JL. When and how to treat chronic lymphocytic leukemia. N Engl J Med. 2000;343(24):1799–801.
Dijkstra MK, van Lom K, Tielemans D, et al. 17p13/TP53 deletion in B-CLL patients is associated with microRNA-34a downregulation. Leukemia. 2009;23(3):625–7.
Dohner H, Stilgenbauer S, Benner A, et al. Genomic aberrations and survival in chronic lymphocytic leukemia. N Engl J Med. 2000;343(26):1910–6.
Efanov A, Zanesi N, Nazaryan N, et al. CD5+CD23+ leukemic cell populations in TCL1 transgenic mice show significantly increased proliferation and Akt phosphorylation. Leukemia. 2010;24(5):970–5.
Fabbri M, Bottoni A, Shimizu M, et al. Association of a microRNA/TP53 feedback circuitry with pathogenesis and outcome of B-cell chronic lymphocytic leukemia. JAMA. 2011;305(1):59–67.
Fabbri M, Ivan M, Cimmino A, et al. Regulatory mechanisms of microRNAs involvement in cancer. Expert Opin Biol Ther. 2007;7(7):1009–19.
Ferracin M, Zagatti B, Rizzotto L, et al. MicroRNAs involvement in fludarabine refractory chronic lymphocytic leukemia. Mol Cancer. 2010;26(9):123.
Ghosh S, May MJ, Kopp EB. NF-kappa B and Rel proteins: evolutionarily conserved mediators of immune responses. Annu Rev Immunol. 1998;16:225–60.
Gryshchenko I, Hofbauer S, Stoecher M, et al. MDM2 SNP309 is associated with poor outcome in B-cell chronic lymphocytic leukemia. J Clin Oncol. 2008;26(14):2252–7.
Haiat S, Billard C, Quiney C, et al. Role of BAFF and APRIL in human B-cell chronic lymphocytic leukaemia. Immunology. 2006;118(3):281–92.
Hammarsund M, Corcoran MM, Wilson W, et al. Characterization of a novel B-CLL candidate gene–DLEU7–located in the 13q14 tumor suppressor locus. FEBS Lett. 2004;556(1–3):75–80.
Harfe BD. MicroRNAs in vertebrate development. Curr Opin Genet Dev. 2005;15:410–5.
He L, He X, Lim LP, et al. A microRNA component of the p53 tumour suppressor network. Nature. 2007;447(7148):1130–4.
Herling M, Patel KA, Khalili J, et al. TCL1 shows a regulated expression pattern in chronic lymphocytic leukemia that correlates with molecular subtypes and proliferative state. Leukemia. 2006;20(2):280–5.
Iorio MV, Croce CM. MicroRNA involvement in human cancer. Carcinogenesis. 2012;33(6):1126–33.
Jemal A, Siegel R, Xu J, et al. Cancer statistics, 2010. CA Cancer J Clin. 2010;60(5):277–300.
Katsumata M, Siegel RM, Louie DC, et al. Differential effects of Bcl-2 on T and B cells in transgenic mice. Proc Natl Acad Sci U S A. 1992;89(23):11376–80.
Kitada S, Andersen J, Akar S, et al. Expression of apoptosis-regulating proteins in chronic lymphocytic leukemia: correlations with in vitro and in vivo chemoresponses. Blood. 1998;91(9):3379–89.
Klein U, Lia M, Crespo M, et al. The DLEU2/miR-15a/16-1 cluster controls B cell proliferation and its deletion leads to chronic lymphocytic leukemia. Cancer Cell. 2010;17(1):28–40.
Laine J, Kunstle G, Obata T, et al. The protooncogene TCL1 is an Akt kinase coactivator. Mol Cell. 2000;6(2):395–407.
Lee SY, Reichlin A, Santana A, et al. TRAF2 is essential for JNK but not NF-kappaB activation and regulates lymphocyte proliferation and survival. Immunity. 1997;7(5):703–13.
Mackay F, Schneider P, Rennert P, et al. BAFF AND APRIL: a tutorial on B cell survival. Annu Rev Immunol. 2003;21:231–64.
Menin C, Scaini MC, De Salvo GL, et al. Association between MDM2-SNP309 and age at colorectal cancer diagnosis according to p53 mutation status. J Natl Cancer Inst. 2006;98(4):285–8.
Merkel O, Asslaber D, Pinon JD, et al. Interdependent regulation of p53 and miR-34a in chronic lymphocytic leukemia. Cell Cycle. 2010;9(14):2764–8.
Mertens D, Wolf S, Schroeter P, et al. Down-regulation of candidate tumor suppressor genes within chromosome band 13q14.3 is independent of the DNA methylation pattern in B-cell chronic lymphocytic leukemia. Blood. 2002;99(11):4116–21.
Messmer BT, Messmer D, Allen SL, et al. In vivo measurements document the dynamic cellular kinetics of chronic lymphocytic leukemia B cells. J Clin Invest. 2005;115(3):755–64.
Migliazza A, Bosch F, Komatsu H, et al. Nucleotide sequence, transcription map, and mutation analysis of the 13q14 chromosomal region deleted in B-cell chronic lymphocytic leukemia. Blood. 2001;97(7):2098–104.
Moreno C, Montserrat E. Genetic lesions in chronic lymphocytic leukemia: what’s ready for prime time use? Haematologica. 2010;95(1):12–5.
Mraz M, Malinova K, Kotaskova J, et al. miR-34a, miR-29c and miR-17-5p are downregulated in CLL patients with TP53 abnormalities. Leukemia. 2009;23(6):1159–63.
Neilson JR, Auer R, White D, et al. Deletions at 11q identify a subset of patients with typical CLL who show consistent disease progression and reduced survival. Leukemia. 1997;11(11):1929–32.
Ohmiya N, Taguchi A, Mabuchi N, et al. MDM2 promoter polymorphism is associated with both an increased susceptibility to gastric carcinoma and poor prognosis. J Clin Oncol. 2006;24(27):4434–40.
Orchard JA, Ibbotson RE, Davis Z, et al. ZAP-70 expression and prognosis in chronic lymphocytic leukaemia. Lancet. 2004;363(9403):105–11.
Ouillette P, Erba H, Kujawski L, et al. Integrated genomic profiling of chronic lymphocytic leukemia identifies subtypes of deletion 13q14. Cancer Res. 2008;68(4):1012–21.
Palamarchuk A, Efanov A, Nazaryan N, et al. 13q14 deletions in CLL involve cooperating tumor suppressors. Blood. 2010;115(19):3916–22.
Palamarchuk A, Yan PS, Zanesi N, et al. Tcl1 protein functions as an inhibitor of de novo DNA methylation in B-cell chronic lymphocytic leukemia (CLL). Proc Natl Acad Sci U S A. 2012;109(7):2555–60.
Pekarsky Y, Calin GA, Aqeilan R. Chronic lymphocytic leukemia: molecular genetics and animal models. Curr Top Microbiol Immunol. 2005;294:51–70.
Pekarsky Y, Croce CM. Is miR-29 an oncogene or tumor suppressor in CLL? Oncotarget. 2010;1(3):224–7.
Pekarsky Y, Koval A, Hallas C, et al. Tcl1 enhances Akt kinase activity and mediates its nuclear translocation. Proc Natl Acad Sci U S A. 2000;97(7):3028–33.
Pekarsky Y, Palamarchuk A, Maximov V, et al. Tcl1 functions as a transcriptional regulator and is directly involved in the pathogenesis of CLL. Proc Natl Acad Sci U S A. 2008;105(50):19643–8.
Pekarsky Y, Santanam U, Cimmino A, et al. Tcl1 expression in chronic lymphocytic leukemia is regulated by miR-29 and miR-181. Cancer Res. 2006;66(24):11590–3.
Pekarsky Y, Zanesi N, Aqeilan RI, et al. Animal models for chronic lymphocytic leukemia. J Cell Biochem. 2007;100(5):1109–18.
Planelles L, Carvalho-Pinto CE, Hardenberg G, et al. APRIL promotes B-1 cell-associated neoplasm. Cancer Cell. 2004;6(4):399–408.
Ramkissoon SH, Mainwaring LA, Ogasawara Y, et al. Hematopoietic-specific microRNA expression in human cells. Leuk Res. 2006;30(5):643–7.
Rassenti LZ, Huynh L, Toy TL, et al. ZAP-70 compared with immunoglobulin heavy-chain gene mutation status as a predictor of disease progression in chronic lymphocytic leukemia. N Engl J Med. 2004;351(9):893–901.
Raveche ES. Possible immunoregulatory role for CD5+ B cells. Clin Immunol Immunopathol. 1990;56(2):135–50.
Raveche ES, Salerno E, Scaglione BJ, et al. Abnormal microRNA-16 locus with synteny to human 13q14 linked to CLL in NZB mice. Blood. 2007;109(12):5079–86.
Rondeau G, Moreau I, Bezieau S, et al. Comprehensive analysis of a large genomic sequence at the putative B-cell chronic lymphocytic leukaemia (B-CLL) tumour suppresser gene locus. Mutat Res. 2001;458(3–4):55–70.
Rossi S, Shimizu M, Barbarotto E, et al. MicroRNA fingerprinting of CLL patients with chromosome 17p deletion identify a miR-21 score that stratifies early survival. Blood. 2010;116(6):945–52.
Sampath D, Liu C, Vasan K, et al. Histone deacetylases mediate the silencing of miR-15a, miR-16, and miR-29b in chronic lymphocytic leukemia. Blood. 2012;119(5):1162–72.
Sanchez-Beato M, Sanchez-Aguilera A, Piris MA. Cell cycle deregulation in B-cell lymphomas. Blood. 2003;101(4):1220–35.
Santanam U, Zanesi N, Efanov A, et al. Chronic lymphocytic leukemia modeled in mouse by targeted miR-29 expression. Proc Natl Acad Sci U S A. 2010;107(27):12210–5.
Schuh K, Avots A, Tony HP, et al. Nuclear NF-ATp is a hallmark of unstimulated B cells from B-CLL patients. Leuk Lymphoma. 1996;23(5–6):583–92.
Sieklucka M, Pozarowski P, Bojarska-Junak A, et al. Apoptosis in B-CLL: the relationship between higher ex vivo spontaneous apoptosis before treatment in III-IV Rai stage patients and poor outcome. Oncol Rep. 2008;19(6):1611–20.
Smonskey MT, Block AW, Deeb G, et al. Monoallelic and biallelic deletions of 13q14.3 in chronic lymphocytic leukemia: FISH vs miRNA RT-qPCR detection. Am J Clin Pathol. 2012;137:641–6.
Stilgenbauer S, Bullinger L, Benner A, et al. Incidence and clinical significance of 6q deletions in B cell chronic lymphocytic leukemia. Leukemia. 1999;13(9):1331–4.
Tarasov V, Jung P, Verdoodt B, et al. Differential regulation of microRNAs by p53 revealed by massively parallel sequencing: miR-34a is a p53 target that induces apoptosis and G1-arrest. Cell Cycle. 2007;6(13):1586–93.
Tsujimoto Y, Cossman J, Jaffe E, et al. Involvement of the bcl-2 gene in human follicular lymphoma. Science. 1985;228(4706):1440–3.
Tsujimoto Y, Finger LR, Yunis J, et al. Cloning of the chromosome breakpoint of neoplastic B cells with the t(14;18) chromosome translocation. Science. 1984;226(4678):1097–9.
Van Antwerp DJ, Martin SJ, Kafri T, et al. Suppression of TNF-alpha-induced apoptosis by NF-kappaB. Science. 1996;274(5288):787–9.
van der Vlag J, Otte AP. Transcriptional repression mediated by the human polycomb-group protein EED involves histone deacetylation. Nat Genet. 1999;23(4):474–8.
Virgilio L, Narducci MG, Isobe M, et al. Identification of the TCL1 gene involved in T-cell malignancies. Proc Natl Acad Sci U S A. 1994;91(26):12530–4.
Visone R, Rassenti LZ, Veronese A, et al. Karyotype-specific microRNA signature in chronic lymphocytic leukemia. Blood. 2009;114(18):3872–9.
Visone R, Veronese A, Rassenti LZ, et al. miR-181b is a biomarker of disease progression in chronic lymphocytic leukemia. Blood. 2011;118(11):3072–9.
Volinia S, Calin GA, Liu CG, et al. A microRNA expression signature of human solid tumors defines cancer gene targets. Proc Natl Acad Sci U S A. 2006;103(7):2257–61.
Wang CY, Mayo MW, Baldwin Jr AS. TNF- and cancer therapy-induced apoptosis: potentiation by inhibition of NF-kappaB. Science. 1996;274(5288):784–7.
Wolf S, Mertens D, Schaffner C, et al. B-cell neoplasia associated gene with multiple splicing (BCMS): the candidate B-CLL gene on 13q14 comprises more than 560 kb covering all critical regions. Hum Mol Genet. 2001;10(12):1275–85.
Zanesi N, Pekarsky Y, Trapasso F, et al. MicroRNAs in mouse models of lymphoid malignancies. J Nucleic Acids Investig. 2010;1(1):36–40.
Zapata JM, Krajewska M, Morse 3rd HC, et al. TNF receptor-associated factor (TRAF) domain and Bcl-2 cooperate to induce small B cell lymphoma/chronic lymphocytic leukemia in transgenic mice. Proc Natl Acad Sci U S A. 2004;101(47):16600–5.
Zenz T, Mertens D, Dohner H, et al. Molecular diagnostics in chronic lymphocytic leukemia—pathogenetic and clinical implications. Leuk Lymphoma. 2008;49(5):864–73.
Zenz T, Mohr J, Eldering E, et al. miR-34a as part of the resistance network in chronic lymphocytic leukemia. Blood. 2009;113(16):3801–8.
Acknowledgements
This work was supported by ACS Research Scholar Award, Swan Family Award, and CLL Global Foundation (to Y. Pekarsky).
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2013 Springer Science+Business Media New York
About this chapter
Cite this chapter
Balatti, V., Pekarky, Y., Rizzotto, L., Croce, C.M. (2013). miR Deregulation in CLL. In: Malek, S. (eds) Advances in Chronic Lymphocytic Leukemia. Advances in Experimental Medicine and Biology, vol 792. Springer, New York, NY. https://doi.org/10.1007/978-1-4614-8051-8_14
Download citation
DOI: https://doi.org/10.1007/978-1-4614-8051-8_14
Published:
Publisher Name: Springer, New York, NY
Print ISBN: 978-1-4614-8050-1
Online ISBN: 978-1-4614-8051-8
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)