
Abstract
The progression of a tumor cell mass beyond 2 mm is critically dependent on neoangiogenesis. Angiogenic factors secreted by tumor cells, infiltrating macrophages, and stromal cells aggressively promote proliferation and migration of endothelial cells. The nascent primitive vasculatures are usually morphologically and functionally abnormal due to several features such as the lack of a vascular smooth muscle cell layer, abrupt change of the blood vessel diameter, tortuosity, and leakiness. Those characteristics which alter the blood flow and the transport of molecules in tumors led to the discovery of the enhanced permeability and retention (EPR) of nanosize molecules in tumor tissues. Following its discovery, various anticancer nanoconstructs have been developed with the EPR effect as a central mechanism for tumor targeting. However, the development of these nanodrugs has been hampered by a slow progress towards the clinic. Only nine nanomedicines have been approved for anticancer treatment for the last 26 years. In this chapter, we discuss various aspects that may explain the limited transition for an efficient anticancer nanomedicine. The specificity of the tumor vasculature, the discrepancy in tumor biology, the role of animal tumor models, and the physicochemical characteristics of nanoconstructs are closely examined. This chapter provides new considerations for successful development of EPR-based anticancer nanomedicine.
Sebastien Taurin and Hayley Nehoff contributed equally to the work.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others

Keywords
- Nitric Oxide
- Vascular Endothelial Growth Factor
- Tumor Vasculature
- Tumor Vessel
- Vascular Endothelial Growth Factor Gene
These keywords were added by machine and not by the authors. This process is experimental and the keywords may be updated as the learning algorithm improves.
Introduction
Angiogenesis is fundamental for many biological processes such as development, reproduction, and wound healing and has been implicated in the progression of a variety of diseases including diabetic retinopathy, rheumatoid arthritis, age-related macular degeneration (AMD), psoriasis, and tumor progression [1–4]. The early stages of the tumor development are characterized by the aberrant activation of oncogenes, inhibition of tumors suppressor genes, and modifications of genes that directly and indirectly control cell proliferation, all as a result of the accumulation of discrete genetic changes and epigenetics alterations [5]. Once the tumor has reached a certain size, the tumor propagation and progression will be dependent on the immediate environment. In 1889, Stephen Paget proposed the “seed and soil” hypothesis based on the concept that the microenvironment of a developing tumor is a crucial regulator of its growth and expansion [6]. The capacity of transplanted tumor cells to promote blood vessel formation was demonstrated by Greenblatt and Shubik [7] and Ehrmann and Knoth [8] who demonstrated that a diffusible factor produced by tumor cells can induce neovascularization. In the early 1970s, Folkman proposed that the tumor growth is essentially dependent on the establishment of its own vascular supply [9, 10]. Independent of the cellular origin of the cancer, angiogenesis is the critical step for the growth of tumours beyond 2 mm as well as the development of metastasis. The activation of tumor angiogenesis relies essentially on the balance between the pro-angiogenic factors and the anti-angiogenic factors.
The induction of the tumor vasculature growth is termed the “angiogenic switch” [11, 12] and is dependent on the increased expression of the pro-angiogenic genes and/or a decreased expression of anti-angiogenic factors. Many potential regulators of angiogenesis have been identified including acidic fibroblast growth factor (aFGF), basic fibroblast growth factor (bFGF), vascular endothelial growth factor (VEGF), transforming growth factor-α (TGF-α), transforming growth factor-β (TGF-β), hepatocyte growth factor (HGF), tumor necrosis factor-α (TNF-α), angiogenin, interleukin (IL)-8, angiopoietins, angiotensin (ANG)-II, bradykinin, and prostaglandins [1, 13–17]. Negative regulators of angiogenesis were also identified and included factors such as thrombospondin [18], the 16 kDa fragment of prolactin [19], angiostatin [20], endosatin [21], and vasostatin [22].
The increased expression of these angiogenic factors has been demonstrated in several types of cancer and has been associated with increased permeability of the tumor vasculature compared to normal blood vessels [23–25]. Further, angiogenic factors have been associated with structural aberrations of the tumor blood vessels (for review see [26]). The higher permeability of the tumor blood vessels favors the accumulation of macromolecules and lipids in the interstitium of the tumor for extended periods of time. This feature of the tumor vasculature led to the characterization of the enhanced permeability and retention (EPR) effect of macromolecular drugs in solid tumors [27]. The EPR effect is the result of the distinctive vascular permeability of solid tumors [28] and inflammatory tissues [29]. The characterization of this phenomenon allowed the development of the first anticancer nanomedicine by Maeda: styrene co-maleic acid conjugated neocarzinostatin (SMANCS) for the treatment of hepatocellular carcinoma [30]. Following this discovery, several laboratories have developed EPR-based nanomedicine. The main advantage of the EPR-based anticancer nanomedicines is their altered pharmacokinetics caused by their hydrodynamic diameter as it exceeds 7 nm, a size sufficient to escape kidney filtration and urinary excretion [31, 32]. These nanoconstructs can exhibit prolonged circulatory half-life, high area under the concentration/time curve (AUC), and higher partition into tumor tissues [33–36]. Since the first nanomedicine was developed in 1986, the Food and Drug Administration (FDA) and several agencies worldwide have approved over 30 nano-therapeutics for clinical use, 11 of which are for the detection and treatment of various cancers. Despite the improvement in the design and targeting efficiency of these nanomedicines to the tumor site, the transition from the bench to the clinic is particularly slow. In this chapter, we will present an overview of the mechanisms involved in neovascularization, as well as the specific characteristics of the tumor vasculature. We will also discuss the critical considerations that might influence nanomedicine targeting efficiency to solid tumors utilizing tumor vasculature permeability.
Mechanism of Tumor Angiogenesis
The origin of the blood vessel formation is different depending on the biological process which it serves. During embryogenesis, the de novo formation of blood vessel originates from the differentiation of angioblasts into mature endothelial cells and their subsequent assembly into tubes, a process called vasculogenesis [37]. Several angiogenic factors such as VEGF, VEGFR-2, bFGF, and TGF-β influence angioblast differentiation into mature endothelial cells [38–40]. Further development of these native vessels is the result of angiogenesis, a process in which new capillaries emerge by sprouting from existing ones [37]. Distinctive signaling mechanisms will promote either venous or arterial differentiation [41]. The recruitment of periendothelial cells such as vascular smooth muscle cells or pericytes is essential for the maturation of the blood vessel by inhibiting the endothelial cell proliferation and promoting the formation of extracellular matrix [42]. The periendothelial cells can also assist the endothelial cells to acquire specialized functions in different vascular beds [42]. In contrast, neovascularization taking place at postembryonic stage involves essentially angiogenesis as a result of the proliferation and migration of differentiated endothelial cells [13]. With the exception of few physiological processes such as wound healing and the female reproductive cycle where endothelial cells are transiently activated and proliferating, the endothelial cells are largely quiescent in mature vessels. The percentage of endothelial cells entering the cell cycle is only 0.45 % for arteries and arterioles and 0.11 % for capillaries [43].
Tumor expansion is marked by a constitutive activation of the “angiogenic switch” in most cases [11]. The newly formed blood vessels will emerge by sprouting from existing ones and sustain tumor growth [11]. However, recent studies have challenged these conceptions and identified several concomitant mechanisms contributing to the neovascularization of tumors. These mechanisms have been mainly characterized in the tumor vasculature but their contribution remains poorly understood. New blood vessels can emerge from vasculogenic mimicry where tumor cells can line and form a vessel-like structure [44] or putative cancer stem cells can differentiate into an endothelial cell lineage and contribute to angiogenesis [45]. Other studies have demonstrated the capacity of tumor cells to hijack an existing blood vessel, a process known as vessel co-option [46]. In other cases, new blood vessels can arise through intussusceptive angiogenesis where one existing vessel splits into two new vessels [47]. Several studies have also demonstrated the involvement of bone marrow-derived cells for the repair of adult vessels and the expansion of tumor ones. Endothelial cell progenitors can be mobilized from the bone marrow and transported through the blood circulation to become incorporated into the vascular walls of the growing blood vessels [48].
Tumor Vasculature as a Target for Selective Delivery of Nanomedicine
Normal vasculature networks consist of arterioles, capillaries, and venules and form a well-organized network with dichotomous branching and hierarchic order [49]. Newly formed tumor vessels are usually abnormal in form and architecture with narrowed, tortuous, and fragmented blood vessels. In addition, tumor vessels usually lack a smooth muscle layer and innervation, with defective endothelial linings and basement membranes [50]. Some structures are dilated, saccular, poorly aligned, and heterogeneous [51]. Many vascular mediators such as bradykinin [52], prostaglandin [53], nitric oxide (NO) [54], peroxynitrite (ONOO−) [29], matrix metalloproteinases (MMP) [29], and VEGF [55] have been shown to play an important role in these alterations.
As a consequence of these defects, tumor vessels usually harbor wide fenestrations [56]. The blood flow is often irregular with vessels having different diameters and abnormal branching patterns. These tumor vessels are leaky and show increased permeability to large circulating molecules with fenestration sizes ranging from 300 nm to 4,700 nm [57–59]. Furthermore, lymphatic drainage of tumor tissues is generally deficient and limits the clearance of macromolecules [9, 27, 60–62]. Evidence for increased endothelial permeability of tumor vessels to large molecules was clearly demonstrated by Maeda, 26 years ago, using Evans blue dye (EBD) injected intravenously into rodents. After injection the dye bound to albumin in the bloodstream and the complex selectively concentrated into tumor tissue [28]. Additional studies using soluble tracers further demonstrated the extravasation of large molecules from the tumor vessel [63, 64]. EBD extravasation and accumulation in the tumor was the first demonstration the EPR effect concept [28]. The accumulation of nanosize drugs in the tumor tissue is time dependent ranging from several hours to several days [60, 65]. Maeda’s work demonstrated that the rate of accumulation of macromolecules and lipids in the tumor was inversely proportional to their clearance rate. Following SMANCS, the first nanomedicine approved and used for the treatment of hepatocellular carcinoma in Japan (see Table 8.1), several laboratories have developed nanosize drug carriers. But, for the past 20 years, few nanomedicines were approved for the chemotherapeutic treatments of various cancers as well as for their detection (see Table 8.1). Among these formulations, liposome nanocarriers achieved significant success such as Doxil, DaunoXome, Depocyt, and Myocet (Table 8.1). Second- and third-generation types of micellar or polymeric drug carriers are currently being developed or evaluated in clinical trials (phase I–III).
To be efficient for cancer treatment, the size and shape of nanoparticles are critical for their accumulation at the tumor site [66]. Several studies have demonstrated that both criteria are essential for the longevity of the nanomedicine in the circulation, their distribution to different organs [66], as well as their recognition and elimination through the reticuloendothelial cells system (RES) [67]. The RES is composed of macrophages present in the liver, spleen, and bone marrow [67]. Generally, particles larger than 100 nm are rapidly eliminated from the circulation by the RES [68, 69]. To decrease their recognition by macrophages, several strategies have been developed, for instance, the addition of synthetic polymers such as polyethylene glycol (PEG) on the surface to sterically hinder interaction with plasma proteins [70] and reduce opsonization [71].
Overall, nanomedicine advantages over conventional drugs rely on the EPR effect and their improved pharmacokinetics that lower their systemic cytotoxicity. A schematic representation of this phenomenon is illustrated in Fig. 8.1. Furthermore drug delivery nanotechnology allows the controlled release of anticancer drugs and might partly circumvent multidrug resistance mechanisms that involve cell-surface protein pumps [72].

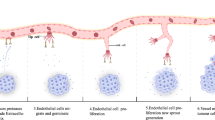
Differences between normal and tumor tissue in relation to the targeting of nanomedicines by the enhanced permeability and retention (EPR) effect. (a) Normal tissue contains tightly connected endothelial cells which prevents the diffusion of the nanomedicine outside the blood vessel. (b) Tumor tissue contains large fenestrates between the endothelial cells allowing the nanomedicines to reach the matrix and the tumor cells by the EPR effect. VEGF and NO secreted by tumor cells, stromal cells, and macrophages increase permeability and stimulate angiogenesis and the migration of endothelial cells towards the tumor. A considerable proportion of the nanomedicine never reaches the tumor either due to entrapment or nonspecific interaction with collagen composing the matrix, or removal through macrophage endocytosis. Nanomedicines tend to concentrate at the periphery of the tumor, only a small proportion will diffuse to the center of the tumor
Factors Contributing to the EPR Effect
Several studies have demonstrated that the EPR effect is dependent on angiogenic factors produced from the tumor cells, stromal cells, or other cell types such as VEGF, bradykinin, nitric oxide, peroxynitrite, and other cytokines [73–75]. All these factors increase blood flow and promote diffusion and retention of nanomedicines inside tumors.
VEGF
The vascular permeability factor or vascular endothelial growth factor (VPF/VEGF) was originally characterized from guinea pig ascites as a secreted protein inducing vascular permeability [76] and was later found in various human tumor cell lines [77]. The same protein was also later identified as a specific and potent vascular endothelial cell mitogen [78]. VEGF is highly expressed in most tumors (2–30-fold higher than normal tissue) and was shown to contribute to the tumor blood vessel structural abnormality [79]. The contribution of VEGF to the EPR effect was demonstrated by Claffey et al. who showed a greater extravasation of large molecules in tumors overexpressing VEGF [80].
VEGF is a homodimeric glycoprotein comprised of two identical subunits [78]. VEGF expression is regulated at the level of transcription by alternative splicing of the VEGF gene and the VEGF165 isoform is the most abundant and assimilated as the native soluble heparin-binding endothelial mitogen activator [78]. Other VEGF isoforms have been identified and arise from different VEGF splicing such as VEGF121, VEGF189 and VEGF206. The VEGF121 isoform is secreted and fully soluble but lacks the heparin binding site, while VEGF189 and VEGF206 are largely sequestered at the cell surface and extracellular matrix and bind avidly to heparin and heparin-like moieties [78, 81]. In addition to the alternative splicing, a proteolytic activation of VEGF has been demonstrated for VEGF165, VEGF189, and VEGF206 following plasmin [82, 83] and matrix metalloproteinase (MMP) activations [84]. These patterns of activation regulate bioavailability and bioactivity and also determine receptor specificities. VEGF acts mainly in a paracrine fashion binding to receptors expressed at the surface of endothelial cells. VEGF165 binds to two receptor tyrosine kinases, VEGFR-1 (Flt-1), VEGFR-2 (KDR or Flk-1), as well as Neuropilin (NRP)-1 and NRP-2, transmembrane glycoproteins [85, 86]. In addition, several VEGF-related genes have been identified including VEGF-B, VEGF-C, VEGF-D, and placenta growth factor (PlGF) [87]. These members of the VEGF gene cluster undergo alternative splicing with the exception of VEGF-C [87]. VEGF-C and VEGF-D were shown to bind to VEGFR-3 (Flt-4) and promote lymphangiogenesis [88].
The expression of VEGF is upregulated by multiple factors including hypoxia. Under hypoxia, hypoxia-inducible factor (HIF)-1α dimerizes with the constitutive HIF-1β to bind to the hypoxic response element (HRE) present in the promoter of the VEGF gene and stimulate its expression [89]. HIF-1α is involved in the activation of transcription of many genes involved in the activation of angiogenesis and other physiologic processes (for review, see [89]).
Bradykinin
Bradykinin (kinin) is a peptide that causes vasodilatation and increased vascular permeability. Bradykinin is generated from its precursor kininogen by limited proteolysis by various serine proteases such as kallikrein, cathepsins, and collagenases [90]. Kininogens are multifunctional glycoproteins mainly synthesized in the liver but also in the kidneys, salivary glands, and endothelial cells [91] and circulate in human plasma in low (50–68 kDa) and high (80–120 kDa) molecular weight forms [92]. Bradykinins’ half-life is a few seconds in the plasma and is rapidly degraded by proteases such as kininase and angiotensin-converting enzyme (ACE) [92]. A greater synthesis of bradykinin has been reported in several types of cancer [52, 93] as well as the expression of bradykinin receptor B2 [94]. The effect of bradykinin appears to be direct as the release of bradykinin triggers vasodilatation and increased vascular permeability as well as indirect as mediated by the production of nitric oxide through the stimulation of the nitric oxide synthase [95], prostaglandins [96], and various cytokines such as interleukin-1, interleukin-6, and interleukin-8 [94]. The permeabilizing action of bradykinin was found to be similar to VEGF but mediated through a different pathway.
Nitric Oxide (NO)
NO is a signaling messenger and contributes to several pathways and biological processes. NO is produced from l-arginine by nitric oxide synthase (NOS) in the presence of oxygen. In pathological conditions such as cancer and inflammatory tissue, NO production is largely increased and plays an important role in the extravasation of large molecules and thus contributes to the EPR effect [54, 60]. Increased NO production was also associated with the overexpression of the inducible form of NOS (iNOS) in the tumor tissues due to the infiltration of leukocytes [97].
Moreover, NO to the same extent as oxidized NO products such as peroxynitrite contributes to the vascular permeability of solid tumor [54]. Peroxynitrite (ONOO−) is a reaction product of NO and anion superoxide O2− [98]. The increased production of peroxynitrite triggers the maturation of pro-matrix metalloproteinases (pro-MMP) into MMPs, which promotes remodeling of the extracellular matrix and contributes to the vascular permeability [29].
The identification of the factors contributing to the EPR effect has resulted in the development of anticancer nanomedicine. However, with the exception of a few clinically approved nanomedicines (see Table 8.1), many nanoconstructs failed to achieve a significant outcome in the clinic. The lack of complete understanding of the EPR effect and its specific biological implications has so far impaired EPR effect-based therapy as a paradigm for cancer treatment. The following describes some of the factors that could account for the slow transition of the EPR-based nanomedicine to an effective cancer treatment.
Animal Models for the EPR Effect
The EPR effect has been repeatedly proven in animal models through the use of large molecules such as the EBD. EBD binds instantly to plasma albumin which results in a large molecular weight complex of about 7 nm diameter that can simulate the effect of a nanomedicine. A diameter larger than 7 nm will escape renal filtration and urinary excretion [31], due to the slit diaphragms at the level of the podocyte foot of the glomerulus which prevent the filtration of globular plasma proteins above this size [32]. Therefore, large particles can exhibit prolonged circulatory half-life, high area under concentration/time curve (AUC), and higher partitioning into tumor tissues [33]. After 6 h, there is usually a distinct accumulation in tumor lesions compared to surrounding tissues. Many nanomedicines have been observed to accumulate in tumor tissue from 2-fold and up to 27-fold more than free drugs depending on the nanocarrier, the drug encapsulated, and the xenograft tumor model used (Fig. 8.2a).

Variability of the different nanocarriers based on their accumulation profile in the tumor, animal models used, and the site of tumor implantation and metastasis in animal model. (a) Comparison of the proportion of the different nanocarriers accumulation in the tumor [184–196]. (b) Comparison of the tumoral accumulation of different nanocarriers based on the animal model used [184, 189, 194, 197–201]. (c) Comparison of accumulation of different nanocarriers based on the site of the tumor implantation either subcutaneous (s.c.), orthotopic, or metastatic [184–197, 202, 203]
The question of whether the results of EPR-based drug targeting in animal models can be faithfully translated to the clinic remains unanswered. Macrophage infiltration has been demonstrated in a large cohort of cancers. The production of VEGF and NO by tumor-associated macrophages (TAM) and their role in cancer development is also well documented [99–101]. To determine the anticancer properties of a given nanomedicine against a specific human cancer, it is necessary to utilize immunocompromised mice to enable the use of human tumor xenografts. However human cancer patients are rarely immunocompromised. A change in macrophage activity in immunocompromised mice [102] can result in less VEGF and NO leading to a tumor with reduced vascular density, which in turn limits the access of the nanoconstructs to the tumor. Furthermore, the results obtained from immunocompromised models differ from results obtained in immunocompetent mice. In various drug delivery systems (conjugates, liposomes, and micelles), the tumor accumulation is a 2-fold higher in immunocompetent mice relative to immunocompromised ones (see Fig. 8.2b). Moreover, immunocompetent mice bear murine tumors and not human cancer cell lines which further complicate interpretation of in vivo animal data and jeopardize its value in predicting the performance of new drugs in clinical trials.
The expression of VEGF and its receptors between commonly used human tumor cell lines and their clinically isolated variants differs. It is clearly evident that tumor cell lines have pronounced expression of VEGF and its receptors with far less variability in comparison to clinical tumors. For example, human breast cancer cell lines MCF-7 and MDA-MB-231 expressed VEGFR-1, VEGFR-2, and VEGFR-3 as well as the ligands VEGF-A, VEGF-C, and VEGF-D with VEGF-B being found only in the MCF-7 cells [103–108]. In contrast, the expression pattern in breast cancer tumors collected from patients is more limited to one specific type of receptor and/or ligand and more importantly not all tumors tested within this cancer type expressed VEGFR and/or its ligand [109–111]. A similar observation was made with prostate cancer and lung cancer.
Relevant to this is the design of nanomedicine targeted to tumors which relies on the conjugation of target ligands that bind strongly to tumor cell-surface receptors to increase cell recognition, cell specificity, and cellular uptake. Galactosamine [112], transferrin [113], and folate [114] have been incorporated in nanomedicine based on the preferential expression of these molecules by cancer cells. Despite promising in vitro studies, these targeted nanomedicines failed to demonstrate significant benefit at the preclinical or clinical level [115]. The discrepancy between the results obtained from testing specific tumor cell lines in tumor models and the clinical trials points further to the sampling errors in generalizing the results of from specific cell line to that of relevant tumors [116].
A substantial difference between tumor models in animals and those of human patients is the progression rate. Animals usually develop a large, clinically relevant tumor (>5 mm) 1 week following subcutaneous (SC) tumor cell inoculation, while such a tumor volume can take years to develop in a human (Table 8.2). This rapid progression rate in animal models results in the overestimation of the targeting role of the EPR effect. Animal tumors developing quickly presumably produce a large quantity of VEGF and vascular mediators to support their rapid growth. In addition a 1 gram tumor mass in a 30 gram mouse is about 3 % of its total weight. In humans, a comparable tumor would weigh 2–5 kg, which is an advanced tumor stage that is not an ideal for utilization of anticancer nanomedicine. Finally, tumors are usually implanted SC in animal models, which allow the developing tumors to take advantage of the extensive cutaneous vascular network for extending their blood supply, a condition that is rarely encountered in human malignancy.
Data collected from available literature to date are plotted in Fig. 8.2c. Although there is a trend towards higher concentration of nanoconstructs in SC models, the results are not conclusive given the limited number of studies. Whether site of tumor development can influence the efficacy of the EPR effect remains an unanswered question.
Tumor Biology Diversity
Tumor Doubling Time (TDT)
Tumor doubling time (TDT) is an important factor to consider when designing EPR-based anticancer nanomedicine. Most cytotoxic drugs selectively target cancers by exploiting differential tumor characteristics such as high proliferation rates, hypoxia, and genome instability. The TDTs provide a selection trait that is exploited by chemotherapeutics that target DNA synthesis and cytoskeleton remodeling. Many chemotherapeutic agents fail to cope with rapidly dividing tumors as the amount of drug necessary to kill a given number of cells will double with each tumor doubling. However, the dose that will elicit dose-limiting toxicity will remain the same. A short TDT is well known to be associated with an unfavorable survival prognosis [117–122]. TDT is a highly heterogeneous, both within and between different tumor types, stages, and grades. There is a large degree of variation of TDT between tumors of different tissue origins. Pituitary adenoma, for example, has an extremely long TDT of 506–5,378 days and within the tumor type the TDT varies by ten times [123], while in meningiomas and neurinomas the TDTs are 6.5 days and 7.67 days, respectively [124]. Some tumor types have a high variation of the TDT, for instance, lung adenocarcinoma has an extremely high variation in TDT of 964-fold [125] followed by breast cancer with a variation of 117.5-fold [126] (see Table 8.2). TDT can also differ according to the specific cellular origin within a given tissue. Bronchoalveolar cancer, for example, has an extremely varied TDT of 36–1,092 days, a variation of 30.3-fold [125], while small cell lung cancer has a TDT of 61.9–120.4, a mere 1.9-fold difference [127]. In addition, TDT can range depending on tumor grade (see Table 8.2). Poorly differentiated hepatocellular carcinomas corresponding to the Edmonson grade III or IV are highly invasive and have a DT of 13–239 days [128], while well-differentiated tumors corresponding to Edmondson grade I or I–II has a significantly extended TDT of 54.7–1,508.3 days [128] (see Table 8.2). Interestingly, hepatocellular carcinomas are highly vascularized and the microvessel density (MVD) is not affected by the tumor grade (see Table 8.2). Astrocytoma also follows this trend with the TDT of grade IV astrocytoma, according to the WHO grading system, varying between 1.4 and 319 days, the TDT of grade III 30–472 days and the TDT of grade I–II tumors 138–1,045 [129, 130]. The tumor grade also correlated with the MVD, with higher grade having a higher MVD (see Table 8.2). The same trend was observed with prostate cancer and breast cancer where a high grade correlates with a lower doubling time and a higher MVD (Table 8.2).
The primary or metastatic status of a tumor can also cause large fluctuations. For example, primary melanoma may have a DT of 50–377 days [131], while metastatic melanoma may have a DT of 8–212 days [132].
EPR-based anticancer nanomedicine should consider doubling time variation when planning the release mechanism of active chemotherapeutic agents from its nanocarrier, as well as the internalization rate of macromolecular complexes into tumor cells. For example, a slow-releasing amide bond between the polymer backbone and the drug, or slowly internalized liposome, could both be a good choice for tumors with a slow DT. In contrast a fast-releasing micelle or an ester bond linkage can be a better fit for rapidly dividing tumors. Generally, EPR-based nanomedicine has a wider therapeutic window [133], an advantage that can be exploited to shape dose regimens based on individual patient conditions. A tumor’s inherent sensitivity to specific chemotherapeutic agents as well as TDT is of the upmost importance in designing EPR-based anticancer nanomedicine.
Microvascular Density (MVD)
The EPR effect is strictly dependent on the vasculature of the tumor with theoretical assumption that all tumors independently of their origin, stage, and organs will behave identically. However, this concept is drastically challenged by a number of reports that show a high diversity in angiogenesis behavior [12, 51, 134, 135]. Nagy et al. have identified six structurally and functionally distinct types of blood vessels in human cancers [134]. Vascular density can provide, in most tumors, a prognostic indication of tumor progression. As shown in Table 8.2, vascular density is largely dependent on the type of cancer and varies largely within each tumor type. For instance, renal cell carcinoma is highly vascularized [136], while the density of microvessels appears low in head and neck squamous cell carcinoma [137] or in ovarian carcinoma [138]. In addition, higher stages of cancer are well correlated with higher microvascular density as observed in astrocytoma and prostate cancer (Table 8.2), while in other types of tumors such as renal cell carcinoma, no direct correlation can be established between tumor stage and vascular density (Table 8.2). Furthermore, metastatic tumors tend to possess higher vascular density compared to non-metastatic tumors [139–143]. Another element regarding the EPR effect is the secretion of angiogenic factors such as VEGF by the tumor. Vascular permeability can be altered by VEGF as well as a wide array of inflammatory mediators [144], which can affect the extent of nanomedicine accumulation driven by the EPR effect and the penetration of the nanoconstruct into the tumor. As mentioned previously, there is a large heterogeneity in the expression of VEGF between different types of cancers. When designing a nanocarrier, the properties of the targeted tumor tissue such as the cancer type, the microvascular density, and the secretion of permeability factors such as VEGF should therefore be taken into account in order to take full advantage of the EPR phenomenon.
Optimization of Drug Nanocarriers for the EPR Effect
To optimize the engineering of nanoparticles for specific delivery, careful consideration should be undertaken regarding the biology of the tissue being targeted. In many instances, the nature of the nanomedicine itself has been a limiting factor that negatively impacted its chance of clinical success. The loading of active drug into a delivery system can be insufficient due to the physical or chemical limitations to achieve the critical dose needed to treat the tumor. For example, the HPMA copolymer-paclitaxel conjugate showed insufficient drug loading (≤10 %) with a particle size in the range of 12–15 nm [145] and lacked stability due to the use of an ester linker [146]. Consequently insufficient tumor tissue accumulation of the drug was evident in phase I clinical trials [147]. Another factor limiting the efficacy of nanomedicines is the fast release rate of drug in the circulation. For instance, low molecular weight HPMA copolymer-camptothecin conjugate showed a rapid release of drug and quick renal filtration and consequent bladder toxicity in phase I clinical trials [148]. The nanomedicine was designed with a labile ester linker, decreasing its stability and therefore its tumor accumulation [149]. In this example, a low molecular weight (below the renal excretory threshold of 7 nm) coupled to the toxicity associated with a fast release rate resulted in the drug failing to achieve EPR-based pharmacokinetics.
Following are a few considerations inherent to the design of nanomedicine that may significantly influence the outcome of EPR-based drug targeting (see Fig. 8.3).

Schematic representation of the variables influencing the clinical application of a nanomedicine. The biocompatibility, internalization, and release should be carefully considered when designing a nanomedicine utilizing tumor vascular abnormalities for targeted cancer treatment
Internalization of the Nanocarrier
The concentration of drug inside the tumor resulting from the EPR effect in a subset of highly vascularized tumors does not guarantee the efficient internalization of the drug within the tumor cells. Multiple factors can influence the cellular internalization process of the nanomedicine. Usually, nanoparticles and polymer-based drug delivery systems are internalized by endocytosis, a multistep process that culminates in the formation of a late endosome which finally fuses with a lysosome [150]. Malignant cells have an accelerated metabolism, a high glucose requirement, and an increased glucose uptake characterized by the elevated expression of glucose transporter proteins (GLUT) [151]. However, recent studies have shown that many cancer cell lines exhibit limited capacity for endocytosis compared to normal cells [152, 153].
Compared to tumor cells, macrophages usually exhibit a higher uptake of nanosized molecules [66, 154] as they can recognize nanomedicine either through their Toll-like receptor 4 (TLR-4) [155] or through scavenger receptors [156]. Much work has therefore been devoted to the development of nanoparticles which can evade macrophage recognition, resulting in longer circulatory time and increased interaction with target tissue. On this basis, polyethylene glycol (PEG) is the polymer most commonly used to enhance in vivo circulatory half-life [157, 158]. Coating nanoparticles with PEG results in the formation of a polymeric layer which sterically hinders the interaction of nanoparticles with plasma proteins and cell membranes [159] preventing opsonization and phagocytosis by components of the RES [160, 161]. PEG-liposome-incorporated doxorubicin (Doxil®) is approved by the FDA for the treatment of ovarian cancer (see Table 8.1). Additional polymers such as N-(2-hydroxypropyl) methylacrylamide (HPMA), polyacrylamide, or poly(vinyl pyrrolidone) have also been used to improve the circulation time and steric hindrance of nanomedicines [162, 163]. The main disadvantage of this strategy is that it limits the interaction of (stealth) nanoconstructs with the tumor cell membrane and subsequently reduced internalization and uptake by tumor cells. To improve specific uptake by endocytosis, several nanoparticles have been coated with receptor ligands such as folate [164] or transferrin [165] to induce receptor-mediated endocytosis. These coatings increased the accumulation of drug inside tumor cells. However, the practical advantages in the management of human tumors in the clinic remain to be proven. Following intracellular internalization, active drug should be liberated from the lysosomal compartment to reach its cellular target. Mechanisms to escape the lysosomal compartment and improve intracellular targeted delivery have been described by Breunig et al. [166]. Another consideration relevant to relatively large sized macromolecular nanomedicine is their nonspecific interactions with the extracellular matrix; to reach tumor cells, nanoconstructs must move through the matrix, a highly interconnected network of collagen fibers that intermingle with proteins such as proteoglycans and glycosaminoglycans. This semisolid barrier could significantly reduce the amount of nanomedicine reaching tumor cells, either through nonspecific interaction (Fig. 8.1) or simply by impeding convection movements of relatively large sized nanoconstructs [167]. This could lead to nanomedicine being locally concentrated in proximity to the capillary that it leaked from without reaching the target tumor cells.
Recently, several studies have developed methods to circumvent these limitations such as using of the tumor penetrating peptide, iRGD, which has been shown to increase the delivery of nanomedicines in solid tumors by improving its interstitial transport [168]. Additional therapeutic strategies aiming to normalize the tumor vasculature and extracellular matrix in order to improve tumoral penetration of the drug have been described (for review [51, 169]).
Release Rate
Conjugates can be synthesized through covalent linking of drugs to polymeric carriers such as SMANCS (Table 8.1) [30]. In comparison, entrapment of drug inside a micellar structure requires either covalent or non-covalent bonds (ionic, hydrogen bonds, or hydrophobic) and involves a block polymer or copolymer. Various chemical bonds such as amide, ester, azide, imine, hydrazone, thioether, and urethane are currently used to prepare nanomedicines [29, 170]. Based on the nature of these chemical bonds, the release of drug from its carrier can depend on either pH, usually acidic pH of the lysosome [170], temperature [70], or on enzymatic cleavage [171]. Furthermore, the nature of this bond will determine the release rate; for example, an ester bond ensures a rapid release of drug due to an abundance of esterases in plasma, whereas an amide bond will show a slower release profile [29, 148]. The comparison of release rates between polymer conjugates, liposomes, and micelles nanomedicines after 24 h incubation (Fig. 8.4a) in different studies showed a distinctive profile. Overall, polymer conjugates and micelles have a comparable release rate which is higher than that of liposomes (Fig. 8.4a). The release profile of liposomes appears relatively homogeneous with a mean value of 24 % (3–39 %), while the release profile of conjugates and micelles appears heterogeneous across several studies with a mean value of 39 % (2.5–100 %) and 41 % (2.5–100 %), respectively.

Variation of nanocarrier release rate and accumulation profile. (a) Determination of the release rate over a 24 h period of different nanocarriers across several studies [162, 204–212]. (b) Comparison of the proportion of the different nanocarriers accumulation in the tumor and various organs [184–197, 202, 203]
In order for a nanocarrier to provide tumor targeting, the carrier should have a stable chemical bond with the cargo drug while in circulation. This prevents the rapid release of free drug and permits a therapeutic effect at the site of action. A rapid drug release from its delivery system in plasma can result in a biodistribution and toxicity profile comparable to its related free drug. In contrast, engineering a stable linkage between the drug and its carrier can result in a slow release rate at the target site and inability to reach the critical therapeutic concentration. The release rate of nanoconstructs needs to be tailored for the treatment of a specified tumor doubling time (see Table 8.2). Thus, the choice of a specific linker is critical for a favorable anticancer outcome of EPR-targeted nanosystems.
Biocompatibility
The EPR-based accumulation of active drug inside the tumor rarely exceeds 5 % of the total dose of nanomedicine administrated by i.v. injection. The majority of the injected dose accumulates in various organs such as liver and spleen and to a minor extent kidneys and lungs [172]. As nanomedicines reach sizes of 7 nm, classical pharmacokinetics cannot be accurately applied due to two drastic changes. Firstly, nanosized drugs cannot be eliminated by renal glomeruli as they exceed the renal threshold of excretion dictated by the pore size in the glomeruli [32, 173]. Secondly, their organ distribution is limited to tissues that have capillaries with large enough endothelial fenestrations to allow macromolecular drugs to pass through [32, 33]. The EPR effect utilizes the unique characteristic of large gaps between endothelial cells that makes up tumor vessels. Usually these gaps can vary from few nanometers to up to 1200 nm in size [174, 175]. At this large size, nanomedicines can preferentially accumulate in tumor tissues. However, tumors are not the only organs with such large fenestrae as the spleen and liver show similar characteristics. Liver sinusoid can have fenestrae of around 100 nm in humans [176], whereas the spleen has large sinusoid lumina of ~ 5 μm that can support extravasation of aged red blood cells [32]. With such a large fenestration size, great amounts of nanosized drugs accumulate in these organs. As shown in Fig. 8.4b meta-analysis of 73 studies over the last 10 years revealed that with all the EPR effect-based nanoconstructs that were used, the liver and spleen were the two major organs competing with tumor for the nanoconstructs (conjugate, micelles, and liposomes). While spleen function can be compensated for by other lymphatic organs, liver damage due to the concentration of cytotoxic nanomedicine remains a challenge to successful anticancer drug targeting. For example, nanoconstructs of cis-platinum have reduced toxicity in the kidney compared to the free drug but result in a dose-limiting liver toxicity [177].
Surface modification of the nanomedicine, such as PEGylation, may increase their retention in the systemic circulation and favor tumor accumulation. However, more than 90 % of PEGylated nanoparticles will still be removed through liver clearance within several hours of administration. Studies have demonstrated that as little as ~2 % of the total i.v. administered dose was found in the tumor after 4 h [172]. Thus there is a legitimate safety concern regarding the off target accumulation of the drug delivery system. Ideally, the drug carrier should be eliminated after drug release. But, unless the nanocarrier is biodegradable, it will remain in the body and be dealt with as a foreign body. The innate elements of the immune system could be stimulated nonspecifically by these foreign bodies through TLR-4 [155]. Activated macrophages will phagocytose and attempt to degrade the nanocarrier in its lysosomal compartment. Failure to do so may lead to the formation of foreign body giant cells caused by fusion of multiple macrophages or monocytes [178] and ultimately to the formation of lesions resembling granulomas [179]. This can potentially result in the pathological formation of a dense fibrous capsule replacing the original functional tissue. Another concern in relation to the accumulation of nondegradable materials is the induction of malignancy resulting from frustrated phagocytosis and prolonged inflammation [180].
To address these issues, recent work has focused on the development of biodegradable and nonimmunologic drug delivery systems containing either enzymatically or reductively degradable spacers such as poly(-d,l-lactide-co-glycoside) (PLGA) [181] or the star HPMA polymer carrier which enable a controlled degradation of the drug carrier [182, 183]. Some of these carriers demonstrate prolonged blood circulation and tumor drug accumulation but difficulties in the reproducibility of their synthesis could hamper further clinical development [182].
To summarize, EPR-related parameters of a nanoparticle delivery platform such as long circulatory half-life, reduced elimination, and altered distribution could be a double-edged sword. Careful consideration of these parameters is essential for effective, safe, and more personalized cancer treatments.
Conclusion
As a general concept of tumor vasculatures, vascular permeability has allowed the development of a variety of anticancer nanomedicines. In theory, the nanomedicine should decrease systemic toxicity and improve the delivery to the tumor site. However, despite high expectations for this targeting strategy, over the last 26 years, only a few nanomedicines have successfully exploited this concept and made the transition to the clinic. Possible reasons for this slow transition are the lack of control of essential parameters for a good delivery such as release rate, internalization, and biocompatibility. Moreover, the variability of tumors biology such as doubling time and microvascular density can influence the targeting potential of EPR-based nanomedicine. Consideration of these variables as well as the development of modular delivery systems of macromolecules can significantly hasten the transition of anticancer nanomedicine towards clinical application.
Abbreviations
- EPR:
-
Enhanced permeability and retention
- VEGF:
-
Vascular endothelial growth factor
- VEGFR:
-
Vascular endothelial growth factor receptor
- bFGF:
-
Basic fibroblast growth factor
- TGF:
-
Tumor growth factor
- MMP:
-
Matrix metalloproteinases
- NO:
-
Nitric oxide
- EBD:
-
Evans blue dye
- SMANCS:
-
Styrene co-maleic acid conjugated neocarzinostatin
- TDT:
-
Tumor doubling time
- HPMA:
-
N-(2-hydroxypropyl)methacrylamide
- RES:
-
Reticuloendothelial system
References
Risau W (1997) Mechanisms of angiogenesis. Nature 386(6626):671–674. doi:10.1038/386671a0
Folkman J (1995) Angiogenesis in cancer, vascular, rheumatoid and other disease. Nat Med 1(1):27–31
Heidenreich R, Rocken M, Ghoreschi K (2009) Angiogenesis drives psoriasis pathogenesis. Int J Exp Pathol 90(3):232–248. doi:10.1111/j.1365-2613.2009.00669.x
Veritti D, Sarao V, Lanzetta P (2012) Neovascular age-related macular degeneration. Ophthalmologica 227(Suppl 1):11–20. doi:10.1159/000337154
Hahn WC, Weinberg RA (2002) Rules for making human tumor cells. N Engl J Med 347(20):1593–1603. doi:10.1056/NEJMra021902
Paget S (1889) The distribution of secondary growths in cancer of the breast. Lancet 133(3421):571–573. doi:10.1016/s0140-6736(00)49915-0
Greenblatt M, Shubi P (1968) Tumor angiogenesis: transfilter diffusion studies in the hamster by the transparent chamber technique. J Natl Cancer Inst 41(1):111–124
Ehrmann RL, Knoth M (1968) Choriocarcinoma. Transfilter stimulation of vasoproliferation in the hamster cheek pouch. Studied by light and electron microscopy. J Natl Cancer Inst 41(6):1329–1341
Folkman J (1971) Tumor angiogenesis: therapeutic implications. N Engl J Med 285(21):1182–1186. doi:10.1056/NEJM197111182852108
Folkman J (1972) Anti-angiogenesis: new concept for therapy of solid tumors. Ann Surg 175(3):409–416
Hanahan D, Folkman J (1996) Patterns and emerging mechanisms of the angiogenic switch during tumorigenesis. Cell 86(3):353–364
Hanahan D, Weinberg Robert A (2011) Hallmarks of cancer: the next generation. Cell 144(5):646–674. doi:10.1016/j.cell.2011.02.013
Folkman J, Shing Y (1992) Angiogenesis. J Biol Chem 267(16):10931–10934
Maisonpierre PC, Suri C, Jones PF, Bartunkova S, Wiegand SJ, Radziejewski C, Compton D, McClain J, Aldrich TH, Papadopoulos N, Daly TJ, Davis S, Sato TN, Yancopoulos GD (1997) Angiopoietin-2, a natural antagonist for tie2 that disrupts in vivo angiogenesis. Science 277(5322):55–60
Escobar E, Rodriguez-Reyna TS, Arrieta O, Sotelo J (2004) Angiotensin ii, cell proliferation and angiogenesis regulator: biologic and therapeutic implications in cancer. Curr Vasc Pharmacol 2(4):385–399
Ishihara K, Kamata M, Hayashi I, Yamashina S, Majima M (2002) Roles of bradykinin in vascular permeability and angiogenesis in solid tumor. Int Immunopharmacol 2(4):499–509. doi:10.1016/s1567-5769(01)00193-x
Dempke W, Rie C, Grothey A, Schmoll HJ (2001) Cyclooxygenase-2: a novel target for cancer chemotherapy? J Cancer Res Clin Oncol 127(7):411–417
Good DJ, Polverini PJ, Rastinejad F, Le Beau MM, Lemons RS, Frazier WA, Bouck NP (1990) A tumor suppressor-dependent inhibitor of angiogenesis is immunologically and functionally indistinguishable from a fragment of thrombospondin. Proc Natl Acad Sci USA 87(17):6624–6628
Ferrara N, Clapp C, Weiner R (1991) The 16k fragment of prolactin specifically inhibits basal or fibroblast growth factor stimulated growth of capillary endothelial cells. Endocrinology 129(2):896–900
O’Reilly MS, Holmgren L, Shing Y, Chen C, Rosenthal RA, Moses M, Lane WS, Cao Y, Sage EH, Folkman J (1994) Angiostatin: a novel angiogenesis inhibitor that mediates the suppression of metastases by a lewis lung carcinoma. Cell 79(2):315–328. doi:10.1016/0092-8674(94)90200-3
O’Reilly MS, Boehm T, Shing Y, Fukai N, Vasios G, Lane WS, Flynn E, Birkhead JR, Olsen BR, Folkman J (1997) Endostatin: an endogenous inhibitor of angiogenesis and tumor growth. Cell 88(2):277–285
Pike SE, Yao L, Jones KD, Cherney B, Appella E, Sakaguchi K, Nakhasi H, Teruya-Feldstein J, Wirth P, Gupta G, Tosato G (1998) Vasostatin, a calreticulin fragment, inhibits angiogenesis and suppresses tumor growth. J Exp Med 188(12):2349–2356
Peterson HI, Appelgren KL (1973) Experimental studies on the uptake and rentention of labelled proteins in a rat tumour. Eur J Cancer 9(8):543–547
Ward JD, Hadfield MG, Becker DP, Lovings ET (1974) Endothelial fenestrations and other vascular alterations in primary melanoma of the central nervous system. Cancer 34(6):1982–1991
Ackerman NB, Hechmer PA (1978) Studies on the capillary permeability of experimental liver metastases. Surg Gynecol Obstet 146(6):884–888
Nagy JA, Chang SH, Dvorak AM, Dvorak HF (2009) Why are tumour blood vessels abnormal and why is it important to know[quest]. Br J Cancer 100(6):865–869
Maeda H, Matsumura Y (1989) Tumoritropic and lymphotropic principles of macromolecular drugs. Crit Rev Ther Drug Carrier Syst 6(3):193–210
Matsumura Y, Maeda H (1986) A new concept for macromolecular therapeutics in cancer chemotherapy: mechanism of tumoritropic accumulation of proteins and the antitumor agent smancs. Cancer Res 46(12 Part 1):6387–6392
Greish K, Fang J, Inutsuka T, Nagamitsu A, Maeda H (2003) Macromolecular therapeutics: advantages and prospects with special emphasis on solid tumour targeting. Clin Pharmacokinet 42(13):1089–1105
Maeda H, Takeshita J, Kanamaru R (1979) A lipophilic derivative of neocarzinostatin. A polymer conjugation of an antitumor protein antibiotic. Int J Pept Protein Res 14(2):81–87
Choi HS, Liu W, Misra P, Tanaka E, Zimmer JP, Itty Ipe B, Bawendi MG, Frangioni JV (2007) Renal clearance of quantum dots. Nat Biotechnol 25(10):1165–1170. doi:10.1038/nbt1340
Sarin H (2010) Physiologic upper limits of pore size of different blood capillary types and another perspective on the dual pore theory of microvascular permeability. J Angiogenes Res 2:14. doi:10.1186/2040-2384-2-14
Greish K (2007) Enhanced permeability and retention of macromolecular drugs in solid tumors: a royal gate for targeted anticancer nanomedicines. J Drug Target 15(7–8):457–464. doi:10.1080/10611860701539584
Zhu Y, Che L, He H, Jia Y, Zhang J, Li X (2011) Highly efficient nanomedicines assembled via polymer–drug multiple interactions: tissue-selective delivery carriers. J Control Release 152(2):317–324. doi:10.1016/j.jconrel.2011.03.013
Immordino ML, Dosio F, Cattel L (2006) Stealth liposomes: review of the basic science, rationale, and clinical applications, existing and potential. Int J Nanomedicine 1(3):297–315
Greish K (2010) Enhanced permeability and retention (epr) effect for anticancer nanomedicine drug targeting. Methods Mol Biol 624:25–37. doi:10.1007/978-1-60761-609-2_3
Risau W, Flamme I (1995) Vasculogenesis. Annu Rev Cell Dev Biol 11:73–91. doi:10.1146/annurev.cb.11.110195.000445
Ferrara N (1999) Role of vascular endothelial growth factor in the regulation of angiogenesis. Kidney Int 56(3):794–814. doi:10.1046/j.1523-1755.1999.00610.x
Carmeliet P, Ferreira V, Breier G, Pollefeyt S, Kieckens L, Gertsenstein M, Fahrig M, Vandenhoeck A, Harpal K, Eberhardt C, Declercq C, Pawling J, Moons L, Collen D, Risau W, Nagy A (1996) Abnormal blood vessel development and lethality in embryos lacking a single vegf allele. Nature 380(6573):435–439. doi:10.1038/380435a0
Patel-Hett S, D’Amore PA (2011) Signal transduction in vasculogenesis and developmental angiogenesis. Int J Dev Biol 55(4–5):353–363. doi:10.1387/ijdb.103213sp
Swift MR, Weinstein BM (2009) Arterial–venous specification during development. Circ Res 104(5):576–588. doi:10.1161/circresaha.108.188805
Carmeliet P, Jain RK (2011) Molecular mechanisms and clinical applications of angiogenesis. Nature 473(7347):298–307
Hobson B, Denekamp J (1984) Endothelial proliferation in tumours and normal tissues: continuous labelling studies. Br J Cancer 49(4):405–413
Hendrix MJC, Seftor EA, Hess AR, Seftor REB (2003) Vasculogenic mimicry and tumour-cell plasticity: lessons from melanoma. Nat Rev Cancer 3(6):411–421
Wang R, Chadalavada K, Wilshire J, Kowalik U, Hovinga KE, Geber A, Fligelman B, Leversha M, Brennan C, Tabar V (2010) Glioblastoma stem-like cells give rise to tumour endothelium. Nature 468(7325):829–833, doi:http://www.nature.com/nature/journal/v468/n7325/abs/nature09624.html#supplementary-information
Holash J, Maisonpierre PC, Compton D, Boland P, Alexander CR, Zagzag D, Yancopoulos GD, Wiegand SJ (1999) Vessel cooption, regression, and growth in tumors mediated by angiopoietins and vegf. Science 284(5422):1994–1998. doi:10.1126/science.284.5422.1994
De Spiegelaere W, Casteleyn C, Van den Broeck W, Plendl J, Bahramsoltani M, Simoens P, Djonov V, Cornillie P (2012) Intussusceptive angiogenesis: a biologically relevant form of angiogenesis. J Vasc Res 49(5):390–404. doi:10.1159/000338278
Rafii S, Heissig B, Hattori K (2002) Efficient mobilization and recruitment of marrow-derived endothelial and hematopoietic stem cells by adenoviral vectors expressing angiogenic factors. Gene Ther 9(10):631–641. doi:10.1038/sj.gt.3301723
Herbert SP, Stainier DY (2011) Molecular control of endothelial cell behaviour during blood vessel morphogenesis. Nat Rev Mol Cell Biol 12(9):551–564. doi:10.1038/nrm3176
Brown JM, Giaccia AJ (1998) The unique physiology of solid tumors: opportunities (and problems) for cancer therapy. Cancer Res 58(7):1408–1416
Goel S, Duda DG, Xu L, Munn LL, Boucher Y, Fukumura D, Jain RK (2011) Normalization of the vasculature for treatment of cancer and other diseases. Physiol Rev 91(3):1071–1121. doi:10.1152/physrev.00038.2010
Maeda H, Matsumura Y, Kato H (1988) Purification and identification of [hydroxyprolyl3]bradykinin in ascitic fluid from a patient with gastric cancer. J Biol Chem 263(31):16051–16054
Greenhough A, Smartt HJM, Moore AE, Roberts HR, Williams AC, Paraskeva C, Kaidi A (2009) The cox-2/pge2 pathway: key roles in the hallmarks of cancer and adaptation to the tumour microenvironment. Carcinogenesis 30(3):377–386. doi:10.1093/carcin/bgp014
Maeda H, Noguchi Y, Sato K, Akaike T (1994) Enhanced vascular permeability in solid tumor is mediated by nitric oxide and inhibited by both new nitric oxide scavenger and nitric oxide synthase inhibitor. Cancer Sci 85(4):331–334. doi:10.1111/j.1349-7006.1994.tb02362.x
Dvorak HF, Nagy JA, Feng D, Brown LF, Dvorak AM (1999) Vascular permeability factor/vascular endothelial growth factor and the significance of microvascular hyperpermeability in angiogenesis. Curr Top Microbiol Immunol 237:97–132
Roberts WG, Palade GE (1997) Neovasculature induced by vascular endothelial growth factor is fenestrated. Cancer Res 57(4):765–772
Dvorak HF, Nagy JA, Dvorak JT, Dvorak AM (1988) Identification and characterization of the blood vessels of solid tumors that are leaky to circulating macromolecules. Am J Pathol 133(1):95–109
Yuan F, Leunig M, Huang SK, Berk DA, Papahadjopoulos D, Jain RK (1994) Mirovascular permeability and interstitial penetration of sterically stabilized (stealth) liposomes in a human tumor xenograft. Cancer Res 54(13):3352–3356
Fang J, Nakamura H, Maeda H (2011) The epr effect: unique features of tumor blood vessels for drug delivery, factors involved, and limitations and augmentation of the effect. Adv Drug Deliv Rev 63(3):136–151
Noguchi Y, Wu J, Duncan R, Strohalm J, Ulbrich K, Akaike T, Maeda H (1998) Early phase tumor accumulation of macromolecules: a great difference in clearance rate between tumor and normal tissues. Jpn J Cancer Res 89(3):307–314
Padera TP, Kadambi A, di Tomaso E, Carreira CM, Brown EB, Boucher Y, Choi NC, Mathisen D, Wain J, Mark EJ, Munn LL, Jain RK (2002) Lymphatic metastasis in the absence of functional intratumor lymphatics. Science 296(5574):1883–1886. doi:10.1126/science.1071420
Leu AJ, Berk DA, Lymboussaki A, Alitalo K, Jain RK (2000) Absence of functional lymphatics within a murine sarcoma: a molecular and functional evaluation. Cancer Res 60(16):4324–4327
Kohn S, Nagy JA, Dvorak HF, Dvorak AM (1992) Pathways of macromolecular tracer transport across venules and small veins. Structural basis for the hyperpermeability of tumor blood vessels. Lab Invest 67(5):596–607
Lichtenbeld HC, Yuan F, Michel CC, Jain RK (1996) Perfusion of single tumor microvessels: application to vascular permeability measurement. Microcirculation 3(4):349–357
Seymour LW, Miyamoto Y, Maeda H, Brereton M, Strohalm J, Ulbrich K, Duncan R (1995) Influence of molecular weight on passive tumour accumulation of a soluble macromolecular drug carrier. Eur J Cancer 31A(5):766–770
Arnida J-AMM, Ray A, Peterson CM, Ghandehari H (2011) Geometry and surface characteristics of gold nanoparticles influence their biodistribution and uptake by macrophages. Eur J Pharm Biopharm 77(3):417–423. doi:10.1016/j.ejpb.2010.11.010
Moghimi SM, Hunter AC, Murray JC (2001) Long-circulating and target-specific nanoparticles: theory to practice. Pharmacol Rev 53(2):283–318
Davis ME, Chen ZG, Shin DM (2008) Nanoparticle therapeutics: an emerging treatment modality for cancer. Nat Rev Drug Discov 7(9):771–782. doi:10.1038/nrd2614
Decuzzi P, Pasqualini R, Arap W, Ferrari M (2009) Intravascular delivery of particulate systems: does geometry really matter? Pharm Res 26(1):235–243. doi:10.1007/s11095-008-9697-x
Torchilin VP (2007) Micellar nanocarriers: pharmaceutical perspectives. Pharm Res 24(1):1–16. doi:10.1007/s11095-006-9132-0
Brigger I, Dubernet C, Couvreur P (2002) Nanoparticles in cancer therapy and diagnosis. Adv Drug Deliv Rev 54(5):631–651
Minko T, Dharap SS, Pakunlu RI, Wang Y (2004) Molecular targeting of drug delivery systems to cancer. Curr Drug Targets 5(4):389–406
Pinto MP, Badtke MM, Dudevoir ML, Harrell JC, Jacobsen BM, Horwitz KB (2010) Vascular endothelial growth factor secreted by activated stroma enhances angiogenesis and hormone-independent growth of estrogen receptor-positive breast cancer. Cancer Res 70(7):2655–2664. doi:10.1158/0008-5472.can-09-4373
Dvorak HF (2002) Vascular permeability factor/vascular endothelial growth factor: a critical cytokine in tumor angiogenesis and a potential target for diagnosis and therapy. J Clin Oncol 20(21):4368–4380
Maeda H, Fang J, Inutsuka T, Kitamoto Y (2003) Vascular permeability enhancement in solid tumor: various factors, mechanisms involved and its implications. Int Immunopharmacol 3(3):319–328. doi:10.1016/s1567-5769(02)00271-0
Senger D, Galli S, Dvorak A, Perruzzi C, Harvey V, Dvorak H (1983) Tumor cells secrete a vascular permeability factor that promotes accumulation of ascites fluid. Science 219(4587):983–985. doi:10.1126/science.6823562
Senger DR, Perruzzi CA, Feder J, Dvorak HF (1986) A highly conserved vascular permeability factor secreted by a variety of human and rodent tumor cell lines. Cancer Res 46(11):5629–5632
Ferrara N, Henzel W (1989) Pituitary follicular cells secrete a novel heparin-binding growth factor specific for vascular endothelial cells. Biochem Biophys Res Commun 161(2):851–858
Jain RK (2005) Normalization of tumor vasculature: an emerging concept in antiangiogenic therapy. Science 307(5706):58–62. doi:10.1126/science.1104819
Claffey KP, Brown LF, del Aguila LF, Tognazzi K, Yeo KT, Manseau EJ, Dvorak HF (1996) Expression of vascular permeability factor/vascular endothelial growth factor by melanoma cells increases tumor growth, angiogenesis, and experimental metastasis. Cancer Res 56(1):172–181
Leung D, Cachianes G, Kuang W, Goeddel D, Ferrara N (1989) Vascular endothelial growth factor is a secreted angiogenic mitogen. Science 246(4935):1306–1309. doi:10.1126/science.2479986
Houck KA, Leung DW, Rowland AM, Winer J, Ferrara N (1992) Dual regulation of vascular endothelial growth factor bioavailability by genetic and proteolytic mechanisms. J Biol Chem 267(36):26031–26037
Park JE, Keller GA, Ferrara N (1993) The vascular endothelial growth factor (vegf) isoforms: differential deposition into the subepithelial extracellular matrix and bioactivity of extracellular matrix-bound vegf. Mol Biol Cell 4(12):1317–1326
Lee S, Jilani SM, Nikolova GV, Carpizo D, Iruela-Arispe ML (2005) Processing of vegf-a by matrix metalloproteinases regulates bioavailability and vascular patterning in tumors. J Cell Biol 169(4):681–691. doi:10.1083/jcb.200409115
Geretti E, Shimizu A, Klagsbrun M (2008) Neuropilin structure governs vegf and semaphorin binding and regulates angiogenesis. Angiogenesis 11(1):31–39. doi:10.1007/s10456-008-9097-1
Soker S, Fidder H, Neufeld G, Klagsbrun M (1996) Characterization of novel vascular endothelial growth factor (vegf) receptors on tumor cells that bind vegf via its exon 7-encoded domain. J Biol Chem 271(10):5761–5767. doi:10.1074/jbc.271.10.5761
Yamazaki Y, Morita T (2006) Molecular and functional diversity of vascular endothelial growth factors. Mol Divers 10(4):515–527. doi:10.1007/s11030-006-9027-3
Alitalo K, Tammela T, Petrova TV (2005) Lymphangiogenesis in development and human disease. Nature 438(7070):946–953. doi:10.1038/nature04480
Semenza GL (2003) Targeting hif-1 for cancer therapy. Nat Rev Cancer 3(10):721–732. doi:10.1038/nrc1187
Maeda H (2012) Vascular permeability in cancer and infection as related to macromolecular drug delivery, with emphasis on the epr effect for tumor-selective drug targeting. Proc Jpn Acad Ser B Phys Biol Sci 88(3):53–71
Bhoola KD, Figueroa CD, Worthy K (1992) Bioregulation of kinins: kallikreins, kininogens, and kininases. Pharmacol Rev 44(1):1–80
Leeb-Lundberg LMF, Marceau F, Müller-Esterl W, Pettibone DJ, Zuraw BL (2005) International union of pharmacology. Xlv. Classification of the kinin receptor family: from molecular mechanisms to pathophysiological consequences. Pharmacol Rev 57(1):27–77. doi:10.1124/pr.57.1.2
Matsumura Y, Maruo K, Kimura M, Yamamoto T, Konno T, Maeda H (1991) Kinin-generating cascade in advanced cancer patients and in vitro study. Jpn J Cancer Res 82(6):732–741
Wu J, Akaike T, Hayashida K, Miyamoto Y, Nakagawa T, Miyakawa K, Muller-Esterl W, Maeda H (2002) Identification of bradykinin receptors in clinical cancer specimens and murine tumor tissues. Int J Cancer 98(1):29–35
Cockcroft JR, Chowienczyk PJ, Brett SE, Ritter JM (1994) Effect of ng-monomethyl-l-arginine on kinin-induced vasodilation in the human forearm. Br J Clin Pharmacol 38(4):307–310
Ignarro LJ, Byrns RE, Buga GM, Wood KS (1987) Mechanisms of endothelium-dependent vascular smooth muscle relaxation elicited by bradykinin and vip. Am J Physiol 253(5 Pt 2):H1074–1082
Doi K, Akaike T, Horie H, Noguchi Y, Fujii S, Beppu T, Ogawa M, Maeda H (1996) Excessive production of nitric oxide in rat solid tumor and its implication in rapid tumor growth. Cancer 77(8 Suppl):1598–1604, doi:10.1002/(sici)1097-0142(19960415)77:8<1598::aid-cncr27>3.0.co;2-u
Beckman JS, Beckman TW, Chen J, Marshall PA, Freeman BA (1990) Apparent hydroxyl radical production by peroxynitrite: implications for endothelial injury from nitric oxide and superoxide. Proc Natl Acad Sci 87(4):1620–1624
Yu JL, Rak JW (2003) Host microenvironment in breast cancer development: inflammatory and immune cells in tumour angiogenesis and arteriogenesis. Breast Cancer Res 5(2):83–88
Pollard JW (2008) Macrophages define the invasive microenvironment in breast cancer. J Leukoc Biol 84(3):623–630. doi:10.1189/jlb.1107762
Siveen KS, Kuttan G (2009) Role of macrophages in tumour progression. Immunol Lett 123(2):97–102. doi:10.1016/j.imlet.2009.02.011
Robinson-Smith TM, Isaacsohn I, Mercer CA, Zhou M, Van Rooijen N, Husseinzadeh N, McFarland-Mancini MM, Drew AF (2007) Macrophages mediate inflammation-enhanced metastasis of ovarian tumors in mice. Cancer Res 67(12):5708–5716. doi:10.1158/0008-5472.can-06-4375
Laakkonen P, Waltari M, Holopainen T, Takahashi T, Pytowski B, Steiner P, Hicklin D, Persaud K, Tonra JR, Witte L, Alitalo K (2007) Vascular endothelial growth factor receptor 3 is involved in tumor angiogenesis and growth. Cancer Res 67(2):593–599. doi:10.1158/0008-5472.can-06-3567
Lee TH, Seng S, Sekine M, Hinton C, Fu Y, Avraham HK, Avraham S (2007) Vascular endothelial growth factor mediates intracrine survival in human breast carcinoma cells through internally expressed vegfr1/flt1. PLoS Med 4(6):e186. doi:10.1371/journal.pmed.0040186
Mezquita B, Mezquita J, Pau M, Mezquita C (2010) A novel intracellular isoform of vegfr-1 activates src and promotes cell invasion in mda-mb-231 breast cancer cells. J Cell Biochem 110(3):732–742. doi:10.1002/jcb.22584
Timoshenko AV, Chakraborty C, Wagner GF, Lala PK (2006) Cox-2-mediated stimulation of the lymphangiogenic factor vegf-c in human breast cancer. Br J Cancer 94(8):1154–1163. doi:10.1038/sj.bjc.6603067
Timoshenko AV, Rastogi S, Lala PK (2007) Migration-promoting role of vegf-c and vegf-c binding receptors in human breast cancer cells. Br J Cancer 97(8):1090–1098. doi:10.1038/sj.bjc.6603993
Weigand M, Hantel P, Kreienberg R, Waltenberger J (2005) Autocrine vascular endothelial growth factor signalling in breast cancer. Evidence from cell lines and primary breast cancer cultures in vitro. Angiogenesis 8(3):197–204. doi:10.1007/s10456-005-9010-0
Hoeben A, Landuyt B, Highley MS, Wildiers H, Van Oosterom AT, De Bruijn EA (2004) Vascular endothelial growth factor and angiogenesis. Pharmacol Rev 56(4):549–580. doi:10.1124/pr.56.4.3
Ryden L, Stendahl M, Jonsson H, Emdin S, Bengtsson NO, Landberg G (2005) Tumor-specific vegf-a and vegfr2 in postmenopausal breast cancer patients with long-term follow-up. Implication of a link between vegf pathway and tamoxifen response. Breast Cancer Res Treat 89(2):135–143. doi:10.1007/s10549-004-1655-7
Schmidt M, Voelker HU, Kapp M, Dietl J, Kammerer U (2008) Expression of vegfr-1 (flt-1) in breast cancer is associated with vegf expression and with node-negative tumour stage. Anticancer Res 28(3A):1719–1724
Seymour LW, Ferry DR, Anderson D, Hesslewood S, Julyan PJ, Poyner R, Doran J, Young AM, Burtles S, Kerr DJ, Committee ftCRCPIICT (2002) Hepatic drug targeting: phase i evaluation of polymer-bound doxorubicin. J Clin Oncol 20(6):1668–1676. doi:10.1200/jco.20.6.1668
Qian ZM, Li H, Sun H, Ho K (2002) Targeted drug delivery via the transferrin receptor-mediated endocytosis pathway. Pharmacol Rev 54(4):561–587. doi:10.1124/pr.54.4.561
Wang S, Low PS (1998) Folate-mediated targeting of antineoplastic drugs, imaging agents, and nucleic acids to cancer cells. J Control Release 53(1–3):39–48
Lammers T, Kiessling F, Hennink WE, Storm G (2011) Drug targeting to tumors: principles, pitfalls and (pre-) clinical progress. J Control Release. doi:10.1016/j.jconrel.2011.09.063
Garcia-Bennett A, Nees M, Fadeel B (2011) In search of the holy grail: folate-targeted nanoparticles for cancer therapy. Biochem Pharmacol 81(8):976–984. doi:10.1016/j.bcp.2011.01.023
Blankenberg FG, Teplitz RL, Ellis W, Salamat MS, Min BH, Hall L, Boothroyd DB, Johnstone IM, Enzmann DR (1995) The influence of volumetric tumor doubling time, DNA ploidy, and histologic grade on the survival of patients with intracranial astrocytomas. AJNR Am J Neuroradiol 16(5):1001–1012
Malaise EP, Chavaudra N, Charbit A, Tubiana M (1974) Relationship between the growth rate of human metastases, survival and pathological type. Eur J Cancer 10(7):451–459
Okazaki N, Yoshino M, Yoshida T, Suzuki M, Moriyama N, Takayasu K, Makuuchi M, Yamazaki S, Hasegawa H, Noguchi M et al (1989) Evaluation of the prognosis for small hepatocellular carcinoma based on tumor volume doubling time. A preliminary report. Cancer 63(11):2207–2210
Tanaka K, Shimada H, Miura M, Fujii Y, Yamaguchi S, Endo I, Sekido H, Togo S, Ike H (2004) Metastatic tumor doubling time: most important prehepatectomy predictor of survival and nonrecurrence of hepatic colorectal cancer metastasis. World J Surg 28(3):263–270. doi:10.1007/s00268-003-7088-3
Usuda K, Saito Y, Sagawa M, Sato M, Kanma K, Takahashi S, Endo C, Chen Y, Sakurada A, Fujimura S (1994) Tumor doubling time and prognostic assessment of patients with primary lung cancer. Cancer 74(8):2239–2244
Weiss W (1974) Tumor doubling time and survival of men with bronchogenic carcinoma. Chest 65(1):3–8
Tanaka Y, Hongo K, Tada T, Sakai K, Kakizawa Y, Kobayashi S (2003) Growth pattern and rate in residual nonfunctioning pituitary adenomas: correlations among tumor volume doubling time, patient age, and mib-1 index. J Neurosurg 98(2):359–365. doi:10.3171/jns.2003.98.2.0359
Nakasu S, Nakasu Y, Nakajima M, Yokoyama M, Matsuda M, Handa J (1996) Potential doubling time and tumour doubling time in meningiomas and neurinomas. Acta Neurochir (Wien) 138(6):763–770
Winer-Muram HT, Jennings SG, Tarver RD, Aisen AM, Tann M, Conces DJ, Meyer CA (2002) Volumetric growth rate of stage I lung cancer prior to treatment: serial ct scanning. Radiology 223(3):798–805
Kuroishi T, Tominaga S, Morimoto T, Tashiro H, Itoh S, Watanabe H, Fukuda M, Ota J, Horino T, Ishida T et al (1990) Tumor growth rate and prognosis of breast cancer mainly detected by mass screening. Jpn J Cancer Res 81(5):454–462
Arai T, Kuroishi T, Saito Y, Kurita Y, Naruke T, Kaneko M (1994) Tumor doubling time and prognosis in lung cancer patients: evaluation from chest films and clinical follow-up study. Japanese lung cancer screening research group. Jpn J Clin Oncol 24(4):199–204
El-Assal ON, Yamanoi A, Soda Y, Yamaguchi M, Igarashi M, Yamamoto A, Nabika T, Nagasue N (1998) Clinical significance of microvessel density and vascular endothelial growth factor expression in hepatocellular carcinoma and surrounding liver: possible involvement of vascular endothelial growth factor in the angiogenesis of cirrhotic liver. Hepatology 27(6):1554–1562. doi:10.1002/hep.510270613
Jaaskelainen J, Haltia M, Laasonen E, Wahlstrom T, Valtonen S (1985) The growth rate of intracranial meningiomas and its relation to histology. An analysis of 43 patients. Surg Neurol 24(2):165–172
James BM, Gillard JH, Antoun NM, Scott IS, Coleman N, Pinto EM, Jefferies SJ, Burnet NG (2007) Glioblastoma doubling time and cellular proliferation markers. Clin Oncol 19(3):S33
Carlson JA (2003) Tumor doubling time of cutaneous melanoma and its metastasis. Am J Dermatopathol 25(4):291–299
Yoshino M (1983) Growth kinetics of hepatocellular carcinoma. Jpn J Clin Oncol 13(1):45–52
Garnett MC, Kallinteri P (2006) Nanomedicines and nanotoxicology: some physiological principles. Occup Med 56(5):307–311. doi:10.1093/occmed/kql052
Nagy JA, Chang SH, Shih SC, Dvorak AM, Dvorak HF (2010) Heterogeneity of the tumor vasculature. Semin Thromb Hemost 36(3):321–331. doi:10.1055/s-0030-1253454
Pasqualini R, Arap W, McDonald DM (2002) Probing the structural and molecular diversity of tumor vasculature. Trends Mol Med 8(12):563–571
Ng IO, Poon RT, Lee JM, Fan ST, Ng M, Tso WK (2001) Microvessel density, vascular endothelial growth factor and its receptors flt-1 and flk-1/kdr in hepatocellular carcinoma. Am J Clin Pathol 116(6):838–845. doi:10.1309/fxnl-qtn1-94fh-ab3a
Dunphy F, Stack BC Jr, Boyd JH, Dunleavy TL, Kim HJ, Dunphy CH (2002) Microvessel density in advanced head and neck squamous cell carcinoma before and after chemotherapy. Anticancer Res 22(3):1755–1758
Hollingsworth HC, Kohn EC, Steinberg SM, Rothenberg ML, Merino MJ (1995) Tumor angiogenesis in advanced stage ovarian carcinoma. Am J Pathol 147(1):33–41
Abulafia O, Triest WE, Sherer DM, Hansen CC, Ghezzi F (1995) Angiogenesis in endometrial hyperplasia and stage I endometrial carcinoma. Obstet Gynecol 86(4 Pt 1):479–485
Bosari S, Lee AK, DeLellis RA, Wiley BD, Heatley GJ, Silverman ML (1992) Microvessel quantitation and prognosis in invasive breast carcinoma. Hum Pathol 23(7):755–761
Graham CH, Rivers J, Kerbel RS, Stankiewicz KS, White WL (1994) Extent of vascularization as a prognostic indicator in thin (< 0.76 mm) malignant melanomas. Am J Pathol 145(3):510–514
Weidner N, Carroll PR, Flax J, Blumenfeld W, Folkman J (1993) Tumor angiogenesis correlates with metastasis in invasive prostate carcinoma. Am J Pathol 143(2):401–409
Weidner N, Semple JP, Welch WR, Folkman J (1991) Tumor angiogenesis and metastasis—correlation in invasive breast carcinoma. N Engl J Med 324(1):1–8. doi:10.1056/nejm199101033240101
Weis SM, Cheresh DA (2005) Pathophysiological consequences of vegf-induced vascular permeability. Nature 437(7058):497–504. doi:10.1038/nature03987
Van S, Das SK, Wang X, Feng Z, Jin Y, Hou Z, Chen F, Pham A, Jiang N, Howell SB, Yu L (2010) Synthesis, characterization, and biological evaluation of poly(l-gamma-glutamyl-glutamine)-paclitaxel nanoconjugate. Int J Nanomedicine 5:825–837. doi:10.2147/ijn.s13482
Duncan R, Vicent MJ, Greco F, Nicholson RI (2005) Polymer–drug conjugates: towards a novel approach for the treatment of endrocrine-related cancer. Endocr Relat Cancer 12(Supplement 1):S189–S199. doi:10.1677/erc.1.01045
Meerum Terwogt JM, ten Bokkel Huinink WW, Schellens JH, Schot M, Mandjes IA, Zurlo MG, Rocchetti M, Rosing H, Koopman FJ, Beijnen JH (2001) Phase i clinical and pharmacokinetic study of pnu166945, a novel water-soluble polymer-conjugated prodrug of paclitaxel. Anticancer Drugs 12(4):315–323
Wachters FM, Groen HJ, Maring JG, Gietema JA, Porro M, Dumez H, de Vries EG, van Oosterom AT (2004) A phase i study with mag-camptothecin intravenously administered weekly for 3 weeks in a 4-week cycle in adult patients with solid tumours. Br J Cancer 90(12):2261–2267. doi:10.1038/sj.bjc.6601811
Schoemaker NE, van Kesteren C, Rosing H, Jansen S, Swart M, Lieverst J, Fraier D, Breda M, Pellizzoni C, Spinelli R, Grazia Porro M, Beijnen JH, Schellens JH, ten Bokkel Huinink WW (2002) A phase i and pharmacokinetic study of mag-cpt, a water-soluble polymer conjugate of camptothecin. Br J Cancer 87(6):608–614. doi:10.1038/sj.bjc.6600516
Zaki N, Tirelli N (2010) Gateways for the intracellular access of nanocarriers: a review of receptor-mediated endocytosis mechanisms and of strategies in receptor targeting. Expert Opin Drug Deliv 7(8):895–913
Macheda ML, Rogers S, Best JD (2005) Molecular and cellular regulation of glucose transporter (glut) proteins in cancer. J Cell Physiol 202(3):654–662. doi:10.1002/jcp.20166
Hu Z, Luo F, Pan Y, Hou C, Ren L, Chen J, Wang J, Zhang Y (2008) Arg-gly-asp (rgd) peptide conjugated poly(lactic acid)-poly(ethylene oxide) micelle for targeted drug delivery. J Biomed Mater Res A 85(3):797–807. doi:10.1002/jbm.a.31615
Zhang Y, Yang M, Portney NG, Cui D, Budak G, Ozbay E, Ozkan M, Ozkan CS (2008) Zeta potential: a surface electrical characteristic to probe the interaction of nanoparticles with normal and cancer human breast epithelial cells. Biomed Microdevices 10(2):321–328. doi:10.1007/s10544-007-9139-2
Yu T, Malugin A, Ghandehari H (2011) Impact of silica nanoparticle design on cellular toxicity and hemolytic activity. ACS Nano 5(7):5717–5728. doi:10.1021/nn2013904
Kedmi R, Ben-Arie N, Peer D (2010) The systemic toxicity of positively charged lipid nanoparticles and the role of toll-like receptor 4 in immune activation. Biomaterials 31(26):6867–6875. doi:10.1016/j.biomaterials.2010.05.027
Dobrovolskaia MA, McNeil SE (2007) Immunological properties of engineered nanomaterials. Nat Nanotechnol 2(8):469–478. doi:10.1038/nnano.2007.223
Klibanov AL, Maruyama K, Torchilin VP, Huang L (1990) Amphipathic polyethyleneglycols effectively prolong the circulation time of liposomes. FEBS Lett 268(1):235–237
Papahadjopoulos D, Allen TM, Gabizon A, Mayhew E, Matthay K, Huang SK, Lee KD, Woodle MC, Lasic DD, Redemann C et al (1991) Sterically stabilized liposomes: improvements in pharmacokinetics and antitumor therapeutic efficacy. Proc Natl Acad Sci USA 88(24):11460–11464
Senior J, Delgado C, Fisher D, Tilcock C, Gregoriadis G (1991) Influence of surface hydrophilicity of liposomes on their interaction with plasma protein and clearance from the circulation: studies with poly(ethylene glycol)-coated vesicles. Biochim Biophys Acta 1062(1):77–82
Senior JH (1987) Fate and behavior of liposomes in vivo: a review of controlling factors. Crit Rev Ther Drug Carrier Syst 3(2):123–193
Torchilin VP, Omelyanenko VG, Papisov MI, Bogdanov AA Jr, Trubetskoy VS, Herron JN, Gentry CA (1994) Poly(ethylene glycol) on the liposome surface: on the mechanism of polymer-coated liposome longevity. Biochim Biophys Acta 1195(1):11–20
Shiah JG, Dvorak M, Kopeckova P, Sun Y, Peterson CM, Kopecek J (2001) Biodistribution and antitumour efficacy of long-circulating n-(2-hydroxypropyl)methacrylamide copolymer-doxorubicin conjugates in nude mice. Eur J Cancer 37(1):131–139
Lasic DD, Martin FJ, Gabizon A, Huang SK, Papahadjopoulos D (1991) Sterically stabilized liposomes: a hypothesis on the molecular origin of the extended circulation times. Biochim Biophys Acta 1070(1):187–192
Dixit V, Van den Bossche J, Sherman DM, Thompson DH, Andres RP (2006) Synthesis and grafting of thioctic acid−peg−folate conjugates onto au nanoparticles for selective targeting of folate receptor-positive tumor cells. Bioconjug Chem 17(3):603–609. doi:10.1021/bc050335b
Bellocq NC, Pun SH, Jensen GS, Davis ME (2003) Transferrin-containing, cyclodextrin polymer-based particles for tumor-targeted gene delivery. Bioconjug Chem 14(6):1122–1132. doi:10.1021/bc034125f
Breunig M, Bauer S, Goepferich A (2008) Polymers and nanoparticles: intelligent tools for intracellular targeting? Eur J Pharm Biopharm 68(1):112–128. doi:10.1016/j.ejpb.2007.06.010
Haley B, Frenkel E (2008) Nanoparticles for drug delivery in cancer treatment. Urol Oncol 26(1):57–64. doi:10.1016/j.urolonc.2007.03.015
Sugahara KN, Teesalu T, Karmali PP, Kotamraju VR, Agemy L, Greenwald DR, Ruoslahti E (2010) Coadministration of a tumor-penetrating peptide enhances the efficacy of cancer drugs. Science 328(5981):1031–1035. doi:10.1126/science.1183057
Jain RK, Stylianopoulos T (2010) Delivering nanomedicine to solid tumors. Nat Rev Clin Oncol 7(11):653–664. doi:10.1038/nrclinonc.2010.139
Howard MD, Ponta A, Eckman A, Jay M, Bae Y (2011) Polymer micelles with hydrazone-ester dual linkers for tunable release of dexamethasone. Pharm Res 28(10):2435–2446. doi:10.1007/s11095-011-0470-1
Duncan R (2003) The dawning era of polymer therapeutics. Nat Rev Drug Discov 2(5):347–360. doi:10.1038/nrd1088
Bae YH, Park K (2011) Targeted drug delivery to tumors: myths, reality and possibility. J Control Release 153(3):198–205. doi:10.1016/j.jconrel.2011.06.001
Longmire M, Choyke PL, Kobayashi H (2008) Clearance properties of nano-sized particles and molecules as imaging agents: considerations and caveats. Nanomedicine (Lond) 3(5):703–717. doi:10.2217/17435889.3.5.703
Yuan A, Yang PC, Yu CJ, Lee YC, Yao YT, Chen CL, Lee LN, Kuo SH, Luh KT (1995) Tumor angiogenesis correlates with histologic type and metastasis in non-small-cell lung cancer. Am J Respir Crit Care Med 152(6 Pt 1):2157–2162
Hobbs SK, Monsky WL, Yuan F, Roberts WG, Griffith L, Torchilin VP, Jain RK (1998) Regulation of transport pathways in tumor vessels: role of tumor type and microenvironment. Proc Natl Acad Sci USA 95(8):4607–4612
Horn T, Henriksen JH, Christoffersen P (1986) The sinusoidal lining cells in “normal” human liver. A scanning electron microscopic investigation. Liver 6(2):98–110
Uchino H, Matsumura Y, Negishi T, Koizumi F, Hayashi T, Honda T, Nishiyama N, Kataoka K, Naito S, Kakizoe T (2005) Cisplatin-incorporating polymeric micelles (nc-6004) can reduce nephrotoxicity and neurotoxicity of cisplatin in rats. Br J Cancer 93(6):678–687. doi:10.1038/sj.bjc.6602772
Anderson JM, Rodriguez A, Chang DT (2008) Foreign body reaction to biomaterials. Semin Immunol 20(2):86–100. doi:10.1016/j.smim.2007.11.004
Mukhopadhyay S, Gal AA (2010) Granulomatous lung disease: an approach to the differential diagnosis. Arch Pathol Lab Med 134(5):667–690. doi:10.1043/1543-2165-134.5.667
O’Neill LAJ (2008) How frustration leads to inflammation. Science 320(5876):619–620. doi:10.1126/science.1158398
Panyam J, Labhasetwar V (2004) Sustained cytoplasmic delivery of drugs with intracellular receptors using biodegradable nanoparticles. Mol Pharm 1(1):77–84
Etrych T, Kovář L, Strohalm J, Chytil P, Říhová B, Ulbrich K (2011) Biodegradable star hpma polymer–drug conjugates: biodegradability, distribution and anti-tumor efficacy. J Control Release 154(3):241–248. doi:10.1016/j.jconrel.2011.06.015
Dvořák M, Kopečková P, Kopeček J (1999) High-molecular weight HPMA copolymer-adriamycin conjugates. J Control Release 60(2–3):321–332. doi:10.1016/s0168-3659(99)00087-5
Cabral H, Nishiyama N, Kataoka K (2007) Optimization of (1,2-diamino-cyclohexane)platinum(ii)-loaded polymeric micelles directed to improved tumor targeting and enhanced antitumor activity. J Control Release 121(3):146–155. doi:10.1016/j.jconrel.2007.05.024
Gabizon AA (1995) Liposome circulation time and tumor targeting: implications for cancer chemotherapy. Adv Drug Deliv Rev 16(2–3):285–294. doi:10.1016/0169-409x(95)00030-b
Lim HJ, Masin D, McIntosh NL, Madden TD, Bally MB (2000) Role of drug release and liposome-mediated drug delivery in governing the therapeutic activity of liposomal mitoxantrone used to treat human a431 and ls180 solid tumors. J Pharmacol Exp Ther 292(1):337–345
Lopes de Menezes DE, Hudon N, McIntosh N, Mayer LD (2000) Molecular and pharmacokinetic properties associated with the therapeutics of bcl-2 antisense oligonucleotide g3139 combined with free and liposomal doxorubicin. Clin Cancer Res 6(7):2891–2902
Newman MS, Colbern GT, Working PK, Engbers C, Amantea MA (1999) Comparative pharmacokinetics, tissue distribution, and therapeutic effectiveness of cisplatin encapsulated in long-circulating, pegylated liposomes (spi-077) in tumor-bearing mice. Cancer Chemother Pharmacol 43(1):1–7
Nishiyama N, Okazaki S, Cabral H, Miyamoto M, Kato Y, Sugiyama Y, Nishio K, Matsumura Y, Kataoka K (2003) Novel cisplatin-incorporated polymeric micelles can eradicate solid tumors in mice. Cancer Res 63(24):8977–8983
Unezaki S, Maruyama K, Ishida O, Suginaka A, J-i H, Iwatsuru M (1995) Enhanced tumor targeting and improved antitumor activity of doxorubicin by long-circulating liposomes containing amphipathic poly(ethylene glycol). Int J Pharm 126(1–2):41–48. doi:10.1016/0378-5173(95)04074-9
Vaage J, Barbera-Guillem E, Abra R, Huang A, Working P (1994) Tissue distribution and therapeutic effect of intravenous free or encapsulated liposomal doxorubicin on human prostate carcinoma xenografts. Cancer 73(5):1478–1484
Xiong XB, Huang Y, Lu WL, Zhang X, Zhang H, Nagai T, Zhang Q (2005) Enhanced intracellular delivery and improved antitumor efficacy of doxorubicin by sterically stabilized liposomes modified with a synthetic rgd mimetic. J Control Release 107(2):262–275. doi:10.1016/j.jconrel.2005.03.030
Yokoyama M, Okano T, Sakurai Y, Fukushima S, Okamoto K, Kataoka K (1999) Selective delivery of adriamycin to a solid tumor using a polymeric micelle carrier system. J Drug Target 7(3):171–186. doi:10.3109/10611869909085500
Yoo HS, Park TG (2004) Folate receptor targeted biodegradable polymeric doxorubicin micelles. J Control Release 96(2):273–283. doi:10.1016/j.jconrel.2004.02.003
Zhang JQ, Zhang ZR, Yang H, Tan QY, Qin SR, Qiu XL (2005) Lyophilized paclitaxel magnetoliposomes as a potential drug delivery system for breast carcinoma via parenteral administration: in vitro and in vivo studies. Pharm Res 22(4):573–583. doi:10.1007/s11095-005-2496-8
Laginha KM, Verwoert S, Charrois GJ, Allen TM (2005) Determination of doxorubicin levels in whole tumor and tumor nuclei in murine breast cancer tumors. Clin Cancer Res 11(19 Pt 1):6944–6949. doi:10.1158/1078-0432.ccr-05-0343
Bae Y, Nishiyama N, Fukushima S, Koyama H, Yasuhiro M, Kataoka K (2005) Preparation and biological characterization of polymeric micelle drug carriers with intracellular ph-triggered drug release property: tumor permeability, controlled subcellular drug distribution, and enhanced in vivo antitumor efficacy. Bioconjug Chem 16(1):122–130. doi:10.1021/bc0498166
Peng CL, Lai PS, Lin FH, Yueh-Hsiu Wu S, Shieh MJ (2009) Dual chemotherapy and photodynamic therapy in an ht-29 human colon cancer xenograft model using sn-38-loaded chlorin-core star block copolymer micelles. Biomaterials 30(21):3614–3625. doi:10.1016/j.biomaterials.2009.03.048
Rafi M, Cabral H, Kano MR, Mi P, Iwata C, Yashiro M, Hirakawa K, Miyazono K, Nishiyama N, Kataoka K (2012) Polymeric micelles incorporating (1,2-diaminocyclohexane)platinum (ii) suppress the growth of orthotopic scirrhous gastric tumors and their lymph node metastasis. J Control Release. doi:10.1016/j.jconrel.2012.01.038
Saito Y, Yasunaga M, Kuroda J, Koga Y, Matsumura Y (2010) Antitumour activity of nk012, sn-38-incorporating polymeric micelles, in hypovascular orthotopic pancreatic tumour. Eur J Cancer 46(3):650–658. doi:10.1016/j.ejca.2009.11.014
Takahashi A, Ohkohchi N, Yasunaga M, Kuroda J, Koga Y, Kenmotsu H, Kinoshita T, Matsumura Y (2010) Detailed distribution of nk012, an sn-38-incorporating micelle, in the liver and its potent antitumor effects in mice bearing liver metastases. Clin Cancer Res 16(19):4822–4831. doi:10.1158/1078-0432.ccr-10-1467
Immordino ML, Brusa P, Arpicco S, Stella B, Dosio F, Cattel L (2003) Preparation, characterization, cytotoxicity and pharmacokinetics of liposomes containing docetaxel. J Control Release 91(3):417–429
Jukanti R, Devraj G, Shashank AS, Devraj R (2011) Biodistribution of ascorbyl palmitate loaded doxorubicin pegylated liposomes in solid tumor bearing mice. J Microencapsul 28(2):142–149. doi:10.3109/02652048.2010.542496
Colombo PE, Boustta M, Poujol S, Pinguet F, Rouanet P, Bressolle F, Vert M (2007) Biodistribution of doxorubicin-alkylated poly(l-lysine citramide imide) conjugates in an experimental model of peritoneal carcinomatosis after intraperitoneal administration. Eur J Pharm Sci 31(1):43–52. doi:10.1016/j.ejps.2007.02.004
Conover CD, Greenwald RB, Pendri A, Gilbert CW, Shum KL (1998) Camptothecin delivery systems: enhanced efficacy and tumor accumulation of camptothecin following its conjugation to polyethylene glycol via a glycine linker. Cancer Chemother Pharmacol 42(5):407–414
Harada M, Murata J, Sakamura Y, Sakakibara H, Okuno S, Suzuki T (2001) Carrier and dose effects on the pharmacokinetics of t-0128, a camptothecin analogue-carboxymethyl dextran conjugate, in non-tumor- and tumor-bearing rats. J Control Release 71(1):71–86
Li C, Newman RA, Wu QP, Ke S, Chen W, Hutto T, Kan Z, Brannan MD, Charnsangavej C, Wallace S (2000) Biodistribution of paclitaxel and poly(l-glutamic acid)-paclitaxel conjugate in mice with ovarian oca-1 tumor. Cancer Chemother Pharmacol 46(5):416–422
Schluep T, Cheng J, Khin KT, Davis ME (2006) Pharmacokinetics and biodistribution of the camptothecin-polymer conjugate it-101 in rats and tumor-bearing mice. Cancer Chemother Pharmacol 57(5):654–662. doi:10.1007/s00280-005-0091-7
Song R, Joo Jun Y, Ik Kim J, Jin C, Sohn YS (2005) Synthesis, characterization, and tumor selectivity of a polyphosphazene-platinum(ii) conjugate. J Control Release 105(1–2):142–150. doi:10.1016/j.jconrel.2005.03.016
Sparreboom A, Scripture CD, Trieu V, Williams PJ, De T, Yang A, Beals B, Figg WD, Hawkins M, Desai N (2005) Comparative preclinical and clinical pharmacokinetics of a cremophor-free, nanoparticle albumin-bound paclitaxel (abi-007) and paclitaxel formulated in cremophor (taxol). Clin Cancer Res 11(11):4136–4143. doi:10.1158/1078-0432.ccr-04-2291
Sugahara S, Kajiki M, Kuriyama H, Kobayashi TR (2007) Complete regression of xenografted human carcinomas by a paclitaxel-carboxymethyl dextran conjugate (az10992). J Control Release 117(1):40–50. doi:10.1016/j.jconrel.2006.10.009
Wang X, Zhao G, Van S, Jiang N, Yu L, Vera D, Howell SB (2010) Pharmacokinetics and tissue distribution of pgg-paclitaxel, a novel macromolecular formulation of paclitaxel, in nu/nu mice bearing nci-460 lung cancer xenografts. Cancer Chemother Pharmacol 65(3):515–526. doi:10.1007/s00280-009-1058-x
Maeda H, Bharate GY, Daruwalla J (2009) Polymeric drugs for efficient tumor-targeted drug delivery based on EPR-effect. Eur J Pharm Biopharm 71(3):409–419. doi:10.1016/j.ejpb.2008.11.010
Gordon AN, Fleagle JT, Guthrie D, Parkin DE, Gore ME, Lacave AJ (2001) Recurrent epithelial ovarian carcinoma: a randomized phase iii study of pegylated liposomal doxorubicin versus topotecan. J Clin Oncol 19(14):3312–3322
James ND, Coker RJ, Tomlinson D, Harris JR, Gompels M, Pinching AJ, Stewart JS (1994) Liposomal doxorubicin (doxil): an effective new treatment for kaposi’s sarcoma in AIDS. Clin Oncol 6(5):294–296
(1996) FDA approves daunoxome as first-line therapy for kaposi’s sarcoma. Food and drug administration. J Int Assoc Physicians AIDS Care 2(5):50–51
Chhikara BS, Parang K (2010) Development of cytarabine prodrugs and delivery systems for leukemia treatment. Expert Opin Drug Deliv 7(12):1399–1414
Swenson CE, Bolcsak LE, Batist G, Guthrie TH Jr, Tkaczuk KH, Boxenbaum H, Welles L, Chow SC, Bhamra R, Chaikin P (2003) Pharmacokinetics of doxorubicin administered i.V. As myocet (tlc d-99; liposome-encapsulated doxorubicin citrate) compared with conventional doxorubicin when given in combination with cyclophosphamide in patients with metastatic breast cancer. Anticancer Drugs 14(3):239–246
Ando K, Mori K, Corradini N, Redini F, Heymann D (2011) Mifamurtide for the treatment of nonmetastatic osteosarcoma. Expert Opin Pharmacother 12(2):285–292
Dinndorf PA, Gootenberg J, Cohen MH, Keegan P, Pazdur R (2007) Fda drug approval summary: pegaspargase (oncaspar) for the first-line treatment of children with acute lymphoblastic leukemia (all). Oncologist 12(8):991–998
Oerlemans C, Bult W, Bos M, Storm G, Nijsen JF, Hennink WE (2010) Polymeric micelles in anticancer therapy: targeting, imaging and triggered release. Pharm Res 27(12):2569–2589
Gradishar WJ (2006) Albumin-bound paclitaxel: a next-generation taxane. Expert Opin Pharmacother 7(8):1041–1053
Morana G, Salviato E, Guarise A (2007) Contrast agents for hepatic MRI. Cancer Imag 7 Spec No A:S24–S27. doi:10.1102/1470-7330.2007.9001
Jung CW (1995) Surface properties of superparamagnetic iron oxide mr contrast agents: ferumoxides, ferumoxtran, ferumoxsil. Magn Reson Imaging 13(5):675–691
Aoki T, Nakata H, Watanabe H, Nakamura K, Kasai T, Hashimoto H, Yasumoto K, Kido M (2000) Evolution of peripheral lung adenocarcinomas. Am J Roentgenol 174(3):763–768
Mucci LA, Powolny A, Giovannucci E, Liao Z, Kenfield SA, Shen R, Stampfer MJ, Clinton SK (2009) Prospective study of prostate tumor angiogenesis and cancer-specific mortality in the health professionals follow-up study. J Clin Oncol 27(33):5627–5633. doi:10.1200/jco.2008.20.8876
Cucchetti A, Vivarelli M, Piscaglia F, Nardo B, Montalti R, Grazi GL, Ravaioli M, La Barba G, Cavallari A, Bolondi L, Pinna AD (2005) Tumor doubling time predicts recurrence after surgery and describes the histological pattern of hepatocellular carcinoma on cirrhosis. J Hepatol 43(2):310–316. doi:10.1016/j.jhep.2005.03.014
Poon RT, Ng IO, Lau C, Yu WC, Yang ZF, Fan ST, Wong J (2002) Tumor microvessel density as a predictor of recurrence after resection of hepatocellular carcinoma: a prospective study. J Clin Oncol 20(7):1775–1785
Daumas-Duport C, Scheithauer B, O’Fallon J, Kelly P (1988) Grading of astrocytomas. A simple and reproducible method. Cancer 62(10):2152–2165
Wang M, Tang J, Liu S, Yoshida D, Teramoto A (2005) Expression of cathepsin b and microvascular density increases with higher grade of astrocytomas. J Neurooncol 71(1):3–7. doi:10.1007/s11060-004-9163-5
Herbst C, Kosmehl H, Stiller KJ, Berndt A, Eiselt M, Schubert J, Katenkamp D (1998) Evaluation of microvessel density by computerised image analysis in human renal cell carcinoma. Correlation to pt category, nuclear grade, proliferative activity and occurrence of metastasis. J Cancer Res Clin Oncol 124(3–4):141–147
Lee JY, Kim CK, Choi D, Park BK (2008) Volume doubling time and growth rate of renal cell carcinoma determined by helical ct: a single-institution experience. Eur Radiol 18(4):731–737. doi:10.1007/s00330-007-0820-x
Tas F, Yavuz E, Aydiner A, Saip P, Disci R, Iplikci A, Topuz E (2000) Angiogenesis and p53 protein expression in breast cancer: prognostic roles and interrelationships. Am J Clin Oncol 23(6):546–553
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2013 Springer Science+Business Media New York
About this chapter
Cite this chapter
Taurin, S., Nehoff, H., van Aswegen, T., Greish, K. (2013). Tumor Vasculature, EPR Effect, and Anticancer Nanomedicine: Connecting the Dots. In: Bae, Y., Mrsny, R., Park, K. (eds) Cancer Targeted Drug Delivery. Springer, New York, NY. https://doi.org/10.1007/978-1-4614-7876-8_8
Download citation
DOI: https://doi.org/10.1007/978-1-4614-7876-8_8
Published:
Publisher Name: Springer, New York, NY
Print ISBN: 978-1-4614-7875-1
Online ISBN: 978-1-4614-7876-8
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)