
Abstract
Steroid receptor coactivators (SRCs), including SRC-1, SRC-2, and SRC-3, mediate transcriptional activities of nuclear receptors and other transcription factors. SRCs’ activities and functions are regulated by multiple signaling pathways, including those of hormones, growth factors, and cytokines, and are determined by post-translational modifications, including phosphorylation, ubiquitination, sumoylation, acetylation, and methylation. SRCs integrate signals from a variety of pathways that regulate multiple cellular processes such as metabolism, reproduction, and growth. For the growth response, they regulate proliferation, survival, migration, and invasion, and promote tumor development and metastasis. SRCs are highly disregulated in many types of cancers at multiple levels including gene amplification, mutation, and mRNA/protein overexpression. Alterations of SRCs are frequently associated with advanced tumor progression and drug resistance. As such, SRCs are important prognostic cancer biomarkers and could serve as therapeutic targets for cancer therapy.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others


Keywords
- Steroid receptor coactivators (SRCs)
- Nuclear receptor
- Gene transcription
- Posttranslational modifications
- Signaling pathways
- Tumorigenesis
- Cancer metastasis
- Drug targeting
1 Introduction
The p160 steroid receptor coactivator (SRC) family, consisting of SRC-1, SRC-2, and SRC-3, were originally identified as transcriptional coactivators of nuclear hormone receptors (NRs) for estrogen, progesterone, and androgen. SRC-1, also known as nuclear receptor coactivator 1(NCOA1), was cloned and characterized as the first NR coactivator in 1995 [1]. SRC-2, also known as NCOA2, GRIP1 (glucocorticoid receptor interacting protein 1), and TIF2 (transcriptional intermediary factor 2), was identified soon after the cloning of SRC-1 [2, 3]. SRC-3 was then identified by several laboratories nearly in the same year of 1997, and was provided with different names [4–7]: AIB1 (amplified in breast cancer 1), p/CIP (p300/CBP interacting protein), RAC3 (RAR-associated coactivator 3), ACTR (activator of thyroid and retinoic acid receptor), and TRAM1 (thyroid receptor activator molecule 1). Since the discovery of the first coactivator (SRC-1) in 1995, a substantial number of studies have been conducted to elucidate the molecular actions of SRCs in normal physiology and pathology. In this chapter, we will focus on the cancer-related functions of SRCs and the underlying molecular mechanisms, thereby highlighting the molecular structures and functional interacting partners of SRCs, the regulation of SRCs’ activities by posttranslational modifications (PTMs), and the integration of multiple oncogenic signaling pathways by SRCs that promote tumor development and progression.
2 Structures and Transcriptional Interacting Partners of SRCs
SRC proteins share a common structure that contains five functional domains/regions (Fig. 1.1): the N-terminal basic helix-loop-helix-Per/ARNT/Sim (bHLH/PAS) domain, the serine/threonine rich (S/T) domain, the nuclear receptor interacting domain (RID), the CBP(cAMP-response element binding protein-binding protein) interacting domain (CID) or activation domain 1 (AD1), and the activation domain 2 (AD2) or the histone acetyltransferase (HAT) domain at the C-terminus [10, 11]. Each domain has different interacting partners that confer various functions (Fig. 1.1). The bHLH/PAS domain harbors nuclear localization signals (NLS) and is the most conserved region. This domain is also termed activation domain 3 (AD3) as it is responsible for the interaction of SRCs with multiple co-coactivators and non-NR transcriptional factors. CoCoA was shown to interact with SRC-2 through the bHLH/PAS domain and work cooperatively with p300/CBP co-coactivators to regulate NR-mediated gene transcription [12]. hBrm-associated factor 57 (BAF57), a core component of SNI/SWF chromatin remodeling complex, binds to the bHLH/PAS domain of SRCs and bridges the SNI/SWF chromatin remodeling complex to ER/SRCs transcription complex to promote estrogen-responsive gene transcription [13]. Melanoma antigen gene protein-A11 (MAGE-11) interacts with both TIF2 and AR and potentiates AR transcriptional activity probably through stabilizing the AR-TIF2 transcription complex on the target gene promoter [14]. The bHLH/PAS domain also mediates the interaction of SRCs with several non-NR transcription factors such as Stat3 [15] and p53 [16] that are important factors in cancer.

Structural domains and transcriptional interacting partners of SRCs. SRC proteins contain five functional domains: the N-terminal bHLH/PAS domain, the serine/threonine rich (S/T) domain, the nuclear receptor interacting domain (RID), the p300/CBP interacting domain (CID) or activation domain 1 (AD1), and the C-terminal activation domain 2 (AD2). SRC-1 and SRC-3 harbor a histone acetyltransferase domain (HAT) in the C-terminus. A representative list of transcriptional interacting partners within each domain of SRCs are indicated above (for interacting transcriptional factors) or below (for interacting coactivators) the structure. Interacting proteins are referenced from [8, 9] and as mentioned in the text
The S/T-rich domains of SRCs are frequently targeted by protein kinases and phosphatases, which regulate SRC protein stability and activity [17]. The S/T-rich domain of SRC-3 mediates its interaction with E2F1, an essential transcription factor in cell cycle control [18]. SRCs bind to NRs through the RID domain that harbors three “LXXLL” NR-binding motifs where “L” represents leucine residue and “X” denotes any amino acid [19, 20]. The interactions of SRCs with NRs are either hormone-dependent or hormone-independent based upon the NRs that SRCs are bound to and the growth conditions. Besides mediating the interaction with NRs, the RID domain of SRC-3 is important for its interaction with NFкB [21]. Following the binding to NRs, SRCs recruit p300/CBP histone acetyltransferases through the CID domain, which promotes chromatin remodeling and the recruitment of general transcription machinery [5, 22–24]. In the C-terminus of SRCs resides the activation domain 2 (AD2) that recruits CARM1 and PRMT1 methyltransferases [25, 26]. Interestingly, the C-terminus of SRC-1 and SRC-3 also contains a HAT domain [6, 27], but its functional substrates remain to be substantiated. In addition, the C-terminus was shown to mediate the interaction of SRCs with AP-1 transcription factors that play critical roles in cancer cell proliferation and invasion [28].
Of note, SRCs also have been shown to interact with other oncogenic transcription factors such as Rb [29] and HIF1α [30], and to potentiate activities of these transcription factors, although it is unclear which precise domains within SRCs are required for these interactions. Taken together, SRCs interact with a variety of transcriptional factors and coregulators through their five functional domains, suggesting that SRCs are important molecules that integrate diverse cellular processes.
3 Molecular Codes of SRCs: PTMs Targeted by Multiple Signaling Pathways
As coactivators of a variety of transcription factors, the activity and functions of SRCs are regulated by multiple signaling pathways. The molecular regulation of SRCs’ activity and functions are determined by post-translational modifications (PTMs, Figs. 1.2 and 1.3), including phosphorylation, ubiquitination, sumoylation, acetylation, and methylation, all of which coordinately regulate SRCs’ cellular localization, stability, and the interactions with their functional partners.

PTMs of SRC-1 and SRC-2. Some identified serine (S) and threonine (T) phosphorylation sites and sumoylated lysine (K) residues of SRC-1 and SRC-2 are indicated in the schematic structure. In the “( )” are shown certain kinases that target the specific phosphorylation residues. The two conserved sumoylation sites within RID domain of SRCs are shown in bold

PTMs of SRC-3 and SRC-3∆4. (a) Selectedphosphorylation sites of serine (S), threonine (T), or tyrosine (Y), lysine (K) residues with ubiquitination (Ub), sumoylation (SUMO), or acetylation (Ac), and arginine (R) residue with methylation (Me) of SRC-3 are indicated in the schematic structures. In the “( )” are also shown certain modifying enzymes for each specific PTM. Both K723 and K786 are sumoylation sites as well that are conserved in SRCs. (b) Phosphorylation codes of SRC-3∆4 that are targeted by PAK1 for EGF signal transduction to FAK. PAK1 phosphorylates SRC-3∆4 at T56, which mediates the interaction with EGFR, and at S569 and S676, which is important for the interaction with FAK. See the text for details (It should be noted that SRC-3 contains over 50 different PTMs that have been identified by a variety of techniques)
4 Phosphorylations
SRCs are phosphorylated by protein kinases in response to multiple signals including hormones, growth factors, and cytokines. These signals work independently or in concert to regulate SRCs’ activities and functions.
5 Hormone-Induced Phosphorylations of SRCs
Hormones stimulate target gene transcription not only by activating hormone receptors via direct binding, but also by activating protein kinases that subsequently phosphorylate hormone receptors and coregulators including SRCs (Fig. 1.4). Hormones such as estrogen, progesterone, androgen, and glucocorticoid stimulate the activation of multiple kinases such as ERK1/2, Akt, p38, and JNK; rapid activations of these kinases by hormones are referred to as non-genomic signaling in contrast to direct actions of the receptors on the nuclear genome [31]. In response to E2 stimulation, SRC-3 is phosphorylated at multiple residues including T24 in the N-terminus, S505 and S543 in the S/T-rich region, and S857, S860, and S867 in the RID region [32]. E2-induced phosphorylation of SRC-3 occurs acutely (within minutes) and is dependent on ERα [33]. Phosphorylations at these residues promote the interaction of SRC-3 with ERα and CBP, and augment SRC-3’s transcriptional activity. Interestingly, while androgen/AR induces SRC-3 phosphorylations similar to E2/ ERα, progesterone/PR is unable to do so, suggesting there is ligand/receptor/coactivator specificity in hormone-induced SRC phosphorylations. In agreement with this notion, SRC-2 is a primary coactivator for glucocorticoid receptor (GR) and is phosphorylated upon the stimulation of dexamethason eat five residues (S469, S487, S493, S499, and S565) in the S/T-rich region and one residue (S736) in the RID region [34]. Phosphorylations of SRC-2 facilitate GR transcriptional activity. Similarly, progesterone/PR stimulates SRC-1 phosphorylations that are important for SRC-1 transcription activity (Weiwen Long and Bert O’Malley, unpublished data). Collectively, these findings demonstrate that phosphorylations of SRCs serve as an integrating link between non-genomic and genomic actions of hormones.

SRCs-mediated hormone signaling and the cross-talk with growth factor and cytokine signals in regulating NR target gene expression. Upon the binding of hormone (H), nuclear receptors (NRs) dimerize and bind to the hormone responsive element (HRE) of target genes. SRCs coactivate gene transcription by interacting with DNA-bound NRs and then recruiting secondary coactivators including p300/CBP histone acetyltransferases and CARM1/PRMT1 histone methyltranseferases. p300/CBP and CARM1/PRMT1 elicit histone acetylation (Ac) and methylation (Me), respectively, both of which facilitates the assembly of the general transcriptional machinery consisting of TBP, TAFIIs, and Pol II, and the subsequent gene transcription. SRCs also are capable of recruiting SWI/SNF chromatin remodeling complex via BAF57 and further potentiate gene transcription. The transcriptional activities of SRCs and NRs are regulated by multiple signaling pathways including those of cytokines (Cy), growth factors (GF), and non-genomic hormone actions, mainly through protein kinase-mediated phosphorylations (P) that are important for the interactions of SRCs with NRs and other coactivators
6 Phosphorylations of SRCs Induced by Cytokines and Growth Factors
SRCs also are phosphorylated upon stimulation by growth factors and/or cytokines, which facilitates their functions in coactivating NR-mediated gene expression in both ligand-dependent and ligand-independent mechanisms; these stimulations serve as important molecular mechanisms for anti-hormone resistance during cancer therapy. SRC-1 is phosphorylated at T1179 and S1185 by MAPK upon stimulation by interleukin 6 (IL-6)and co-activates AR in a ligand-independent manner in prostate cancer cells [35]. Interestingly IL-4promotes PP2A-directed dephosphorylation of SRC-1 which is important for SRC-1/Stat6-regulated IL-4 target gene expression in Ramos B lymphoma cells [36]. Cytokines such as TNFa stimulate IKK-directed phosphorylation of SRC-3 which potentiates NFкB-mediated gene expression in breast cancer cells [32, 37].
SRC-1, SRC-2, and SRC-3 all are known to be regulated by growth factor signals. Epidermal growth factor (EGF) stimulates ERK2-directed phosphorylations of SRC-1 that are important for the interaction of SRC-1 with CBP in PR-dependent transactivation [38]. T1426 in AD2 of SRC-1 is targeted by Cdk1 and Cdk2, and this phosphorylation is important for PR/SRC-1-mediated cell cycle control [39]. SRC-2 is phosphorylated at S736 by EGF stimulation, and S736-phosphorylated SRC-2 promotes AR-dependent but ligand-independent transactivation, suggesting a potential role of SRC-2-mediated cross-talk between growth factor signaling and AR signaling in recurrent prostate cancer progression [40]. SRC-3 activity is tightly regulated by growth factor signaling. Both EGF and IGF-1 stimulate SRC-3 phosphorylation at tyrosine 1357 (Y1357) that is directed by AbI kinase in breast cancer and lung cancer cells [41]. Phosphorylation of Y1357 is critical for SRC-3 oncogenic activity in co-activating ERα and NFкB and promoting anchorage-independent cancer cell growth. Heregulin 1β, an EGF-like growth factor, stimulates SRC-3 phosphorylation through ERBB2 oncogenic kinase [42] which is implicated in the breast cancer tamoxifen resistance. cAMP/PKA signaling also induces SRC-1 and SRC-2 phosphorylation and potentiates NR-mediated transactivation in a ligand-independent manner [43, 44].
7 Phosphorylation Codes for SRC-3∆4 Acting as an EGF Signaling Adaptor
SRC-3∆4 is a splicing variant of SRC-3 with the deletion of exon 4 (Fig. 1.3b) [45, 46]. In comparison with full-length SRC-3 protein, SRC-3∆4 lacks an N-terminal bHLH/PAS region that harbors the NLS. Consequently, SRC-3∆4 primarily localizes in the cell cytosol due to its absence of an NLS. Upon EGF stimulation, PAK1 phosphorylates SRC-3Δ4 on T56 at the N-terminus and S659 and S676 within the RID region. Phosphorylations of SRC-3Δ4 promote its localization to the plasma membrane region where it interacts with EGFR through the N-terminus, which is mediated by T56 phosphorylation, and with FAK through the RID region, which is mediated by phosphorylations of S659 and S676(Fig. 1.3b). SRC-3Δ4 mediates the interaction between EGFR and FAK, thereby promoting EGF-induced c-Src activation and FAK phosphorylation on Y925, which in turn drives cancer cell migration and metastasis.
8 Ubiquitination and Its Regulation by Phosphorylation
Ubiquitination plays an essential role in regulating the stability and functions of SRC proteins. Ubiquitination of SRCs is frequently dependent on phosphorylations of specific residues that mediate the interaction of SRCs with the ubiquitin E3 ligases. The stability and activity of SRC-3 have been shown to be regulated by several protein kinase signals that are coupled to different E3 ligase complexes. GSK3β phosphorylates SRC-3 at S505 in the S/T-rich region, which is required for the binding of Fbw7α, a component of the SCF (Skp, Cullin, F-box protein containing) E3 ligase complex. SRC-3 is then ubiquitinated by SCFFbw7α at lysine 723 (K723) and K786 that are within the receptor-interacting domain. Interestingly, mono-ubiquitinations at K723 and K786 enhance the interaction of SRC-3 with ERα, ERα’s phosphorylation at S118, and expression of the target genes in response to E2 stimulation [47]. Subsequent poly-ubiquitinations then lead to the degradation of SRC-3 and termination of SRC-3/ERα-regulated gene transcription. Ubiquitination-coupled activation of SRC-3 is also manifested by retinoic acid (RA) signaling, which involves the phosphorylation of SRC-3 at S860 by RA-activated p38, the subsequent phospho-S860-mediated recruitment of Cullin 3-based E3 ligase, ubiquitination of SRC-3, and finally activation of SRC-3/RAR-regulated gene transcription [48, 49]. In addition, SRC-3 protein stability and activity are regulated by a phospho-dependent degron that is located in the N-terminal bHLH/PAS region [50, 51]. Ser102 in the degron is phosphorylated by CKI (casein kinase I), which is required for the recruitment of a speckle-type POZ protein (SPOP)-based E3 ligase complex and the subsequent ubiquitination and turnover of SRC-3. Interestingly, phosphatases PP2A and PP1 target phosphorylated Ser102 and inhibit SRC-3 protein ubiquitination [50].
In addition to ubiquitin-dependent proteasomal degradation pathways, SRC-3 protein turnover is regulated by an ubiquitin-independent mechanism [52]. Importantly, PKCζ phosphorylates multiple residues in an acidic fragment that is important for the interaction of SRC-3 with the C8 subunit of the 20S proteasome, and enhances SRC-3 protein stability by inhibiting both ubiquitin-dependent and ubiquitin-independent proteolytic pathways [53].
In contrast with the extensive study on SRC-3, much less is known about the regulation of ubiquitination and stability of SRC-1 and SRC-2. However, a single nucleotide polymorphism (SNP) P1272S was shown to increase SRC-1 protein stability by disrupting a potential GSK3β-directed phospho-dependent degradation code in the AD2 region of SRC-1 [54]. Although no specific phosphorylation and ubiquitination sites were revealed, an ubiquitination-coupled activation mechanism for SRC-2 was shown to be regulated by cAMP/PKA signaling [44].
9 Sumoylation
Sumoylation is a type of PTM that involves an addition of small ubiquitin-like modifier (SUMO) to the lysine residues of proteins [55]. In spite of its structural and enzymatic processing similarities to ubiquitination, sumoylation often alters a protein’s binding affinity with the associating partners or its subcellular localization rather than degradation. All of the SRCs were shown to be sumoylated on two conserved lysine residues of the nuclear receptor-interacting domains (Figs. 1.2 and 1.3); sumoylations alter the interaction of SRCs with NRs and their transcriptional activities [56]. However, while sumoylations enhance the interaction of SRC-1 and SRC-2 with PR and AR respectively [57, 58], and their retention and transactivity in the nucleus, SRC-3 transactivity is attenuated by sumoylations at K723 and K786 [47]. As aforementioned, K723 and K786 of SRC-3 are ubiquitination sites as well, and the ubiquitinations on these sites are important for the interaction of SRC-3 with ERα and their transactivity. As such, the attenuation of SRC-3 transactivity by sumoylations at K723 and K786 is likely due to the competitive blocking of ubiquitinations on these two sites. Although the regulation of SRCs’ sumoylations by phosphorylations have not been reported to date, both SRC-2 and SRC-3 harbor a phosphorylation-dependent sumoylation motif (PDSF) ψKxExxSP (where ψ is a large hydrophobic residue)that has been characterized in heat shock factor 1 (HSF-1) and MEF2A transcription factors [59, 60]. S736 within the PDSF of SRC-2 is a phosphorylation site targeted by EGF/ERK1/2 signaling [40], and K731 is a sumoylation site that enhances SRC-2 transactivity [58]. It would be of interest to determine whether this is a bona fide PDSF that plays a role in SRC-2 activity.
10 Acetylation and Methylation
SRCs act as bridging factors to recruit histone acetyltransferases such as p300/CBP and histone methyltransferases such as CARM1/PRMT1 to DNA-bound NRs to remodel chromatin and regulate gene transcription (Fig. 1.4). Interestingly, these histone modifying enzymes target not only histones but also SRCs and NRs [61–64]. SRCs were shown to be acetylated by p300 [61] and methylated by CARM1 upon E2 stimulation [63, 64]. While histone acetylation and methylation facilitate the assembly of the transcription machinery and subsequent gene transcription, acetylation or methylation of SRC-3 leads to the dissociation of NRs/cofactors complex and the termination of gene transcription.
11 Molecular Actions of SRCs in Cancer Cells In Vitro and in Mouse Tumor Models In Vivo: SRCs as Integrators of Multiple Signaling Pathways
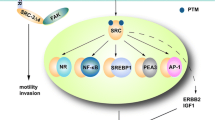
SRCs, in particular SRC-3, function as important mediators and integrators of a variety of oncogenic signaling (e.g., hormones, growth factors, and cytokines) pathways to regulate virtually every aspect of cellular processes: proliferation, survival, migration, and invasion (Figs. 1.4 and 1.5). Hormone signaling acts independently or synergistically with growth factor or cytokine signaling to regulate these cellular processes in which SRCs play important, integrating roles (Fig. 1.4). The actions of SRCs are exquisitely regulated by PTMs (mainly phosphorylations) that are stimulated by the oncogenic signals. Increased growth factor and/or cytokine signaling hijack SRCs to drive the progression of cancer cells from hormone-dependent to hormone-independent growth and elicit anti-hormone resistance, for example, anti-estrogen resistance in breast cancer. Based on the expression status of hormone receptors (mainly ER and PR) and growth factor tyrosine kinase receptors (mainly ERBB2 and EGFR) and the origin of cancer cells, breast cancers can be classified as 4 subtypes [65, 66]: luminal A (ER+ PR+ERBB2-), luminal B (ER+ PR+ERBB2+), ERBB2-enriched (ER−PR−ERBB2++), and basal-like (also known as triple-negative, ER−PR−ERBB2−). Accompanied by enhanced growth factor signaling, luminal A subtype can progress to luminal B and further to ERBB2-enriched subtype, which leads to advanced cancer phenotype and increased anti-estrogen resistance. Basal-like breast cancer cells often have upregulated EGFR expression and high aggressiveness. As discussed below, SRC-1 and SRC-3 play critical functions in all four subtypes of breast cancer, whereas the roles of SRC-2 are minor.

SRC-3 integrates multiple signaling pathways to promote tumorigenesis and metastasis. Besides its regulation of hormone signaling as illustrated in Fig. 1.4, SRC-3 integrates multiple growth factor and cytokine signaling pathways to regulate a variety of cancer cell processes. Only a subset of these pathways are illustrated in Fig 1.5. In response to TNFα/IL-1 signals, SRC-3 is phosphorylated by IKKs and coactivates NFкB-mediated Bcl-2 expression for cancer cell survival. SRC-3 upregulates the expression of multiple components of IGF1-IGFR-PI3K/Akt pathway that is important for both cancer cell proliferation and survival. Upon EGF or IGF-1 stimulation, SRC-3 is targeted by protein kinases including c-AB1, and then coactivates E2F1-mediated expression of cell cycle genes including cyclins E and A. SRC-3 itself is a target gene of E2F-1 (indicated by a dashed arrow), and upregulation of SRC-3 by E2F1 might boost other signaling pathways regulated by SRC-3, for example, the IGF1/Akt pathway. SRC-3 is also important for the activation (phosphorylations) of ERBB2 and EGFR and the downstream kinases such as JNK, and plays a role in tumor angiogenesis, likely by coactivating HIF1. In addition, SRC-3∆4 acts as an EGF signaling adaptor by bridging EGFR to FAK, and promotes EGF-induced FAK phosphorylations and cancer cell migration. Furthermore, SRC-3 is targeted by ERK3 kinase and coactivates PEA-3/AP-1-mediated MMP gene expression for promoting cell invasion
12 SRCs with Hormones/NRs-Mediated Signaling
12.1 Estrogen/ER Signaling in Breast Cancer
Both SRC-1 and SRC-3 act as coactivators of ERα to mediate estradiol signaling in promoting breast cancer cell proliferation and survival. SRC-1 is important for estradiol-induced cell proliferation ofMCF-7, a breast cancer cell line of luminal A subtype [67–69]. Depletion of SRC-1 differentially affected E2-inducible genes: with a significant decrease in the expression of pS2 and stromal cell-derived factor 1 (SDF-1) but little effect on c-Myc [69]. Both MCF-7 and T47D (another breast cancer cell line of luminal A subtype) overexpress SRC-3. Depletion of SRC-3in these cells diminishes the expression of estrogen/ER target genes including cyclin D1, c-Myc, and Bcl-2, and inhibits cell proliferation but increases apoptosis [70, 71]. Consequently, depletion of SRC-3 in MCF-7 cells inhibits estrogen-induced colony formation in soft agar and xenograft tumor growth in nude mice [72]. E2 exerts non-genomic signaling by activating multiple kinases including ERK1/2 and IKKα. E2-induced phosphorylations of SRC-3 via ERK1/2 and IKKα are critical for SRC-3’s activity in promoting the expression of E2 target genes such as cyclin D1 and c-Myc [32, 73]. The functional relationship between SRC-3 and ERα in tumorigenesis was revealed in a mouse mammary tumor virus (MMTV)-SRC-3 transgenic mouse model [74]. Transgenic overexpression of SRC-3 induced tumors primarily in mammary glands but also in other organs including uterus and lung, suggesting SRC-3 is a bona fide oncogene. Ovariectomy in MMTV-SRC-3 mice greatly decreased mammary tumor formation, and genetic deletion of ERα in MMTV-SRC-3 mice by crossing with ERα-null mice completely abolished mammary tumor formation [75]. Taken together, these findings suggest that SRC-3 and possibly SRC-1 are critical for E2/ER signaling in promoting breast cancer progression.
12.2 Androgen/AR Signaling in Prostate Cancer
While estrogen/ER signaling is critical for the progression of breast cancer, androgen/AR signaling plays an essential role in hormone responsive prostate cancer. All of three SRC members have been shown to coactivate AR to meditate androgen signaling in prostate cancer cells. Depletion of either SRC-1 [76], SRC-2 [77, 78], or SRC-3 [79, 80] significantly decreases androgen-dependent AR transcriptional activity and LNCaP prostate cancer cell growth in culture and in xenograft tumor mouse models. On the contrary, overexpression of SRC-3 or SRC-2 in LNCap greatly enhances the responsiveness of AR to androgen or to ligand-independent stimulation and promotes cell growth.
13 Interplay of SRCs with ERBB2 Signaling: Anti-estrogen Resistance in Breast Cancer
ERBB2, a member of the EGFR family, is an oncogenic protein that is frequently overexpressed in advanced breast cancer. ERBB2 overexpression promotes tumor progression, and is highly associated with the progression of ER-positive breast cancer cells from estrogen-dependent to estrogen-independent growth; the overexpression is concomitant with the gain of resistance to anti-estrogen drugs [81]. Both SRC-1 and SRC-3, in concert with ERBB2, play a critical role during these processes. Genetic loss of SRC-3 completely suppressed MMTV-ERBB2 induced mouse mammary tumor formation in association with a remarkable decrease in phosphorylations of ERBB2 and the downstream kinases JNK and Akt [82]. Interestingly, tumor angiogenesis also was curtailed significantly due to the loss of SRC-3. SRC-3 was confirmed as a pro-angiogenic factor recently [83] and acts as a coactivator of HIF1α [30]. While loss of SRC-1 did not significantly decrease the average tumor number formed in MMTV-ERBB2 transgenic mice, it increased tumor latency and greatly inhibited tumor metastasis to the lung [84]. Under cell culture conditions, stable exogenous expression of ERBB2 in MCF-7 elicits an estrogen-independent cell growth and resistance to tamoxifen, both of which were significantly inhibited by depletion of SRC-3 [42]. A similar synergistic role of ERBB2 with SRC-3 or SRC-1 was shown in BT474, a luminal-B subtype of breast cancer cell line with overexpression of ERBB2 [85, 86]. ERBB2 overexpression activates the downstream kinases ERK1/2, JNK, and Akt that phosphorylate ERα and SRCs, which leads to activation of ERα under low concentration or even in the absence of estrogen [42, 87]. In addition to ERα, SRC-3 and SRC-1 interact with and coactivate other transcription factors such as Ets and PEA3 to promote cancer cell growth and invasion in response to enhanced ERBB2 signaling [88, 89]. Conversely, SRC-3 positively regulates ERBB2 expression by competing with PAX-2 (a repressor of ERBB2 gene expression) for the binding to the ERα-bound site of the ERBB2 gene [90]. These findings suggest a positive feedback between ERBB2 and SRC-3 that regulates breast cancer cell proliferation and anti-estrogen resistance.
14 Interplay Between SRCs and EGFR Signaling: From the Membrane to the Nucleus
EGF/EGFR signaling is implicated in multiple cancers including those of breast, prostate, and lung. EGF signaling stimulates SRCs’ phosphorylation and activation; activated SRCs then work with transcriptional factors including E2F-1, ETS, and AP-1 to regulate cell proliferation, migration, and invasion (Fig. 1.5).
In breast cancer, increased EGFR signaling is frequently associated with the invasive and triple-negative phenotype. In a triple negative breast cancer cell line MDA-MB231, SRC-3 and its N-terminus-deleted isoform SRC-3∆4 elegantly regulate distinct aspects of the EGF signaling at different cellular locations (Fig. 1.5). SRC-3∆4 acts as a signaling adaptor bridging EGFR to FAK at the plasma membrane to mediate activation of FAK and promote cell migration upon the EGF signal [46]. SRC-3 is phosphorylated by Ab1 kinase atY1357 in response to EGF stimulation, and this phosphorylation is important for SRC-3’s function in coactivating AP-1 and E2F1 in the nucleus and promoting cell growth [41]. Intriguingly, EGFR tyrosine phosphorylation and activity is regulated by SRC-3. Depletion of SRC-3 greatly decreased EGF-induced EGFR tyrosine phosphorylation, which led to decreased JNK kinase activity and growth inhibition of MDAMB231 cells [91]. While the detailed molecular mechanism is unclear, downregulation of tyrosine phosphatases by SRC-3 partly contributes to an increase of EGFR phosphorylation.
In prostate cancer, enhanced EGFR signaling is highly associated with castration-resistant (androgen-independent) cancer progression. On one hand, EGF signaling stimulates AR tyrosine phosphorylation (Y534) via c-Src kinase [92], and AR tyrosine phosphorylation promotes its nuclear localization and transactivity. On the other hand, EGF signaling targets SRCs via MAPK-directed phosphorylations. For example, SRC-2 is phosphorylated at S736 by EGF stimulation, and S736-phosphorylated SRC-2 promotes AR-dependent but androgen-independent transactivation [40]. Together, EGF signaling stimulates AR/SRCs’ transactivity and promotes prostate cancer cell growth in culture and tumor growth in castrated mice [40, 92].
15 Regulation of IGF-1/Akt Signaling by SRC-3 and SRC-1
IGF-1/Akt signaling is another molecular pathway that is regulated by SRCs and is critical for SRCs’ oncogenic functions (Fig. 1.5). The IGF-1/Akt signaling pathway initiates with the binding of IGF-1 to its receptor (IGF-1R) on the cell membrane and the subsequent activation of IGF-1R by auto-tyrosine phosphorylation, followed by the recruitment of insulin receptor substrate (IRS) proteins and IRS-mediated activation of the phosphatidylinositol 3-kinase (PI3K)/Akt pathway [93]. SRC-3 regulates the expression levels of multiple components of the IGF-1/Akt pathway, includingIGF-1/IGF-2, IGF-1R, IRS-1/IRS-2, and Akt in breast cancer cells [94] and prostate cancer cells [95], thereby regulating the activities/phosphorylations of IGF-1R and Akt and promoting cancer cell proliferation and anchorage-independent cell growth. The regulation of IGF-1/Akt signaling by SRC-3 was corroborated in vivo. Increased IGF-1 expression and activation of IGF-1R, Akt, and mTOR was seen in MMTV-AIB1 (SRC-3) transgenic mice, which led to spontaneous mammary tumor formation [74]. In contrast, genetic depletion of SRC-3 in v-Ha-Ras transgenic mice caused a remarkable decrease in tumor initiation and metastasis to the lung, partly due to decreased expression of IRS-1 and IRS-2 and the attenuated Akt activity [96]. Loss of SRC-1 did not affect primary mammary tumor growth, but greatly decreased tumor metastasis to the lung in MMTV-polyoma middle T antigen (PyMT) transgenic mice by downregulating ERBB2 expression and Akt phosphorylation [97].
16 Interplay of SRCs with Cytokine Signaling in Promoting Cancer Cell Aggressiveness
IL-6 signaling promotes castration-resistant prostate cancer progression by activating AR in an androgen-independent mechanism [98–100]. Besides the direct effect on AR, IL-6 signaling modulates SRCs’ activity as well. SRC-1 phosphorylation by MAPK in response to IL-6 is important for ligand-independent activation of AR [35]. In addition, upregulation of SRC-2 in LNCaP cells upon long-term treatment with IL-6 is associated with acquired resistance to bicalutamide, an anti-androgen drug [101]. Conversely, cytokine production can be regulated by SRCs. For example, SRC-1 upregulates colony-stimulating factor-1 (CSF-1) expression and promotes the recruitment of macrophages to the tumor site, which contributes to tumor metastasis [97].
SRC-3 is phosphorylated upon the stimulation of cytokines such as TNFα and IL-1β [32, 37]. Phosphorylations of SRC-3 stimulate the interactions of SRC-3 with ERα and NFкB, which are important for TNFα-induced cyclin D1 expression and cell proliferation [102]. Interestingly, SRC-3 was shown to interact with translational repressors TIA1 (T cell restricted antigen 1) and TIAR (TIA1 related homologue) and regulate the translation of TNFα and interlukin 1 mRNAs [103], for which the implication in cancer is unclear at present.
A recent study revealed an intriguing interplay between a cleaved isoform of SRC-1 and TNFα signaling during endometriosis [104] which shed new light on the roles of SRCs in inflammation-associated diseases such as cancers. Endometriosis is an inflammation-driven disease that is initiated by the migration of endometrial cells to distal sites. While TNFα is well-known as a critical driving factor for endometriosis, it is less clear how the intrinsic pro-apoptotic activity of TNFα is silenced during this process. It is shown in this interesting study that TNFα-induced MMP9 cleaves SRC-1 at Pro-790, and the c-terminus of cleaved SRC-1 promotes endometriosis by blocking caspase8-mediated apoptosis and by stimulating epithelial-to-mesenchymal transition (EMT) for increased invasiveness. Given the positive associations of both SRC-1 and inflammation with advanced and metastatic tumor stages, cytokine signaling and SRC-1 conceivably could synergistically promote cancer progression and metastasis following similar mechanisms as shown in endometriosis. Indeed, SRC-1 was shown to positively regulate TWIST, a master regulator of EMT, thereby promoting tumor cell migration/invasion and metastasis [89]. It would be interesting to determine whether SRC-1 undergoes proteolytic cleavage to produce the cleaved isoform during cancer progression and whether this cleaved form of SRC-1 is responsible for the upregulation of TWIST and EMT-associated tumor cell migration and invasion.
17 Phospho-dependent Regulation of SRC-3 by an Atypic MAPK for Cancer Cell Invasion
SRC-3 promotes cancer cell invasion by coactivating PEA-3- and AP-1-regulated matrix metalloproteinase (MMP) expression [105–107], but the invasive signals to SRC-3 and the molecular regulation of SRC-3 proinvasive activity were not elucidated until a recent study that revealed a phospho-dependent regulation of SRC-3 proinvasive activity by an atypical MAPK ERK3 (Fig. 1.5) [108]. ERK3 was identified as an interacting partner of SRC-3 by immunoprecipitation-mass spectrometry (IP-MS) analyses. ERK3 phosphorylates SRC-3 at serine 857 (S857), and this ERK3-mediated phosphorylation at S857 is essential for SRC-3’s interaction with the ETS transcription factor PEA3, promoting upregulation of matrix metalloproteinase (MMP) gene expression and proinvasive activity in lung cancer cells. ERK3/SRC-3 signaling drives cancer cells to invade and form tumors in the lung. As such, this study not only revealed a molecular mechanism for regulating SRC-3 proinvasive activity, but also identified a novel oncogenic function for ERK3 in promoting lung cancer invasiveness.
18 Alterations of SRCs and the Clinical Implications in Cancers
As transcriptional coactivators integrating multiple signaling pathways to regulate cancer cell proliferation, survival, migration, and invasion, SRCs are highly dysregulated in a variety of cancers including breast cancer, prostate cancer, endometrial cancer, ovarian cancer, lung cancer, colorectal cancer, liver cancer, and pancreatic cancer. Alterations of SRCs, including primarily gene amplification, mRNA or protein overexpression, are implicated in cancer progression and metastasis and tumor resistance to therapeutic interventions.
19 Gene Amplification
Gene amplification is one of the fundamental features for defining an oncogene. SRC genes, in particular SRC-3 (also known as AIB1-Amplified in breast cancer gene 1), are amplified in multiple human cancers. Gain of copy numbers of SRC-1 and SRC-2 genes were reported in a recent study with a cohort of 218 prostate tumors consisting of 181 primaries and 37 metastases [109]. Of particular note, SRC-2 gene amplification was detected in 8 % of primary tumors and 37 % of metastases. Gain of SRC-2 expression is associated with increased rates of prostate tumor recurrence. SRC-3 gene amplification has been shown in multiple cancers including breast cancer (with 5–10 % frequency [4, 110]), ovarian cancer (with 7–25 % frequency [110, 111]), colorectal cancer (with 10–32 % frequency [112, 113], lung cancer (with 8.2–27 % frequency [114, 115]), and hepatocellular cancer (40 % frequency, [116]). SRC-3 gene amplification contributes to upregulation of SRC-3 mRNA and protein in cancers, and is positively correlated with advanced tumor stages.
20 Mutations
In contrast to high frequencies of gene amplification and mRNA/protein overexpression, gene mutation of SRCs is rarely detected in cancers. Even though a few point mutations of SRC-1 [117] and SRC-2 [109] were identified in tumors, the frequency is very low (~1 %) and the pathological association is undetermined. Interestingly, a fusion between MOZ (monocytic leukemia zinc finger) gene and TIF2 (SRC-2) gene has been repeatedly detected in acute myeloid leukemia [118–120]. MOZ-TIF2 fusion protein retains the PHD zinc finger domain and the MYST domain of MOZ and the CBP-interacting domain (CID) and activation domain 2 (AD2) of TIF2. The recruitment of p300/CBP via the CID of MOZ-TIF2 is essential for leukemogenesis of the fusion gene [120].
21 Upregulation of mRNA and Proteins
Expression of SRCs, in particular SRC-3 and SRC-1, are frequently upregulated in a variety of cancers. SRC-3 is the second most overexpressed oncogene among all human cancers, second only to c-myc. Upregulation of SRCs is often co-current with elevated protein kinase signaling, which indicates high tumor grade, increased tumor invasiveness, and tumor resistance to therapeutic treatments. A substantial number of studies have investigated the expression and clinical implication of SRCs in a variety of cancers, with focus mostly on breast cancer and prostate cancer.
22 Breast Cancer
Breast cancer progression relies on two major signaling pathways: hormone (e.g. E2/ERα) signaling and epidermal growth factor signaling pathways [81]. Antihormonal drugs (e.g. tamoxifen, an estradiol antagonist, and letrozole, an anti-aromatase inhibitor) have been commonly used for treating ER/PR-positive breast cancers. However, antihormone resistance (either naïve or acquired resistance after therapy) and associated cancer recurrence have been major obstacles for curing breast cancer patients. The acquisition of resistance to antihormonal drugs is often associated with a transition of hormone signaling-dependent to growth factor signaling-dependent tumor growth [121, 122]. SRC-1 and SRC-3 are overexpressed at high frequency in breast cancer, and their overexpression is implicated both in hormone-dependent and in hormone-independent breast cancer progression and metastasis.
SRC-1 expression has been shown to be significantly increased in around 19–30 % breast tumors [123–125]. In line with its primary role in promoting cancer cell migration and invasion in vitro and tumor metastasis in animal models, upregulation of SRC-1 is highly correlated with lymph node metastasis and poor disease-free survival (DFS) of breast cancer patients [88, 126]. Another important role for SRC-1 is the regulation of cancer cell sensitivity to anti-hormone drugs. Indeed, SRC-1 expression has been demonstrated as a predictor of anti-estrogen resistance and/or tumor recurrence following therapy [125, 126].
The implication of SRC-3 expression in breast cancer progression has been extensively studied. From various separate studies [4, 72, 124, 127, 128], overexpression of SRC-3 mRNA and protein has been shown in the range of from 13 % to 74 % of breast tumors, with an average of around 50 % overexpression rate. In agreement with the broad roles of SRC-3 in regulating cancer cell proliferation, survival, migration, and invasion in cultured cells and in animal models, overexpression of SRC-3 is positively associated with advanced tumor grade, increased tumor invasiveness and metastasis, and worse DFS in both ERα-positive and ERα-negative breast tumors [4, 87, 124, 127]. SRC-3 overexpression is frequently associated with enhanced protein kinase signaling. Tyrosine kinase receptor ERBB2 is frequently amplified (gene copy) and overexpressed (mRNA and protein) in cancers [129], with the highest frequency in breast cancer (~25 %). Simultaneous overexpression of SRC-3 and ERBB2 are reported in several studies [87, 128, 130, 131], and their co-overexpression indicates increased tumor resistance to tamoxifen treatment and increased tumor recurrence. Although the regulation of the IGF1R/Akt signaling pathway by SRC-3 has been well demonstrated by cell culture studies and mouse mammary gland tumor models, the correlation between these two has not been shown in tumor studies of breast cancer patients. Given the frequent alterations of both of these two factors in breast cancer, it is of high clinical significance to investigate the association of SRC-3 and IGF1R/Akt signaling pathways and their implications in cancer progression in more detail.
In contrast with ample evidence for the critical roles of SRC-1 and SRC-3 in breast cancer progression and metastasis, few studies have provided conclusive data to support a definite role for SRC-2 in breast cancer.
23 Prostate Cancer
Androgen/AR signaling plays a critical role in the initiation and progression of prostate cancer, and has been a major therapeutic target for treating this disease [132, 133]. Androgen ablation therapy (mainly by chemical castration) effectively inhibits tumor growth during the initial treatment. Unfortunately, most tumors relapse and become resistant to androgen ablation therapy. Castration-resistant cancer progression or recurrence is frequently associated with an advanced and metastatic tumor phenotype and has been a major obstacle for curing prostate cancer. AR and its target genes are commonly expressed in and are believed to drive recurrent prostate cancers [134]. In addition to AR gene mutations and AR overexpression, alterations in AR coactivators including SRCs and upregulation of growth factor signaling are two other major molecular mechanisms for castration-resistant tumor progression [135]. While genetic alterations of the SRC-1 gene are rare in prostate tumors, upregulation of SRC-1 has been shown by a few studies [76, 117, 136]. Upregulation of SRC-1 protein is associated with lymph node metastasis [76] and tumor recurrence after androgen deprivation therapy [117, 136]. Like SRC-1, SRC-2 expression is highly increased in recurrent prostate tumors following androgen deprivation therapy [117, 136]. More importantly, a recent study showed that elevated SRC-2 expression, probably due to SRC-2 gene amplification, was detected in both primary and metastatic prostate tumors [109]. Similar findings were reported for SRC-3 in prostate cancer [79, 106, 137, 138]. Upregulation of SRC-3 is positively correlated with increased Akt activity in prostate tumors [79, 138], which affirms a positive regulatory role of SRC-3 in Akt signaling in prostate cancer.
24 Lung Cancer
SRC-3 functions as an oncogene in lung cancer. Transgenic overexpression of SRC-3 in mice causes spontaneous lung tumor formation [74]. SRC-3 gene amplification and protein overproduction were shown in as high as 27 % of non-small cell lung cancers (NSCLCs) in one study [115]; overexpression correlates with poor disease free and overall survival. Interestingly, ERK3, a kinase that phosphorylates SRC-3 and confers SRC-3 pro-invasive activity in lung cancer cells, was shown to be highly upregulated in lung cancer [108]. Overexpression of SRC-3 protein in lung cancer also was reported in two other studies [139, 140]. The implications of SRC-1 and SRC-2 in lung cancer are not known.
In addition to the cancer types discussed above, SRC-3 has been shown to be overexpressed in many other cancers: a 64 % overexpression rate in high grade ovarian tumors [141], a 67 % overexpression rate in hepatocellular carcinomas [116], a 35 % overexpression rate in colorectal carcinomas [112], and a nearly 70 % overexpression frequency in pancreatic tumors [142].
25 Tumor Suppressor Functions of SRC-3 and SRC-2 in Specific Tissue Context
In contrast with the ample evidence for SRC-3′s function as an oncogene in a multitude of cancers, deletion of the SRC-3 gene promotes proliferation of lymphocytes and induces spontaneous malignant B-cell lymphoma upon aging in mice [143]. Similarly, while SRC-2 has been identified as an oncogene in prostate cancer, a recent study revealed a tumor suppressor role for SRC-2 in liver cancer [144]. Although the molecular mechanisms underlying the unexpected tumor-suppressing roles of SRC-2 and SRC-3 are not understood, these findings suggest that SRCs, as transcriptional coactivators, can regulate cell proliferation and survival either positively or negatively, depending upon the cell- and tissue context.
26 SRCs as Prognostic Biomarkers and Therapeutic Drug Targets
Increased expression and activities of SRCs not only are implicated in cancer progression and metastasis, but also are positively associated with drug resistance, including anti-estrogen resistance in breast cancer [87, 123, 125, 145, 146], resistance to EGFR inhibitors in lung cancer [115]), and resistance to chemotherapeutic drugs such as cisplatin and doxorubicin [30, 147, 148]. Hence, novel therapeutic drugs targeting SRCs might be utilized individually or in combination with other therapeutic drugs for treating cancers in different subtypes and at different stages.
27 Targeting SRCs by Intervening NR-SRC Interactions
The binding of SRCs with NRs via SRCs’ ‘LXXLL’ NR interacting motifs is critical for their transcriptional activity. In addition, the flanking amino acids of the ‘LXXLL’ motif are shown to confer an order of specificity on differential NR/SRCs interactions. On the basis of these molecular mechanisms, there have been efforts to develop peptides containing LXXLL motifs or identifying small molecules that can disrupt the interactions between NRs and SRCs. A screening of the phage display library identified peptides that specifically and effectively inhibit ERα or ERβ transactivity [149–151], but it was unclear what specific ER/SRC interaction(s)was affected by these peptides. Soon after this, a peptide specifically blocking the interaction of SRC-2 with TRβ [152] and other peptides preventing the binding of SRC-1 with ERα or ERβ [153, 154] were identified utilizing a similar strategy. In addition, small molecule inhibitors (SMIs) were identified for targeting SRCs’ interaction with ERα [155] and TRβ [156]. Although these identified peptides or SMIs were shown to efficiently inhibit NRs’ transactivity, their efficacy on NR/SRCs-mediated cell functions were not evaluated in animals.
28 Small Molecule Inhibitors Targeting SRCs for Degradation
Since overexpression of SRC proteins, in particular SRC-3, is highly associated with advanced tumor progression and drug resistance, a potentially more effective strategy would be to identify small molecules that directly target SRCs and downregulate SRCs’ protein stability and activities. Based on this idea, a recent study identified a SMI for SRC-1 and SRC-3 by both activity-based and stability-based screening assays [157]. This proof-of-principle drug, gossypol, downregulates the stability and activities of both SRC-1 and SRC-3 via direct binding, but is less selective forSRC-2 and other cofactors. Importantly, gossypol greatly increases the response of cancer cells to inhibitors of growth factor signaling, including MEK inhibitor AZD6244, EGFR inhibitor AG1478, and IGF-1R inhibitor AG1024. This study demonstrates that SRCs are accessible therapeutic targets to SMIs and encourages additional high throughout screenings for identifying drugs targeting SRCs.
SRCs also can serve as diagnostic and prognostic biomarkers, as they are altered in cancers at multiple levels including gene amplification, mRNA/protein overexpression, and protein posttranslational modifications. Of particular interest is the phosphorylation of SRC-3 at S857. S857 appears to be a hotspot that is targeted by multiple kinases and confers to SRC-3 and SRC-3∆4 a variety of oncogenic functions: augmentation of cancer cell motility when targeted by PAK-1 [46], increase of cancer cell invasion when targeted by ERK3 [108], and gain of chemotherapeutic drug resistance when targeted by IKK [30]. It is of significant clinical interest to test whether phosphorylation on this site is positively associated with human cancer progression and metastasis, thereby serving as a diagnostic and/or prognostic tumor biomarker.
29 Conclusion and Perspective
Cancer cells must acquire a variety of capabilities for tumor initiation, uncontrolled outgrowth, invasion of surroundings, and metastasis to the distant organs. Hanahan and Weinberg have summarized these required capabilities as eight hallmarks of cancer: sustaining proliferative signaling, evading growth suppressors, resisting cell death, enabling replicative immortality, inducing angiogenesis, activating invasion and metastasis, and two more recently recognized hallmarks-reprogramming of energy metabolism and evading immune destruction [158]. Despite little information on the roles of SRCs in those two emerging hallmarks, substantial evidence exists that SRCs act as integrators for the other hallmarks, thereby making great contributions to cancer cells for acquisition of these hallmarks. It has been recognized that cancer cells reprogram energy metabolism to provide fuels and biosynthetic intermediates (nucleosides and amino acids) for uncontrolled cell growth and/or invasion by enhancing energy consumption from glycolysis [159]. Although the roles of SRCs in reprogramming energy metabolism of cancer cells have not been revealed, a number of studies, mostly by using SRC knock-out animals, have shown that SRCs are critical regulators of energy metabolism of glucose and lipids [160–166]. SRC-1 has distinct functions in cell metabolism of white and brown adipose tissues: loss of SRC-1 renders animals more susceptible to high fat diet-induced obesity, whereas loss of SRC-2 or SRC-3 confers protection against it. As such, future work on SRCs in cancer cell metabolism is warranted and should provide new insights on the molecular mechanisms by which SRCs alter the cues for uncontrolled growth and aggressiveness of cancers.
Evading immune destruction and harnessing tumor-associated inflammation is another hallmark that is important for cancer progression. The roles of SRCs in this process have been scarce but surely are worthy of investigations. Although their exact roles in tumor-associated inflammation remain to be determined, SRCs are engaged in cytokine signaling and inflammation. As mentioned above, SRCs are activated (phosphorylated) by cytokine signaling and function as coactivators of NFкB and/or Stats to positively regulate cytokine signaling in cancer cells [32, 35, 37, 101, 102] and in animal models of inflammation-associated disease [104, 167–169]. Hence, it would be of clinical significance to determine in more detail the roles of SRCs in the underlying molecular mechanisms of tumor-associated inflammation. Finally, as our recent proof-of-principle study demonstrates that SRCs are accessible therapeutic targets for SMIs, more effort should be put on future high throughput screenings for therapeutic drugs targeting SRCs for cancers.
References
Onate SA, Tsai SY, Tsai MJ, O’Malley BW (1995) Sequence and characterization of a coactivator for the steroid hormone receptor superfamily. Science 270:1354–1357
Voegel JJ, Heine MJ, Zechel C, Chambon P, Gronemeyer H (1996) TIF2, a 160 kDa transcriptional mediator for the ligand-dependent activation function AF-2 of nuclear receptors. EMBO J 15:3667–3675
Hong H, Kohli K, Garabedian MJ, Stallcup MR (1997) GRIP1, a transcriptional coactivator for the AF-2 transactivation domain of steroid, thyroid, retinoid, and vitamin D receptors. Mol Cell Biol 17:2735–2744
Anzick SL, Kononen J, Walker RL, Azorsa DO, Tanner MM, Guan XY, Sauter G, Kallioniemi OP, Trent JM, Meltzer PS (1997) AIB1, a steroid receptor coactivator amplified in breast and ovarian cancer. Science 277:965–968
Torchia J, Rose DW, Inostroza J, Kamei Y, Westin S, Glass CK, Rosenfeld MG (1997) The transcriptional co-activator p/CIP binds CBP and mediates nuclear-receptor function. Nature 387:677–684
Chen H, Lin RJ, Schiltz RL, Chakravarti D, Nash A, Nagy L, Privalsky ML, Nakatani Y, Evans RM (1997) Nuclear receptor coactivator ACTR is a novel histone acetyltransferase and forms a multimeric activation complex with P/CAF and CBP/p300. Cell 90:569–580
Takeshita A, Cardona GR, Koibuchi N, Suen CS, Chin WW (1997) TRAM-1, A novel 160-kDa thyroid hormone receptor activator molecule, exhibits distinct properties from steroid receptor coactivator-1. J Biol Chem 272:27629–27634
Johnson AB, O’Malley BW (2012) Steroid receptor coactivators 1, 2, and 3: critical regulators of nuclear receptor activity and steroid receptor modulator (SRM)-based cancer therapy. Mol Cell Endocrinol 348:430–439
York B, O’Malley BW (2010) Steroid receptor coactivator (SRC) family: masters of systems biology. J Biol Chem 285:38743–38750
Xu J, Wu RC, O’Malley BW (2009) Normal and cancer-related functions of the p160 steroid receptor co-activator (SRC) family. Nat Rev Cancer 9:615–630
Leo C, Chen JD (2000) The SRC family of nuclear receptor coactivators. Gene 245:1–11
Kim JH, Li H, Stallcup MR (2003) CoCoA, a nuclear receptor coactivator which acts through an N-terminal activation domain of p160 coactivators. Mol Cell 12:1537–1549
Belandia B, Orford RL, Hurst HC, Parker MG (2002) Targeting of SWI/SNF chromatin remodelling complexes to estrogen-responsive genes. EMBO J 21:4094–4103
Askew EB, Bai S, Hnat AT, Minges JT, Wilson EM (2009) Melanoma antigen gene protein-A11 (MAGE-11) F-box links the androgen receptor NH2-terminal transactivation domain to p160 coactivators. J Biol Chem 284:34793–34808
Giraud S, Bienvenu F, Avril S, Gascan H, Heery DM, Coqueret O (2002) Functional interaction of STAT3 transcription factor with the coactivator NcoA/SRC1a. J Biol Chem 277:8004–8011
Lee SK, Kim HJ, Kim JW, Lee JW (1999) Steroid receptor coactivator-1 and its family members differentially regulate transactivation by the tumor suppressor protein p53. Mol Endocrinol 13:1924–1933
Wu RC, Smith CL, O’Malley BW (2005) Transcriptional regulation by steroid receptor coactivator phosphorylation. Endocr Rev 26:393–399
Louie MC, Revenko AS, Zou JX, Yao J, Chen HW (2006) Direct control of cell cycle gene expression by proto-oncogene product ACTR, and its autoregulation underlies its transforming activity. Mol Cell Biol 26:3810–3823
Heery DM, Kalkhoven E, Hoare S, Parker MG (1997) A signature motif in transcriptional co-activators mediates binding to nuclear receptors. Nature 387:733–736
Darimont BD, Wagner RL, Apriletti JW, Stallcup MR, Kushner PJ, Baxter JD, Fletterick RJ, Yamamoto KR (1998) Structure and specificity of nuclear receptor-coactivator interactions. Genes Dev 12:3343–3356
Gao Z, Chiao P, Zhang X, Zhang X, Lazar MA, Seto E, Young HA, Ye J (2005) Coactivators and corepressors of NF-kappaB in IkappaB alpha gene promoter. J Biol Chem 280:21091–21098
Dilworth FJ, Chambon P (2001) Nuclear receptors coordinate the activities of chromatin remodeling complexes and coactivators to facilitate initiation of transcription. Oncogene 20:3047–3054
Kraus WL, Manning ET, Kadonaga JT (1999) Biochemical analysis of distinct activation functions in p300 that enhance transcription initiation with chromatin templates. Mol Cell Biol 19:8123–8135
Demarest SJ, Martinez-Yamout M, Chung J, Chen H, Xu W, Dyson HJ, Evans RM, Wright PE (2002) Mutual synergistic folding in recruitment of CBP/p300 by p160 nuclear receptor coactivators. Nature 415:549–553
Koh SS, Chen D, Lee YH, Stallcup MR (2001) Synergistic enhancement of nuclear receptor function by p160 coactivators and two coactivators with protein methyltransferase activities. J Biol Chem 276:1089–1098
Chen D, Ma H, Hong H, Koh SS, Huang SM, Schurter BT, Aswad DW, Stallcup MR (1999) Regulation of transcription by a protein methyltransferase. Science 284:2174–2177
Spencer TE, Jenster G, Burcin MM, Allis CD, Zhou J, Mizzen CA, McKenna NJ, Onate SA, Tsai SY, Tsai MJ, O’Malley BW (1997) Steroid receptor coactivator-1 is a histone acetyltransferase. Nature 389:194–198
Lee SK, Kim HJ, Na SY, Kim TS, Choi HS, Im SY, Lee JW (1998) Steroid receptor coactivator-1 coactivates activating protein-1-mediated transactivations through interaction with the c-Jun and c-Fos subunits. J Biol Chem 273:16651–16654
Batsche E, Desroches J, Bilodeau S, Gauthier Y, Drouin J (2005) Rb enhances p160/SRC coactivator-dependent activity of nuclear receptors and hormone responsiveness. J Biol Chem 280:19746–19756
Wu MY, Fu J, Xu J, O’Malley BW, Wu RC (2012) Steroid receptor coactivator 3 regulates autophagy in breast cancer cells through macrophage migration inhibitory factor. Cell Res 22:1003–1021
Ordonez-Moran P, Munoz A (2009) Nuclear receptors: genomic and non-genomic effects converge. Cell Cycle 8:1675–1680
Wu RC, Qin J, Yi P, Wong J, Tsai SY, Tsai MJ, O’Malley BW (2004) Selective phosphorylations of the SRC-3/AIB1 coactivator integrate genomic responses to multiple cellular signaling pathways. Mol Cell 15:937–949
Zheng FF, Wu RC, Smith CL, O’Malley BW (2005) Rapid estrogen-induced phosphorylation of the SRC-3 coactivator occurs in an extranuclear complex containing estrogen receptor. Mol Cell Biol 25:8273–8284
Dobrovolna J, Chinenov Y, Kennedy MA, Liu B, Rogatsky I (2012) Glucocorticoid-dependent phosphorylation of the transcriptional coregulator GRIP1. Mol Cell Biol 32:730–739
Ueda T, Mawji NR, Bruchovsky N, Sadar MD (2002) Ligand-independent activation of the androgen receptor by interleukin-6 and the role of steroid receptor coactivator-1 in prostate cancer cells. J Biol Chem 277:38087–38094
Munz T, Litterst CM, Pfitzner E (2011) Interaction of STAT6 with its co-activator SRC-1/NCoA-1 is regulated by dephosphorylation of the latter via PP2A. Nucleic Acids Res 39:3255–3266
Wu RC, Qin J, Hashimoto Y, Wong J, Xu J, Tsai SY, Tsai MJ, O’Malley BW (2002) Regulation of SRC-3 (pCIP/ACTR/AIB-1/RAC-3/TRAM-1) Coactivator activity by I kappa B kinase. Mol Cell Biol 22:3549–3561
Rowan BG, Weigel NL, O’Malley BW (2000) Phosphorylation of steroid receptor coactivator-1. Identification of the phosphorylation sites and phosphorylation through the mitogen-activated protein kinase pathway. J Biol Chem 275:4475–4483
Moore NL, Weigel NL (2011) Regulation of progesterone receptor activity by cyclin dependent kinases 1 and 2 occurs in part by phosphorylation of the SRC-1 carboxyl-terminus. Int J Biochem Cell Biol 43:1157–1167
Gregory CW, Fei X, Ponguta LA, He B, Bill HM, French FS, Wilson EM (2004) Epidermal growth factor increases coactivation of the androgen receptor in recurrent prostate cancer. J Biol Chem 279:7119–7130
Oh AS, Lahusen JT, Chien CD, Fereshteh MP, Zhang X, Dakshanamurthy S, Xu J, Kagan BL, Wellstein A, Riegel AT (2008) Tyrosine phosphorylation of the nuclear receptor coactivator AIB1/SRC-3 is enhanced by Abl kinase and is required for its activity in cancer cells. Mol Cell Biol 28:6580–6593
Shou J, Massarweh S, Osborne CK, Wakeling AE, Ali S, Weiss H, Schiff R (2004) Mechanisms of tamoxifen resistance: increased estrogen receptor-HER2/neu cross-talk in ER/HER2-positive breast cancer. J Natl Cancer Inst 96:926–935
Rowan BG, Garrison N, Weigel NL, O’Malley BW (2000) 8-Bromo-cyclic AMP induces phosphorylation of two sites in SRC-1 that facilitate ligand-independent activation of the chicken progesterone receptor and are critical for functional cooperation between SRC-1 and CREB binding protein. Mol Cell Biol 20:8720–8730
Hoang T, Fenne IS, Cook C, Borud B, Bakke M, Lien EA, Mellgren G (2004) cAMP-dependent protein kinase regulates ubiquitin-proteasome-mediated degradation and subcellular localization of the nuclear receptor coactivator GRIP1. J Biol Chem 279:49120–49130
Reiter R, Wellstein A, Riegel AT (2001) An isoform of the coactivator AIB1 that increases hormone and growth factor sensitivity is overexpressed in breast cancer. J Biol Chem 276:39736–39741
Long W, Yi P, Amazit L, LaMarca HL, Ashcroft F, Kumar R, Mancini MA, Tsai SY, Tsai MJ, O’Malley BW (2010) SRC-3Delta4 mediates the interaction of EGFR with FAK to promote cell migration. Mol Cell 37:321–332
Wu RC, Feng Q, Lonard DM, O’Malley BW (2007) SRC-3 coactivator functional lifetime is regulated by a phospho-dependent ubiquitin time clock. Cell 129:1125–1140
Gianni M, Parrella E, Raska I Jr, Gaillard E, Nigro EA, Gaudon C, Garattini E, Rochette-Egly C (2006) P38MAPK-dependent phosphorylation and degradation of SRC-3/AIB1 and RARalpha-mediated transcription. EMBO J 25:739–751
Ferry C, Gaouar S, Fischer B, Boeglin M, Paul N, Samarut E, Piskunov A, Pankotai-Bodo G, Brino L, Rochette-Egly C (2011) Cullin 3 mediates SRC-3 ubiquitination and degradation to control the retinoic acid response. Proc Natl Acad Sci USA 108:20603–20608
Li C, Liang YY, Feng XH, Tsai SY, Tsai MJ, O’Malley BW (2008) Essential phosphatases and a phospho-degron are critical for regulation of SRC-3/AIB1 coactivator function and turnover. Mol Cell 31:835–849
Li C, Ao J, Fu J, Lee DF, Xu J, Lonard D, O’Malley BW (2011) Tumor-suppressor role for the SPOP ubiquitin ligase in signal-dependent proteolysis of the oncogenic co-activator SRC-3/AIB1. Oncogene 30:4350–4364
Li X, Lonard DM, Jung SY, Malovannaya A, Feng Q, Qin J, Tsai SY, Tsai MJ, O’Malley BW (2006) The SRC-3/AIB1 coactivator is degraded in a ubiquitin- and ATP-independent manner by the REGgamma proteasome. Cell 124:381–922
Yi P, Feng Q, Amazit L, Lonard DM, Tsai SY, Tsai MJ, O’Malley BW (2008) Atypical protein kinase C regulates dual pathways for degradation of the oncogenic coactivator SRC-3/AIB1. Mol Cell 29:465–476
Hartmaier RJ, Richter AS, Gillihan RM, Sallit JZ, McGuire SE, Wang J, Lee AV, Osborne CK, O’Malley BW, Brown PH, Xu J, Skaar TC, Philips S, Rae JM, Azzouz F, Li L, Hayden J, Henry NL, Nguyen AT, Stearns V, Hayes DF, Flockhart DA, Oesterreich S (2012) A SNP in steroid receptor coactivator-1 disrupts a GSK3beta phosphorylation site and is associated with altered tamoxifen response in bone. Mol Endocrinol 26:220–227
Gareau JR, Lima CD (2010) The SUMO pathway: emerging mechanisms that shape specificity, conjugation and recognition. Nat Rev Mol Cell Biol 11:861–871
Li S, Shang Y (2007) Regulation of SRC family coactivators by post-translational modifications. Cell Signal 19:1101–1112
Chauchereau A, Amazit L, Quesne M, Guiochon-Mantel A, Milgrom E (2003) Sumoylation of the progesterone receptor and of the steroid receptor coactivator SRC-1. J Biol Chem 278:12335–12343
Kotaja N, Karvonen U, Janne OA, Palvimo JJ (2002) The nuclear receptor interaction domain of GRIP1 is modulated by covalent attachment of SUMO-1. J Biol Chem 277:30283–30288
Hietakangas V, Anckar J, Blomster HA, Fujimoto M, Palvimo JJ, Nakai A, Sistonen L (2006) PDSM, a motif for phosphorylation-dependent SUMO modification. Proc Natl Acad Sci USA 103:45–50
Shalizi A, Gaudilliere B, Yuan Z, Stegmuller J, Shirogane T, Ge Q, Tan Y, Schulman B, Harper JW, Bonni A (2006) A calcium-regulated MEF2 sumoylation switch controls postsynaptic differentiation. Science 311:1012–1017
Chen H, Lin RJ, Xie W, Wilpitz D, Evans RM (1999) Regulation of hormone-induced histone hyperacetylation and gene activation via acetylation of an acetylase. Cell 98:675–686
Fu M, Wang C, Zhang X, Pestell RG (2004) Acetylation of nuclear receptors in cellular growth and apoptosis. Biochem Pharmacol 68:1199–1208
Feng Q, Yi P, Wong J, O’Malley BW (2006) Signaling within a coactivator complex: methylation of SRC-3/AIB1 is a molecular switch for complex disassembly. Mol Cell Biol 26:7846–7857
Naeem H, Cheng D, Zhao Q, Underhill C, Tini M, Bedford MT, Torchia J (2007) The activity and stability of the transcriptional coactivator p/CIP/SRC-3 are regulated by CARM1-dependent methylation. Mol Cell Biol 27:120–134
Eroles P, Bosch A, Perez-Fidalgo JA, Lluch A (2012) Molecular biology in breast cancer: intrinsic subtypes and signaling pathways. Cancer Treat Rev 38:698–707
Neve RM, Chin K, Fridlyand J, Yeh J, Baehner FL, Fevr T, Clark L, Bayani N, Coppe JP, Tong F, Speed T, Spellman PT, DeVries S, Lapuk A, Wang NJ, Kuo WL, Stilwell JL, Pinkel D, Albertson DG, Waldman FM, McCormick F, Dickson RB, Johnson MD, Lippman M, Ethier S, Gazdar A, Gray JW (2006) A collection of breast cancer cell lines for the study of functionally distinct cancer subtypes. Cancer Cell 10:515–527
Tai H, Kubota N, Kato S (2000) Involvement of nuclear receptor coactivator SRC-1 in estrogen-dependent cell growth of MCF-7 cells. Biochem Biophys Res Commun 267:311–316
Cavarretta IT, Mukopadhyay R, Lonard DM, Cowsert LM, Bennett CF, O’Malley BW, Smith CL (2002) Reduction of coactivator expression by antisense oligodeoxynucleotides inhibits ERalpha transcriptional activity and MCF-7 proliferation. Mol Endocrinol 16:253–270
Kishimoto H, Wang Z, Bhat-Nakshatri P, Chang D, Clarke R, Nakshatri H (2005) The p160 family coactivators regulate breast cancer cell proliferation and invasion through autocrine/paracrine activity of SDF-1alpha/CXCL12. Carcinogenesis 26:1706–1715
Planas-Silva MD, Shang Y, Donaher JL, Brown M, Weinberg RA (2001) AIB1 enhances estrogen-dependent induction of cyclin D1 expression. Cancer Res 61:3858–3862
Karmakar S, Foster EA, Smith CL (2009) Unique roles of p160 coactivators for regulation of breast cancer cell proliferation and estrogen receptor-alpha transcriptional activity. Endocrinology 150:1588–1596
List HJ, Lauritsen KJ, Reiter R, Powers C, Wellstein A, Riegel AT (2001) Ribozyme targeting demonstrates that the nuclear receptor coactivator AIB1 is a rate-limiting factor for estrogen-dependent growth of human MCF-7 breast cancer cells. J Biol Chem 276:23763–23768
Park KJ, Krishnan V, O’Malley BW, Yamamoto Y, Gaynor RB (2005) Formation of an IKKalpha-dependent transcription complex is required for estrogen receptor-mediated gene activation. Mol Cell 18:71–82
Torres-Arzayus MI, Font de Mora J, Yuan J, Vazquez F, Bronson R, Rue M, Sellers WR, Brown M (2004) High tumor incidence and activation of the PI3K/AKT pathway in transgenic mice define AIB1 as an oncogene. Cancer Cell 6:263–274
Torres-Arzayus MI, Zhao J, Bronson R, Brown M (2010) Estrogen-dependent and estrogen-independent mechanisms contribute to AIB1-mediated tumor formation. Cancer Res 70:4102–4111
Agoulnik IU, Vaid A, Bingman WE III, Erdeme H, Frolov A, Smith CL, Ayala G, Ittmann MM, Weigel NL (2005) Role of SRC-1 in the promotion of prostate cancer cell growth and tumor progression. Cancer Res 65:7959–7967
Agoulnik IU, Vaid A, Nakka M, Alvarado M, Bingman WE III, Erdem H, Frolov A, Smith CL, Ayala GE, Ittmann MM, Weigel NL (2006) Androgens modulate expression of transcription intermediary factor 2, an androgen receptor coactivator whose expression level correlates with early biochemical recurrence in prostate cancer. Cancer Res 66:10594–10602
Shi XB, Xue L, Zou JX, Gandour-Edwards R, Chen H, deVere White RW (2008) Prolonged androgen receptor loading onto chromatin and the efficient recruitment of p160 coactivators contribute to androgen-independent growth of prostate cancer cells. Prostate 68:1816–1826
Zhou HJ, Yan J, Luo W, Ayala G, Lin SH, Erdem H, Ittmann M, Tsai SY, Tsai MJ (2005) SRC-3 is required for prostate cancer cell proliferation and survival. Cancer Res 65:7976–7983
Zou JX, Zhong Z, Shi XB, Tepper CG, deVere White RW, Kung HJ, Chen H (2006) ACTR/AIB1/SRC-3 and androgen receptor control prostate cancer cell proliferation and tumor growth through direct control of cell cycle genes. Prostate 66:1474–1486
Higgins MJ, Baselga J (2011) Targeted therapies for breast cancer. J Clin Invest 121:3797–3803
Fereshteh MP, Tilli MT, Kim SE, Xu J, O’Malley BW, Wellstein A, Furth PA, Riegel AT (2008) The nuclear receptor coactivator amplified in breast cancer-1 is required for Neu (ErbB2/HER2) activation, signaling, and mammary tumorigenesis in mice. Cancer Res 68:3697–3706
Al-Otaiby M, Tassi E, Schmidt MO, Chien CD, Baker T, Salas AG, Xu J, Furlong M, Schlegel R, Riegel AT, Wellstein A (2012) Role of the nuclear receptor coactivator AIB1/SRC-3 in angiogenesis and wound healing. Am J Pathol 180:1474–1484
Han JS, Crowe DL (2010) Steroid receptor coactivator 1 deficiency increases MMTV-neu mediated tumor latency and differentiation specific gene expression, decreases metastasis, and inhibits response to PPAR ligands. BMC Cancer 10:629
Chen B, Wang Y, Kane SE, Chen S (2008) Improvement of sensitivity to tamoxifen in estrogen receptor-positive and herceptin-resistant breast cancer cells. J Mol Endocrinol 41:367–377
Su Q, Hu S, Gao H, Ma R, Yang Q, Pan Z, Wang T, Li F (2008) Role of AIB1 for tamoxifen resistance in estrogen receptor-positive breast cancer cells. Oncology 75:159–168
Osborne CK, Bardou V, Hopp TA, Chamness GC, Hilsenbeck SG, Fuqua SA, Wong J, Allred DC, Clark GM, Schiff R (2003) Role of the estrogen receptor coactivator AIB1 (SRC-3) and HER-2/neu in tamoxifen resistance in breast cancer. J Natl Cancer Inst 95: 353–361
Fleming FJ, Myers E, Kelly G, Crotty TB, McDermott EW, O’Higgins NJ, Hill AD, Young LS (2004) Expression of SRC-1, AIB1, and PEA3 in HER2 mediated endocrine resistant breast cancer; a predictive role for SRC-1. J Clin Pathol 57:1069–1074
Qin L, Liu Z, Chen H, Xu J (2009) The steroid receptor coactivator-1 regulates twist expression and promotes breast cancer metastasis. Cancer Res 69:3819–3827
Hurtado A, Holmes KA, Geistlinger TR, Hutcheson IR, Nicholson RI, Brown M, Jiang J, Howat WJ, Ali S, Carroll JS (2008) Regulation of ERBB2 by oestrogen receptor-PAX2 determines response to tamoxifen. Nature 456:663–666
Lahusen T, Fereshteh M, Oh A, Wellstein A, Riegel AT (2007) Epidermal growth factor receptor tyrosine phosphorylation and signaling controlled by a nuclear receptor coactivator, amplified in breast cancer 1. Cancer Res 67:7256–7265
Guo Z, Dai B, Jiang T, Xu K, Xie Y, Kim O, Nesheiwat I, Kong X, Melamed J, Handratta VD, Njar VC, Brodie AM, Yu LR, Veenstra TD, Chen H, Qiu Y (2006) Regulation of androgen receptor activity by tyrosine phosphorylation. Cancer Cell 10:309–319
Pollak M (2012) The insulin and insulin-like growth factor receptor family in neoplasia: an update. Nat Rev Cancer 12:159–169
Oh A, List HJ, Reiter R, Mani A, Zhang Y, Gehan E, Wellstein A, Riegel AT (2004) The nuclear receptor coactivator AIB1 mediates insulin-like growth factor I-induced phenotypic changes in human breast cancer cells. Cancer Res 64:8299–8308
Yan J, Yu CT, Ozen M, Ittmann M, Tsai SY, Tsai MJ (2006) Steroid receptor coactivator-3 and activator protein-1 coordinately regulate the transcription of components of the insulin-like growth factor/AKT signaling pathway. Cancer Res 66:11039–11046
Kuang SQ, Liao L, Zhang H, Lee AV, O’Malley BW, Xu J (2004) AIB1/SRC-3 deficiency affects insulin-like growth factor I signaling pathway and suppresses v-Ha-ras-induced breast cancer initiation and progression in mice. Cancer Res 64:1875–1885
Wang S, Yuan Y, Liao L, Kuang SQ, Tien JC, O’Malley BW, Xu J (2009) Disruption of the SRC-1 gene in mice suppresses breast cancer metastasis without affecting primary tumor formation. Proc Natl Acad Sci USA 106:151–156
Drachenberg DE, Elgamal AA, Rowbotham R, Peterson M, Murphy GP (1999) Circulating levels of interleukin-6 in patients with hormone refractory prostate cancer. Prostate 41: 127–133
Adler HL, McCurdy MA, Kattan MW, Timme TL, Scardino PT, Thompson TC (1999) Elevated levels of circulating interleukin-6 and transforming growth factor-beta1 in patients with metastatic prostatic carcinoma. J Urol 161:182–187
Lee SO, Lou W, Hou M, de Miguel F, Gerber L, Gao AC (2003) Interleukin-6 promotes androgen-independent growth in LNCaP human prostate cancer cells. Clin Cancer Res 9:370–376
Feng S, Tang Q, Sun M, Chun JY, Evans CP, Gao AC (2009) Interleukin-6 increases prostate cancer cells resistance to bicalutamide via TIF2. Mol Cancer Ther 8:665–671
Rubio MF, Werbajh S, Cafferata EG, Quaglino A, Colo GP, Nojek IM, Kordon EC, Nahmod VE, Costas MA (2006) TNF-alpha enhances estrogen-induced cell proliferation of estrogen-dependent breast tumor cells through a complex containing nuclear factor-kappa B. Oncogene 25:1367–1377
Yu C, York B, Wang S, Feng Q, Xu J, O’Malley BW (2007) An essential function of the SRC-3 coactivator in suppression of cytokine mRNA translation and inflammatory response. Mol Cell 25:765–778
Han SJ, Hawkins SM, Begum K, Jung SY, Kovanci E, Qin J, Lydon JP, DeMayo FJ, O’Malley BW (2012) A new isoform of steroid receptor coactivator-1 is crucial for pathogenic progression of endometriosis. Nat Med 18:1102–1111
Qin L, Liao L, Redmond A, Young L, Yuan Y, Chen H, O’Malley BW, Xu J (2008) The AIB1 oncogene promotes breast cancer metastasis by activation of PEA3-mediated matrix metalloproteinase 2 (MMP2) and MMP9 expression. Mol Cell Biol 28:5937–5950
Yan J, Erdem H, Li R, Cai Y, Ayala G, Ittmann M, Yu-Lee LY, Tsai SY, Tsai MJ (2008) Steroid receptor coactivator-3/AIB1 promotes cell migration and invasiveness through focal adhesion turnover and matrix metalloproteinase expression. Cancer Res 68:5460–5468
Li LB, Louie MC, Chen HW, Zou JX (2008) Proto-oncogene ACTR/AIB1 promotes cancer cell invasion by up-regulating specific matrix metalloproteinase expression. Cancer Lett 261:64–73
Long W, Foulds CE, Long W, Foulds CE, Qin J, Liu J, Ding C, Lonard DM, Solis LM, Wistuba II, Qin J, Tsai SY, Tsai MJ, O’Malley BW (2012) ERK3 signals through SRC-3 coactivator to promote human lung cancer cell invasion. J Clin Invest 122:1869–1880
Taylor BS, Schultz N, Hieronymus H, Gopalan A, Xiao Y, Carver BS, Arora VK, Kaushik P, Cerami E, Reva B, Antipin Y, Mitsiades N, Landers T, Dolgalev I, Major JE, Wilson M, Socci ND, Lash AE, Heguy A, Eastham JA, Scher HI, Reuter VE, Scardino PT, Sander C, Sawyers CL, Gerald WL (2010) Integrative genomic profiling of human prostate cancer. Cancer Cell 18:11–22
Bautista S, Valles H, Walker RL, Anzick S, Zeillinger R, Meltzer P, Theillet C (1998) In breast cancer, amplification of the steroid receptor coactivator gene AIB1 is correlated with estrogen and progesterone receptor positivity. Clin Cancer Res 4:2925–2929
Tanner MM, Grenman S, Koul A, Johannsson O, Meltzer P, Pejovic T, Borg A, Isola JJ (2000) Frequent amplification of chromosomal region 20q12-q13 in ovarian cancer. Clin Cancer Res 6:1833–1839
Xie D, Sham JS, Zeng WF, Lin HL, Bi J, Che LH, Hu L, Zeng YX, Guan XY (2005) Correlation of AIB1 overexpression with advanced clinical stage of human colorectal carcinoma. Hum Pathol 36:777–783
Lassmann S, Weis R, Makowiec F, Roth J, Danciu M, Hopt U, Werner M (2007) Array CGH identifies distinct DNA copy number profiles of oncogenes and tumor suppressor genes in chromosomal- and microsatellite-unstable sporadic colorectal carcinomas. J Mol Med (Berl) 85:293–304
He LR, Zhao HY, Li BK, Zhang LJ, Liu MZ, Kung HF, Guan XY, Bian XW, Zeng YX, Xie D (2010) Overexpression of AIB1 negatively affects survival of surgically resected non-small-cell lung cancer patients. Ann Oncol 21:1675–1681
Cai D, Shames DS, Raso MG, Xie Y, Kim YH, Pollack JR, Girard L, Sullivan JP, Gao B, Peyton M, Nanjundan M, Byers L, Heymach J, Mills G, Gazdar AF, Wistuba I, Kodadek T, Minna JD (2010) Steroid receptor coactivator-3 expression in lung cancer and its role in the regulation of cancer cell survival and proliferation. Cancer Res 70:6477–6485
Wang Y, Wu MC, Sham JS, Zhang W, Wu WQ, Guan XY (2002) Prognostic significance of c-myc and AIB1 amplification in hepatocellular carcinoma. A broad survey using high-throughput tissue microarray. Cancer 95:2346–2352
Maki HE, Waltering KK, Wallen MJ, Martikainen PM, Tammela TL, van Weerden WM, Vessella RL, Visakorpi T (2006) Screening of genetic and expression alterations of SRC1 gene in prostate cancer. Prostate 66:1391–1398
Carapeti M, Aguiar RC, Goldman JM, Cross NC (1998) A novel fusion between MOZ and the nuclear receptor coactivator TIF2 in acute myeloid leukemia. Blood 91:3127–3133
Carapeti M, Aguiar RC, Watmore AE, Goldman JM, Cross NC (1999) Consistent fusion of MOZ and TIF2 in AML with inv(8)(p11q13). Cancer Genet Cytogenet 113:70–72
Deguchi K, Ayton PM, Carapeti M, Kutok JL, Snyder CS, Williams IR, Cross NC, Glass CK, Cleary ML, Gilliland DG (2003) MOZ-TIF2-induced acute myeloid leukemia requires the MOZ nucleosome binding motif and TIF2-mediated recruitment of CBP. Cancer Cell 3:259–271
Osborne CK, Schiff R (2011) Mechanisms of endocrine resistance in breast cancer. Annu Rev Med 62:233–247
Ariazi EA, Ariazi JL, Cordera F, Jordan VC (2006) Estrogen receptors as therapeutic targets in breast cancer. Curr Top Med Chem 6:181–202
Fleming FJ, Hill AD, McDermott EW, O’Higgins NJ, Young LS (2004) Differential recruitment of coregulator proteins steroid receptor coactivator-1 and silencing mediator for retinoid and thyroid receptors to the estrogen receptor-estrogen response element by beta-estradiol and 4-hydroxytamoxifen in human breast cancer. J Clin Endocrinol Metab 89:375–383
Hudelist G, Czerwenka K, Kubista E, Marton E, Pischinger K, Singer CF (2003) Expression of sex steroid receptors and their co-factors in normal and malignant breast tissue: AIB1 is a carcinoma-specific co-activator. Breast Cancer Res Treat 78:193–204
McBryan J, Theissen SM, Byrne C, Hughes E, Cocchiglia S, Sande S, O’Hara J, Tibbitts P, Hill AD, Young LS (2011) Metastatic progression with resistance to aromatase inhibitors is driven by the steroid receptor coactivator SRC-1. Cancer Res 72:548–559
Myers E, Fleming FJ, Crotty TB, Kelly G, McDermott EW, O’Higgins JN, Hill AD, Young LS (2004) Inverse relationship between ER-beta and SRC-1 predicts outcome in endocrine-resistant breast cancer. Br J Cancer 91:1687–1693
Zhao C, Yasui K, Lee CJ, Kurioka H, Hosokawa Y, Oka T, Inazawa J (2003) Elevated expression levels of NCOA3, TOP1, and TFAP2C in breast tumors as predictors of poor prognosis. Cancer 98:18–23
Bouras T, Southey MC, Venter DJ (2001) Overexpression of the steroid receptor coactivator AIB1 in breast cancer correlates with the absence of estrogen and progesterone receptors and positivity for p53 and HER2/neu. Cancer Res 61:903–907
Arteaga CL, Sliwkowski MX, Osborne CK, Perez EA, Puglisi F, Gianni L (2011) Treatment of HER2-positive breast cancer: current status and future perspectives. Nat Rev Clin Oncol 9:16–32
Kirkegaard T, McGlynn LM, Campbell FM, Muller S, Tovey SM, Dunne B, Nielsen KV, Cooke TG, Bartlett JM (2007) Amplified in breast cancer 1 in human epidermal growth factor receptor – positive tumors of tamoxifen-treated breast cancer patients. Clin Cancer Res 13:1405–1411
Lahusen T, Henke RT, Kagan BL, Wellstein A, Riegel AT (2009) The role and regulation of the nuclear receptor co-activator AIB1 in breast cancer. Breast Cancer Res Treat 116:225–237
Agoulnik IU, Weigel NL (2006) Androgen receptor action in hormone-dependent and recurrent prostate cancer. J Cell Biochem 99:362–372
Tsao CK, Small AC, Galsky MD, Oh WK (2012) Overcoming castration resistance in prostate cancer. Curr Opin Urol 22:167–174
Pienta KJ, Bradley D (2006) Mechanisms underlying the development of androgen-independent prostate cancer. Clin Cancer Res 12:1665–1671
Heemers HV, Tindall DJ (2007) Androgen receptor (AR) coregulators: a diversity of functions converging on and regulating the AR transcriptional complex. Endocr Rev 28:778–808
Gregory CW, He B, Johnson RT, Ford OH, Mohler JL, French FS, Wilson EM (2001) A mechanism for androgen receptor-mediated prostate cancer recurrence after androgen deprivation therapy. Cancer Res 61:4315–4319
Gnanapragasam VJ, Leung HY, Pulimood AS, Neal DE, Robson CN (2001) Expression of RAC 3, a steroid hormone receptor co-activator in prostate cancer. Br J Cancer 85:1928–1936
Ayala G, Yan J, Li R, Ding Y, Thompson TC, Mims MP, Hayes TG, MacDonnell V, Lynch RG, Frolov A, Miles BJ, Wheeler TM, Harper JW, Tsai MJ, Ittmann MM, Kadmon D (2008) Bortezomib-mediated inhibition of steroid receptor coactivator-3 degradation leads to activated Akt. Clin Cancer Res 14:7511–7518
Marquez-Garban DC, Chen HW, Fishbein MC, Goodglick L, Pietras RJ (2007) Estrogen receptor signaling pathways in human non-small cell lung cancer. Steroids 72:135–143
Wang H, Zhang D, Wu W, Zhang J, Guo D, Wang Q, Jing T, Xu C, Bian X, Yang K (2010) Overexpression and gender-specific differences of SRC-3 (SRC-3/AIB1) immunoreactivity in human non-small cell lung cancer: an in vivo study. J Histochem Cytochem 58:1121–1127
Yoshida H, Liu J, Samuel S, Cheng W, Rosen D, Naora H (2005) Steroid receptor coactivator-3, a homolog of Taiman that controls cell migration in the Drosophila ovary, regulates migration of human ovarian cancer cells. Mol Cell Endocrinol 245:77–85
Henke RT, Haddad BR, Kim SE, Rone JD, Mani A, Jessup JM, Wellstein A, Maitra A, Riegel AT (2004) Overexpression of the nuclear receptor coactivator AIB1 (SRC-3) during progression of pancreatic adenocarcinoma. Clin Cancer Res 10:6134–6142
Coste A, Antal MC, Chan S, Kastner P, Mark M, O’Malley BW, Auwerx J (2006) Absence of the steroid receptor coactivator-3 induces B-cell lymphoma. EMBO J 25:2453–2464
O’Donnell KA, Keng VW, York B, Reineke EL, Seo D, Fan D, Silverstein KA, Schrum CT, Xie WR, Mularoni L, Wheelan SJ, Torbenson MS, O’Malley BW, Largaespada DA, Boeke JD (2012) A sleeping beauty mutagenesis screen reveals a tumor suppressor role for Ncoa2/Src-2 in liver cancer. Proc Natl Acad Sci USA 109:E1377–E1386
Smith CL, Nawaz Z, O’Malley BW (1997) Coactivator and corepressor regulation of the agonist/antagonist activity of the mixed antiestrogen, 4-hydroxytamoxifen. Mol Endocrinol 11:657–666
Redmond AM, Bane FT, Stafford AT, McIlroy M, Dillon MF, Crotty TB, Hill AD, Young LS (2009) Coassociation of estrogen receptor and p160 proteins predicts resistance to endocrine treatment; SRC-1 is an independent predictor of breast cancer recurrence. Clin Cancer Res 15:2098–2106
He LR, Liu MZ, Li BK, Rao HL, Deng HX, Guan XY, Zeng YX, Xie D (2009) Overexpression of AIB1 predicts resistance to chemoradiotherapy and poor prognosis in patients with primary esophageal squamous cell carcinoma. Cancer Sci 100:1591–1596
Chen Q, Li W, Wan Y, Xia X, Wu Q, Chen Y, Lai Z, Yu C, Li W (2012) Amplified in breast cancer 1 enhances human cholangiocarcinoma growth and chemoresistance by simultaneous activation of Akt and Nrf2 pathways. Hepatology 55:1820–1829
Norris JD, Paige LA, Christensen DJ, Chang CY, Huacani MR, Fan D, Hamilton PT, Fowlkes DM, McDonnell DP (1999) Peptide antagonists of the human estrogen receptor. Science 285:744–746
Chang C, Norris JD, Gron H, Paige LA, Hamilton PT, Kenan DJ, Fowlkes D, McDonnell DP (1999) Dissection of the LXXLL nuclear receptor-coactivator interaction motif using combinatorial peptide libraries: discovery of peptide antagonists of estrogen receptors alpha and beta. Mol Cell Biol 19:8226–8239
Hall JM, Chang CY, McDonnell DP (2000) Development of peptide antagonists that target estrogen receptor beta-coactivator interactions. Mol Endocrinol 14:2010–2023
Geistlinger TR, Guy RK (2001) An inhibitor of the interaction of thyroid hormone receptor beta and glucocorticoid interacting protein 1. J Am Chem Soc 123:1525–1526
Galande AK, Bramlett KS, Burris TP, Wittliff JL, Spatola AF (2004) Thioether side chain cyclization for helical peptide formation: inhibitors of estrogen receptor-coactivator interactions. J Pept Res 63:297–302
Galande AK, Bramlett KS, Trent JO, Burris TP, Wittliff JL, Spatola AF (2005) Potent inhibitors of LXXLL-based protein-protein interactions. Chembiochem 6:1991–1998
Rodriguez AL, Tamrazi A, Collins ML, Katzenellenbogen JA (2004) Design, synthesis, and in vitro biological evaluation of small molecule inhibitors of estrogen receptor alpha coactivator binding. J Med Chem 47:600–611
Arnold LA, Estebanez-Perpina E, Togashi M, Jouravel N, Shelat A, McReynolds AC, Mar E, Nguyen P, Baxter JD, Fletterick RJ, Webb P, Guy RK (2005) Discovery of small molecule inhibitors of the interaction of the thyroid hormone receptor with transcriptional coregulators. J Biol Chem 280:43048–43055
Wang Y, Lonard DM, Yu Y, Chow DC, Palzkill TG, O’Malley BW (2012) Small molecule inhibition of the steroid receptor coactivators, SRC-3 and SRC-1. Mol Endocrinol 25:2041–2053
Hanahan D, Weinberg RA (2011) Hallmarks of cancer: the next generation. Cell 144:646–674
Dang CV (2012) Links between metabolism and cancer. Genes Dev 26:877–890
Picard F, Gehin M, Annicotte J, Rocchi S, Champy MF, O’Malley BW, Chambon P, Auwerx J (2002) SRC-1 and TIF2 control energy balance between white and brown adipose tissues. Cell 111:931–941
Wang Z, Qi C, Krones A, Woodring P, Zhu X, Reddy JK, Evans RM, Rosenfeld MG, Hunter T (2006) Critical roles of the p160 transcriptional coactivators p/CIP and SRC-1 in energy balance. Cell Metab 3:111–122
Louet JF, Coste A, Amazit L, Tannour-Louet M, Wu RC, Tsai SY, Tsai MJ, Auwerx J, O’Malley BW (2006) Oncogenic steroid receptor coactivator-3 is a key regulator of the white adipogenic program. Proc Natl Acad Sci USA 103:17868–17873
Coste A, Louet JF, Lagouge M, Lerin C, Antal MC, Meziane H, Schoonjans K, Puigserver P, O’Malley BW, Auwerx J (2008) The genetic ablation of SRC-3 protects against obesity and improves insulin sensitivity by reducing the acetylation of PGC-1{alpha}. Proc Natl Acad Sci USA 105:17187–17192
Chopra AR, Louet JF, Saha P, An J, Demayo F, Xu J, York B, Karpen S, Finegold M, Moore D, Chan L, Newgard CB, O’Malley BW (2008) Absence of the SRC-2 coactivator results in a glycogenopathy resembling Von Gierke’s disease. Science 322:1395–1399
Louet JF, Chopra AR, Sagen JV, An J, York B, Tannour-Louet M, Saha PK, Stevens RD, Wenner BR, Ilkayeva OR, Bain JR, Zhou S, DeMayo F, Xu J, Newgard CB, O’Malley BW (2010) The coactivator SRC-1 is an essential coordinator of hepatic glucose production. Cell Metab 12:606–618
York B, Reineke EL, Sagen JV, Nikolai BC, Zhou S, Louet JF, Chopra AR, Chen X, Reed G, Noebels J, Adesina AM, Yu H, Wong LJ, Tsimelzon A, Hilsenbeck S, Stevens RD, Wenner BR, Ilkayeva O, Xu J, Newgard CB, O’Malley BW (2012) Ablation of steroid receptor coactivator-3 resembles the human CACT metabolic myopathy. Cell Metab 15:752–763
Ma X, Xu L, Wang S, Cui B, Li X, Xu J, Ning G (2011) Deletion of steroid receptor coactivator-3 gene ameliorates hepatic steatosis. J Hepatol 55:445–452
Li J, Liu YH, Ou S, Dai XM, Wang JP, Su YP (2012) Steroid receptor coactivator-3 differentially regulates the inflammatory response in peritoneal macrophages. Mol Med Report 5:1099–1105
Li J, Niu J, Ou S, Ye ZY, Liu DQ, Wang FC, Su YP, Wang JP (2012) Effects of SCR-3 on the immunosuppression accompanied with the systemic inflammatory response syndrome. Mol Cell Biochem 364:29–37
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2014 Springer Science+Business Media New York
About this chapter
Cite this chapter
Long, W., O’Malley, B.W. (2014). Steroid Receptor Coactivators (SRCs) as Integrators of Multiple Signaling Pathways in Cancer Progression. In: Kumar, R. (eds) Nuclear Signaling Pathways and Targeting Transcription in Cancer. Cancer Drug Discovery and Development. Humana Press, New York, NY. https://doi.org/10.1007/978-1-4614-8039-6_1
Download citation
DOI: https://doi.org/10.1007/978-1-4614-8039-6_1
Published:
Publisher Name: Humana Press, New York, NY
Print ISBN: 978-1-4614-8038-9
Online ISBN: 978-1-4614-8039-6
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)