
Abstract
Cancer is the leading cause of death worldwide. Despite improvements in diagnosis and treatment over the past two decades, cancer continues to present a serious challenge to oncologists, especially when the disease has already spread to a distant site at the time of diagnosis. The high degree of variation in gene expression, observed not only in tumors arising from different tissues but also in tumors arising from the same tissue, and sometimes in distinct areas of the same tumor, is likely to be responsible for evolutionary adaptation and consequently tumor survival.
Cellular heterogeneity has historically been viewed solely as the result of genetic instability. However, it has now become increasingly clear that changes in gene expression that occur without altering the DNA sequence—better known as epigenetic changes—can likewise contribute to tumorigenesis. Elucidating the mechanisms that account for cancer heterogeneity will be essential to the design of new drugs capable of overcoming the major limitations of current therapies. These limitations include the treatment of cancers able to escape immune surveillance or adapt to chemotherapy regimens as well as invasive and metastatic cancers.
Here, we review recent progress in the understanding of tumor genetics and epigenetics and translate these findings into potential clinical practice.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others


Keywords
- Adenomatous Polyposis Coli
- Lynch Syndrome
- Genetic Instability
- Mixed Lineage Leukemia
- Aberrant Crypt Focus
These keywords were added by machine and not by the authors. This process is experimental and the keywords may be updated as the learning algorithm improves.
Introduction
The word cancer comes from the Latin translation of karkinoma; the term was derived by Hippocrates (460–370 B.C.) from the Greek word for crab, karkinos. Karkinoma was used by the Greek physician to describe a malignant growth because veins spreading outward from the tumor mass reminded him of crab claws. Due to these angiogenesis observations, Hippocrates is considered the first person to clearly recognize the difference between malignant and benign tumors. We now know that apart from their histological features, other substantial differences occur between these two groups of tumors, including the presence in malignant tumors and the absence/infrequency in benign tumors of phenotypic instability [1, 2]. Inherent instability of tumor cells is a widespread phenomenon in cancer that drives tumor progression through the generation of more aggressive subtypes undergoing a positive Darwinian selection. Starting from Boveri’s suggestions of genetic instability in cancer [3], many groundbreaking discoveries have been made in recent decades in the field of molecular biology, making it increasingly clear that genetic instability is not the only driving force for tumor progression. Epigenetic modification of DNA or of chromatin-associated proteins, a heritable change in gene expression or cellular phenotype caused by mechanisms other than changes in the underlying DNA sequence, can lead to critical changes in gene function and drive tumor progression to an invasive cancer. It has also been proposed that cancer-initiating mutations could even follow an epigenetic disruption of progenitor cells [4]. Thus, epigenetics might play an important role in both cancer pre-initiation and progression.
Understanding cancer diversity is crucial to achieve improved diagnosis and patient treatment. Indeed, the elucidation of the mechanisms that allow cancer cells to constantly adapt and evolve during the course of the disease will help prevent cancer growth and progression. Importantly, due to their potential reversible outcome, epigenetic changes are being investigated as potential therapeutic targets, and this has led to the development of new anticancer drugs.
In the first part of this chapter, we will summarize major genetic and epigenetic pathways involved in the pathogenesis of human cancer. In the second part, we will focus on one of the best-defined models for genetic and epigenetic progression, colorectal cancer (CRC). Finally, we will discuss how emerging information about genetic and molecular diversity can be used to assess cancer risk and/or guide therapy.
Genetic Instability
Chromosome instability (CIN) and microsatellite instability (MSI) are the major genetic instability pathways that can lead to cancer pathogenesis. In the following paragraphs, we will consider the most important molecular contributors toward the progressive loss of a stable karyotype thereby initiating and sustaining cancer.
Chromosome Instability
CIN refers to an increased rate of the loss or gain of whole or large sections of chromosomes during cell division. This increased rate of unbalanced chromosomal rearrangement eventually leads to a multistep accumulation of genetic abnormalities, including amplification of proto-oncogenes and inactivation of tumor suppressor genes, which may directly promote tumor cell growth. For instance, loss of tumor suppressor genes often results from the loss of genetic information inherited from one parent, a phenomenon known as the loss of heterozygosity (LOH) [5].
An imbalance in chromosome number is also referred to as aneuploidy. Although aneuploidy can be detected at early steps of malignant transformation, and even in certain premalignant lesions, the number of chromosomal aberrations usually increases with tumor progression [6–8]. Whether chromosome abnormalities can be both the cause and the effect of cancer is still under investigation. Similarly, the scientific community is divided over the assignment of the origin of chromosomal abnormalities. Many studies suggest that aneuploidy arises from the inability to faithfully ensure equal segregation of chromosomes during mitosis [9, 10]. This mitotic chromosomal instability has been mainly correlated to numerical and functional abnormalities of centrosomes. Indeed, the presence of multiple centrosomes can lead to multipolar mitosis, enabling the survival of tetraploid cells and the generation of an aneuploid population that evolves to become genetically unstable and tumorigenic [11]. However, it should be considered that centrosome abnormalities effectively destabilize chromosomes only in cells with a compromised spindle checkpoint function. Usually, cell cycle checkpoint activation slows or arrests cell cycle progression, thereby allowing for efficient repair and thus preventing transmission of DNA damage to the progeny [12, 13]. The fate of damaged cells mainly depends on the status of the p53-dependent G1 cell cycle checkpoint pathway [14]. In the presence of p53, mutant cells are rapidly eliminated through cell cycle arrest and/or apoptosis, whereas a defective p53 pathway permits their propagation. Consistent with this, loss of p53 function is associated with increased aneuploidy [15–17], gene amplification [18], point mutation [19], and homologous recombination [20].
Cyclin-dependent kinases (CDKs) are targets of checkpoints that control entry into the next phase of the cell cycle. The activity of CDKs is frequently deregulated in tumor cells due to genetic or epigenetic alterations of CDK–cyclin complexes or to downregulation of several CDK inhibitors including p21CIP/WAF, p27KIP, and p16INK4A [21]. Centrosome amplification can be correlated with multiple genes of the cell cycle engine. For instance, centrosome duplication is controlled by CDK2/cyclin E complex, which is inhibited by p21CIP/WAF [22, 23]. Thus, overexpression of cyclin E or p21CIP/WAF inhibition results in centrosome amplification. Mutational inactivation of p21CIP/WAF is infrequent [24]; however, aberrant p21CIP/WAF promoter gene methylation is common in cancer and results in strikingly reduced expression of its regulated protein [25]. These findings lead to the idea that aneuploidy may not be only genetic in origin.
In addition to defects in either cell cycle machinery or checkpoints as potential causes of CIN, other mechanisms, such as telomere erosion, may be involved in the generation of unstable cells. Telomeres are specialized DNA structures located at the end of chromosomes with an important role in the prevention of chromosome fusion [26]. Normal somatic cells show a progressive loss of telomeres during DNA proliferation due to end replication problems of DNA polymerase, eventually leading to replicative senescence. Telomere erosion has been linked to both tumor suppression and genetic instability. Dysfunctional telomeres activate DNA damage response. In the setting of a competent p53 pathway, this initiates senescence and apoptotic programs to inhibit tumorigenesis, whereas in cells with mutant p53, dysfunctional telomeres promote genome instability and progression to cancer [27, 28]. Telomere-related CIN results from repeated breakage–fusion–bridge cycles (BFBCs), and this is thought to be a key event in tumorigenesis of different tissues, including colon [29], cervix [30], and blood [31].
Like telomere erosion, DNA palindrome formation can lead to genetic instability by initiating BFBCs [32]. However, it is unknown how palindromes form, although they appear early in cancer progression.
Every cell division presents a chance for mutations. Because stem cells have the property of self-renewal, any mutation conferring a selective growth advantage occurring in the stem cell compartment will be perpetuated into its progeny. This genetic lesion, in turn, can lead the daughter cells to acquire new properties through additional cycles of genetic aberrations. This concept has been well demonstrated for chronic myeloid leukemia (CML). Following radiation exposure, the BCR/ABL oncogene is likely to induce genetic instability in CSCs that predisposes the progeny to increased BFBCs [33]. Such important findings can also be applied to chemotherapy and explain why sequential treatment with multiple tyrosine kinase inhibitors still fails to completely eradicate the disease [34].
The Opposing Roles of Aneuploidy
Although the so-called aneuploidy hypothesis postulates that an abnormal chromosome number can drive tumor progression, some researchers have argued that aneuploidy is only a benign side effect of transformation [35]. Indeed, several lines of evidence demonstrate that an altered karyotype can decrease the rate of cell proliferation or even cause cell death. Using centromere-associated protein E (CENPE) heterozygous animals, which develop whole chromosome aneuploidy in the absence of mutations that compromise chromosome segregation fidelity, Weaver et al. have found that aneuploidy promotes tumorigenesis in some contexts and inhibits it in others [36]. Specifically, low rates of CIN promote tumors, whereas high rates of CIN cause cell death. Thus, aneuploidy can act both as a tumor inducer and a tumor suppressor. Such an effect is also analogous to chemotherapy-induced genetic instability, in which high levels of DNA damage lead to cellular death and tumor regression. The most probable explanation for the impairment of cell fitness is the gene dosage hypothesis in which gains or losses of whole chromosomes immediately alter the dosage of hundreds of genes in a cell, leading to imbalances in critical proteins [37]. The possible resulting changes include the alteration of the function of a specific protein, the defect of stoichiometric-sensitive complexes, the favoring of promiscuous molecular interactions, and the accumulation of improperly folded or aggregated proteins negatively affecting cell proliferation. However, aneuploid cells are often able to trigger adaptive dosage compensation responses at the proteome level which may be accelerated by aneuploid-induced genetic instability, suggesting the existence of a functional and destabilizing positive feedback loop of aneuploidy in cancer.
The role of aneuploidy in tumorigenesis remains poorly understood. It is conceivable that cellular outcome is dependent on the extent of aneuploidy induced. This could explain why aneuploidy can be compatible with normal growth and development. Polyploidy is common, for example, in the liver, where frequent multipolar mitosis yield diverse hepatocyte populations, some with aneuploidy [38]. Interestingly, the genetic variation found in hepatocytes is postulated to be an advantage for liver function by allowing the cellular selection of discrete hepatocyte populations to expand and protect the organ from certain injury and poisonous substances [38].
Microsatellite Instability
MSI refers to length alterations of mononucleotide or dinucleotide repeats (e.g., TTTT or CACACA) located mostly in intronic DNA sequences. MSI is mainly due to errors during DNA replication and to a defective post-replicative repair system. Indeed, defects in both DNA mismatch repair (MMR) and base-excision repair (BER) systems have been identified in MSI-positive tumors. The DNA sequences repaired by the MMR system are residual mismatches that have evaded proofreading during replication. Base mispairs, if not corrected by the MMR system, may cause nucleotide transitions or transversions, allowing a novel base to alter the authentic genetic sequence. Importantly, the role of MMR proteins in the repair process can be uncoupled from the MMR-dependent cell-killing response, the latter being based on the ability of MMR proteins to trigger checkpoint activation and apoptosis in response to DNA damage [39, 40].
In late 1993 [41], altered CA repeats in colon cancer were correlated for the first time to a mutation in a gene which codes for a factor essential for replication fidelity or repair. At the same time, Lynch syndrome (also termed hereditary nonpolyposis CRC, HNPCC) was associated with germ-line mutations to one of two MMR genes, human mutL homologue 1 (hMLH1) or human mutS homologue 2 (hMSH2), with mutations of other MMR genes being rare [42–45]. hMLH1 and hMSH2 genes were also reported as inactivated via promoter DNA methylation in a sporadic subset of MSI-positive tumors [46, 47]. In the remaining tumors, no identifiable MMR gene mutations were found, indicating that additional factor(s) could have been responsible for the MSI phenotype [48–52].
Although CIN and MSI can be distinguished from one another by their molecular characteristics, evidence suggests that there might be some degree of overlap. In a study by Goel et al., 3.4 % of the analyzed CRCs showed the coincidence of MSI-high (MSI-H) and LOH events [53], and in the poorly metastatic KM12C cell line, both patterns of genetic instability were found to coexist [54].
Epigenetic Instability
The term epigenetics is defined as the heritable but potentially reversible changes in gene expression that occur without alterations in the DNA sequence [55–58]. Epigenetic modifications include DNA methylation, histone modifications, and microRNAs (miRNAs). Accumulating evidence indicates that these modifications are profoundly altered in human cancers. The key players of such complex processes comprise a long list of enzymes cooperating together and include DNA methyltransferases (DNMTs), methyl-CpG binding proteins, histone modifying enzymes, chromatin remodeling factors, transcription factors, and chromosomal proteins.
DNA Methylation
DNA methylation involves the addition of a methyl group to the 5′ position of the cytosine pyrimidine ring. In mammals, this phenomenon occurs exclusively at a cytosine followed by guanine (CpG). About 70–80 % of CpG sites contain methylated cytosines in somatic cells [59]. Although the CG dinucleotides are present along all chromosomes, the CG density is higher in some areas than others [60]. These so-called CpG islands are present in the promoter and exon regions of approximately 40 % of mammalian genes and regulate gene expression. Several experiments have shown that methylation of promoter CpG islands plays an important role in gene silencing [61], genomic imprinting [62], X-chromosome inactivation [63], the silencing of intragenomic parasites [64], and carcinogenesis [65, 66].
Although the origin of aberrant DNA methylation patterns remains to be established, several studies have suggested that alterations in the DNA methylome could be directly affected by diets that are deficient in folate and methionine; exposure to metals, such as arsenic, lead, and chromium; and inflammation or viral/bacterial infection, i.e., chronic inflammatory bowel disease (IBD) and Helicobacter pylori infection of gastric epithelial cells [67].
Epigenetic factors play a critical role in development, dictating the rules that establish and maintain stem cell identity. Loss of cellular identity leads to an increased ability to grow and proliferate, ultimately causing the onset of cancer. Oct4, Nanog, and Sox2 transcription factors are expressed by embryonic stem cells (ESCs) during development, conferring pluripotency [68–71], but are repressed through promoter hypermethylation during adulthood [72, 73]. In the context of cancer, expression of these ESC-associated genes occurs [74] in accordance with the idea that cancer arises through the dedifferentiation of fully committed and specialized cells or from “maturation arrest” of stem cells [75]. Specifically, DNA hypomethylation has been found in a variety of human cancers [76–84] and affects not only Oct4, Nanog, and Sox2 but a long list of genes. The extent of hypomethylation has been correlated with tumor grade and prognosis in liver, breast, and ovarian cancers [85–87], but not in prostate cancer [88]. Thus, the inappropriate epigenetic (re)activation of tissue-specific genes plays a critical role in cancer.
DNA hypomethylation in tumors also occurs at repetitive sequences. Half of the human genome consists of highly repeated, interspersed DNA sequences, and recent studies have highlighted that their hypermethylation represents a mechanism to prevent chromosomal instability, translocation, and gene disruption caused by the reactivation of transposable elements, such as SINE (short interspersed elements), LINE (long interspersed elements), and HERV (human endogenous retroviruses) sequences. Indeed, loss of methylation at these elements contributes to oncogenic transformation or tumor progression [89–91].
Besides DNA hypomethylation, de novo methylation within the promoter region of tumor suppressor genes has also been observed in cancer. The retinoblastoma gene (Rb) was the first classic tumor suppressor gene in which CpG island hypermethylation was detected [92, 93]. Following this discovery, other tumor suppressor proteins including von Hippel–Lindau (VHL), INK4A, E-cadherin, MLH1, and breast cancer 1, early onset (BRCA1) were found to be silenced in cancer through hypermethylation of their promoters [46, 94–100]. The so-called CpG island methylator phenotype (CIMP) was first described by Toyota et al. in 1999 [101]. In their study, two distinct types of hypermethylation were found: one appearing as a result of the aging process and the other, specific for cancer. Age-related methylation was shown to be very frequent in primary CRCs, while cancer-related methylation was relatively infrequent and never observed in normal colon mucosa. Detailed analysis of this latter type of methylation revealed a prominent pattern, suggesting the presence of a hypermethylator phenotype in a subset of CRCs. The authors concluded that through its ability to silence multiple genes simultaneously, CIMP can be considered functionally equivalent to genetic instability, resulting in the rapid accumulation of multiple molecular aberrations with a potential to trigger the neoplastic process. Additional work from other groups has suggested that promoter hypermethylation of tumor suppressor genes can follow the formation of transcriptionally inactive chromatin [102]. From this point of view, hypermethylation could be held responsible for maintaining gene silencing, rather than initiating it. Importantly, hypomethylation or hypermethylation may not result in gross changes in gene expression per se, as cancer appears to be linked to a global epigenetic disequilibrium [103].
DNA Methyltransferase and Polycomb Genes: Key Players in Epigenetic Silencing
The enzymes directly responsible for CpG island hypermethylation of tumor suppressor genes are the DNMTs. Both increased expression and increased activity of DNMTs have been found in human cancers, including colon cancer [104–107]. Polycomb group (PcG) proteins have been suggested to serve as recruitment platforms for DNMTs [108, 109], helping maintain the transcriptional repression of target genes through many cycles of cell division. PcG genes are organized in two multiprotein complexes, Polycomb repressive complex 2 (PRC2) and 1 (PRC1), which have been implicated in silencing initiation and stable maintenance of gene repression, respectively [110].
Among the most studied PRC1 members is B-cell-specific Moloney murine leukemia virus integration site 1 (Bmi-1), which contributes to CSC self-renewal in part by inactivating the INK4A-ARF locus-encoded p16INK4A and p14ARF proteins, thus delaying the onset of senescence [111]. However, Bmi-1 can also act in an INK4A independent manner, for example, modulating Wnt and Notch pathways [112]. Enhancer of zeste homologue 2 (Ezh2), the histone methyltransferase of PRC2, plays a master regulatory role in controlling stem cell differentiation [113], cell proliferation [114], early embryogenesis [115], and X-chromosome inactivation [116]. Moreover, a functional link between dysregulation of Ezh2 and repression of E-cadherin during cancer progression has been reported, suggesting a critical role for this PcG gene in the invasive process [117]. A correlation between the cell cycle machinery and Ezh2-mediated epigenetic gene silencing has also been demonstrated. Specifically, CDKs have been found to phosphorylate Ezh2, maintaining its oncogenic and gene-silencing functions, and ultimately contributing to the aggressive phenotype of tumors [118]. Briefly, many cancer types show an overexpression of Ezh2, predicting poor prognosis, metastasis, and chemoresistance [119–124]. A significant association between polymorphisms of the Ezh2 gene and cancer risk/outcome has been reported for the first time in lung cancer [125] and more recently in CRC patients [126], thus introducing the concept of epigenetic polymorphism testing for cancer therapy. However, our comprehension of the precise role of PcGs in tumorigenesis and mechanisms of their regulation remains incomplete. While there are about 15 unique PcG genes in Drosophila [127], in mammals there are multiple orthologues of many PcGs, making possible hundreds of different combinations to assemble multiprotein complexes. Further studies are needed to complete this puzzle and obtain useful information to develop new ways to treat, cure, or even prevent cancer.
Histone Modifications
The histones constitute a family of small basic proteins that are involved in the packaging of eukaryotic DNA. Histone N-terminal tails may undergo many enzymatic posttranslational modifications, including acetylation, methylation, phosphorylation, ubiquitylation, and sumoylation. These modifications provide an important regulatory platform for processes such as gene transcription and DNA damage repair. For instance, acetylation of the lysine residues at the N terminus of histone proteins leads to chromatin relaxation by reducing the affinity between histones and DNA. Decompaction of the chromatin structure allows accessibility of the DNA by RNA polymerase II (Pol II), stimulating gene transcription.
The combination of histone posttranslational modifications is thought to give rise to a histone code that is interpreted by an array of diverse proteins. These proteins can be divided into three classes: “readers,” “writers,” and “erasers.” Misreading, miswriting, and mis-erasing of histone methylation marks can be associated with oncogenesis and progression [128]. Mixed lineage leukemia (MLL) is an example of cancer driven by epigenetic alterations involving histone modifications [129]. These leukemias are characterized by translocations of the MLL gene, which normally methylates histone H3 on lysine 4 (H3K4), a mark typically associated with gene activation. MLL translocations encode MLL fusion proteins that have lost H3K4 methyltransferase activity and possess the ability to reprogram differentiated myeloid cells into multipotent CSCs. Changes in global histone modification patterns have also been observed in other cancers, including lymphoma, breast, colon, bone, cervix, lung, testis, neuroblastoma, osteosarcoma, and prostate [130–132]. Particularly, global loss of monoacetylation and trimethylation of histone H4 has been reported as a common hallmark of human tumor cells [130].
miRNAs
miRNAs are short noncoding RNAs that bind to complementary mRNA molecules, promoting their degradation and/or translation into a protein. Studies suggest that the human genome may encode over 1,000 miRNAs, a limited number compared with the number of mRNAs, typically estimated at ~30,000 [133]. However, miRNAs may regulate hundreds of mRNAs, affecting a range of processes, including organismal development and the establishment and maintenance of tissue differentiation [134, 135]. Importantly, an epigenetic crosstalk between miRNAs and DNA methylation has been reported. Specifically, a wide range of tumor suppressor miRNAs has aberrant methylation profiles in human cancers. Mir-127 and mir-124 are examples of the first two miRNAs identified that undergo transcriptional inactivation by CpG island hypermethylation [136, 137]. Epigenetic repression of these molecules leads to changes in histone modifications; thus, epigenetic modifications are profoundly linked to each other. Figure 14.1 shows a summary of both genetic and epigenetic mechanisms that drive cell transformation and promote cancer development and progression.

Scheme illustrating the mechanisms that drive cell transformation and promote cancer development and progression. Both genetic and epigenetic mechanisms are depicted
The Genetic and Molecular Diversity of Colorectal Carcinoma
CRC is a leading cause of cancer deaths worldwide. Roughly, three molecular subtypes of CRCs have been described: CIN, MSI, and CIMP. A small subgroup of tumors also exists in which none of these phenotypes have been detected [138].
According to the CIN pathway, the classical multistep pathway of colon carcinogenesis proposed by Vogelstein et al. in 1988, CRC develops as a result of the pathologic transformation of a normal colonic epithelium into a dysplastic epithelium and ultimately into an invasive cancer through an adenomatous polyp. Aberrant crypt foci (ACF), microscopic surface abnormalities first identified in carcinogen-treated rodents [139] and later in human colon [140], are postulated to be a precursor to the adenoma due to the presence of molecular and genetic abnormalities, i.e., MSI [141]. Particularly, ACF formation is initiated by mutations in the adenomatous polyposis coli (APC) tumor suppressor gene [142]. APC is considered a strong negative regulator of the Wnt pathway, being part of the β-catenin destruction complex, which also includes the scaffold proteins axin or conductin/axin2, casein kinase I (protein kinase CKI), and glycogen synthase kinase 3β (GSK3β). In normal cells, this complex phosphorylates β-catenin, leading to its ubiquitination and destruction by proteasome 26 S [143]. Loss of APC leads to β-catenin accumulation in the cytosol, binding to cytosolic T cell-factor/lymphoid-enhancer-factor (Tcf/Lef) proteins, translocation of the resulting complex to the nucleus, and activation of transcription [144]. Target genes include c-myc and cyclin D1 [145, 146]. Thus, one effect of APC inactivation is proliferation of the affected cells.
The importance of APC dysfunction in colon cancer is well established. Individuals who inherit a defective allele of the APC gene suffer from familial adenomatous polyposis (FAP), an autosomal dominant disease in which thousands of colonic polyps, many of which will progress to cancer if not removed, are developed during childhood and adolescence [147]. Furthermore, somatic mutation of the APC gene is found in the majority of sporadic CRC [148]. APC has usually been implicated in CIN, but this is still a matter of debate. Michor et al. have developed a mathematical approach for the cellular dynamics of colon cancer initiation, showing that genetic instability is an early event and thus a driving force of tumorigenesis, since a small number of CIN genes are sufficient to initiate colorectal tumorigenesis before APC inactivation [149].
ACF are considered microadenomas. In Vogelstein’s model, the progression from microadenoma to intermediate adenoma is accompanied by K-ras activation [150]. The K-ras gene encodes a 21-kD protein (p21ras) involved in G protein-mediated signal transduction. Ras mutations usually lead to constitutive activation of the signaling pathways controlling cell proliferation and differentiation [151]. After the formulation of Vogelstein’s theory, K-ras mutations were actually reported to occur in every step of colon carcinogenesis. Such an idea was supported by two observations: (1) both small and large adenomas sometimes have the same incidence of K-ras mutations and (2) K-ras mutations can be heterogeneous within the same carcinoma [152–154], suggesting a correlation to late tumorigenesis. By using a different sampling method to collect tumor DNA, Ishii et al. showed that K-ras mutations are instead homogeneous within the same carcinoma, and therefore they do not occur in late carcinogenesis [155].
The transition from an intermediate adenoma to a late adenoma is characterized by the loss of the deleted in colorectal cancer (DCC) tumor suppressor gene. Identified in 1990 by Fearon et al. within a previously described LOH region at 18q, the DCC gene encodes a protein which has been suggested to allow intestinal cell migration from the base to the top of the glandular crypts by reducing cell–matrix contacts and reinforcing cell–cell contacts through association with ezrin/radixin/moesin and merlin (ERM-M) proteins [156, 157]. Mutations of both DCC alleles contribute to tumor development by disrupting such contacts. In addition to DCC, SMAD2 and SMAD4 tumor suppressor genes are the targets of 18q LOH [158, 159]. Whereas mutations of DCC and SMAD2 seem to be very rare in CRC [160, 161], SMAD4 inactivation is likely to be involved in advanced stages such as distant metastasis [162].
Finally, allelic loss of the p53 tumor suppressor gene allows a growing tumor with multiple genetic alterations to evade cell cycle arrest and apoptosis, thus permitting a late adenoma to progress to carcinoma [150].
In summary, Vogelstein’s colon carcinogenesis model includes five key steps: (1) APC gene mutation leads to hyper-proliferation and (2), in succession, the formation of a class I adenoma; (3) a class II adenoma forms following K-ras activation; (4) loss of DCC is then responsible for class III adenoma formation; and (5) invasive cancer requires mutation of the p53 gene [150].
Our understanding of the molecular pathogenesis of CRC has advanced significantly since Vogelstein’s model was initially proposed, resulting in several reconsiderations of the so-called Vogelgram. We now know that many more genes and steps may be involved. Some of the early evidence that there were multiple molecular pathways to CRC came from identification of different histological and genetic features between CRCs in Lynch syndrome and CRCs developing through the Vogelstein’s adenoma–carcinoma sequence. Lynch-associated CRCs are more commonly right sided, often poorly differentiated or mucin-producing, and have a dense lymphocytic infiltrate and a Crohn’s-like reaction. Genetically, as we have already discussed, Lynch-associated CRCs are characterized by mutations in the DNA MMR system which are likely responsible for MSI. As shown in 1999 by Salahshor et al., mutations in APC and p53 are not necessary for initiation and progression of such MSI-positive CRC [163]. These types of tumors carry instead a mutation in the type II TGF beta receptor (TGFβR2) resulting in the inhibition of the TGFβ signaling pathway and a low metastatic rate. In accordance, Warusavitarne et al. have demonstrated that restoring TGFβ signaling reduces tumorigenicity and increases invasion and metastasis in MSI-H CRC cell lines [164].
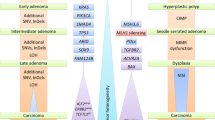
Additional evidence of the existence of multiple adenoma–carcinoma sequences came from the classification of colorectal polyps into two major groups: conventional adenomas and serrated polyps, the latter including hyperplastic polyps (HP), sessile serrated adenoma (SSA), sessile adenomas (SA), and mixed polyps [165]. Serrated polyps are usually found in the left colon, are smaller in size than adenomatous polyps, and have erroneously been considered as benign in nature. However, an equivalent to the adenoma–carcinoma sequence has recently been suggested for adenomas arising from those polyps, which includes an activating mutation in the BRAF gene as the initiating event triggering the malignant transformation of the polyp [166]. Somatic molecular alterations associated with serrated polyps also include K-ras mutations, hMLH1, and MGMT methylation, the prevalence of which varies according to the subtype of serrated polyp [167]. The evidence that serrated polyposis is a genetic predisposition is accumulating. Its genetic basis is yet to be fully determined, though a small number of patients have reported mutations in mutY homolog (E. coli) (MUTYH) [168], phosphatase and tensin homolog (PTEN) [169], and ephrinB2 (EPHB2) genes [170]. Figure 14.2 illustrates how different pathogenic pathways can be involved in initiation and progression of right- versus left-sided colon cancers.

A simplified scheme illustrating how different pathogenic pathways can be involved in initiation and progression of right- versus left-sided colon cancers
One of the intriguing questions is whether the three above-described pathways of colon carcinogenesis initiate in identical cells or whether three different cells are the targets of multiple mutations. Over the last decade, the opinion on cancer biology has drastically changed. Contrary to the longstanding clonal evolution model described by Nowell in the late 1970s [171], the CSC hypothesis has recently proposed that not every cell of the body could be the target of malignant transformation. The limited lifespan of a committed cell is likely shorter than the time necessary to accumulate tumor-inducing genetic changes. Therefore, cancer-initiating capability could be a unique feature of the long-lived, self-renewing stem cells [172]. The CSC hypothesis is neither a universal model for all cancers nor for all patients with the same disease. While some cancers have been hypothesized to initiate as a stem cell disease, they may then progress by clonal evolution of their CSCs, as CRC has been suggested to do through CIN [173]. The aforementioned pathways of colon carcinogenesis could be derived from three different CSCs. Importantly, epigenetic modifications are likely to occur in these cells prior to the first gatekeeper mutation. Indeed, five lines of evidence suggest the existence of an epigenetically disrupted progenitor-cell population from which tumors arise: (1) tumor-related properties are stable but reversible; (2) global epigenetic changes must precede the earliest genetic alterations as they are always found, even in benign neoplasms; (3) cloned mouse melanoma nuclei can differentiate into normal mouse cells, indicating tumor properties can be reprogrammed and therefore are epigenetically controlled; (4) neoplastic clones can be maintained solely by a small population of cells with stem cell properties; and (5) the tumor microenvironment can affect the epigenetic state of progenitor cells [4]. Consistently, aberrant promoter methylation of several genes (p16, MINT31, MINT2, MINT1, MGMT, hMLH1 HLTF, and SLC5A8) has been observed in ACF, thus confirming that epigenetic disruption is a primary rather than a secondary event in colon tumorigenesis [174–176]. From this point of view, tumor heterogeneity and progression could be explained independently of genetic clonal evolution. This means that the ability to metastasize may not require subsequent mutation and clonal selection within a large tumor mass but could be an intrinsic feature of the progenitor cell from which the tumor arises. Unfortunately, no unifying theory has emerged to explain cancer origin and progression. This is an urgent challenge to address in the future in order to achieve targeted cancer therapies.
Cancer Diversity: From Players to Clinical Application
Early FAP and Lynch syndrome diagnoses and appropriate CRC follow-up care can improve survival. Genetic tests for both diseases have been developed. These detect mutations in the APC and MMR genes (MSH2 and MLH1), respectively, and can be used to assess risk and guide treatment decisions. Unfortunately, the accuracy of tests to detect germ-line mutations in candidate genes continues to be challenging [177, 178] and triggers debate over the ability of a proposed test to predict responsiveness to chemotherapy. For instance, a few research groups have recently evaluated classical MMR genes as predictive or prognostic biomarkers for colon cancer, and according to the most recent study, they are independent predictors of disease-free survival (DFS) in patients with stage III colon cancer receiving adjuvant 5-FU–oxaliplatin combination therapy (FOLFOX) [179–183]. Important findings about the utility of knowing the MSI status of non-MMR genes to select patients for chemotherapeutic treatment have recently came from Dorard et al., which have considered in their study a previously unknown mutation in the gene encoding the chaperone heat shock protein (HSP) 110. HSP110 T17 intronic DNA microsatellite mutations in MSI CRC result in the loss of HSP110 exon 9 and expression of a truncated protein, HSP110ΔE9, increasing tumor sensitivity to anticancer agents such as oxaliplatin and 5-FU [184].
Throughout this chapter, we have provided evidence to support the epigenetic origin of cancer. Importantly, as we gain insight into the functional significance of global changes in chromatin structure, and as new tools for specific and efficient detection of epigenetic marks become available, there will be an enormous opportunity to develop markers for disease diagnosis and drug response, as well as strategies to prevent further disease progression. In this context, the recent advent of microarray technologies has allowed the identification of epigenetic signatures for different cancers. Each tumor type has been suggested to have a specific “hypermethylome” [185], thus defining CpG hypermethylation maps for a growing list of primary tumors, including glioblastoma [186], acute myeloid leukemia [187], ovarian carcinoma [188], astrocytoma [189], and colon cancer [190]. As the list of tumor suppressor genes that are silenced through promoter hypermethylation grows, a correlation with response to therapy is investigated. For instance, transcription factor AP-2 epsilon (activating enhancer binding protein 2 epsilon), also known as TFAP2E, has recently been found to be hypermethylated in CRC patients correlating with the overexpression of the Wnt antagonist Dickkopf-related protein 4 (Dkk4) and chemoresistance [191]. Thus, the importance of epigenetic modifications in predicting patient prognosis and response to chemotherapy is increasingly recognized by several studies. Epigenetic markers may be detected easily in circulating DNA (cirDNA) in the plasma or other bodily fluids. For instance, circulating methylated septin (SEPT) 9 DNA in plasma is considered a biomarker for CRC [192]. However, further studies are needed to clearly define specific markers for accurate cancer detection and risk assessment. Consistently, the first epigenome-wide DNA modification profiling of plasma or other bodily fluids from cancer patients has been provided only recently by Cortese et al. in the context of prostate cancer [193].
Importantly, due to their reversibility, epigenetic changes are being investigated as potential therapeutic targets, leading to the development of new anticancer drugs. The first generation of Food and Drug Administration (FDA)-approved epigenetic-based drugs includes two DNA-demethylating agents, 5-azacytidine (AZA) and decitabine (DAC), and two histone deacetylase (HDAC) inhibitors, vorinostat (Vo) and valproic acid (VA). These drugs were developed for the treatment of blood diseases, in particular myelodysplastic syndromes (MDS), against which they were reported to be highly effective, leading to significant improvements in patient quality of life and survival [194]. Although epigenetic drugs in clinical trials for hematological malignancies have been successful, results were much more disappointing for solid tumors, probably because CSCs in solid tumors are confined to a niche that is less reachable by these drugs. Moreover, epigenetic drugs were reported to be toxic, triggering common side effects including nausea, vomiting, diarrhea, and myelosuppression. Nevertheless, the observation that low doses of DNMT and HDAC inhibitors together are able to reverse gene silencing associated with promoter methylation has created much interest. Particularly, the combination of HDAC and DNMT inhibition has been reported to be very effective (and synergistic) in inducing apoptosis, differentiation, and/or cell growth arrest in human lung, breast, thoracic, leukemic, and colon cancer cell lines [195]. Combining current cancer treatments with distinct chromatin remodeling factors may reduce the effective drug concentration and related systemic toxicity; however, other questions remain to be addressed. Specifically, pleiotropic effects and the lack of specificity of epigenetic drugs continue to pose important implications for clinical treatment. Indeed, epigenetic drugs have recently been reported to be able to wake up metastasis-related genes [196]. This finding strongly highlights the need to accurately assess the clinical effectiveness and side effects of putative epigenetic treatments before human testing. This can only be achieved through a full comprehension of cancer dynamics at the cellular and molecular level.
Concluding Remarks
One of the main unresolved problems of current available therapeutic treatments for cancer is the lack of selectivity combined with the lack of efficacy. To design a more successful approach and possibly achieve complete tumor regression, it will be necessary to identify the genetic as well as the epigenetic alterations underlying cancer etiology and progression, not only for each cancer, but probably for each patient. In conclusion, cancer can be viewed as a complex adaptive system [197]. Cancer cells evolve and adapt to resist the death-inducing stimuli they are subject to. As opposed to old-fashioned chemotherapy, emerging and future personalized therapies will help controlling the occurrence of unstable cells with acquired multidrug resistance by targeting only tumor cells while sparing normal cells and tissues.
References
Volpe JP (1988) Genetic instability of cancer. Why a metastatic tumor is unstable and a benign tumor is stable. Cancer Genet Cytogenet 34(1):125–134
Berman JJ (2008) The mystery of tumor diversity and of type-specific tumor uniformity. Neoplasms: Principles of Development and Diversity, Jones and Bartlett publishers, Sudbury, MA, p 42–50
Boveri T (2008) Concerning the origin of malignant tumours by theodor boveri. Translated and annotated by henry harris. J Cell Sci 121(Suppl 1):1–84
Feinberg AP, Ohlsson R, Henikoff S (2006) The epigenetic progenitor origin of human cancer. Nat Rev Genet 7(1):21–33
Lasko D, Cavenee W, Nordenskjold M (1991) Loss of constitutional heterozygosity in human cancer. Annu Rev Genet 25:281–314
Hopman AH, Moesker O, Smeets AW, Pauwels RP, Vooijs GP, Ramaekers FC (1991) Numerical chromosome 1, 7, 9, and 11 aberrations in bladder cancer detected by in situ hybridization. Cancer Res 51(2):644–651
Alers JC, Krijtenburg PJ, Rosenberg C, Hop WC, Verkerk AM, Schroder FH, van der Kwast TH, Bosman FT, van Dekken H (1997) Interphase cytogenetics of prostatic tumor progression: specific chromosomal abnormalities are involved in metastasis to the bone. Lab invest 77(5):437–448
Placer J, Espinet B, Salido M, Sole F, Gelabert-Mas A (2005) Correlation between histologic findings and cytogenetic abnormalities in bladder carcinoma: a fish study. Urology 65(5):913–918
Jallepalli PV, Lengauer C (2001) Chromosome segregation and cancer: cutting through the mystery. Nat Rev Cancer 1(2):109–117
Cahill DP, Lengauer C, Yu J, Riggins GJ, Willson JK, Markowitz SD, Kinzler KW, Vogelstein B (1998) Mutations of mitotic checkpoint genes in human cancers. Nature 392(6673):300–303
Shi Q, King RW (2005) Chromosome nondisjunction yields tetraploid rather than aneuploid cells in human cell lines. Nature 437(7061):1038–1042
Hartwell LH, Weinert TA (1989) Checkpoints: controls that ensure the order of cell cycle events. Science 246(4930):629–634
Elledge SJ (1996) Cell cycle checkpoints: preventing an identity crisis. Science 274(5293):1664–1672
Kastan MB, Onyekwere O, Sidransky D, Vogelstein B, Craig RW (1991) Participation of p53 protein in the cellular response to DNA damage. Cancer Res 51(23 Pt 1):6304–6311
Bischoff FZ, Yim SO, Pathak S, Grant G, Siciliano MJ, Giovanella BC, Strong LC, Tainsky MA (1990) Spontaneous abnormalities in normal fibroblasts from patients with li-fraumeni cancer syndrome: aneuploidy and immortalization. Cancer Res 50(24):7979–7984
Bouffler SD, Kemp CJ, Balmain A, Cox R (1995) Spontaneous and ionizing radiation-induced chromosomal abnormalities in p53-deficient mice. Cancer Res 55(17):3883–3889
Fukasawa K, Choi T, Kuriyama R, Rulong S, Vande Woude GF (1996) Abnormal centrosome amplification in the absence of p53. Science 271(5256):1744–1747
Yin Y, Tainsky MA, Bischoff FZ, Strong LC, Wahl GM (1992) Wild-type p53 restores cell cycle control and inhibits gene amplification in cells with mutant p53 alleles. Cell 70(6):937–948
Havre PA, Yuan J, Hedrick L, Cho KR, Glazer PM (1995) P53 inactivation by hpv16 e6 results in increased mutagenesis in human cells. Cancer Res 55(19):4420–4424
Mekeel KL, Tang W, Kachnic LA, Luo CM, DeFrank JS, Powell SN (1997) Inactivation of p53 results in high rates of homologous recombination. Oncogene 14(15):1847–1857
Malumbres M, Barbacid M (2001) To cycle or not to cycle: a critical decision in cancer. Nat Rev Cancer 1(3):222–231
Hinchcliffe EH, Li C, Thompson EA, Maller JL, Sluder G (1999) Requirement of cdk2-cyclin e activity for repeated centrosome reproduction in xenopus egg extracts. Science 283(5403):851–854
Adams PD, Sellers WR, Sharma SK, Wu AD, Nalin CM, Kaelin WG Jr (1996) Identification of a cyclin-cdk2 recognition motif present in substrates and p21-like cyclin-dependent kinase inhibitors. Mol Cell Biol 16(12):6623–6633
Hu YX, Watanabe H, Ohtsubo K, Yamaguchi Y, Ha A, Motoo Y, Okai T, Sawabu N (1998) Infrequent expression of p21 is related to altered p53 protein in pancreatic carcinoma. Clin Cancer Res 4(5):1147–1152
Roman-Gomez J, Castillejo JA, Jimenez A, Gonzalez MG, Moreno F, Rodriguez Mdel C, Barrios M, Maldonado J, Torres A (2002) 5′ cpg island hypermethylation is associated with transcriptional silencing of the p21(cip1/waf1/sdi1) gene and confers poor prognosis in acute lymphoblastic leukemia. Blood 99(7):2291–2296
Cervantes RB, Lundblad V (2002) Mechanisms of chromosome-end protection. Curr Opin Cell Biol 14(3):351–356
Maser RS, DePinho RA (2002) Connecting chromosomes, crisis, and cancer. Science 297(5581):565–569
Deng Y, Chan SS, Chang S (2008) Telomere dysfunction and tumour suppression: the senescence connection. Nat Rev Cancer 8(6):450–458
Rampazzo E, Bertorelle R, Serra L, Terrin L, Candiotto C, Pucciarelli S, Del Bianco P, Nitti D, De Rossi A (2010) Relationship between telomere shortening, genetic instability, and site of tumour origin in colorectal cancers. Br J Cancer 102(8):1300–1305
Zhang A, Wang J, Zheng B, Fang X, Angstrom T, Liu C, Li X, Erlandsson F, Bjorkholm M, Nordenskjord M, Gruber A, Wallin KL, Xu D (2004) Telomere attrition predominantly occurs in precursor lesions during in vivo carcinogenic process of the uterine cervix. Oncogene 23(44):7441–7447
Lin TT, Letsolo BT, Jones RE, Rowson J, Pratt G, Hewamana S, Fegan C, Pepper C, Baird DM (2010) Telomere dysfunction and fusion during the progression of chronic lymphocytic leukemia: evidence for a telomere crisis. Blood 116(11):1899–1907
Butler DK, Yasuda LE, Yao MC (1996) Induction of large DNA palindrome formation in yeast: implications for gene amplification and genome stability in eukaryotes. Cell 87(6):1115–1122
Chakraborty S, Stark JM, Sun CL, Modi H, Chen W, O’Connor TR, Forman SJ, Bhatia S, Bhatia R (2012) Chronic myelogenous leukemia stem and progenitor cells demonstrate chromosomal instability related to repeated breakage-fusion-bridge cycles mediated by non-homologous end joining. Blood 119(26):6187–6197
Cortes J, Jabbour E, Kantarjian H, Yin CC, Shan J, O’Brien S, Garcia-Manero G, Giles F, Breeden M, Reeves N, Wierda WG, Jones D (2007) Dynamics of bcr-abl kinase domain mutations in chronic myeloid leukemia after sequential treatment with multiple tyrosine kinase inhibitors. Blood 110(12):4005–4011
Hede K (2005) Which came first? Studies clarify role of aneuploidy in cancer. J Natl Cancer Inst 97(2):87–89
Weaver BA, Silk AD, Montagna C, Verdier-Pinard P, Cleveland DW (2007) Aneuploidy acts both oncogenically and as a tumor suppressor. Cancer Cell 11(1):25–36
Sheltzer JM, Amon A (2011) The aneuploidy paradox: costs and benefits of an incorrect karyotype. Trends Genet 27(11):446–453
Duncan AW, Taylor MH, Hickey RD, Hanlon Newell AE, Lenzi ML, Olson SB, Finegold MJ, Grompe M (2010) The ploidy conveyor of mature hepatocytes as a source of genetic variation. Nature 467(7316):707–710
Modrich P (1997) Strand-specific mismatch repair in mammalian cells. J Biol Chem 272(40):24727–24730
Duckett DR, Drummond JT, Murchie AI, Reardon JT, Sancar A, Lilley DM, Modrich P (1996) Human mutsalpha recognizes damaged DNA base pairs containing o6-methylguanine, o4-methylthymine, or the cisplatin-d(gpg) adduct. Proc Natl Acad Sci USA 93(13):6443–6447
Ionov Y, Peinado MA, Malkhosyan S, Shibata D, Perucho M (1993) Ubiquitous somatic mutations in simple repeated sequences reveal a new mechanism for colonic carcinogenesis. Nature 363(6429):558–561
Fishel R, Lescoe MK, Rao MR, Copeland NG, Jenkins NA, Garber J, Kane M, Kolodner R (1993) The human mutator gene homolog msh2 and its association with hereditary nonpolyposis colon cancer. Cell 75(5):1027–1038
Leach FS, Nicolaides NC, Papadopoulos N, Liu B, Jen J, Parsons R, Peltomaki P, Sistonen P, Aaltonen LA, Nystrom-Lahti M et al (1993) Mutations of a muts homolog in hereditary nonpolyposis colorectal cancer. Cell 75(6):1215–1225
Papadopoulos N, Lindblom A (1997) Molecular basis of hnpcc: mutations of mmr genes. Hum Mutat 10(2):89–99
Peltomaki P, de la Chapelle A (1997) Mutations predisposing to hereditary nonpolyposis colorectal cancer. Adv Cancer Res 71:93–119
Kane MF, Loda M, Gaida GM, Lipman J, Mishra R, Goldman H, Jessup JM, Kolodner R (1997) Methylation of the hmlh1 promoter correlates with lack of expression of hmlh1 in sporadic colon tumors and mismatch repair-defective human tumor cell lines. Cancer Res 57(5):808–811
Herman JG, Umar A, Polyak K, Graff JR, Ahuja N, Issa JP, Markowitz S, Willson JK, Hamilton SR, Kinzler KW, Kane MF, Kolodner RD, Vogelstein B, Kunkel TA, Baylin SB (1998) Incidence and functional consequences of hmlh1 promoter hypermethylation in colorectal carcinoma. Proc Natl Acad Sci USA 95(12):6870–6875
Liu B, Nicolaides NC, Markowitz S, Willson JK, Parsons RE, Jen J, Papadopolous N, Peltomaki P, de la Chapelle A, Hamilton SR et al (1995) Mismatch repair gene defects in sporadic colorectal cancers with microsatellite instability. Nat Genet 9(1):48–55
Borresen AL, Lothe RA, Meling GI, Lystad S, Morrison P, Lipford J, Kane MF, Rognum TO, Kolodner RD (1995) Somatic mutations in the hmsh2 gene in microsatellite unstable colorectal carcinomas. Hum Mol Genet 4(11):2065–2072
Bubb VJ, Curtis LJ, Cunningham C, Dunlop MG, Carothers AD, Morris RG, White S, Bird CC, Wyllie AH (1996) Microsatellite instability and the role of hmsh2 in sporadic colorectalcancer. Oncogene 12(12):2641–2649
Thibodeau SN, French AJ, Roche PC, Cunningham JM, Tester DJ, Lindor NM, Moslein G, Baker SM, Liskay RM, Burgart LJ, Honchel R, Halling KC (1996) Altered expression of hmsh2 and hmlh1 in tumors with microsatellite instability and genetic alterations in mismatch repair genes. Cancer Res 56(21):4836–4840
Wu Y, Nystrom-Lahti M, Osinga J, Looman MW, Peltomaki P, Aaltonen LA, de la Chapelle A, Hofstra RM, Buys CH (1997) Msh2 and mlh1 mutations in sporadic replication error-positive colorectal carcinoma as assessed by two-dimensional DNA electrophoresis. Genes Chromosomes Cancer 18(4):269–278
Goel A, Arnold CN, Niedzwiecki D, Chang DK, Ricciardiello L, Carethers JM, Dowell JM, Wasserman L, Compton C, Mayer RJ, Bertagnolli MM, Boland CR (2003) Characterization of sporadic colon cancer by patterns of genomic instability. Cancer Res 63(7):1608–1614
Camps J, Morales C, Prat E, Ribas M, Capella G, Egozcue J, Peinado MA, Miro R (2004) Genetic evolution in colon cancer km12 cells and metastatic derivates. Int J Cancer 110(6):869–874
Henikoff S, Matzke MA (1997) Exploring and explaining epigenetic effects. Trends Genet 13(8):293–295
Wolffe AP, Matzke MA (1999) Epigenetics: regulation through repression. Science 286(5439):481–486
Jablonka E, Lamb MJ (2002) The changing concept of epigenetics. Ann N Y Acad Sci 981:82–96
Jaenisch R, Bird A (2003) Epigenetic regulation of gene expression: how the genome integrates intrinsic and environmental signals. Nat Genet 33(Suppl):245–254
Ehrlich M, Gama-Sosa MA, Huang LH, Midgett RM, Kuo KC, McCune RA, Gehrke C (1982) Amount and distribution of 5-methylcytosine in human DNA from different types of tissues of cells. Nucleic Acids Res 10(8):2709–2721
Gardiner-Garden M, Frommer M (1987) Cpg islands in vertebrate genomes. J Mol Biol 196(2):261–282
Bird A (2002) DNA methylation patterns and epigenetic memory. Genes Dev 16(1):6–21
Feil R, Khosla S (1999) Genomic imprinting in mammals: an interplay between chromatin and DNA methylation? Trends Genet 15(11):431–435
Panning B, Jaenisch R (1998) Rna and the epigenetic regulation of x chromosome inactivation. Cell 93(3):305–308
Yoder JA, Walsh CP, Bestor TH (1997) Cytosine methylation and the ecology of intragenomic parasites. Trends Genet 13(8):335–340
Baylin SB, Herman JG, Graff JR, Vertino PM, Issa JP (1998) Alterations in DNA methylation: a fundamental aspect of neoplasia. Adv Cancer Res 72:141–196
Jones PA, Laird PW (1999) Cancer epigenetics comes of age. Nat Genet 21(2):163–167
Berdasco M, Esteller M (2010) Aberrant epigenetic landscape in cancer: how cellular identity goes awry. Dev Cell 19(5):698–711
Nichols J, Zevnik B, Anastassiadis K, Niwa H, Klewe-Nebenius D, Chambers I, Scholer H, Smith A (1998) Formation of pluripotent stem cells in the mammalian embryo depends on the pou transcription factor oct4. Cell 95(3):379–391
Chambers I, Colby D, Robertson M, Nichols J, Lee S, Tweedie S, Smith A (2003) Functional expression cloning of nanog, a pluripotency sustaining factor in embryonic stem cells. Cell 113(5):643–655
Mitsui K, Tokuzawa Y, Itoh H, Segawa K, Murakami M, Takahashi K, Maruyama M, Maeda M, Yamanaka S (2003) The homeoprotein nanog is required for maintenance of pluripotency in mouse epiblast and es cells. Cell 113(5):631–642
Boyer LA, Lee TI, Cole MF, Johnstone SE, Levine SS, Zucker JP, Guenther MG, Kumar RM, Murray HL, Jenner RG, Gifford DK, Melton DA, Jaenisch R, Young RA (2005) Core transcriptional regulatory circuitry in human embryonic stem cells. Cell 122(6):947–956
Hattori N, Nishino K, Ko YG, Hattori N, Ohgane J, Tanaka S, Shiota K (2004) Epigenetic control of mouse oct-4 gene expression in embryonic stem cells and trophoblast stem cells. J Biol Chem 279(17):17063–17069
Yeo S, Jeong S, Kim J, Han JS, Han YM, Kang YK (2007) Characterization of DNA methylation change in stem cell marker genes during differentiation of human embryonic stem cells. Biochem Biophys Res Commun 359(3):536–542
Guo Y, Liu S, Wang P, Zhao S, Wang F, Bing L, Zhang Y, Ling EA, Gao J, Hao A (2011) Expression profile of embryonic stem cell-associated genes oct4, sox2 and nanog in human gliomas. Histopathology 59(4):763–775
Li Y, Laterra J (2012) Cancer stem cells: distinct entities or dynamically regulated phenotypes? Cancer Res 72(3):576–580
Gama-Sosa MA, Slagel VA, Trewyn RW, Oxenhandler R, Kuo KC, Gehrke CW, Ehrlich M (1983) The 5-methylcytosine content of DNA from human tumors. Nucleic Acids Res 11(19):6883–6894
Goelz SE, Vogelstein B, Hamilton SR, Feinberg AP (1985) Hypomethylation of DNA from benign and malignant human colon neoplasms. Science 228(4696):187–190
Bedford MT, van Helden PD (1987) Hypomethylation of DNA in pathological conditions of the human prostate. Cancer Res 47(20):5274–5276
Feinberg AP, Gehrke CW, Kuo KC, Ehrlich M (1988) Reduced genomic 5-methylcytosine content in human colonic neoplasia. Cancer Res 48(5):1159–1161
Kim YI, Giuliano A, Hatch KD, Schneider A, Nour MA, Dallal GE, Selhub J, Mason JB (1994) Global DNA hypomethylation increases progressively in cervical dysplasia and carcinoma. Cancer 74(3):893–899
Cravo M, Pinto R, Fidalgo P, Chaves P, Gloria L, Nobre-Leitao C, Costa Mira F (1996) Global DNA hypomethylation occurs in the early stages of intestinal type gastric carcinoma. Gut 39(3):434–438
Bernardino J, Roux C, Almeida A, Vogt N, Gibaud A, Gerbault-Seureau M, Magdelenat H, Bourgeois CA, Malfoy B, Dutrillaux B (1997) DNA hypomethylation in breast cancer: an independent parameter of tumor progression? Cancer Genet Cytogenet 97(2):83–89
Shen L, Fang J, Qiu D, Zhang T, Yang J, Chen S, Xiao S (1998) Correlation between DNA methylation and pathological changes in human hepatocellular carcinoma. Hepatogastroenterology 45(23):1753–1759
Santourlidis S, Florl A, Ackermann R, Wirtz HC, Schulz WA (1999) High frequency of alterations in DNA methylation in adenocarcinoma of the prostate. Prostate 39(3):166–174
Lin CH, Hsieh SY, Sheen IS, Lee WC, Chen TC, Shyu WC, Liaw YF (2001) Genome-wide hypomethylation in hepatocellular carcinogenesis. Cancer Res 61(10):4238–4243
Soares J, Pinto AE, Cunha CV, Andre S, Barao I, Sousa JM, Cravo M (1999) Global DNA hypomethylation in breast carcinoma: correlation with prognostic factors and tumor progression. Cancer 85(1):112–118
Widschwendter M, Jiang G, Woods C, Muller HM, Fiegl H, Goebel G, Marth C, Muller-Holzner E, Zeimet AG, Laird PW, Ehrlich M (2004) DNA hypomethylation and ovarian cancer biology. Cancer Res 64(13):4472–4480
Yang B, Sun H, Lin W, Hou W, Li H, Zhang L, Li F, Gu Y, Song Y, Li Q, Zhang F (2011) Evaluation of global DNA hypomethylation in human prostate cancer and prostatic intraepithelial neoplasm tissues by immunohistochemistry. Urol Oncol doi:10.1016/j urolonc.2011.05.009
Jurgens B, Schmitz-Drager BJ, Schulz WA (1996) Hypomethylation of l1 line sequences prevailing in human urothelial carcinoma. Cancer Res 56(24):5698–5703
Takai D, Yagi Y, Habib N, Sugimura T, Ushijima T (2000) Hypomethylation of line1 retrotransposon in human hepatocellular carcinomas, but not in surrounding liver cirrhosis. Jpn J Clin Oncol 30(7):306–309
Roman-Gomez J, Jimenez-Velasco A, Agirre X, Castillejo JA, Navarro G, San Jose-Eneriz E, Garate L, Cordeu L, Cervantes F, Prosper F, Heiniger A, Torres A (2008) Repetitive DNA hypomethylation in the advanced phase of chronic myeloid leukemia. Leuk Res 32(3):487–490
Greger V, Passarge E, Hopping W, Messmer E, Horsthemke B (1989) Epigenetic changes may contribute to the formation and spontaneous regression of retinoblastoma. Hum Genet 83(2):155–158
Sakai T, Toguchida J, Ohtani N, Yandell DW, Rapaport JM, Dryja TP (1991) Allele-specific hypermethylation of the retinoblastoma tumor-suppressor gene. Am J Hum Genet 48(5):880–888
Gonzalez-Zulueta M, Bender CM, Yang AS, Nguyen T, Beart RW, Van Tornout JM, Jones PA (1995) Methylation of the 5′ cpg island of the p16/cdkn2 tumor suppressor gene in normal and transformed human tissues correlates with gene silencing. Cancer Res 55(20):4531–4535
Graff JR, Herman JG, Lapidus RG, Chopra H, Xu R, Jarrard DF, Isaacs WB, Pitha PM, Davidson NE, Baylin SB (1995) E-cadherin expression is silenced by DNA hypermethylation in human breast and prostate carcinomas. Cancer Res 55(22):5195–5199
Herman JG, Latif F, Weng Y, Lerman MI, Zbar B, Liu S, Samid D, Duan DS, Gnarra JR, Linehan WM et al (1994) Silencing of the vhl tumor-suppressor gene by DNA methylation in renal carcinoma. Proc Natl Acad Sci USA 91(21):9700–9704
Merlo A, Herman JG, Mao L, Lee DJ, Gabrielson E, Burger PC, Baylin SB, Sidransky D (1995) 5′ cpg island methylation is associated with transcriptional silencing of the tumour suppressor p16/cdkn2/mts1 in human cancers. Nat Med 1(7):686–692
Cunningham JM, Christensen ER, Tester DJ, Kim CY, Roche PC, Burgart LJ, Thibodeau SN (1998) Hypermethylation of the hmlh1 promoter in colon cancer with microsatellite instability. Cancer Res 58(15):3455–3460
Veigl ML, Kasturi L, Olechnowicz J, Ma AH, Lutterbaugh JD, Periyasamy S, Li GM, Drummond J, Modrich PL, Sedwick WD, Markowitz SD (1998) Biallelic inactivation of hmlh1 by epigenetic gene silencing, a novel mechanism causing human msi cancers. Proc Natl Acad Sci USA 95(15):8698–8702
Catteau A, Harris WH, Xu CF, Solomon E (1999) Methylation of the brca1 promoter region in sporadic breast and ovarian cancer: correlation with disease characteristics. Oncogene 18(11):1957–1965
Toyota M, Ahuja N, Ohe-Toyota M, Herman JG, Baylin SB, Issa JP (1999) Cpg island methylator phenotype in colorectal cancer. Proc Natl Acad Sci USA 96(15):8681–8686
Clark SJ, Melki J (2002) DNA methylation and gene silencing in cancer: which is the guilty party? Oncogene 21(35):5380–5387
Esteller M (2008) Epigenetics in cancer. N Engl J Med 358(11):1148–1159
Issa JP, Vertino PM, Wu J, Sazawal S, Celano P, Nelkin BD, Hamilton SR, Baylin SB (1993) Increased cytosine DNA-methyltransferase activity during colon cancer progression. J Natl Cancer Inst 85(15):1235–1240
Schmidt WM, Sedivy R, Forstner B, Steger GG, Zochbauer-Muller S, Mader RM (2007) Progressive up-regulation of genes encoding DNA methyltransferases in the colorectal adenoma-carcinoma sequence. Mol Carcinog 46(9):766–772
Nosho K, Shima K, Irahara N, Kure S, Baba Y, Kirkner GJ, Chen L, Gokhale S, Hazra A, Spiegelman D, Giovannucci EL, Jaenisch R, Fuchs CS, Ogino S (2009) Dnmt3b expression might contribute to cpg island methylator phenotype in colorectal cancer. Clin Cancer Res 15(11):3663–3671
Ibrahim AE, Arends MJ, Silva AL, Wyllie AH, Greger L, Ito Y, Vowler SL, Huang TH, Tavare S, Murrell A, Brenton JD (2011) Sequential DNA methylation changes are associated with dnmt3b overexpression in colorectal neoplastic progression. Gut 60(4):499–508
Esteller M (2007) Epigenetic gene silencing in cancer: the DNA hypermethylome. Hum mol genet 16(Spec 1):R50–59
Vire E, Brenner C, Deplus R, Blanchon L, Fraga M, Didelot C, Morey L, Van Eynde A, Bernard D, Vanderwinden JM, Bollen M, Esteller M, Di Croce L, de Launoit Y, Fuks F (2006) The polycomb group protein ezh2 directly controls DNA methylation. Nature 439(7078):871–874
Lund AH, van Lohuizen M (2004) Polycomb complexes and silencing mechanisms. Curr Opin Cell Biol 16(3):239–246
Jacobs JJ, Kieboom K, Marino S, DePinho RA, van Lohuizen M (1999) The oncogene and polycomb-group gene bmi-1 regulates cell proliferation and senescence through the ink4a locus. Nature 397(6715):164–168
Douglas D, Hsu JH, Hung L, Cooper A, Abdueva D, van Doorninck J, Peng G, Shimada H, Triche TJ, Lawlor ER (2008) Bmi-1 promotes ewing sarcoma tumorigenicity independent of cdkn2a repression. Cancer Res 68(16):6507–6515
Ezhkova E, Pasolli HA, Parker JS, Stokes N, Su IH, Hannon G, Tarakhovsky A, Fuchs E (2009) Ezh2 orchestrates gene expression for the stepwise differentiation of tissue-specific stem cells. Cell 136(6):1122–1135
Bracken AP, Pasini D, Capra M, Prosperini E, Colli E, Helin K (2003) Ezh2 is downstream of the prb-e2f pathway, essential for proliferation and amplified in cancer. EMBO J 22(20):5323–5335
O’Carroll D, Erhardt S, Pagani M, Barton SC, Surani MA, Jenuwein T (2001) The polycomb-group gene ezh2 is required for early mouse development. Mol Cell Biol 21(13):4330–4336
Plath K, Fang J, Mlynarczyk-Evans SK, Cao R, Worringer KA, Wang H, de la Cruz CC, Otte AP, Panning B, Zhang Y (2003) Role of histone h3 lysine 27 methylation in x inactivation. Science 300(5616):131–135
Cao Q, Yu J, Dhanasekaran SM, Kim JH, Mani RS, Tomlins SA, Mehra R, Laxman B, Cao X, Yu J, Kleer CG, Varambally S, Chinnaiyan AM (2008) Repression of e-cadherin by the polycomb group protein ezh2 in cancer. Oncogene 27(58):7274–7284
Chen S, Bohrer LR, Rai AN, Pan Y, Gan L, Zhou X, Bagchi A, Simon JA, Huang H (2010) Cyclin-dependent kinases regulate epigenetic gene silencing through phosphorylation of ezh2. Nat Cell Biol 12(11):1108–1114
Fluge O, Gravdal K, Carlsen E, Vonen B, Kjellevold K, Refsum S, Lilleng R, Eide TJ, Halvorsen TB, Tveit KM, Otte AP, Akslen LA, Dahl O (2009) Expression of ezh2 and ki-67 in colorectal cancer and associations with treatment response and prognosis. Br J Cancer 101(8):1282–1289
Richter GH, Plehm S, Fasan A, Rossler S, Unland R, Bennani-Baiti IM, Hotfilder M, Lowel D, von Luettichau I, Mossbrugger I, Quintanilla-Martinez L, Kovar H, Staege MS, Muller-Tidow C, Burdach S (2009) Ezh2 is a mediator of ews/fli1 driven tumor growth and metastasis blocking endothelial and neuro-ectodermal differentiation. Proc Natl Acad Sci USA 106(13):5324–5329
Rao ZY, Cai MY, Yang GF, He LR, Mai SJ, Hua WF, Liao YJ, Deng HX, Chen YC, Guan XY, Zeng YX, Kung HF, Xie D (2010) Ezh2 supports ovarian carcinoma cell invasion and/or metastasis via regulation of tgf-beta1 and is a predictor of outcome in ovarian carcinoma patients. Carcinogenesis 31(9):1576–1583
Chen Y, Xie D, Yin Li W, Man Cheung C, Yao H, Chan CY, Chan CY, Xu FP, Liu YH, Sung JJ, Kung HF (2010) Rnai targeting ezh2 inhibits tumor growth and liver metastasis of pancreatic cancer in vivo. Cancer Lett 297(1):109–116
Hu SP, Zhou GB, Luan JA, Chen YP, Xiao DW, Deng YJ, Huang LQ, Cai KL (2010) Polymorphisms of hla-a and hla-b genes in genetic susceptibility to esophageal carcinoma in chaoshan han chinese. Dis Esophagus 23(1):46–52
Ren G, Baritaki S, Marathe H, Feng J, Park S, Beach S, Bazeley PS, Beshir AB, Fenteany G, Mehra R, Daignault S, Al-Mulla F, Keller E, Bonavida B, de la Serna I, Yeung KC (2012) Polycomb protein ezh2 regulates tumor invasion via the transcriptional repression of the metastasis suppressor rkip in breast and prostate cancer. Cancer Res 72(12):3091–3104
Yoon KA, Gil HJ, Han J, Park J, Lee JS (2010) Genetic polymorphisms in the polycomb group gene ezh2 and the risk of lung cancer. J Thorac Oncol 5(1):10–16
Crea F, Fornaro L, Paolicchi E, Masi G, Frumento P, Loupakis F, Salvatore L, Cremolini C, Schirripa M, Graziano F, Ronzoni M, Ricci V, Farrar WL, Falcone A, Danesi R (2012) An ezh2 polymorphism is associated with clinical outcome in metastatic colorectal cancer patients. Ann Oncol 23(5):1207–1213
Simon J (1995) Locking in stable states of gene expression: transcriptional control during drosophila development. Curr Opin Cell Biol 7(3):376–385
Chi P, Allis CD, Wang GG (2010) Covalent histone modifications–miswritten, misinterpreted and mis-erased in human cancers. Nat Rev Cancer 10(7):457–469
Krivtsov AV, Armstrong SA (2007) Mll translocations, histone modifications and leukaemia stem-cell development. Nat Rev Cancer 7(11):823–833
Fraga MF, Ballestar E, Villar-Garea A, Boix-Chornet M, Espada J, Schotta G, Bonaldi T, Haydon C, Ropero S, Petrie K, Iyer NG, Perez-Rosado A, Calvo E, Lopez JA, Cano A, Calasanz MJ, Colomer D, Piris MA, Ahn N, Imhof A, Caldas C, Jenuwein T, Esteller M (2005) Loss of acetylation at lys16 and trimethylation at lys20 of histone h4 is a common hallmark of human cancer. Nat Genet 37(4):391–400
Seligson DB, Horvath S, Shi T, Yu H, Tze S, Grunstein M, Kurdistani SK (2005) Global histone modification patterns predict risk of prostate cancer recurrence. Nature 435(7046):1262–1266
Song JS, Kim YS, Kim DK, Park SI, Jang SJ (2012) Global histone modification pattern associated with recurrence and disease-free survival in non-small cell lung cancer patients. Pathol Int 62(3):182–190
Pritchard CC, Cheng HH, Tewari M (2012) Microrna profiling: approaches and considerations. Nat Rev Genet 13(5):358–369
Wienholds E, Kloosterman WP, Miska E, Alvarez-Saavedra E, Berezikov E, de Bruijn E, Horvitz HR, Kauppinen S, Plasterk RH (2005) Microrna expression in zebrafish embryonic development. Science 309(5732):310–311
Alvarez-Garcia I, Miska EA (2005) Microrna functions in animal development and human disease. Development 132(21):4653–4662
Saito Y, Liang G, Egger G, Friedman JM, Chuang JC, Coetzee GA, Jones PA (2006) Specific activation of microrna-127 with downregulation of the proto-oncogene bcl6 by chromatin-modifying drugs in human cancer cells. Cancer Cell 9(6):435–443
Lujambio A, Ropero S, Ballestar E, Fraga MF, Cerrato C, Setien F, Casado S, Suarez-Gauthier A, Sanchez-Cespedes M, Git A, Spiteri I, Das PP, Caldas C, Miska E, Esteller M (2007) Genetic unmasking of an epigenetically silenced microrna in human cancer cells. Cancer Res 67(4):1424–1429
Walther A, Johnstone E, Swanton C, Midgley R, Tomlinson I, Kerr D (2009) Genetic prognostic and predictive markers in colorectal cancer. Nat Rev Cancer 9(7):489–499
Bird RP (1987) Observation and quantification of aberrant crypts in the murine colon treated with a colon carcinogen: preliminary findings. Cancer Lett 37(2):147–151
Pretlow TP, Barrow BJ, Ashton WS, O’Riordan MA, Pretlow TG, Jurcisek JA, Stellato TA (1991) Aberrant crypts: putative preneoplastic foci in human colonic mucosa. Cancer Res 51(5):1564–1567
Greenspan EJ, Cyr JL, Pleau DC, Levine J, Rajan TV, Rosenberg DW, Heinen CD (2007) Microsatellite instability in aberrant crypt foci from patients without concurrent colon cancer. Carcinogenesis 28(4):769–776
Jen J, Powell SM, Papadopoulos N, Smith KJ, Hamilton SR, Vogelstein B, Kinzler KW (1994) Molecular determinants of dysplasia in colorectal lesions. Cancer Res 54(21):5523–5526
Liu C, Li Y, Semenov M, Han C, Baeg GH, Tan Y, Zhang Z, Lin X, He X (2002) Control of beta-catenin phosphorylation/degradation by a dual-kinase mechanism. Cell 108(6):837–847
Behrens J, von Kries JP, Kuhl M, Bruhn L, Wedlich D, Grosschedl R, Birchmeier W (1996) Functional interaction of beta-catenin with the transcription factor lef-1. Nature 382(6592):638–642
He TC, Sparks AB, Rago C, Hermeking H, Zawel L, da Costa LT, Morin PJ, Vogelstein B, Kinzler KW (1998) Identification of c-myc as a target of the apc pathway. Science 281(5382):1509–1512
Shtutman M, Zhurinsky J, Simcha I, Albanese C, D’Amico M, Pestell R, Ben-Ze’ev A (1999) The cyclin d1 gene is a target of the beta-catenin/lef-1 pathway. Proc Natl Acad Sci USA 96(10):5522–5527
Friedrich A, Kullmann F (2003) familial adenomatous polyposis syndrome (fap): pathogenesis and molecular mechanisms. Med Klin (Munich) 98(12):776–782
Segditsas S, Tomlinson I (2006) Colorectal cancer and genetic alterations in the wnt pathway. Oncogene 25(57):7531–7537
Michor F, Iwasa Y, Rajagopalan H, Lengauer C, Nowak MA (2004) Linear model of colon cancer initiation. Cell Cycle 3(3):358–362
Vogelstein B, Fearon ER, Hamilton SR, Kern SE, Preisinger AC, Leppert M, Nakamura Y, White R, Smits AM, Bos JL (1988) Genetic alterations during colorectal-tumor development. N Engl J Med 319(9):525–532
Shapiro P (2002) Ras-map kinase signaling pathways and control of cell proliferation: relevance to cancer therapy. Crit Rev Clin Lab Sci 39(4–5):285–330
Al-Mulla F, Going JJ, Sowden ET, Winter A, Pickford IR, Birnie GD (1998) Heterogeneity of mutant versus wild-type ki-ras in primary and metastatic colorectal carcinomas, and association of codon-12 valine with early mortality. J Pathol 185(2):130–138
Giaretti W, Monaco R, Pujic N, Rapallo A, Nigro S, Geido E (1996) Intratumor heterogeneity of k-ras2 mutations in colorectal adenocarcinomas: association with degree of DNA aneuploidy. Am J Pathol 149(1):237–245
Shibata D, Schaeffer J, Li ZH, Capella G, Perucho M (1993) Genetic heterogeneity of the c-k-ras locus in colorectal adenomas but not in adenocarcinomas. J Natl Cancer Inst 85(13):1058–1063
Ishii M, Sugai T, Habano W, Nakamura S (2004) Analysis of ki-ras gene mutations within the same tumor using a single tumor crypt in colorectal carcinomas. J Gastroenterol 39(6):544–549
Fearon ER, Cho KR, Nigro JM, Kern SE, Simons JW, Ruppert JM, Hamilton SR, Preisinger AC, Thomas G, Kinzler KW et al (1990) Identification of a chromosome 18q gene that is altered in colorectal cancers. Science 247(4938):49–56
Martin M, Simon-Assmann P, Kedinger M, Mangeat P, Real FX, Fabre M (2006) Dcc regulates cell adhesion in human colon cancer derived ht-29 cells and associates with ezrin. Eur J Cell Biol 85(8):769–783
Hahn SA, Schutte M, Hoque AT, Moskaluk CA, da Costa LT, Rozenblum E, Weinstein CL, Fischer A, Yeo CJ, Hruban RH, Kern SE (1996) Dpc4, a candidate tumor suppressor gene at human chromosome 18q21.1. Science 271(5247):350–353
Eppert K, Scherer SW, Ozcelik H, Pirone R, Hoodless P, Kim H, Tsui LC, Bapat B, Gallinger S, Andrulis IL, Thomsen GH, Wrana JL, Attisano L (1996) Madr2 maps to 18q21 and encodes a tgfbeta-regulated mad-related protein that is functionally mutated in colorectal carcinoma. Cell 86(4):543–552
Rashid A, Hamilton SR (1997) Genetic alterations in sporadic and crohn’s-associated adenocarcinomas of the small intestine. Gastroenterology 113(1):127–135
Takenoshita S, Tani M, Mogi A, Nagashima M, Nagamachi Y, Bennett WP, Hagiwara K, Harris CC, Yokota J (1998) Mutation analysis of the smad2 gene in human colon cancers using genomic DNA and intron primers. Carcinogenesis 19(5):803–807
Miyaki M, Iijima T, Konishi M, Sakai K, Ishii A, Yasuno M, Hishima T, Koike M, Shitara N, Iwama T, Utsunomiya J, Kuroki T, Mori T (1999) Higher frequency of smad4 gene mutation in human colorectal cancer with distant metastasis. Oncogene 18(20):3098–3103
Salahshor S, Kressner U, Pahlman L, Glimelius B, Lindmark G, Lindblom A (1999) Colorectal cancer with and without microsatellite instability involves different genes. Genes Chromosomes Cancer 26(3):247–252
Warusavitarne J, McDougall F, de Silva K, Barnetson R, Messina M, Robinson BG, Schnitzler M (2009) Restoring tgfbeta function in microsatellite unstable (msi-h) colorectal cancer reduces tumourigenicity but increases metastasis formation. Int J Colorectal Dis 24(2):139–144
Snover DC, Jass JR, Fenoglio-Preiser C, Batts KP (2005) Serrated polyps of the large intestine: a morphologic and molecular review of an evolving concept. Am J Clin Pathol 124(3):380–391
O’Brien MJ, Yang S, Mack C, Xu H, Huang CS, Mulcahy E, Amorosino M, Farraye FA (2006) Comparison of microsatellite instability, cpg island methylation phenotype, braf and kras status in serrated polyps and traditional adenomas indicates separate pathways to distinct colorectal carcinoma end points. Am J Surg Pathol 30(12):1491–1501
Rosty C, Parry S, Young JP (2011) Serrated polyposis: an enigmatic model of colorectal cancer predisposition. Patholog Res Int 2011:157073
Boparai KS, Dekker E, Van Eeden S, Polak MM, Bartelsman JF, Mathus-Vliegen EM, Keller JJ, van Noesel CJ (2008) Hyperplastic polyps and sessile serrated adenomas as a phenotypic expression of myh-associated polyposis. Gastroenterology 135(6):2014–2018
Heald B, Mester J, Rybicki L, Orloff MS, Burke CA, Eng C (2010) Frequent gastrointestinal polyps and colorectal adenocarcinomas in a prospective series of pten mutation carriers. Gastroenterology 139(6):1927–1933
Kokko A, Laiho P, Lehtonen R, Korja S, Carvajal-Carmona LG, Jarvinen H, Mecklin JP, Eng C, Schleutker J, Tomlinson IP, Vahteristo P, Aaltonen LA (2006) Ephb2 germline variants in patients with colorectal cancer or hyperplastic polyposis. BMC Cancer 6:145
Nowell PC (1976) The clonal evolution of tumor cell populations. Science 194(4260):23–28
Wicha MS, Liu S, Dontu G (2006) Cancer stem cells: an old idea–a paradigm shift. Cancer Res 66(4):1883–1890, discussion 1895–1886
Odoux C, Fohrer H, Hoppo T, Guzik L, Stolz DB, Lewis DW, Gollin SM, Gamblin TC, Geller DA, Lagasse E (2008) A stochastic model for cancer stem cell origin in metastatic colon cancer. Cancer Res 68(17):6932–6941
Chan AO, Broaddus RR, Houlihan PS, Issa JP, Hamilton SR, Rashid A (2002) Cpg island methylation in aberrant crypt foci of the colorectum. Am J Pathol 160(5):1823–1830
Moinova HR, Chen WD, Shen L, Smiraglia D, Olechnowicz J, Ravi L, Kasturi L, Myeroff L, Plass C, Parsons R, Minna J, Willson JK, Green SB, Issa JP, Markowitz SD (2002) Hltf gene silencing in human colon cancer. Proc Natl Acad Sci USA 99(7):4562–4567
Li H, Myeroff L, Smiraglia D, Romero MF, Pretlow TP, Kasturi L, Lutterbaugh J, Rerko RM, Casey G, Issa JP, Willis J, Willson JK, Plass C, Markowitz SD (2003) Slc5a8, a sodium transporter, is a tumor suppressor gene silenced by methylation in human colon aberrant crypt foci and cancers. Proc Natl Acad Sci USA 100(14):8412–8417
Chen S, Watson P, Parmigiani G (2005) Accuracy of msi testing in predicting germline mutations of msh2 and mlh1: a case study in bayesian meta-analysis of diagnostic tests without a gold standard. Biostatistics 6(3):450–464
Scott RJ, Meldrum C, Crooks R, Spigelman AD, Kirk J, Tucker K, Koorey D (2001) Familial adenomatous polyposis: more evidence for disease diversity and genetic heterogeneity. Gut 48(4):508–514
Zaanan A, Cuilliere-Dartigues P, Guilloux A, Parc Y, Louvet C, de Gramont A, Tiret E, Dumont S, Gayet B, Validire P, Flejou JF, Duval A, Praz F (2010) Impact of p53 expression and microsatellite instability on stage iii colon cancer disease-free survival in patients treated by 5-fluorouracil and leucovorin with or without oxaliplatin. Ann Oncol 21(4):772–780
Sargent DJ, Marsoni S, Monges G, Thibodeau SN, Labianca R, Hamilton SR, French AJ, Kabat B, Foster NR, Torri V, Ribic C, Grothey A, Moore M, Zaniboni A, Seitz JF, Sinicrope F, Gallinger S (2010) Defective mismatch repair as a predictive marker for lack of efficacy of fluorouracil-based adjuvant therapy in colon cancer. J Clin Oncol 28(20):3219–3226
Kim ST, Lee J, Park SH, Park JO, Lim HY, Kang WK, Kim JY, Kim YH, Chang DK, Rhee PL, Kim DS, Yun H, Cho YB, Kim HC, Yun SH, Lee WY, Chun HK, Park YS (2010) Clinical impact of microsatellite instability in colon cancer following adjuvant folfox therapy. Cancer Chemother Pharmacol 66(4):659–667
Des Guetz G, Lecaille C, Mariani P, Bennamoun M, Uzzan B, Nicolas P, Boisseau A, Sastre X, Cucherousset J, Lagorce C, Schischmanoff PO, Morere JF (2010) Prognostic impact of microsatellite instability in colorectal cancer patients treated with adjuvant folfox. Anticancer Res 30(10):4297–4301
Zaanan A, Flejou JF, Emile JF, Des GG, Cuilliere-Dartigues P, Malka D, Lecaille C, Validire P, Louvet C, Rougier P, de Gramont A, Bonnetain F, Praz F, Taieb J (2011) Defective mismatch repair status as a prognostic biomarker of disease-free survival in stage iii colon cancer patients treated with adjuvant folfox chemotherapy. Clin Cancer Res 17(23):7470–7478
Dorard C, de Thonel A, Collura A, Marisa L, Svrcek M, Lagrange A, Jego G, Wanherdrick K, Joly AL, Buhard O, Gobbo J, Penard-Lacronique V, Zouali H, Tubacher E, Kirzin S, Selves J, Milano G, Etienne-Grimaldi MC, Bengrine-Lefevre L, Louvet C, Tournigand C, Lefevre JH, Parc Y, Tiret E, Flejou JF, Gaub MP, Garrido C, Duval A (2011) Expression of a mutant hsp110 sensitizes colorectal cancer cells to chemotherapy and improves disease prognosis. Nat Med 17(10):1283–1289
Costello JF, Fruhwald MC, Smiraglia DJ, Rush LJ, Robertson GP, Gao X, Wright FA, Feramisco JD, Peltomaki P, Lang JC, Schuller DE, Yu L, Bloomfield CD, Caligiuri MA, Yates A, Nishikawa R, Su Huang H, Petrelli NJ, Zhang X, O’Dorisio MS, Held WA, Cavenee WK, Plass C (2000) Aberrant cpg-island methylation has non-random and tumour-type-specific patterns. Nat Genet 24(2):132–138
Martinez R, Martin-Subero JI, Rohde V, Kirsch M, Alaminos M, Fernandez AF, Ropero S, Schackert G, Esteller M (2009) A microarray-based DNA methylation study of glioblastoma multiforme. Epigenetics 4(4):255–264
Figueroa ME, Lugthart S, Li Y, Erpelinck-Verschueren C, Deng X, Christos PJ, Schifano E, Booth J, van Putten W, Skrabanek L, Campagne F, Mazumdar M, Greally JM, Valk PJ, Lowenberg B, Delwel R, Melnick A (2010) DNA methylation signatures identify biologically distinct subtypes in acute myeloid leukemia. Cancer Cell 17(1):13–27
Houshdaran S, Hawley S, Palmer C, Campan M, Olsen MN, Ventura AP, Knudsen BS, Drescher CW, Urban ND, Brown PO, Laird PW (2010) DNA methylation profiles of ovarian epithelial carcinoma tumors and cell lines. PLoS One 5(2):e9359
Wu X, Rauch TA, Zhong X, Bennett WP, Latif F, Krex D, Pfeifer GP (2010) Cpg island hypermethylation in human astrocytomas. Cancer Res 70(7):2718–2727
Xu Y, Hu B, Choi AJ, Gopalan B, Lee BH, Kalady MF, Church JM, Ting AH (2012) Unique DNA methylome profiles in cpg island methylator phenotype colon cancers. Genome Res 22(2):283–291
Ebert MP, Tanzer M, Balluff B, Burgermeister E, Kretzschmar AK, Hughes DJ, Tetzner R, Lofton-Day C, Rosenberg R, Reinacher-Schick AC, Schulmann K, Tannapfel A, Hofheinz R, Rocken C, Keller G, Langer R, Specht K, Porschen R, Stohlmacher-Williams J, Schuster T, Strobel P, Schmid RM (2012) Tfap2e-dkk4 and chemoresistance in colorectal cancer. N Engl J Med 366(1):44–53
deVos T, Tetzner R, Model F, Weiss G, Schuster M, Distler J, Steiger KV, Grutzmann R, Pilarsky C, Habermann JK, Fleshner PR, Oubre BM, Day R, Sledziewski AZ, Lofton-Day C (2009) Circulating methylated sept9 DNA in plasma is a biomarker for colorectal cancer. Clin Chem 55(7):1337–1346
Cortese R, Kwan A, Lalonde E, Bryzgunova O, Bondar A, Wu Y, Gordevicius J, Park M, Oh G, Kaminsky Z, Tverkuviene J, Laurinavicius A, Jankevicius F, Sendorek DH, Haider S, Wang SC, Jarmalaite S, Laktionov P, Boutros PC, Petronis A (2012) Epigenetic markers of prostate cancer in plasma circulating DNA. Hum Mol Genet 21(16):3619–3631
Musolino C, Sant’antonio E, Penna G, Alonci A, Russo S, Granata A, Allegra A (2010) Epigenetic therapy in myelodysplastic syndromes. Eur J Haematol 84(6):463–473
Zhu WG, Otterson GA (2003) The interaction of histone deacetylase inhibitors and DNA methyltransferase inhibitors in the treatment of human cancer cells. Curr Med Chem Anticancer Agents 3(3):187–199
Yu Y, Zeng P, Xiong J, Liu Z, Berger SL, Merlino G (2010) Epigenetic drugs can stimulate metastasis through enhanced expression of the pro-metastatic ezrin gene. PLoS One 5(9):e12710
Schwab ED, Pienta KJ (1996) Cancer as a complex adaptive system. Med hypotheses 47(3):235–241
Acknowledgements
This work was supported by Ri.MED foundation (M. G. F.) and NIH grant DK085711 (E. L.). The authors are grateful to Julie Chandler, Lynda Guzik, and Aaron DeWard for editorial assistance.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2013 Springer Science+Business Media New York
About this chapter
Cite this chapter
Francipane, M.G., Lagasse, E. (2013). A Study of Cancer Heterogeneity: From Genetic Instability to Epigenetic Diversity in Colorectal Cancer. In: Bae, Y., Mrsny, R., Park, K. (eds) Cancer Targeted Drug Delivery. Springer, New York, NY. https://doi.org/10.1007/978-1-4614-7876-8_14
Download citation
DOI: https://doi.org/10.1007/978-1-4614-7876-8_14
Published:
Publisher Name: Springer, New York, NY
Print ISBN: 978-1-4614-7875-1
Online ISBN: 978-1-4614-7876-8
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)