
Abstract
In this chapter, we will consider only those inborn errors of glycogen and fatty acid metabolism that cause exclusively or predominantly neuromuscular disorders. Because this is meant to be a practical book, we have subdivided these disorders into two groups according to their typical clinical presentation. Thus, we first consider diseases characterized by dynamic symptoms and then review disorders characterized by static symptoms. Although the mitochondrial myopathies are reviewed in a separate chapter (see Chap. 64), we discuss here some defects of the mitochondrial respiratory chain that are dominated by exercise intolerance, and, sometimes, myoglobinuria. To understand the dynamic versus the static clinical presentation of these disorders, a brief review of muscle metabolism at rest and during exercise is helpful.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others


Keywords
- Glycogen storage disease
- lipid storage disease
- metabolic myopathy
- McArdle disease
- Pompe disease
- acid maltase deficiency
- polygucosan disease
- myophosphorylase deficiency
Introduction
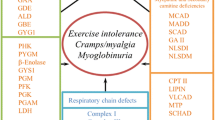
Inborn errors of glycogen and fatty acid metabolism that cause exclusively or predominantly neuromuscular disorders are characterized by dynamic or static symptoms. Dynamic symptoms are acute, recurrent, reversible muscle dysfunctions, manifesting as exercise intolerance, myalgia with or without painful cramps (contractures), and often culminating in muscle breakdown and myoglobinuria. In contrast, static symptoms are manifested by fixed, often progressive weakness, sometimes simulating dystrophic or neurogenic processes (Fig. 63.1).

The two major clinical syndromes seen in metabolic myopathies. Deficient enzymes are denoted by abbreviations or gene symbols as follows: GAA acid maltase (acid α-glucosidase) (GSD II), GDE glycogen debranching (GSD III), ALD aldolase (GSD IX), GBE glycogen branching (GSD IV), GYG1 glycogenin (GSD 0), PHK phosphorylase kinase (GSD VIII), PYGM myophosphorylase (GSD V), GYS1 glycogen synthetase, PGM phosphoglucomutase (GSD IV), PFK phosphofructokinase (GSD VII), PGK phosphoglycerate kinase (GSD IX), PGAM phosphoglycerate mutase (GSD X), LDH lactate dehydrogenase (GSD XI), MTP mitochondrial trifunctional enzyme, MCAD medium-chain acyl-CoA dehydrogenase, MADD multiple acyl-CoA dehydrogenase, SCAD short-chain acyl-CoA dehydrogenase, GAII glutaric aciduria type II, NLSDI neutral lipid storage diseases with ichthyosis (Chanarin-Dorfman disease), NLSDM neutral storage disease with myopathy, CPT II carnitine palmitoyltransferase II, VLCAD very-long-chain acyl-CoA dehydrogenase, SCHAD short-chain 3-hydroxyacyl-CoA dehydrogenase, ISCU nonheme iron-sulfur (Fe-S) protein, CoQ 10 coenzyme Q10
To understand glycogen and lipid storage disorders, a brief review of muscle metabolism at rest and during exercise is helpful.
The “fuel” utilized by muscle depends on several factors, most importantly the type, intensity, and duration of exercise but also diet and physical conditioning. At rest, muscle utilizes predominantly fatty acids. At the opposite end of the spectrum, the energy for extremely intense exercise (close to one’s maximal oxygen uptake, or VO2max, in dynamic exercise or close to maximal force generation in isometric exercise) derives from anaerobic glycolysis, especially when there is a “burst” of activity with rapid acceleration to maximal exercise. During submaximal exercise, the type of fuel utilized by muscle depends on the relative intensity of exertion. At low intensity (below 50 % VO2max), blood glucose and free fatty acids (FFA) are the primary sources of energy. At higher intensities, the proportion of energy derived from carbohydrate oxidation increases, and muscle glycogen becomes an important fuel; at 70–80 % VO2max, aerobic metabolism of glycogen is the crucial source of energy, and fatigue appears to set in when glycogen is exhausted. The type of circulating substrate utilized during mild exercise varies with time, and there is a gradual increase in the utilization of FFA over glucose until, a few hours into exercise, lipid oxidation becomes the major source of energy. Because the availability of FFA from adipose tissue is virtually unlimited, a normal person can perform moderate dynamic exercise for many hours.
Figure 63.2 illustrates schematically mitochondrial energy metabolism, including the “points of entry” of carbohydrate and lipid fuel. Pyruvate, the terminal product of aerobic glycolysis, is carried across the inner mitochondrial membrane by an incompletely known transporter system. Transport of FFA requires a more complex system, which includes two enzymes (carnitine palmitoyltransferase [CPT] I and CPT II), a carrier molecule (l-carnitine), and a translocase (carnitine-acylcarnitine translocase, CACT). After oxidation of pyruvate through the pyruvate dehydrogenase complex (PDHC) and of fatty acyl-CoAs through ß-oxidation, both carbohydrate and lipid fuels converge into a common central metabolite, acetyl-CoA, which is further oxidized in the Krebs cycle. The reducing equivalents produced in the Krebs cycle and in the ß-oxidation spirals are passed along a chain of proteins embedded in the inner mitochondrial membrane (the electron transport chain) to molecular oxygen with production of water. The electron transport chain consists of four multimeric complexes (I to IV) plus two small electron carriers, coenzyme Q (or ubiquinone) and cytochrome c. The energy generated by these reactions is used to pump protons from the mitochondrial matrix into the space between inner and outer mitochondrial membranes. This creates an electrochemical proton gradient across the inner membrane. A fifth multimeric complex (complex V or adenosine triphosphate [ATP] synthase), a tiny rotary engine, converts the energy of the electrochemical proton gradient into ATP, in a process known as oxidation/phosphorylation coupling. The terminal pathway of oxidative metabolism, comprising the electron transport chain and complex V, is known as the respiratory chain.

Schematic representation of mitochondrial metabolism. Abbreviations: PDHC pyruvate dehydrogenase complex, CPT carnitine palmitoyltransferase, VLCAD very-long-chain acyl-CoA dehydrogenase, TP trifunctional protein, LCAD long-chain acyl-CoA dehydrogenase, MCAD medium-chain acyl-CoA dehydrogenase, SCAD short-chain acyl-CoA dehydrogenase, HAD 3-hydroxyacyl-CoA dehydrogenase, KT 3-ketothiolase, ETFox oxidized form of electron transfer flavoprotein, ETFred reduced form of electron transfer flavoprotein, ETFDH ETF-coenzyme Q oxidoreductase, CG!-58 is the protein that activates ATGL, ATGL adipocyte triglyceride lipase (Reproduced from DiMauro and Haller [198], with permission)
Disorders Causing Exercise Intolerance and Myoglobinuria
In general, there is a good correlation between the circumstances leading to clinical problems and the different roles of glycogen and lipid metabolism in the provision of energy. Thus, the complaints of patients with glycogenoses are almost invariably related to an identifiable, and usually strenuous, bout of exertion, and the muscles that hurt, swell, or cramp up are those that have been engaged in that particular type of exercise.
In contrast, patients with disorders of lipid metabolism, such as CPT II or very-long-chain acyl-CoA dehydrogenase (VLCAD) deficiency, often have no warning of an impending episode of myoglobinuria, which usually follows prolonged moderate exercise and may be heralded by myalgia of exercising muscles but is never accompanied by painful cramps. In addition, prolonged fasting in and by itself may cause myoglobinuria, in which case any muscle group may be affected, including respiratory muscles; a few patients with CPT II deficiency have been taken to the emergency room in respiratory distress during an episode of myoglobinuria [1]. The deleterious effect of fasting in CPT II deficiency is easily explained by the increased dependence of muscle on FFA oxidation, which is partially blocked. Conversely, some patients with myophosphorylase deficiency, a glycogen storage disease, note a beneficial effect of fasting on their exercise ability. This is explained by the mobilization of FFA, which facilitates the physiological switch from carbohydrate to lipid utilization.
The respiratory chain is indeed the “business end” of mitochondrial metabolism, where the energy generated by carbohydrate and lipid oxidation is released as ATP. This raises an interesting question: if disturbances of glycogen and lipid metabolism cause exercise intolerance, cramps, and myoglobinuria by impairing ATP production, why did it take so long to associate this syndrome with defects in the respiratory chain, the energetic pathway “par excellence”? The answer is that these patients often stay below the clinical radar because they contradict the rules of mitochondrial genetics (see below) [2].
Glycogenoses
In reviewing the glycogenoses causing exercise intolerance and myoglobinuria, we follow the metabolic “flow” in the glycogenolytic and glycolytic pathways rather than the historical numeration (Fig. 63.3).

Scheme of glycogen metabolism and glycolysis. Roman numerals indicate enzymes whose deficiencies are associated with muscle glycogenoses: II acid maltase (GAA, Pompe disease), III debrancher (GDE, Cori-Forbes disease), IV brancher (GBE, Andersen disease), V myophosphorylase (PYGM, McArdle’s disease), VII phosphofructokinase (PFK, Tarui disease), VIII phosphorylase kinase (PHK), IX phoshoglycerate kinase (PGK), X phosphoglycerate mutase (PGAM), XI lactate dehydrogenase (LDH), XII aldolase A. Normal numerals indicate glycogenoses causing exercise intolerance, cramps, and myoglobinuria; italic numerals indicate glycogenoses causing fixed weakness. Abbreviations: UDPG uridine diphosphate glucose, PLD phosphorylase-limit dextrin, AMP adenosine monophosphate, ADP adenosine diphosphate, ATP adenosine triphosphate, P i inorganic phosphate
Phosphorylase Kinase (PhK) Deficiency (Glycogenosis Type VIII)
PhK is a key regulatory enzyme in glycogen metabolism because it activates glycogen phosphorylase by phosphorylating a specific serine in response to neuronal or hormonal stimuli. PhK deficiency is associated with four distinct clinical presentations, which are distinguished on the basis of tissue involvement (liver, muscle, heart, or liver and muscle) and mode of inheritance (autosomal or X-linked). This clinical and genetic heterogeneity is explained by the complexity of the enzyme, a decahexameric protein composed of four subunits (αβγδ)4: the α and β subunits are regulatory, the γ is catalytic, and the δ- is bound to calmodulin and confers calcium sensitivity to the enzyme. In addition, there are two isoforms for the α subunit (muscle and liver, A1 and A2), both encoded by genes on the X-chromosome, and two isoforms for the γ subunit (muscle and testis, G1 and liver, G2). Both G isozymes and the B-subunit are encoded by autosomal genes.
The purely myopathic variant of PhK deficiency usually manifests as a milder form of myophosphorylase deficiency (McArdle’s disease), with exercise intolerance, cramps, and, infrequently, myoglobinuria. One distinguishing laboratory feature is the lactate response to the ischemic forearm exercise test (see Chap. 6), which is usually flat in patients with McArdle’s disease, while it is normal or blunted in patients with PhK deficiency. Only about 15 patients with myopathic PhK deficiency have been reported, most of whom were men, suggesting X-linked inheritance. In agreement with this concept, all molecular defects identified thus far were in the PHKA1 gene [3].
The liver and muscle variant of PhK is an autosomal recessive disorder dominated by hepatomegaly and fasting hypoglycemia with minimal muscle involvement: in one female and four male patients, five distinct nonsense mutations have been identified in the PHKB gene [4].
The cardiac phenotype of PhK deficiency is secondary to mutations in the gene (PRKAG2) encoding the γ2 subunit of AMP-activated protein kinase (AMPK) [5]. The mechanism of this AMPK-mediated PhK inhibition is unknown.
Myophosphorylase Deficiency (Glycogenosis Type V; McArdle’s Disease)
Introduction
Although McArdle did not identify the biochemical defect, never was an eponym more appropriate than in this case. In 1951, on the basis of clinical observations and a few critical lab tests, Brian McArdle gave a remarkably precise description of the metabolic problem. He noted that ischemic exercise resulted in painful cramps of forearm muscles and that no electrical activity was recorded from the shortened muscles, indicating that they were in a state of contracture. He also noted that oxygen consumption and ventilation were normal at rest but increased more than normal with exercise. The astute observation that venous lactate and pyruvate did not increase after exercise led McArdle to conclude that his patient’s disorder was “characterized by a gross failure of the breakdown of glycogen to lactic acid.” Nor was the specific involvement of muscle lost to McArdle, who noted that epinephrine elicited a normal rise of blood glucose and “shed blood” in vitro accumulated lactate normally, leading him to conclude that “the disorder of carbohydrate metabolism affected chiefly, if not entirely, the skeletal muscles.”
Etiology and Pathogenesis
Myophosphorylase deficiency is transmitted as an autosomal recessive trait, and the gene for the muscle isoenzyme of phosphorylase has been localized to chromosome 11 [7]. The apparently autosomal dominant transmission in a few families may be explained by at least two mechanisms: (1) the presence in subsequent generations of homozygous and manifesting heterozygous individuals [8–10] or (2) the presence of one homozygous and one heterozygous parent [11].
The molecular heterogeneity of myophosphorylase deficiency is striking: in the 19 years from the description of the first molecular defects [11, 12], more than 100 different mutations have been reported [13]. The most common mutation in North America and Northern Europe appears to be a cytosine-to-thymine substitution in codon 50 of exon 1, converting an arginine to a stop codon (R50X). This has allowed the use of molecular genetic analysis in leukocytes for diagnostic purposes, thus avoiding the obligatory need of a muscle biopsy [14]. However, as more and more mutations in the myophosphorylase gene are described and leukocytes are increasingly used for diagnosis, it becomes important to establish the relative frequency of the different mutations in ethnic groups. For example, the R50X mutation has never been observed in Japan, where the most common mutation appears to be a 3-bp deletion, TTC at codon 708/709 [15].
The genotype/phenotype relationship in McArdle’s disease remains unclear: the most common genetic defect in typical McArdle patients, R50X, was also found in an infant who had the fatal myopathic variant [11] and in a child who died of sudden infant death syndrome (SIDS) [16]. Of course, one cannot exclude that these unusual presentations may be due to additional gene defects. Unlikely as this situation may appear, it has been verified in two children with McArdle’s disease (both homozygous for the common mutation), who were also homozygous for the most common mutation associated with adenylate deaminase (AMPD) deficiency [17, 18]. One patient had myoglobinuria at the unusually early age of 2 years; the other was a young man with multiple episodes of myoglobinuria and early onset of weakness. Although AMPD deficiency per se is an inconsistent cause of myopathy, it might have worsened the phenotypic expression of myophosphorylase deficiency.
Phosphorylase activity is virtually absent in muscle biopsies when determined either histochemically or biochemically. Biochemical studies show that the enzyme is lacking in most patients, which is consistent with the most common genetic error, a nonsense mutation (see above). As a consequence, glycogen of normal structure accumulates in muscle, reaching concentrations two or three times higher than normal. Morphologically, the stored glycogen is mostly visible at the periphery of muscle fibers, where it forms subsarcolemmal “blebs.” However, glycogen accumulation may be mild enough to escape morphological detection.
Phosphorylase catalyzes the first step in glycogen breakdown by removing 1,4-glucosyl residues phosphorolytically with liberation of glucose-1-phosphate. Hence, lack of phosphorylase impedes glycolysis, as shown by the flat venous lactate response to the ischemic forearm exercise test. Similarly, 31P-nuclear magnetic resonance spectroscopy (MRS) shows lack of acidification during aerobic or ischemic exercise and a greater than normal drop of the phosphocreatine/inorganic phosphate ratio [19].
Two main pathophysiological mechanisms underlie the exercise intolerance of myophosphorylase deficiency: (1) block of anaerobic glycolysis deprives muscle of the energy needed for isometric exercise and (2) block of aerobic glycogen utilization, with the attending shortage of pyruvate and acetyl-CoA, impairs dynamic exercise above certain intensity (approximately 50 % VO2max). In agreement with the concept that oxidative phosphorylation is curtailed by decreased substrate availability, oxygen extraction and maximal oxygen uptake are decreased in myophosphorylase deficiency but may be at least partially restored by intravenous glucose infusion [20]. Haller et al. have also documented lower concentrations of Na+/K+ pumps in needle muscle biopsies from patients with McArdle’s disease than in controls [21]. This finding explains several previous observations in McArdle patients, including the excessive rise of plasma potassium with exercise, the decline in the compound muscle action potential with repetitive stimulation (probably due to extracellular potassium-mediated membrane inexcitability), and the exaggerated increase in heart rate during exercise.
The pathogenesis of contracture and myoglobinuria remains unknown: depletion of high-energy phosphate compounds, especially ATP, has long been postulated to occur, but experimental evidence is lacking.
Resting serum creatine kinase (CK) is consistently elevated in patients with McArdle’s disease, indicating that individual fiber necrosis probably occurs even with daily activities, a concept supported by morphological observations. The cumulative effect of this focal muscle damage along the years may explain the appearance of fixed weakness in older individuals; we found that 28 of 52 patients had fixed weakness, with a mean age of 41.5 years, while the mean age of patients without fixed weakness was 28.1 years [22].
Clinical Presentation
The clinical picture of McArdle’s disease is rather stereotypical, dominated by exercise intolerance manifested by myalgia, premature fatigue, and stiffness or weakness of exercising muscles, which is relieved by rest. The type and amount of exercise needed to precipitate these symptoms vary considerably from patient to patient, possibly in relation to training and diet, but two types of exertion are likely to cause problems: brief intense isometric exercise, such as lifting heavy weights, or less intense but sustained dynamic exercise, such as walking uphill. Moderate exercise, such as walking on level ground, is usually well tolerated. On the other hand, strenuous exercise often results in painful cramps and muscle swelling, which can last for hours. Myoglobinuria is seen in about half of the patients. In fact, McArdle’s disease is the second most common metabolic cause of recurrent myoglobinuria after CPT II deficiency [23]. An interesting phenomenon, almost invariably described by McArdle patients, is the “second wind” that they experience when, at the first appearance of exercise-induced myalgia, they slow down or rest briefly before resuming their activity.
The severity of symptoms may vary markedly in different patients, some of whom have neither cramps nor myoglobinuria but complain only of excessive fatigue and poor stamina, symptoms that are likely to be dismissed as “psychogenic.” On the other hand, about 20 % of patients have progressive weakness, usually starting late in life [24]. A distinct clinical variant reported in four children is characterized by severe generalized weakness at or soon after birth, respiratory insufficiency, and death in infancy [25, 26].
Differential Diagnosis
Differential diagnosis includes other metabolic myopathies, especially glycogenoses and disorders of lipid metabolism.
On purely clinical grounds, myophosphorylase deficiency is indistinguishable from defects of glycolytic enzymes, such as phosphoglucomutase (PGM), phosphofructokinase (PFK), phosphoglycerate kinase (PGK), phosphoglycerate mutase (PGAM), β-enolase (EN), and lactate dehydrogenase (LDH). Laboratory data may offer useful clues. Patients with PFK deficiency have a compensated hemolytic trait, with hyperbilirubinemia and increased reticulocyte count. With the ischemic forearm exercise test, patients with defects of terminal glycolysis (PGK, PGAM, and LDH deficiencies) have an abnormally low but not absent venous lactate response, and patients with LDH deficiency show a low lactate but excessive pyruvate response.
McArdle’s disease should be distinguished from the most common metabolic cause of recurrent myoglobinuria in adults, i.e., CPT II deficiency. In patients with myophosphorylase deficiency, myoglobinuria is invariably triggered by exercise, usually of high intensity. Patients with CPT II deficiency, however, have episodes of myoglobinuria not only after exercise (which can be of moderate intensity but is usually prolonged) but also after prolonged fasting without exertion or after a combination of exercise and fasting. Furthermore, the ischemic forearm exercise test is normal in patients with CPT II deficiency.
Exercise-related myoglobinuria may occur in dystrophinopathies and in malignant hyperthermia. In patients with dystrophinopathies, serum CK levels between attacks are usually much higher than in patients with myophosphorylase deficiency, and transmission is X-linked recessive. In patients with malignant hyperthermia, there is usually a family history of characteristic attacks related to the administration of volatile anesthetics, and inheritance is autosomal dominant.
Intolerance to exercise without myoglobinuria can be due to adenylate deaminase (AMPD) deficiency. In patients with AMPD deficiency, the ischemic forearm exercise test causes a normal rise of venous lactate, in contrast with a lack in the rise of ammonia. Because of the high frequency of the adenylate deaminase deficiency trait, its association with myophosphorylase deficiency may occur and this “double trouble” may aggravate the clinical phenotype [17, 18].
Exercise intolerance without myoglobinuria is a common complaint of malingering and hysterical patients and is the cardinal symptom of the “chronic fatigue syndrome.” However, these diagnoses should be used prudently and only after finding normal serum CK and lactate levels and a normal ischemic forearm exercise test.
Evaluation and Diagnosis
The ischemic forearm exercise test is a valuable but not specific diagnostic test, because absent or abnormally low rise of venous lactate is seen in all defects of muscle glycogenolysis or glycolysis.
Until recently, definitive diagnosis required muscle biopsy showing lack of myophosphorylase immunostaining. Our present knowledge of multiple genetic defects allows us to identify many suspected patients using genomic DNA from white blood cells. However, it is important to keep in mind the ethnic origin of the patient in order to screen for the most likely mutations.
Treatment and Management
Most patients learn to adapt their lifestyles to their limited exercise tolerance, and, within this framework, they can lead nearly normal lives. Late in the course of the disease, fixed weakness, usually moderate and affecting proximal more than distal muscles, may become a problem.
Acute renal insufficiency is the most important complication, occurring in about 50 % of patients with myoglobinuria. In those cases, abundant fluid intake to induce diuresis may suffice, but renal dialysis is often necessary. Uncomplicated episodes of myoglobinuria are followed by complete functional recovery.
There is no specific therapy for McArdle’s disease. Regular moderate aerobic training (resulting in a heart rate of no more than 60–70 % of maximal) is effective because it optimizes alternate fuel delivery and utilization [27]. Another promising therapeutic agent, at least in cases with residual phosphorylase activity, is vitamin B6, because the overall body stores of pyridoxal phosphate (PLP) are depleted in McArdle’s disease due to the frequent lack of enzyme protein (to which PLP is bound) [28–30]. Naturally, patients should be warned about the risks of strenuous exercise and advised to seek medical attention at the first appearance of pigmenturia, especially if accompanied by oliguria. Sucrose ingestion shortly before exercise is beneficial but must be used sparingly to avoid weight gain [31]. The ketogenic diet improved exercise tolerance in one patient but has not been subjected to a formal therapeutic trial [32].
There are two spontaneous animal models of McArdle’s disease, Charolais cattle and Merino sheep. Recently, a knock-in mouse model of the R50X mutation has been obtained that recapitulates faithfully the human disease [33]. This model will provide valuable information on the pathophysiology and allow therapeutic experimentation.
Phosphoglucomutase (PGM) Deficiency (Glycogenosis Type XIV)
A single patient with biochemically and genetically proven PGM deficiency had two exercise-induced episodes of myoglobinuria [34]. Nonischemic forearm exercise caused normal rise of venous lactate but excessive rise of ammonia. Muscle biopsy showed moderate increase of normal-looking glycogen.
Phosphofructokinase (PFK) Deficiency (Glycogenosis Type VII; Tarui Disease)
Introduction
In its typical presentation, this disorder, first described by Tarui et al. in a Japanese family [35] and soon thereafter by Layzer et al. in an Ashkenazi Jewish American patient [36], is clinically indistinguishable from McArdle’s disease. Minor clinical differences include lack of a typical “second wind” phenomenon; more common report of nausea and vomiting accompanying the exercise-induced crises of myalgia, cramps, and weakness; and lower frequency of myoglobinuria attacks. Much more useful in distinguishing PFK deficiency from McArdle’s disease are a few simple laboratory tests, such as increased bilirubin concentration and reticulocyte count (reflecting compensated hemolytic anemia).
Etiology and Pathogenesis
The reason for the hemolytic trait in PFK deficiency is that PFK is a tetrameric enzyme under the control of three autosomal loci: a locus on chromosome 1 encodes the muscle (M) subunit; a locus on chromosome 21 encodes the liver (L) subunit; and a locus on chromosome 10 encodes the platelet (P) isozyme [37]. The three subunits are variably expressed in different tissues. Mature human muscle expresses only the M subunit and contains only the homotetramer M4, while erythrocytes express both the M and L subunits and contain five isozymes, the two homotetramers M4 and L4, and three hybrid isoforms. In patients with typical PFK deficiency, genetic defects of the M subunit cause total lack of activity in muscle but only partial PFK deficiency in red blood cells, where the residual activity (approximately 50 % of normal) is accounted for by the L4 isozyme.
The first molecular defect in PFK deficiency (a splice junction mutation resulting in a large deletion) was identified in the Japanese family originally described by Tarui and coworkers [38]. Soon thereafter Raben and her coworkers described two mutations, a splicing defect and a nucleotide deletion, which are common among Ashkenazi Jewish patients [39, 40]. About 20 distinct mutations have been identified in patients of different ethnic origins. The molecular basis of PFK deficiency in patients with infantile or childhood onset remains unknown.
Genetic defects of PFK-M cause virtual absence of PFK in muscle biopsies, when the activity is determined either histochemically [41] or biochemically. The same defects cause partial enzyme deficiencies in erythrocytes, where the L4 homotetramer accounts for the residual activity (about 50 % of the total). Tissues such as liver and platelets express predominantly or exclusively the non-M PFK subunits and are not affected. The lack of clinical cardiomyopathy or encephalopathy, however, is more difficult to explain, because PFK-M accounts for over 90 % of heart PFK and over 50 % of brain PFK [42].
As a consequence of PFK deficiency, glycogen accumulates in muscle, reaching concentrations two to three times greater than normal. Morphologically, the stored glycogen is mostly seen at the periphery of the fibers, where it is revealed by the periodic acid-Schiff (PAS) histochemical reaction and is normally digested by preincubation with diastase. However, a peculiarity of PFK deficiency is the finding, in muscle fibers of some, usually older, patients, of an abnormal polysaccharide, which stains intensely with PAS but is not digested by diastase (polyglucosan, PG). Ultrastructurally, PG is composed of finely granular and filamentous material, similar to the amylopectin-like polysaccharide that accumulates in branching enzyme deficiency. The presence of this material has been attributed to the accumulation in muscle of glucose-6-phosphate, an activator of the enzyme glycogen synthetase, which alters the delicate ratio between the two main glycogenosynthetic enzymes, synthetase and branching enzyme [43, 44].
PFK is the rate-limiting enzyme of glycolysis and PFK deficiency blocks glycolysis, thus explaining the flat venous lactate response to forearm ischemic exercise. The accumulation in muscle of phosphorylated monoesters, observed by 31P-nuclear MRS in PFK but not in myophosphorylase deficiency, is easily explained by the sites of the two metabolic blocks: midway in the glycolytic pathway for PFK deficiency and preceding glycolysis for myophosphorylase deficiency.
As in myophosphorylase deficiency, PFK deficiency also impairs both anaerobic and aerobic glycogen metabolism and blocks the fall in muscle pH that normally accompanies heavy exercise. It also results in high levels of adenosine diphosphate (ADP) and increased adenine nucleotide degradation with exaggerated production of ammonia and myogenic hyperuricemia during exercise [45]. It causes substrate-limited oxidative metabolism with fluctuations in exercise and oxidative capacity related to the availability of blood-borne fuels [46]. Finally, PFK deficiency causes exaggerated sympathetic neural responses to exercise associated with enhanced mobilization of extramuscular fuels [47] and exaggerated heart rate, cardiac output, and blood flow relative to the muscle capacity to use oxygen [48].
The negative effect of high-carbohydrate meals on exercise tolerance is attributed to the fact that glucose lowers the blood concentration of free fatty acids and ketones, which are alternative fuels in PFK-deficient patients. Thus, not only is glucose ineffective in alleviating exercise intolerance (in contrast to myophosphorylase deficiency), but it is actually harmful, a situation dubbed “out-of-wind” phenomenon [46].
The pathogenesis of contracture and myoglobinuria in PFK remains unknown: depletion of high-energy phosphate compounds, especially adenosine triphosphate, has long been postulated to occur, but experimental evidence is lacking. Abnormal accumulation of metabolites such as adenosine diphosphate may be an important trigger of premature fatigue.
Clinical Presentation
Typically, there is intolerance to intense exercise, often accompanied by cramps of exercising muscles, which is relieved by rest [37]. Although careful history reveals that exercise intolerance is present since childhood, patients usually do not come to medical attention until adolescence, and the diagnosis is most commonly established in young people. Symptoms are more likely to occur with isometric exercise (such as pushing a stalled car) or intense dynamic exercise (such as walking uphill). The exercise intolerance appears to worsen with high-carbohydrate intake [46]. In contrast to patients with McArdle’s disease, patients with PFK deficiency do not experience a “second wind” [49].
A few patients may be jaundiced as a consequence of the hemolytic trait that accompanies PFK-M deficiency, and a few may suffer from gouty arthritis due to hyperuricemia.
Fixed weakness of late onset has been described in a few patients who suffered from exercise intolerance earlier in life [50–54]. A strikingly different clinical presentation consists of severe myopathy in infancy or early childhood, with respiratory failure and death before 2 years of age. This variant has been reported in several children [37]. Although they all had severe myopathy with muscle PFK deficiency, clinical and biochemical data were rather heterogeneous, possibly reflecting different molecular etiologies.
In patients with typical muscle disease, serum CK is usually increased. There is moderate reticulocytosis and increased serum bilirubin, reflecting the hemolytic trait. Uric acid is increased in most patients.
The ischemic forearm exercise test is a useful but not specific test. In patients with PFK-M deficiency (but also in patients with myophosphorylase deficiency or other defects of muscle glycolysis), the increase of venous lactate is absent or inadequate. It is important to keep in mind that the test depends on the patient’s ability and willingness to exercise vigorously.
Needle electromyography (EMG) may be normal or show “myopathic” abnormalities (small and short-duration motor unit action potentials). Furthermore, no electrical activity is recorded from maximally shortened muscles during contractures induced by ischemic exercise.
Studies of 31P-nuclear MRS show the accumulation, even with mild exercise, of glycolytic intermediates in the form of phosphorylated monoesters, which occurs also in other defects of glycolysis but not in myophosphorylase deficiency [55].
Differential Diagnosis
The differential diagnosis is similar to that outlined above for McArdle’s disease. Few laboratory data may offer useful clues because of the compensated hemolytic anemia that accompanies PFK and aldolase deficiency, with consequent hyperbilirubinemia and increased reticulocyte count. Of all the other glycogenoses, only PGK and aldolase deficiency causes a hemolytic trait [56].
Muscle PFK deficiency should be included in the differential diagnosis of late-onset proximal limb weakness. Usually these patients have a history of lifelong exercise intolerance and cramps. Laboratory signs of a hemolytic trait, an abnormal ischemic forearm exercise test, and abundant polyglucosan in the muscle biopsy are clues to the correct diagnosis.
Evaluation and Diagnosis
The ischemic forearm exercise test is a valuable but not specific diagnostic test. In patients with an abnormal ischemic forearm exercise test (i.e., no rise of venous lactate), laboratory evidence of hemolytic anemia suggests PFK deficiency. Documentation of partial PFK deficiency in erythrocytes bolsters this conclusion, but, until recently, definitive diagnosis required biochemical documentation of the enzyme defect in muscle (keeping in mind that PFK is notoriously labile and PFK deficiency is often a spurious finding if muscle is not flash frozen at the time of biopsy). Present knowledge of multiple molecular defects in the PFK-M gene allows identification of many suspected patients using genomic DNA isolated from white blood cells.
Treatment and Management
Most patients learn to adapt their lifestyles to the limited exercise, and, within this framework, they may lead nearly normal lives. In the fifth or sixth decade, some patients develop proximal limb weakness, which can limit their functional independence but is rarely disabling.
Although myoglobinuria is rare in this condition, acute renal insufficiency may occur in patients with myoglobinuria, requiring forced diuresis or renal dialysis. Uncomplicated episodes of myoglobinuria are followed by complete functional recovery.
There is no specific therapy. Glucose administration is not only useless because the metabolic block is halfway in the glycolytic pathway but is in fact detrimental because glucose lowers the blood concentration of alternative fuels, such as free fatty acids and ketone bodies [46].
High-protein diet and aerobic training [57] have proved beneficial in patients with myophosphorylase deficiency and should be tried in patients with PFK deficiency.
A 2-year-old boy with the infantile form of PFK deficiency benefited remarkably – as had a patient with McArdle’s disease [32] – from a ketogenic diet, which was instituted to provide muscle and brain with ketone bodies as alternative fuels [58]. There was clear improvement in strength, EMG features, and electroencephalogram (EEG) pattern. Unfortunately, the child worsened suddenly at 35 months of age and died of complications of pneumonia. Still, a ketogenic diet should be considered in children with the more severe infantile variant of PFK deficiency.
Aldolase (ALD) Deficiency (Glycogenosis Type XII)
Two children with muscle and erythrocyte aldolase deficiency have been reported [59, 60]. Both had transfusion-requiring nonspherocytic hemolytic anemia, muscle weakness, exercise-induced myalgia, and increased serum CK, especially during febrile illnesses (2,620 U/L in one patient and 13,800 U/L in the other). One patient was alive at 4 and ½ years [59], the other died at 4 years during an episode of myoglobinuria and hyperkalemia [60]. Both children were compound heterozygous for mutations in ALDOA, the gene that encodes aldolase A, the only aldolase isozyme present in muscle and erythrocytes. The mutant enzyme is more thermolabile than normal, thus probably explaining the vulnerability of patients to febrile illnesses.
Phosphoglycerate Kinase (PGK) Deficiency (Glycogenosis Type IX)
Primary myopathy is not a common presentation of PGK deficiency, an X-linked recessive disorder most commonly (11 of 33 reported patients) presenting as nonspherocytic hemolytic anemia and central nervous system (CNS) dysfunction. However, purely myopathic presentation is a close second (9 of 33 patients), while isolated blood dyscrasia was reported in 6 patients and the association of myopathy and CNS dysfunction in 4 patients [56]. All myopathic patients complained of exercise intolerance, with cramps and myoglobinuria. Molecular defects were documented in several recent patients, and one, T378P (PGK Afula), caused the peculiar association of myopathy and severe juvenile Parkinsonism [61, 62].
The wide spectrum of clinical phenotypes in PGK deficiency is difficult to explain because PGK is a monomeric enzyme encoded by a single gene on Xq13 and expressed in all tissues except the testis (a testicular isozyme, PGK2, is encoded by a gene on chromosome 19). Different amounts of residual activities in different tissues do not fully explain the clinical heterogeneity [56]. While lack of myoglobinuria in patients with severe hemolytic anemia and brain dysfunction may be attributed to their inability to exercise, it is more difficult to explain the converse situation, lack of blood dyscrasia, or brain disease in patients with myopathy.
Phosphoglycerate Mutase (PGAM) Deficiency (Glycogenosis Type X)
In contrast to PGK deficiency, PGAM deficiency affects only muscle, causing exercise intolerance, cramps, and recurrent myoglobinuria [63]. This is because PGAM is a dimeric enzyme composed of a muscle-specific (M) and a brain-specific (B) subunit, and normal muscle contains predominantly the MM homodimer, which accounts for 95 % of the total activity. The only other tissues containing substantial amounts of the M subunit are heart and sperm, but there is no evidence of cardiomyopathy or male infertility in patients with PGAM deficiency. A dozen patients have been reported, most of them from the United States [64]. All US patients have been black and they harbor one common mutation (W78X) in the PGAM-M gene (encoded by a gene on chromosome 7), suggesting a founder effect [64]. Different mutations were found in two Italian families [65, 66], in a Japanese family [67], and in a Pakistani patient [64, 68].
Despite the abundance of PGAM in muscle, we have observed exercise intolerance in heterozygous relatives of PGAM-deficient patients [64, 67]. A second unusual feature of PGAM deficiency is the frequent (33 % of patients) occurrence of tubular aggregates, which have never been associated with other, more common glycogen storage diseases: the relationship between this morphological abnormality and the enzyme defect remains unexplained.
Cycle exercise responses in two patients were markedly different from those of patients with clinically similar McArdle’s disease: the PGAM-deficient patients had virtually normal cycle exercise and oxidative capacity, no second wind, and no improvement of their exercise capacity with lipid or lactate supplements [69].
The first mutation in the PGAM-B subunit of PGAM has been reported in a patient with hereditary spherocytosis [70].
Beta-Enolase Deficiency (Glycogenosis Type XIII)
A single patient with adult-onset exercise intolerance and generalized weakness but without episodes of myoglobinuria had a flat lactate response to forearm ischemic exercise. Muscle ultrastructure showed subsarcolemmal accumulations of glycogen, and muscle biochemistry revealed an isolated severe deficiency of enolase activity (5 % of the normal mean). Over 90 % of the muscle enzyme is accounted for by the β-enolase form, which is encoded by the ENO3 gene. The patient was compound heterozygous for two missense mutations, probably reducing the stability of the mutant enzyme [71].
Lactate Dehydrogenase (LDH) Deficiency (Glycogenosis Type XI)
The discovery of this glycogenosis was due to the astute observation that a patient with myoglobinuria had predictably sky-high values of serum CK but extremely low values of LDH [72]. LDH is a tetrameric enzyme composed of various proportions of a muscle-specific subunit (LDH-A) and a cardiac subunit (LDH-B). LDH-A is encoded by a gene on chromosome 11, and three different mutations have been identified in Japanese patients [73–75], while the only two described white patients had two distinct mutations [76]. In addition to muscle symptoms, three affected Japanese women suffered from dystocia necessitating cesarian sections, and a few patients had dermatologic problems [77].
Muscle Glycogen Synthetase (GS) Deficiency (Glycogenosis Type 0)
Although glycogen synthetase (GS) deficiency of the liver was described almost 50 years ago and aptly called “glycogenosis” (i.e., lack of glycogen), the first cases of muscle glycogenosis were reported only 5 years ago in a Swedish family, and the condition was dubbed glycogenosis type 0 [78]. Of three siblings, a boy died of sudden cardiac arrest at 10 years of age, his brother suffered from exercise intolerance and hypertrophic cardiomyopathy, and his sister was asymptomatic. Muscle biopsies from the younger siblings showed conspicuous lack of glycogen by periodic acid-Schiff histochemistry and by electron microscopy, as well as increased numbers of mitochondria. All three children harbored a homozygous nonsense mutation in the muscle GS gene (GYS1), which encodes the muscle and heart isoform of the enzyme.
The fourth patient was an 8-year-old previously asymptomatic boy who unexpectedly collapsed and died while climbing up and down the stairs at school [79]. A sister had died at 6 days of age from unknown cause. Muscle morphology showed mitochondrial proliferation and – in retrospect – lack of glycogen by EM. The diagnosis of glycogenosis type 0 is complicated by the mild muscle symptoms (exercise intolerance), the scarce attention paid by morphologists to lack of glycogen (as opposed to glycogen storage), and the misleading clue of the reactive mitochondrial proliferation.
The fifth patient was a Japanese girl who died at age 12 of cardiac arrest [80]. She had had normal early development but suffered from two generalized tonic-clonic seizures at 2 and 4 years. Since age 5, she had recurrent rather stereotypical syncopal episodes after brief exercise. Loss of consciousness lasted a few hours and was followed by muscle weakness and pain. Muscle biopsy showed lack of glycogen by PAS and negative histochemical stain for myophosphorylase, which depends on the presence of endogenous glycogen. Glycogen synthetase activity was 2 % of normal, and glycogen concentration was barely detectable. The modified Gomori trichrome stain showed abundant subsarcolemmal mitochondria. Molecular genetic analysis revealed a compound heterozygous mutation in GYS1.
Glycogenin Deficiency (Glycogenosis Type XV)
The primer of glycogen synthesis is a glycosyltransferase that uses uridine diphosphate glucose as a substrate in an autoglycosylation reaction to generate a short (about 10 glucosyl units) glucose polymer, the kernel of the new glycogen molecule. There are two isoforms of glycogenin, the muscle isozyme glycogenin-1 and the liver isozyme glycogenin-2, which are also expressed partially in the heart.
The first pathogenic mutation in the gene encoding glycogenin-1 (GYG1) was described 2 years ago in a young man who was slower than his peers as a child, suffered from exertional dyspnea, and at age 27 had a life-threatening episode of ventricular fibrillation and was equipped with a permanent defibrillator [81]. Skeletal muscle was devoid of glycogen, whereas the heart showed large accumulation of a poorly structured PAS-positive material, presumably a novel form of abnormal polysaccharide due to the presence in the heart of some glycogenin.
Disorders of Lipid Metabolism
Carnitine Palmitoyltransferase II (CPT II) Deficiency
Introduction
CPT II deficiency (rather, CPT deficiency, because at the time we could not distinguish CPT I and CPT II activities) was identified in 1973 in two brothers with recurrent exercise-induced myoglobinuria whose muscle biopsies had shown no glycogen storage and normal phosphorylase and PFK activities [82, 83]. A hindsight reevaluation of their clinical histories revealed interesting differences from patients with glycogenoses: (1) neither brother had any weakness nor any problem with brief intense exercise; (2) neither complained of cramps but described muscle tenderness preceding myoglobinuria; (3) both had problems with prolonged and not necessarily strenuous exercise; (4) both identified fasting as a precipitating factor, usually in combination with exercise. Their histories summarize the main clinical features of CPT II deficiency, except that additional precipitating factors may also include emotional stress, lack of sleep, and cold exposure.
Etiology and Pathogenesis
CPT II is a key enzyme in the carnitine cycle, needed for the transport of long-chain fatty acids from the cytosol into the mitochondrion. The carnitine cycle is comprised of four elements: (1) carnitine palmitoyltransferase I (CPT I), on the inner aspect of the outer mitochondrial membrane, which catalyzes the esterification of palmitoyl-CoA to palmitoyl-carnitine; (2) the carrier molecule l-carnitine; (3) carnitine palmitoyltransferase II (CPT II), on the inner aspect of the inner mitochondrial membrane, which catalyzes the reverse reaction of CPT I, regenerating palmitoyl-CoA and liberating carnitine; and (4) a carnitine-acylcarnitine translocase (CACT), capable of exchanging acylcarnitine and carnitine across the inner mitochondrial membrane (Fig. 63.2).
Although the existence of two CPT enzymes, one outside and the other inside the inner mitochondrial membrane, was never questioned, for many years it was uncertain whether the two enzymes were distinct proteins under separate genetic control or a single protein with different milieus. It is now known that CPT I and CPT II are different proteins: CPT I is encoded by a gene on chromosome 11q [84], while CPT II is encoded by a gene on chromosome 1p32 [85].
Although by 1990 it was apparent that CPT deficiency was an important cause of recurrent myoglobinuria [23], a vexing question was why the defect of such a key enzyme in lipid metabolism should affect skeletal muscle selectively, especially since there was no evidence for the existence of tissue-specific CPT isozymes. The situation is now clearer. CPT I deficiency does, in fact, cause life-threatening hypoketotic hypoglycemia of infancy induced by fasting and often accompanied by lethargy, coma, and seizures [86]. CPT II deficiency can also cause two different and severe infantile phenotypes: (1) a rapidly lethal neonatal form with hypoketotic hypoglycemia, generalized steatosis, and multiple malformations and (2) an infantile hepatomuscular form characterized by episodes of hypoketotic hypoglycemia, lethargy, seizures, hepatomegaly, cardiomegaly, and cardiac arrhythmias [86]. However, these variants are rare compared to the adult myopathic phenotype. Molecular genetic analysis has revealed more than 30 pathogenic mutations in patients with myopathic CPT II deficiency. Of these, the S113L mutation is common in American patients [87], and screening genomic DNA from white blood cells in suspected patients may eliminate the need for a muscle biopsy. The large predominance of affected men in CPT II deficiency has been perplexing and has suggested a hormonal influence; it is, therefore, interesting that studies of the CPT II promoter region do, in fact, suggest that gene expression may be hormonally regulated [88].
Clinical Presentation
The more common, myopathic form of CPT II deficiency usually presents in adolescents or young adults, predominantly males, with recurrent myoglobinuria following prolonged, though not necessarily strenuous, exercise, prolonged fasting, or a combination of the two conditions. Other precipitating factors include cold exposure, lack of sleep, and, especially in children, intercurrent illnesses with high fever. Between attacks, these patients have normal physical and neurological exams. At difference from the glycogenoses, the attacks of myoglobinuria are not heralded by painful cramps. In addition, exercising muscles are not necessarily the only ones undergoing acute necrosis, and a few patients have been admitted to the hospital in respiratory distress. Another distinguishing feature from the glycogenoses is the normal level of serum CK between attacks of myoglobinuria. Muscle biopsy is usually normal.
Differential Diagnosis
The differential diagnosis includes other inborn errors of metabolism characterized by recurrent myoglobinuria, especially glycogenoses and defects of the mitochondrial respiratory chain. Several features distinguish CPT II deficiency from the glycogenoses: (1) whereas in the glycogenoses, myoglobinuria is always triggered by exercise, in CPT II deficiency, myoglobinuria may follow prolonged fasting or, less frequently, cold exposure, lack of sleep, emotional stress, or intercurrent illness; (2) painful cramps of exercising muscles are not described by patients with CPT II deficiency, who, therefore, lack a useful warning sign of impending myoglobinuria; (3) fixed weakness is much less common in CPT II deficiency than in the glycogenoses; (4) between episodes of myoglobinuria, serum CK is usually normal in CPT II deficiency while it is variably but consistently increased in the glycogenoses. Patients with defects in the mitochondrial respiratory chain typically complain of exercise intolerance and premature fatigue, whereas patients with CPT II deficiency have no problem with brief exercise, even when this is strenuous, and their everyday life is usually normal. CPT II deficiency is a “silent disease” that manifests itself only in extreme situations because the presence of 15–20 % residual enzyme activity in muscle is sufficient to support fat oxidation at rest but does not allow the increase in fat oxidation needed to supply muscle energy during dynamic exercise [89]. Also, patients with CPT II deficiency have no central nervous system involvement, unlike patients with CoQ10 deficiency, in whom recurrent myoglobinuria is usually associated with seizures, ataxia, or mental retardation [90].
Therapy is based on a low-fat, high-carbohydrate dietary regimen, medium-chain triglyceride (MCT) instead of long-chain oil, or triheptanoin as an anaplerotic compound [91]. Another therapeutic approach is based on the upregulation of mitochondrial biogenesis by bezafibrate [92].
Very Long-Chain Acyl-CoA Dehydrogenase (VLCAD) Deficiency
The clinical picture of CPT II deficiency may be indistinguishable from that of some defects in the first step of ß-oxidation, including very-long-chain acyl-CoA dehydrogenase (VLCAD) deficiency and trifunctional protein deficiency.
VLCAD is associated with the inner mitochondrial membrane and is specific for C14–C24 fatty acyl-CoAs. Age at onset and clinical severity of VLCAD deficiency depend on the amount of residual enzyme activity. A severe infantile form is characterized by cardiomyopathy and high mortality. A milder childhood form with hypoketotic hypoglycemia and rare cardiac involvement has a more benign outcome. The adult myopathic form causes recurrent myoglobinuria triggered by prolonged exercise or fasting, thus closely simulating CPT II deficiency [86, 93]. The diagnosis can be made by tandem mass spectroscopy detecting long-chain acylcarnitines (especially C14) in blood or in fibroblasts incubated with long-chain acylcarnitines [94].
The muscle biopsy is usually normal or nonspecifically altered as in patients with CPT II deficiency, but the diagnosis can be established by immunohistochemistry [93].
Mitochondrial Trifunctional Protein (MTP) Deficiency
Mitochondrial trifunctional protein (MTP) comprises three enzyme reactions, enoyl-CoA hydratase, long-chain 3-hydroxyacyl-CoA dehydrogenase (LCHAD), and acyl thiolase, and generates acetyl-CoA and an acyl-CoA shortened by two carbon atoms, which is ready to enter a new β-oxidation cycle. Most patients with MTP have isolated LCHAD deficiency, usually due to one mutation (E510Q) in the HDHA gene encoding the α subunit of the enzyme [95]. A fatal infantile presentation is dominated by encephalopathy and hepatopathy. The childhood presentation is characterized by recurrent myoglobinuria, often accompanied by cardiomyopathy, pigmentary retinopathy, and sensorimotor axonal neuropathy. The episodes of myoglobinuria may be precipitated by exercise, fasting, or intercurrent illnesses and are often accompanied by life-threatening respiratory distress. Because of the concurrent neuropathy, the muscle biopsy shows features of denervation but only rarely lipid storage.
Phosphatidic Acid Phosphatase (LIPIN) Deficiency
It has long been known that many cases of recurrent myoglobinuria, especially in children, go undiagnosed at the molecular level [96]. It is, therefore, important that a new cause of childhood myoglobinuria has now been added, mutations in the LPIN1 gene that encodes the muscle-specific phosphatidic acid phosphatase: this enzyme converts phosphatidate to diacylglycerol in the triacylglycerol pathway [97]. Affected children had episodes of myoglobinuria between 15 months and 7 years of age, usually precipitated by febrile illnesses. Muscle biopsy was essentially normal. It has been proposed that the enzyme defect during stress periods causes accumulation of lysophospholipids, which act as detergents, thus favoring muscle breakdown.
Respiratory Chain Defects
While exercise intolerance is a common complaint in patients with mitochondrial encephalomyopathies, it is often overshadowed by other symptoms and signs [98]. Only relatively recently we have come to appreciate that exercise intolerance, myalgia, and myoglobinuria may be the sole presentation of respiratory chain defects. These may affect complex I, complex III, or complex IV, although they seem to be more commonly associated with complex III deficiency [2, 99].
Complex I Deficiency
Increased blood lactic acid at rest (5.2 mEq/L; normal, less than 2.2) was the only objective indication for a muscle biopsy in a 38-year-old man who complained of severe lifelong exercise intolerance [100]. He was otherwise completely normal, including muscle strength and bulk, resting serum CK, and EMG. He had never noticed pigmenturia, and his mother and siblings had no muscle problems. The muscle biopsy showed intensely cytochrome c oxidase (COX)-positive ragged-red fibers (RRF) and an isolated moderately severe defect of complex I activity. Molecular analysis identified a heteroplasmic (54 % of muscle mitochondrial DNA) nonsense mutation (G11832A) in the gene encoding subunit 4 of complex I (ND4). Single-fiber polymerase chain reaction (PCR) showed that the mutation was much more abundant in RRF than in normal fibers, thus confirming the pathogenicity of this base change.
In 1990, Bet et al. [101] had reported a similar case: a 43-year-old man with lifelong exercise intolerance, myalgia, mild proximal limb weakness, and negative family history. His muscle biopsy also showed abundant RRF and an isolated defect of complex I activity (40 % of normal). Molecular analysis of his muscle revealed a novel pathogenic mutation in the ND1 gene of mtDNA, a seven nucleotide intragenic inversion that alters three highly conserved amino acids [102].
Complex III Deficiency
In 1993, Bouzidi et al. [99] found low complex III activity in the muscle of a 25-year-old man with exercise intolerance in whom they later identified a missense mutation (G15615A) in the cytochrome b gene (the only mtDNA-encoded subunit of complex III) [100]. In the years that followed, 14 more patients were reported with different mutations in the cytochrome b gene of mtDNA (MTCYB) but with the same clinical picture: isolated myopathy with exercise intolerance and sometimes myoglobinuria [99]. The muscle biopsy in all patients showed complex III deficiency and “ragged-blue” fibers with the SDH stain, which were also COX positive. The lack of maternal inheritance and of multisystem involvement keeps these patients under the radar for the diagnosis of mtDNA-related disorders.
Complex IV (COX) Deficiency
Starting at age 15, a 16-year-old woman had four episodes of muscle cramps and myoglobinuria precipitated by prolonged exercise or viral illness [103]. Rather surprisingly, she had only “mildly reduced endurance.” Between attacks, both physical and neurological exams were normal, as were routine laboratory tests, including serum CK and lactate. Interestingly, no tissue other than muscle was clinically affected, and family history was entirely negative. A muscle biopsy at age 16 showed numerous ragged-red fibers, which stained intensely for the succinate dehydrogenase (SDH) reaction but not at all for the cytochrome c oxidase reaction. Biochemical analysis showed a marked and isolated defect of COX activity (14 % of normal), and molecular genetic analysis identified a 15-bp microdeletion in the mtDNA gene encoding subunit III of COX (COX III). The deletion affected 92 % of the mtDNA in muscle and 0.7 % in leukocytes but was not detected in the patient’s fibroblasts or in her mother’s leukocytes.
We studied a very similar case [104], except that our patient was a 34-year-old man who had suffered from exercise intolerance since childhood. He also had an isolated myopathy with recurrent episodes of exercise-induced myoglobinuria, normal neurological exam between attacks, and negative family history. Muscle biopsy showed scattered RRF but numerous COX-negative fibers severely reduced COX activity (13 % of normal), and molecular studies revealed a heteroplasmic nonsense mutation (G5920A) in the COX I gene of mtDNA.
A third case of isolated myopathy due to a mutation in COX II was described in a 14-year-old boy with mild weakness of shoulder and pelvic girdle muscle and exercise intolerance without myoglobinuria [105]. Family history was negative, and although lactate in the CSF was mildly elevated, there was no evidence of brain dysfunction and brain MRI was negative.
A fourth case of isolated mitochondrial myopathy with weakness, myalgia, and one episode of myoglobinuria regarded a 34-year-old woman with no family history of similar disorders. Muscle histochemistry showed over 90 % COX-deficient fibers, and molecular genetic analysis identified a heteroplasmic nonsense mutation in COX I [106].
Reevaluation of a 30-year-old man with exercise-related recurrent myoglobinuria and no family history revealed a mutation (m.7989T>C) in subunit II of COX. Single-fiber PCR showed that COX-negative fibers contained >90 % mutant mtDNAs compared to 52 % in COX-positive fibers [107].
The clinical and molecular characteristics of all reported patients with exercise intolerance and mutations in mtDNA protein-coding genes are summarized in Table 63.1. All patients had pure myopathy, with no evidence of multisystem involvement. The myopathy was dominated by exercise intolerance with premature fatigue and myalgia. Myoglobinuria occurred in a few patients. Fixed weakness was also rare and appeared nonprogressive. All patients were sporadic cases with no family history of similar disorders.
Iron-Sulfur Cluster Scaffold Protein (ISCU) Deficiency
In 1991 and 1993, Haller and coworkers described a young man with lifelong exercise intolerance, dyspnea, cardiac palpitations, and episodes of myoglobinuria [108, 109]. The syndrome, which is common in northern Sweden, was dubbed “mitochondrial myopathy with succinate dehydrogenase and aconitase deficiency” and correctly attributed to altered metabolism of iron-sulfur (Fe-S) cluster proteins, which are prosthetic groups present in complexes I, II, and III and in the Krebs cycle enzyme aconitase. Accordingly, histochemical analysis of muscle showed SDH deficiency, and biochemical analysis showed deficiencies of complex II, complex III, and aconitase. In 2008, homozygosity mapping revealed a single pathogenic mutation in the ISCU gene in three Swedish families [110]. Two years later, Kollberg et al. [111] defined SDH deficiency and accumulation of iron in muscle as the morphological hallmarks of the disease. They also observed that these features transiently disappeared after rhabdomyolysis, warning that false-negative histochemical results may be obtained if muscle biopsies are taken soon after episodes of myoglobinuria.
Coenzyme Q10 (CoQ10) Deficiency
CoQ10, which is encoded by the nuclear genome, transfers electrons from complex I and complex II to complex III (Fig. 63.2). CoQ10 is also the final acceptor of electrons derived from ß-oxidation via the electron transfer flavoprotein (ETF) and the ETF dehydrogenase (Fig. 63.2), and, in this sense, it is part of lipid metabolism. The coexistence of ragged-red fibers and lipid storage in muscle biopsies of patients with the myopathic variant of CoQ10 deficiency underscores the dual metabolic nature of this disorder. Also, CoQ10 deficiency, though first described in 1989 [112], was “rediscovered” in recent years and may have been underestimated as a cause of recurrent myoglobinuria [113, 114]. In the five patients described thus far, primary CoQ10 deficiency in muscle was characterized by the triad: (1) exercise intolerance and recurrent myoglobinuria; (2) central nervous system dysfunction, with seizures or mental retardation; and (3) RRF and markedly increased lipid droplets in the muscle biopsy. Biochemical analysis of muscle shows a partial block at the level of complex III and variably severe deficiency of CoQ10.
Isolated lipid storage myopathy without myoglobinuria has been described in a few patients [115–118]. The report by Gempel et al. [118] implied that myopathic CoQ10 deficiency could be secondary to multiple acyl-CoA dehydrogenase deficiency (MADD) due to mutations in the gene encoding the electron-transferring-flavoprotein dehydrogenase (ETFDH), but this generalization has been contradicted [119, 120].
More common and severe presentations of primary CoQ10 deficiency include infantile encephalomyopathy, childhood-onset cerebellar atrophy and ataxia, and glomerulopathy [90].
Miscellaneous
Adenylate Deaminase Deficiency
Adenylate deaminase (AMPD) deficiency is a muscle disease with variable manifestations [121]. Some patients have no symptoms. The most common complaints are muscle cramping, stiffness, or pain after exercise. In others, fixed weakness, hyporeflexia, paresthesias, periodic paralysis, and repeated infections in childhood have been reported. The serum CK concentration may be increased, but myoglobinuria is extremely rare. Muscle biopsy is usually normal, but a specific histochemical stain facilitates the diagnosis and shows that the enzyme defect is often encountered in virtually asymptomatic subjects. In keeping with this observation, molecular genetic analysis has shown that a common mutation (Q12X) in the AMPD-1 gene, which encodes the muscle isozyme, can explain the 2 % incidence of myoadenylate deficiency encountered in the general population. As mentioned above, two children, who were homozygous for pathogenic mutations in the myophosphorylase gene (glycogenosis type V, McArdle’s disease) or in the muscle phosphofructokinase gene (glycogenosis type VII, Tarui disease), were also homozygous for the AMPD-1 mutation. Interestingly, both children had unusually severe phenotypes, with early episodes of myoglobinuria. This suggests that the per se mild AMPD-1 mutation may have worsened the clinical expression of the two glycogenoses. Thus, it is important to keep in mind the possibility of genetic “double trouble” between adenylate deaminase deficiency and other metabolic errors.
Disorders Causing Fixed Weakness
All but one of the glycogenoses causing exercise intolerance and myoglobinuria are due to muscle-specific enzyme defects, whereas all but one of those causing fixed weakness are due to generalized enzyme defects (Fig. 63.3). This suggests the possibility that factors other than defective substrate utilization may play a role in the etiology of weakness. One such obvious factor is the severe involvement of spinal motor neurons in the infantile form of acid maltase deficiency (Pompe disease) [122, 123]. A more subtle neurogenic involvement may occur in debrancher deficiency, where glycogen storage has been documented in intramuscular nerves [124] and in both Schwann cells and axons of sural nerve biopsies [125, 126]. Subclinical cardiomyopathy may contribute to weakness in both debrancher [127] and, possibly, brancher deficiencies. Similarly, liver dysfunction with hypoglycemia in debrancher deficiency, and with chronic hepatic failure in brancher deficiency, will undoubtedly contribute to the lack of stamina of these patients.
Whereas glycogen storage is mild (sometimes hardly detectable) in the glycogenoses associated with exercise intolerance, it tends to be more severe in the glycogenoses associated with weakness, especially in the infantile and juvenile forms of acid maltase deficiency and in debrancher deficiency.
Glycogenoses
Acid Maltase (α-Glucosidase, GAA deficiency), Glycogenosis Type II
Introduction
The fatal infantile form of acid maltase (acid α-glucosidase, GAA) deficiency was first described in 1932 in separate papers by Pompe and Putschar, who called attention to the severe glycogen storage in the heart [128, 129]. In 1963, Hers documented the defect of GAA in liver, heart, and skeletal muscle of children with “cardiomegaly glycogenosis” [130] and, together with Lejeune and Tines-Sempoux, showed that GAA was a lysosomal enzyme [131]. Thus, GAA deficiency became the prototype of inborn lysosomal diseases [132]. In the years that followed, GAA deficiency was recognized in both children and adults with myopathy but without cardiac involvement, and, by 1973, the three main clinical variants of GAA deficiency were clearly defined [133]. The eponym Pompe disease should be limited to the infantile form, but it is now used to indicate the disease as a whole.
Etiology and Pathogenesis
Acid maltase deficiency is a hereditary condition transmitted as an autosomal recessive trait. The gene encoding GAA is localized on the long arm of chromosome 17, and a variety of molecular defects have been described in both infants and adults [134].
The genotype/phenotype correlation is hard to establish, partly because of the frequency of compound heterozygotes. In general, there is good correlation between the severity of the mutation and the severity of the clinical phenotype. Thus, deletions and nonsense mutations are usually associated with infantile GAA deficiency, whereas “leaky” mutations, such as the common IVS1(−13T->G) splice site mutation, are associated with the adult-onset variant of the disease.
GAA has both α-1,4-glucosidase and α-1,6-glucosidase activity and is, therefore, capable of digesting glycogen completely to glucose. Like other lysosomal enzymes, GAA, a glycoprotein, is synthesized as a high-molecular-weight precursor, which is extensively modified posttranslationally as the protein travels from ribosomes to primary lysosomes through endoplasmic reticulum and Golgi apparatus. Posttranslational processing includes glycosylation, acquisition of a mannose-6-phosphate recognition marker, phosphorylation, and proteolytic trimming. Through these steps, in cultured fibroblasts, a 95-kd precursor is converted into two enzymatically active 76- and 70-kDa glycoproteins [135, 136].
One intriguing question is: why non-muscle tissues in childhood and adult acid maltase deficiency are spared? In agreement with the concept that there are no tissue-specific isozymes of acid maltase, GAA activity is markedly decreased in all tissues not only in infantile acid maltase deficiency but also in the adult form [137]. The difference in clinical expression and pathology between infantile and later-onset forms of GAA deficiency has been attributed to the presence of a small but crucial amount of residual GAA activity in childhood and adult cases but not in infantile cases. The difference in residual activity, first observed in muscle specimens [138], is more evident in fibroblast and muscle cultures from patients with the different variants [139].
A second question concerns the pathogenesis of weakness. Muscle biopsy shows a vacuolar myopathy in all three forms of AMD. In the infantile form, all muscles and all fibers contain many, often confluent vacuoles, resulting in a “lacework” appearance. In childhood and adult GAA deficiency, vacuoles are less numerous and tend to be smaller. Furthermore, in the adult variant, biopsies from clinically unaffected muscles may appear normal, despite the marked decrease of GAA activity. The vacuoles contain PAS-positive material and stain intensely for acid phosphatase, another lysosomal enzyme. The positive acid phosphatase stain is a useful diagnostic clue in otherwise normal biopsy specimens. In agreement with morphological appearance, glycogen content is massively increased in muscle from patients with infantile GAA deficiency, often reaching a level ten times higher than normal. Muscle glycogen concentrations are generally lower in the childhood form and may be normal in the adult variant.
A central riddle regards the pathogenesis of weakness: is it simply due to the mechanical disarray of the contractile system caused by the glycogen-laden lysosomes or rather to an energy defect? The energetic hypothesis is supported by the notion that GAA activity normally releases glucose into the cytoplasm. A compelling scenario for the pathogenesis of acid maltase deficiency, as well as other lysosomal diseases [140], involves a disruption of the vital autophagic process, with accumulation of autophagosomes resulting from defective autophagosome-lysosome fusion [140, 141]. In fact, Nishino and colleagues went as far as stating that “Pompe disease can no longer be viewed simply as a glycogen storage disease,” but rather as a problem in handling excessive numbers of autophagosomes [142].
In infantile GAA deficiency, accumulation of free and intralysosomal glycogen occurs in all tissues and is especially severe in the heart. In the CNS, anterior horn cells of the spinal cord and neurons of brainstem nuclei are more severely affected than neurons of the cerebral cortex [122, 123, 143]. This probably contributes to the flaccid quadriplegia of these infants and may explain why mental retardation does not become apparent within their short life span.
Clinical Presentation
Infantile Pompe disease manifests in the first weeks or months of life with diffuse hypotonia and weakness, giving these infants a “rag doll” appearance (floppy infant syndrome). However, muscle bulk may be increased, and macroglossia is common. There is massive cardiomegaly and less severe hepatomegaly. Despite their extreme weakness, these infants are usually alert and interested in their environment. Respiratory muscle weakness increases susceptibility to pulmonary infections, and death due to cardiac or respiratory failure occurs invariably before 2 years of age and usually within the first year.
Glycemia and the response of blood glucose to epinephrine or glucagon administration are normal. Serum CK is markedly increased. Needle EMG shows myopathic motor unit action potentials (MUAPs) associated with fibrillation potentials, positive waves, complex repetitive discharges, and myotonic discharges. Electrocardiography shows a short P-R interval, giant QRS complexes, and signs of left ventricular or biventricular hypertrophy. Chest radiography shows massive cardiac enlargement.
In childhood GAA deficiency, weakness starts in infancy or early childhood but is less severe than in the infantile form, and progression is slower. Motor milestones may be delayed. Some patients have calf enlargement and, in boys, the clinical picture may suggest the diagnosis of Duchenne muscular dystrophy. Respiratory muscles are affected early and respiratory failure is the usual cause of death within the second or third decade. There is no cardiomegaly, and both hepatomegaly and tongue enlargement are much less frequent than in infantile AMD.
Serum CK is variably but consistently increased. On needle EMG, the association of myopathic MUAPs with signs of abnormal irritability and myotonic discharges should raise the suspicion of acid maltase deficiency.
The clinical hallmark of adult-onset GAA deficiency is a slowly progressive myopathy, starting in the third or fourth decade, but occasionally later, including a few patients with onset in the sixth or seventh decade. Weakness predominates in truncal and proximal muscles, sometimes involving bulbar muscles and causing dysphagia. Respiratory muscles are selectively affected, and respiratory insufficiency may be the presenting complaint, with morning headache or exertional dyspnea [144, 145]. The initial diagnosis in most cases is limb-girdle dystrophy or polymyositis. There is no visceromegaly. Glycogen accumulation in the smooth muscle of cerebral arteries may lead to the formation of intracranial aneurysms, which have been described in a few families [146].
Serum CK is variably increased in most patients. The ischemic forearm exercise test causes a normal rise of venous lactate, indicating that phosphorolytic glycogen breakdown and glycolysis are normal. On needle EMG, fibrillation potentials, positive waves, and myotonic discharges are useful clues to the diagnosis and may be more evident in paraspinal muscles. Studies of pulmonary function show restrictive ventilatory insufficiency, with reduced maximal static inspiratory and expiratory pressures, and early diaphragmatic fatigue. Whole-body muscle MRI can provide an accurate picture of the muscles involved in the course of the disease [147].
Differential Diagnosis
Infantile GAA deficiency has to be distinguished from other causes of the floppy infant syndrome, including spinal muscular atrophy type I (Werdnig-Hoffmann disease) and other metabolic or congenital myopathies. The massive cardiomegaly distinguishes acid maltase deficiency from most of these disorders. Some infants with cytochrome c oxidase deficiency have both myopathy and cardiomyopathy, but cardiomegaly is usually less marked and there is lactic acidosis. Muscle biopsy is virtually pathognomonic for acid maltase deficiency.
AMP-dependent protein kinase (AMP) deficiency can present with massive glycogen storage and cardiomegaly in infancy, but muscle weakness is modest and muscle biopsy shows only minor glycogen storage [5].
Childhood GAA deficiency may simulate Duchenne muscular dystrophy in boys with calf pseudohypertrophy. In Duchenne muscular dystrophy, however, family history may suggest X-linked transmission, serum CK tends to be higher, and needle EMG does not show myotonic discharges. Muscle biopsy clearly differentiates the two conditions.
Other metabolic myopathies of childhood include debrancher deficiency, phosphorylase b kinase deficiency, and carnitine deficiency. Debrancher deficiency myopathy is often accompanied by hepatomegaly and fasting hypoglycemia; there is no response of blood glucose to epinephrine or glucagon administration, and if feasible to do, the ischemic forearm exercise test shows no rise of venous lactate. Differential diagnosis from phosphorylase b kinase deficiency and from carnitine deficiency requires morphological and biochemical studies of muscle.
Muscle biopsy is also required to distinguish childhood GAA deficiency from congenital myopathies, because several patients with Pompe disease have a thin body habitus, elongated facies, and high-arched palate that are commonly seen in congenital myopathies, such as myotubular myopathy, nemaline myopathy, and central core disease.
Adult GAA deficiency is an important consideration in patients thought to have limb-girdle dystrophy or polymyositis. The early and often selective involvement of respiratory muscles and the EMG features, especially in paraspinal muscles (fibrillation potentials, positive waves, complex repetitive discharges, and myotonic discharges), are useful clues to acid maltase deficiency [145].
A disorder resembling acid maltase deficiency but not due to acid maltase deficiency is characterized by X-linked dominant “cardiomyopathy, mental retardation, and autophagic vacuolar myopathy” [148]. The molecular defects in this disorder involve the LAMP-2 gene, which encodes a major lysosomal membrane protein [149].
Evaluation and Diagnosis
Diagnostic workup includes serum CK (variably increased in all three forms of GAA deficiency), electrocardiogram (EKG) and chest radiogram in the infantile form, pulmonary function tests in childhood and adult acid maltase deficiency, MRI of spine/paraspinal muscles and needle EMG of paraspinal muscles in the adult variant (looking for fibrillation potentials and myotonic discharges), ischemic forearm exercise test (to exclude other glycogenolytic or glycolytic defects), and muscle biopsy.
In all three forms of acid maltase deficiency, short of a muscle biopsy, the diagnosis may be aided by determination of acid and neutral maltase activities (and calculation of the acid/neutral activity ratio) in lymphocytes [150].
Treatment and Management
A generally effective enzyme replacement therapy (ERT) with recombinant human GAA (rhGAA) is now available and widely utilized. There is already a vast literature on the subject particularly in the infantile form of the disease [151]. A few problems have emerged recently, such as the immune reaction to rhGAA in infants with null mutations and no GAA protein (i.e., no cross-reactive immunological material, CRIM) [152]. A multicentric, randomized, placebo-controlled trial of alglucosidase alfa, a recombinant human GAA, was conducted in 90 patients (8 years of age or older) with the late-onset form of Pompe disease [153]. The two primary end points were motor (6-min walk test) and respiratory (percentage of predicted forced vital capacity). Secondary and tertiary end points were quantitative muscle testing and maximum inspiratory and expiratory pressures. Treatment with rhGAA for 78 weeks was associated with improved walking distance and stabilization of pulmonary function over a period of 18 months. The results were modest but important considering the slow but inexorable progression of the disease [154]. One question remains unanswered: whether to start ERT in presymptomatic late-onset patients [155].
The response of patients with infantile Pompe disease has been much more patchy, as illustrated by the first four children treated [156, 157]. All four children survived until 4 years or longer, thus meeting the provisional end point of the trial. Three of the children are still alive more than 11 years later, but the two who started therapy late (7 and 8 months of age) are paralytic and ventilator dependent. The child who started therapy at 3 months is still able to walk at age 11 and goes to school, although he has some weakness [151]. The UK experience with 20 infants treated between 2000 and 2009 is equally tattered: overall ventilator-free survival was 35 %; 35 % died at a median age of 10 months; 30 % were alive but ventilator dependent. Again, age and clinical severity at the beginning of treatment were of paramount importance. The authors wisely conclude that ERT for infants with Pompe disease has to be pondered carefully [158].
Patients with childhood or adult AMD and respiratory insufficiency may need intermittent or permanent mechanically assisted ventilation [159]. Controversial results have been obtained with high-protein diet and aerobic exercise, which appears to improve strength and respiratory function in some, but not all patients [160].
Debrancher Deficiency (Glycogenosis Type III)
This is usually a benign disease of childhood characterized by hepatomegaly, growth retardation, and fasting hypoglycemia, which tend to resolve around puberty [161]. However, later in life (third or fourth decade), a small proportion of patients develops a myopathy, which is often more distal than proximal. Wasting of leg muscles and intrinsic hand muscles often leads to the diagnosis of motor neuron disease or peripheral neuropathy. This clinical picture, the “mixed” myopathic and neurogenic EMG pattern, and the often slowed nerve conduction velocity reinforce the impression that weakness in these patients may have a neurogenic component [162, 163]. It is, however, surprising that an enzyme that acts “hand in hand” with muscle phosphorylase in the degradation of glycogen should not cause exercise intolerance and myoglobinuria, at least in those patients that are not severely weak.
The debranching enzyme is a single protein that catalyzes two enzymatic reactions, an oligo-1,4-1,4-glucantransferase and an amylo-1,6-glucosidase, and is encoded by a gene on chromosome 1p21. There are three biochemical variants: a rare deficiency of the transferase activity alone (type IIId), a common deficiency of both enzyme activities in both muscle and liver (type IIIa), and a less frequent deficiency of both enzyme activities in liver but not in muscle (type IIIb) [37]. Numerous mutations have been identified: although the molecular basis for the differential tissue involvement in patients with the IIIa and IIIb variants remains unclear, it is interesting that most patients with the IIIb variant (but none with the IIIa variant) have mutations in exon 3 of the debrancher gene, which are expected to result in truncated proteins [164].
Glycogen Branching Enzyme (GBE) Deficiency (Glycogenosis Type IV)
This glycogenosis has a surprising spectrum of clinical phenotypes, considering that GBE is a single polypeptide (encoded by a gene on chromosome 3). The enzyme defect can be silent or affect predominantly the liver, the heart, skeletal muscle, or the brain [37]. GBE deficiency results in the deposition of an amylopectin-like polysaccharide that has fewer branching points and longer outer chains than normal glycogen and is known as polyglucosan (PG). PG is PAS positive and only partially digested by diastase, which makes it easily recognizable in various tissues and offers an important clue to the correct diagnosis.
The “typical” presentation described by most authors is in infancy with hepatosplenomegaly, progressive cirrhosis, and chronic hepatic failure. Cardiomyopathy dominates the clinical picture in a few older children. We now realize that we have vastly underestimated the infantile neuromuscular presentation.
As recognized in a seminal paper published in 2004 [165], there are two main infantile presentations. The first is a perinatal disorder dubbed “fetal akinesia deformation sequence” (FADS), characterized by multiple congenital contractures (arthrogryposis multiplex congenita), hydrops fetalis, pulmonary hypoplasia, craniofacial abnormalities, intrauterine growth retardation, abnormal amniotic fluid volume, and perinatal death. The second form, labeled “congenital,” should probably be called “fatal infantile” because it presents at or soon after birth with hypotonia, muscle wasting, neuronal involvement, inconsistent cardiomyopathy, and early death. Of the eight patients reported by Bruno et al. [165], three had FADS, three had the congenital form, and two had childhood myopathy. Interestingly, there was a good correlation between molecular severity and clinical severity, which has been confirmed in several subsequent patients.
It is becoming increasingly clear that patients with congenital GBE deficiency present a clinical continuum from FADS to a rapidly fatal congenital multisystem disorder dominated by profound hypotonia, respiratory failure, and inconsistent cardiomyopathy [165–172]. Detailed neuropathology in a few infants showed PG inclusions in neurons of the basal ganglia and thalamus, oculomotor and pontine nuclei, and periaqueductal neurons [169]. In the medulla, PG deposits were found in the hypoglossal nucleus, the dorsal motor nucleus of the vagus, and the nucleus ambiguous [166, 168]. The motor neurons of the spinal cord are also severely affected [173], thus explaining how one of the patients we studied was initially diagnosed as spinal muscular atrophy type I until mutations in the SMN1 gene were ruled out [166].
Brain involvement dominates the clinical picture also in a characteristic late-onset (fifth or sixth decade) form of GBE deficiency known as “adult polyglucosan body disease” (APBD) manifested by progressive upper and lower motor neuron involvement, sensory loss, sphincter problems, and dementia (see Chap. 22). Although APBD is seen in various ethnic groups, this disorder predominates among Ashkenazi Jews, in whom one common mutation (Y329S) probably reflects a founder effect [174].
There is no specific therapy for GBE deficiency, but the recent creation of transgenic mice models that faithfully recapitulate both the fatal infantile form and APBD will facilitate the development of therapeutic modalities [175].
Disorders of Lipid Metabolism
Primary Systemic Carnitine Deficiency (PCD)
The mean age at onset of this autosomal recessive condition ranges from 1 month and 7 years. Progressive cardiomyopathy is the most common presentation: echocardiography and EKG show dilated cardiomyopathy, peaked T waves, and signs of ventricular hypertrophy. Endomyocardial biopsies or postmortem studies show massive lipid storage, and, when measured, carnitine concentration in the myocardium is less than 5 % of normal. Cardiac function responds poorly to inotropics and diuretics but responds dramatically to carnitine supplementation, with progressive normalization of cardiac function indices within a few months. Some infants may present with acute encephalopathy together with hypoketotic hypoglycemia and hepatomegaly with liver steatosis. Myopathy is usually associated with cardiomyopathy or encephalopathy and is manifested by mild motor delay, hypotonia, or slowly progressive proximal weakness. Serum CK is normal or slightly elevated. Muscle biopsy shows lipid storage myopathy and very low levels of total and free carnitine (below 10 % of normal).
Defective carnitine transport in kidney causes defective carnitine reabsorption and excessive carnitine excretion. Deficient intestinal transport results in poor and delayed carnitine absorption. The combination of defective renal and intestinal carnitine handling causes carnitine levels to fall in blood. Hence, it is important to measure blood carnitine concentrations in all infants and young children with unexplained cardiomyopathy. In vitro, deficient carnitine transport has been documented in cultured skin fibroblasts [176] and in cultured muscle cells [177].
PCD is due to mutations in the SLC22A5 gene that encodes the high-affinity, sodium-dependent plasma membrane carnitine transporter (organic cation transporter, OCTN2) [178].
Primary Myopathic Carnitine Deficiency
This disorder, characterized by decreased muscle carnitine with normal serum carnitine, was the first example of carnitine deficiency described by A.G. Engel and Angelini in 1973 in a young woman with progressive proximal weakness and lipid storage myopathy responsive to corticosteroids [179]. The existence of this entity, however, is controversial because there is no definitive documentation of an isolated defect of carnitine uptake in muscle. It is possible that patients with the myopathic form of carnitine deficiency have other fatty acid oxidation defects, either generalized or muscle specific. In some of the patients described, symptoms appeared in the first years of life, but in most the onset was between the second and third decade. There was progressive and sometimes fluctuating weakness of proximal limb and axial muscles of variable severity. A few of these patients had associated cardiomyopathy. Muscle biopsy showed accumulation of triglycerides, especially in type I fibers. Muscle carnitine levels were 20 % of normal or less, while plasma carnitine levels were normal or slightly reduced. Some of the patients improved with carnitine administration.
Secondary Carnitine Deficiency
This condition, characterized by decreased levels of carnitine in blood and, often, in tissues, can accompany diverse disorders, including inborn errors of metabolism, acquired medical disorders, and iatrogenic states.
Examples of inborn errors of metabolism include numerous defects of fatty acid metabolism affecting both the carnitine cycle and β-oxidation, disorders of branched-chain amino acid metabolism, and defects of the mitochondrial respiratory chain.
Examples of acquired medical conditions include those causing decreased carnitine biosynthesis (e.g., hepatic cirrhosis or extreme prematurity), those causing decreased carnitine intake (e.g., malnutrition, chronic total parenteral nutrition, strict vegetarian diet, soy protein infant formula, malabsorption), those causing decreased body stores of carnitine in the face of increased requirements (e.g., pregnancy and lactation, extreme prematurity, infant of carnitine-deficient mother), and those causing increased carnitine loss, such as Fanconi syndrome.
Examples of iatrogenic factors include valproate therapy [180], hemodialysis, and zidovudine administration [181]. It is important to keep in mind these diverse causes of carnitine deficiency because carnitine replacement often results in marked improvement.
Medium-Chain Acyl-CoA Dehydrogenase (MCAD) Deficiency
This relatively common condition presents in childhood as an episodic acute illness with hypoketotic hypoglycemia following intercurrent infection and fasting. The presenting symptoms and signs, in order of decreasing frequency, include lethargy, vomiting, encephalopathy, respiratory arrest, hepatomegaly, seizures, apnea, cardiac arrest, and sudden death. Once the correct diagnosis is established, subsequent crises can generally be prevented, but survivors are at risk for developmental disabilities. In a large retrospective analysis of 120 MCAD patients, weakness was present in 16 % of the survivors [182]. The most effective laboratory test in this, as in other β-oxidation disorders, is the profile of acylcarnitines in plasma as determined by tandem mass spectrometry. The profile is characteristically abnormal both in sick and in well children with MCAD deficiency.
The gene for MCAD has been cloned and assigned to chromosome 1p31. A single mutation (K304E) is present in over 90 % of patients.
Multiple Acyl-CoA Dehydrogenase Deficiency (MADD)
MADD can be due to mutations in either one of two enzymes: electron-transfer flavoprotein (ETF) or ETF/CoQ10 oxidoreductase (ETFDH). Another name for MADD is glutaric aciduria type II. The clinical spectrum ranges from a fatal multisystem disorder of infancy to a less severe disorder of adolescence or adult life. Late-onset MADD often causes severe lipid storage myopathy with proximal and axial weakness. Of considerable practical importance is the riboflavin-responsive form of MADD, which is predominantly associated with ETFDH deficiency [120, 183]. Some of these patients had CoQ10 deficiency in muscle and responded to CoQ10 supplementation alone or together with riboflavin [118], but secondary CoQ10 deficiency is not a consistent finding [120].
Neutral Lipid Storage Disease with Ichthyosis (NLSDI, Chanarin-Dorfman Syndrome)
This disorder was first reported by Chanarin in a Ugandan woman [184], then recognized in several patients from the Mediterranean area [185]. Patients suffer from ichthyosis, steatorrhea, and a neurological syndrome that includes ataxia, nystagmus, neurosensory hearing loss, and slowly progressive proximal limb weakness. The pathological hallmark is massive triglyceride storage in all tissues, including muscle, liver, gastrointestinal epithelium, endometrium, skin, bone marrow, and both fibroblast and muscle cells in culture [186]. The presence of lipid droplets in granulocytes of a blood smear (Jordan anomaly) facilitates diagnosis. The biochemical basis of this condition is a deficiency of the protein (CGI-58) that activates the adipocyte triglyceride lipase (ATGL), which catalyzes the first step in triglyceride hydrolysis, thus liberating fatty acids from triglyceride stores in skeletal muscle and all other tissues. Several mutations in the CGI-58 gene have been identified [185].
Neutral Lipid Storage Disease with Myopathy (NLSDM)
This generalized lipid storage disease is due to mutations in the gene (PNPLA2) encoding the ubiquitous adipocyte triglyceride lipase (ATGL). Although all tissues can be affected, some patients present with “triglyceride deposit cardiomyovasculopathy (TGCV)” or “obesity of the heart” [187], but many patients present with a myopathy that does not start until adulthood and progresses slowly but steadily, such that many patients are confined to a wheelchair late in life. Some patients may also develop severe cardiomyopathy. There is no ichthyosis, but the Jordan anomaly is present in blood smears [120, 188]. Serum CK is increased and was the only consistent abnormality in a young preclinical woman with severe lipid myopathy and a retrotransposal mutation in PNPLA2 [189].
References
Bertorini T, Yeh Y-Y, Trevisan CP, et al. Carnitine palmitoyltransferase deficiency: myoglobinuria and respiratory failure. Neurology. 1980;30:263–71.
Andreu AL, Hanna MG, Reichmann H, et al. Exercise intolerance due to mutations in the cytochrome b gene of mitochondrial DNA. N Engl J Med. 1999;341:1037–44.
Preisler N, Orngreen MC, Echaniz-Laguna A, et al. Muscle phosphorylase kinase deficiency. A neutral metabolic variant or a disease? Neurology. 2012;78:265–8.
Burwinkel B, Maichele AJ, Aagenaes O, et al. Autosomal glycogenosis of liver and muscle due to phosphorylase kinase deficiency is caused by mutations in the phosphorylase kinase beta subunit (PHKB). Hum Mol Genet. 1997;6:1109–15.
Akman HO, Sampayo JN, Ross FA, et al. Fatal infantile cardiac glycogenosis with phosphorylase kinase deficiency and a mutation in the gamma2-subunit of AMP-activated protein kinase. Pediatr Res. 2007;62:499–504.
McArdle B. Myopathy due to a defect in muscle glycogen breakdown. Clin Sci. 1951;10:13–33.
Lebo RV, Gorin F, Fletterick RJ, et al. High-resolution chromosome sorting and DNA spot-blot analysis assign McArdle’s syndrome to chromosome 11. Science. 1984;225:57–9.
Schmidt B, Servidei S, Gabbai AA, et al. McArdle’s disease in two generations: autosomal recessive transmission with manifesting heterozygote. Neurology. 1987;37:1558–61.
Papadimitriou A, Manta P, Divari R, et al. McArdle’s disease: two clinical expressions in the same pedigree. J Neurol. 1990;237:267–70.
Manfredi G, Silvestri G, Servidei S, et al. Manifesting heterozygosis in McArdle’s disease: clinical, morphological, and biochemical studies in a family. J Neurol Sci. 1993;115:91–4.
Tsujino S, Shanske S, DiMauro S. Molecular genetic heterogeneity of myophosphorylase deficiency (McArdle’s disease). N Engl J Med. 1993;329:241–5.
Bartram C, Edwards R, Clague J, Beynon RJ. McArdle’s disease: a nonsense mutation in exon 1 of the muscle glycogen phosphorylase gene explains some but not all cases. Hum Mol Genet. 1993;2:1291–3.
Lucia A, Nogales-Gadea G, Perez M, et al. McArdle disease: what do neurologists need to know? Nat Clin Pract Neurol. 2008;4:568–77.
El-Schahawi M, Tsujino S, Shanske S, DiMauro S. Diagnosis of McArdle’s disease by molecular genetic analysis of blood. Neurology. 1996;47:579–80.
Sugie H, Sugie Y, Ito M, et al. Genetic analysis of Japanese patients with myophosphorylase deficiency (McArdle’s disease): single-codon deletion in exon 17 is the predominant mutation. Clin Chim Acta. 1995;236:81–6.
El-Schahawi M, Bruno C, Tsujino S, et al. Sudden infant death syndrome (SIDS) in a family with myophosphorylase deficiency. Neuromuscul Disord. 1997;7:81–3.
Tsujino S, Shanske S, Carroll JE, et al. Double trouble: combined myophosphorylase and AMP deaminase deficiency in a child homozygous for nonsense mutations at both loci. Neuromuscul Disord. 1995;5:263–6.
Rubio JC, Martin MA, Bautista J, et al. Association of genetically proven deficiencies of myophosphorylase and AMP deaminase: a second case of “double trouble”. Neuromuscul Disord. 1997;7:387–9.
Haller RG, Bertocci LA. Exercise evaluation of metabolic myopathies. In: Engel AG, Franzini-Armstrong C, editors. Myology, vol. 1. 2nd ed. New York: McGraw-Hill; 1994. p. 807–21.
Haller RG, Lewis SF, Cook JD, Blomqvist CG. Myophosphorylase deficiency impairs muscle oxidative metabolism. Ann Neurol. 1985;17:196–9.
Haller RG, Clausen T, Vissing J. Reduced levels of skeletal muscle Na+K+−ATPase in McArdle disease. Neurology. 1998;50:37–40.
DiMauro S, Bresolin N. Phosphorylase deficiency. In: Engel AG, Banker BQ, editors. Myology, vol. 2. New York: McGraw-Hill; 1986. p. 1585–601.
Tonin P, Lewis P, Servidei S, DiMauro S. Metabolic causes of myoglobinuria. Ann Neurol. 1990;27:181–5.
Quinlivan R, Buckley J, James M, et al. McArdle disease: a clinical review. J Neurol Neurosurg Psychiatry. 2010;81:1182–8.
DiMauro S, Hartlage P. Fatal infantile form of muscle phosphorylase deficiency. Neurology. 1978;28:1124–9.
Milstein JM, Herron TM, Haas JE. Fatal infantile muscle phosphorylase deficiency. J Child Neurol. 1989;4:186–8.
Haller RG, Wyrick P, Taivassalo T, Vissing J. Aerobic conditioning: an effective therapy in McArdle’s disease. Ann Neurol. 2006;59:922–8.
Phoenix J, Hopkins P, Bartram C, et al. Effect of vitamin B6 supplementation in McArdle’s disease: a strategic case study. Neuromuscul Disord. 1998;8:210–2.
Izumi R, Suzuki N, Kato K, et al. A case of McArdle disease: efficacy of vitamin B6 on fatigability and impaired glycogenolysis. Intern Med. 2010;49:1623–5.
Sato S, Ohi T, Nishino I, Sugie H. Confirmation of the efficacy of vitamin B6 supplementation for McArdle disease by follow-up muscle biopsy. Muscle Nerve. 2012;45:436–40.
Andersen ST, Haller RG, Vissing J. Effect of oral sucrose shortly before exercise on work capacity in McArdle disease. Arch Neurol. 2008;65:786–9.
Busch V, Gempel K, Hack A, et al. Treatment of glycogenosis type V with ketogenic diet. Ann Neurol. 2005;58:341 [Letter].
Nogales-Gadea G, Pinos T, Lucia A, et al. Knock-in mice for the R50X mutation in the PYGM gene present with McArdle disease. Brain. 2012;135:2048–57.
Stojkovic T, Vissing J, Petit F, et al. Muscle glycogenosis due to phosphoglucomutase 1 deficiency. N Engl J Med. 2009;361:425–7.
Tarui S, Okuno G, Ikua Y, et al. Phosphofructokinase deficiency in skeletal muscle. A new type of glycogenosis. Biochem Biophys Res Comm. 1965;19:517–23.
Layzer RB, Rowland LP, Ranney HM. Muscle phosphofructokinase deficiency. Arch Neurol. 1967;17:512–23.
DiMauro S, Hays AP, Tsujino S. Nonlysosomal glycogenoses. In: Engel AG, Franzini-Armstrong C, editors. Myology, vol. II. New York: McGraw-Hill; 2004. p. 1535–58.
Nakajima H, Kono N, Yamasaki T, et al. Genetic defect in muscle phosphofructokinase deficiency. Abnormal splicing of the muscle phosphofructokinase gene due to a point mutation at the 5′-splice site. J Biol Chem. 1990;265:9392–5.
Raben N, Sherman J, Miller F, et al. A 5′ splice junction mutation leading to exon deletion in an Ashkenazi Jewish family with phosphofructokinase deficiency (Tarui disease). J Biol Chem. 1993;268:4963–7.
Sherman JB, Raben N, Nicastri C, et al. Common mutations in the phosphofructokinase-M gene in Ashkenazi Jewish patients with glycogenosis VII – and their population frequency. Am J Hum Genet. 1994;55:305–13.
Bonilla E, Schotland DL. Histochemical diagnosis of muscle phosphofructokinase deficiency. Arch Neurol. 1970;22:8–12.
Dunaway GA, Kasten TP, Sebo T, Trapp R. Analysis of the phosphofructokinase subunits and isoenzymes in human tissues. Biochem J. 1988;251:677–83.
Agamanolis DP, Askari AD, DiMauro S, et al. Muscle phosphofructokinase deficiency: two cases with unusual polysaccharide accumulation and immunologically active enzyme protein. Muscle Nerve. 1980;3:456–67.
Raben N, Danon MJ, Lu N, et al. Surprises of genetic engineering: a possible model of polyglucosan body disease. Neurology. 2001;56:1739–45.
Mineo I, Kono N, Hara N, et al. Myogenic hyperuricemia. A common pathophysiologic feature of glycogenosis types III, V, and VII. N Engl J Med. 1987;317:75–80.
Haller RG, Lewis SF. Glucose-induced exertional fatigue in muscle phosphofructokinase deficiency. New Engl J Med. 1991;324:364–9.
Vissing J, Vissing SF, MacLean DA. Sympathetic activation in exercise is not dependent on muscle acidosis. Direct evidence from studies in metabolic myopathies. J Clin Invest. 1998;101:1654–60.
Lewis SF, Vora S, Haller RG. Abnormal oxidative metabolism and O2 transport in muscle phosphofructokinase deficiency. J Appl Physiol. 1991;70:391–8.
Haller RG, Vissing J. No spontaneous second wind in muscle phosphofructokinase deficiency. Neurology. 2004;62:82–6.
Serratrice G, Monges A, Roux H, et al. Forme myopathique du deficit en phosphofructokinase. Rev Neurol. 1969;34:271–7.
Hays AP, Hallett M, Delfs J, et al. Muscle phosphofructokinase deficiency: abnormal polysaccharide in a case of late-onset myopathy. Neurology. 1981;31:1077–86.
Danon MJ, Servidei S, DiMauro S, Vora S. Late-onset muscle phosphofructokinase deficiency. Neurology. 1988;38:955–60.
Argov Z, Barash V, Soffer D, et al. Late-onset muscular weakness in phosphofructokinase deficiency due to exon5/intron5 junction point mutation: a unique disorder or the natural course of this glycolytic disorder? Neurology. 1994;44:1097–100.
Malfatti E, Birouk N, Romero NB, et al. Juvenile-onset permanent weakness in muscle phosphofructokinase deficiency. Neuromuscul Disord. 2012;316:173–7.
Argov Z, Bank WJ. Phosphorus magnetic resonance spectroscopy (31P MRS) in neuromuscular disorders. Ann Neurol. 1991;30:90–7.
Spiegel R, Area Gomez E, Akman HO, et al. Myopathic form of phosphoglycerate kinase (PGK) deficiency: a new case and pathogenic considerations. Neuromuscul Disord. 2009;19:207–11.
Haller RG, Wyrick P, Cavender D, et al. Aerobic conditioning: an effective therapy in McArdle’s disease. Neurology. 1998;50:A369.
Swoboda KJ, Specht L, Jones HR, et al. Infantile phosphofructokinase deficiency with arthrogryposis: clinical benefit of a ketogenic diet. J Pediatr. 1997;131:932–4.
Kreuder J, Borkhardt A, Repp R, et al. Inherited metabolic myopathy and hemolysis due to a mutation in aldolase A. N Engl J Med. 1996;334:1100–4.
Yao DC, Tolan DR, Murray MF, et al. Hemolytic anemia and severe rhabdomyolysis caused by compound heterozygous mutations of the gene for erythrocyte/muscle isozyme of aldolase. Blood. 2004;103:2401–3.
Sotiriou E, Greene P, Krishna S, et al. Myopathy and Parkinsonism in phosphoglycerate kinase deficiency. Muscle Nerve. 2010;41:707–10.
DiMauro S, Spiegel R. Progress and problems in muscle glycogenoses. Acta Myologica. 2011;30:96–102.
DiMauro S, Miranda AF, Khan S, et al. Human muscle phosphoglycerate mutase deficiency: newly discovered metabolic myopathy. Science. 1981;212:1277–9.
Naini A, Toscano A, Musumeci O, et al. Muscle phosphoglycerate mutase deficiency revisited. Arch Neurol. 2009;66:394–8.
Toscano A, Tsujino S, Vita G, et al. Molecular basis of muscle phosphoglycerate mutase (PGAM-M) deficiency in the Italian kindred. Muscle Nerve. 1996;19:1134–7.
Tonin P, Bruno C, Cassandrini D, et al. Unusual presentation of phosphoglycerate mutase deficiency due to two different mutations in PGAM-M gene. Neuromuscul Disord. 2009;19:776–8.
Hadjigeorgiou GM, Kawashima N, Bruno C, et al. Manifesting heterozygotes in a Japanese family with a novel mutation in the muscle-specific phosphoglycerate mutase (PGAM-M) gene. Neuromuscul Disord. 1999;9:399–402.
Vissing J, Schmalbruch H, Haller RG, Clausen T. Muscle phosphoglycerate mutase deficiency with tubular aggregates: effect of dantrolene. Ann Neurol. 1999;46:274–7.
Vissing J, Quistorff B, Haller RG. Effects of fuels on exercise capacity in muscle phosphoglycerate mutase deficiency. Arch Neurol. 2005;62:1440–3.
de Atauri P, Repiso A, Oliva B, et al. Characterization of the first described mutation of human red blood cell phosphoglycerate mutase. Biochim Biophys Acta. 2005;1740:403–10.
Comi GP, Fortunato F, Lucchiari S, et al. B-enolase deficiency, a new metabolic myopathy of distal glycolysis. Ann Neurol. 2001;50:202–7.
Kanno T, Sudo K, Takeuchi I, et al. Hereditary deficiency of lactate dehydrogenase M-subunit. Clin Chim Acta. 1980;108:267–76.
Maekawa M, Sudo K, Kanno T, Li S. Molecular characterization of genetic mutation in human lactate dehydrogenase-A (M) deficiency. Biochem Biophys Res Comm. 1990;168:677–82.
Maekawa M, Sudo K, Kanno T, et al. A novel mutation of lactate dehydrogenase A (M) gene in the fifth family with the enzyme deficiency. Hum Mol Genet. 1994;3:825–6.
Maekawa M, Sudo K, Li S, Kanno T. Analysis of genetic mutation in human lactate dehydrogenase-A (M) deficiency using DNA conformation polymorphism in combination with polyacrylamide gradient gel and silver staining. Biochem Biophys Res Comm. 1991;180:1083–90.
Tsujino S, Shanske S, Brownell A, et al. Molecular genetic studies of muscle lactate dehydrogenase deficiency in white patients. Ann Neurol. 1994;36:661–5.
Kanno T, Maekawa M. Lactate dehydrogenase M-subunit deficiency: clinical features, metabolic background, and genetic heterogeneities. Muscle Nerve. 1995;Suppl. 3:S54–60.
Kollberg G, Tulinius M, Gilljam T, et al. Cardiomyopathy and exercise intolerance in muscle glycogen storage disease 0. N Engl J Med. 2007;357:1507–14.
Cameron JM, Levandovskiy V, MacKay N, et al. Identification of a novel mutation in GSY1 (muscle-specific glycogen synthase) resulting in sudden cardiac death, that is diagnosable from skin fibroblasts. Mol Genet Metab. 2009;98:378–82.
Sukigara S, Liang W-C, Komaki H, et al. Muscle glycogen storage disease 0 presenting recurrent syncope with weakness and myalgia. Neuromuscul Disord. 2012;22:162–5.
Moslemi A-R, Lindberg C, Nilsson J, et al. Glycogenin-1 deficiency and inactivated priming of glycogen synthesis. N Engl J Med. 2010;362:1203–10.
DiMauro S, DiMauro-Melis PM. Muscle carnitine palmitoyltransferase deficiency and myoglobinuria. Science. 1973;182:929–31.
Bank WJ, DiMauro S, Bonilla E, et al. A disorder of muscle lipid metabolism and myoglobinuria. Absence of carnitine palmitoyltransferase. N Engl J Med. 1975;292:443–9.
Britton CH, Schultz RA, Zhang B, et al. Human liver mitochondrial carnitine palmitoyltransferase I: characterization of its cDNA and chromosomal localization and partial analysis of the gene. Proc Natl Acad Sci USA. 1995;92:1984–8.
Gellera C, Verderio E, Floridia G, et al. Assignment of the human carnitine palmitoyltransferase II gene (CPT1) to chromosome 1p32. Genomics. 1994;24:195–7.
DiDonato S, Taroni F. Disorders of lipid metabolism. In: Engel AG, Franzini-Armstrong C, editors. Myology, vol. 2. New York: McGraw-Hill; 2004. p. 1587–621.
Kaufmann P, El-Schahawi M, DiMauro S. Carnitine palmitoyltransferase II deficiency: diagnosis by molecular analysis of blood. Mol Cell Biochem. 1997;174:237–9.
Montermini L, Wang H, Verderio E, et al. Identification of 5′ regulatory regions of the human carnitine palmitoyltransferase II gene. Biochim Biophys Acta. 1994;1219:237–40.
Orngreen MC, Duno M, Ejstrup R, et al. Fuel utilization ib subjects with carnitine palmitoyltransferase 2 gene mutations. Ann Neurol. 2005;57:60–6.
Quinzii CM, Hirano M. Coenzyme Q and mitochondrial disease. Dev Disabil Res Rev. 2010;16:183–8.
Roe CR, Yang B-Z, Brunengraber H, et al. Carnitine palmitoyltransferase II deficiency: successful anaplerotic diet therapy. Neurology. 2008;71:260–4.
Bonnefont J-P, Bastin J, Behin A, Djouadi F. Bezafibrate for an inborn mitochondrial beta-oxidation defect. N Engl J Med. 2009;360:838840.
Ohashi Y, Hasegawa Y, Murayama K, et al. A new diagnostic test for VLCAD deficiency using immunohistochemistry. Neurology. 2004;62:2209–13.
Laforet P, Acquaviva-Bourdain C, Rigal O, et al. Diagnostic assessment and long-term follow-up of 13 patients with very long-chain acyl-Coenzyme A dehydrogenase (VLCAD) deficiency. Neuromuscul Disord. 2009;19:324–9.
Laforet P, Vianey-Saban C. Disorders of muscle lipid metabolism: diagnostic and therapeutic challenges. Neuromuscul Disord. 2010;20:693–700.
Tein I, DiMauro S, DeVivo DC. Recurrent childhood myoglobinuria. Adv Pediatr. 1990;37:77–117.
Zeharia A, Shaag A, Houtkooper RH, et al. Mutations in LPIN cause recurrent childhood myoglobinuria in childhood. Am J Hum Genet. 2008;83:489–94.
DiMauro S. Mitochondrial myopathies. Curr Opin Rheumatol. 2006;18:636–41.
Emmanuele V, Sotiriou E, Gutierrez Rios P, et al. A novel mutation in the mitochondrial DNA cytochrome b gene (MTCYB) in a patient with MELAS syndrome. J Child Neurol. 2013;28:236–42.
Andreu AL, Tanji K, Bruno C, et al. Exercise intolerance due to a nonsense mutation in the mtDNA ND4 gene. Ann Neurol. 1999;45:820–3.
Bet L, Bresolin N, Moggio M, et al. A case of mitochondrial myopathy, lactic acidosis and complex I deficiency. J Neurol. 1990;237:399–404.
Musumeci O, Andreu AL, Shanske S, et al. Intragenic inversion of mtDNA: a new type of pathogenic mutation in a patient with mitochondrial myopathy. Am J Hum Genet. 2000;66:1900–4.
Keightley JA, Hoffbuhr KC, Burton MD, et al. A microdeletion in cytochrome c oxidase (COX) subunit III associated with COX deficiency and recurrent myoglobinuria. Nat Genet. 1996;12:410–5.
Karadimas CL, Greenstein P, Sue CM, et al. Recurrent myoglobinuria due to a nonsense mutation in the COX I gene of mtDNA. Neurology. 2000;55:644–9.
Rahman S, Taanman J-W, Cooper M, et al. A missense mutation of cytochrome oxidase subunit II causes defective assembly and myopathy. Am J Hum Genet. 1999;65:1030–9.
Kollberg G, Moslemi A-R, Lindberg C, et al. Mitochondrial myopathy and rhabdomyolysis associated with a novel nonsense mutation in the gene encoding cytochrome c oxidase subunit I. J Neuropathol Exp Neurol. 2005;64:123–8.
McFarland R, Taylor RW, Chinnery PF, et al. A novel sporadic mutation in cytochrome c oxidase subunit II as a cause of rhabdomyolysis. Neuromuscul Disord. 2004;14:162–6.
Haller RG, Henriksson KG, Jorfeldt L, et al. Deficiency of skeletal muscle succinate dehydrogenase and aconitase. J Clin Invest. 1991;88:1197–206.
Hall RE, Henriksson KG, Lewis SF, et al. Mitochondrial myopathy with succinate dehydrogenase and aconitase deficiency. Abnormalities of several iron-sulfur proteins. J Clin Invest. 1993;92:2660–6.
Mochel F, Knight MA, Tong W-H, et al. Splice mutation in the iron-sulfur cluster scaffold protein ISCU causes myopathy with exercise intolerance. Am J Hum Genet. 2008;82:652–60.
Kollberg G, Melberg A, Holme E, Oldfors A. Transient restoration of succinate dehydrogenase activity after rhabdomyolysis in iron-sulfur cluster deficiency myopathy. Neuromuscul Disord. 2010;21:115–20.
Ogasahara S, Engel AG, Frens D, Mack D. Muscle coenzyme Q deficiency in familial mitochondrial encephalomyopathy. Proc Natl Acad Sci USA. 1989;86:2379–82.
Sobreira C, Hirano M, Shanske S, et al. Mitochondrial encephalomyopathy with coenzyme Q10 deficiency. Neurology. 1997;48:1238–43.
DiGiovanni S, Mirabella M, Papacci M, et al. Apoptosis and ROS detoxification enzymes correlate with cytochrome c oxidase deficiency in mitochondrial encephalomyopathies. Mol Cell Neurosci. 2001;17:696–705.
Lalani S, Vladutiu GD, Plunkett K, et al. Isolated mitochondrial myopathy associated with muscle coenzyme Q10 deficiency. Arch Neurol. 2005;62:317–20.
Auré K, Benoist JF, Ogier de Baulny H, et al. Progression despite replacement of a myopathic form of coenzyme Q10 defect. Neurology. 2004;63:727–9.
Horvath R, Scneiderat P, Schoser BGH, et al. Coenzyme Q10 deficiency and isolated myopathy. Neurology. 2006;66:253–5.
Gempel K, Topaloglu H, Talim B, et al. The myopathic form of coenzyme Q10 deficiency is caused by mutations in the electron-transferring-flavoprotein dehydrogenase (ETFDH) gene. Brain. 2007;130:2037–44.
Liang W-C, Ohkuma A, Hayashi YK, et al. ETFDH mutations, CoQ10 levels, and respiratory chain activities in patients with riboflavin-responsive multiple acyl-CoA dehydrogenase deficiency. Neuromuscul Disord. 2009;19:212–6.
Ohkuma A, Noguchi S, Sugie H, et al. Clinical and genetic analysis of lipid storage myopathies. Muscle Nerve. 2009;39:333–42.
Sabina RL. Myoadenylate deficiency. Neurol Clin. 2000;18:185–94.
Gambetti PL, DiMauro S, Baker L. Nervous system in Pompe disease. Ultrastructure and biochemistry. J Neuropathol Exp Neurol. 1971;30:412–30.
Martin JJ, DeBarsy T, Van Hoof F. Pompe disease: an inborn lysosomal disorder with storage of glycogen: a study of brain and striated muscle. Acta Neuropathol. 1973;23:229–44.
Powell HC, Haas R, Hall CL, et al. Peripheral nerve in type III glycogenosis: selective involvement of unmyelinated fiber Schwann cells. Muscle Nerve. 1985;8:667–71.
Moses SW, Gadoth N, Ben-David E, et al. Neuromuscular involvement in glycogen storage disease type III. Acta Paediatr Scand. 1986;7:289–96.
Ugawa Y, Inoue K, Takemura T, Iwamasa T. Accumulation of glycogen in sural nerve axons in adult-onset type III glycogenosis. Ann Neurol. 1986;19:294–7.
Moses SW, Wanderman KL, Myroz A, Frydman M. Cardiac involvement in glycogen storage disease type III. Eur J Paediatr. 1989;148:764–6.
Pompe JC. Over Idiopatische Hypertrophie van het Hart. Ned T Geneesk. 1932;76:304–11.
Putschar W. Uber angeborene Glykogenspeicherkrankenheit des Herzens: “Thesaurismosis glykogenica”. Beitr Path Anat. 1932;90:222–3.
Hers HG. Alpha-glucosidase deficiency in generalized glycogen storage disease (Pompe disease). Biochem J. 1963;86:11–6.
Lejeune N, Thines-Sempoux D, Hers HG. Tissue fractionation studies: 16. Intracellular distribution and properties of alpha-glucosidases in rat liver. Biochem J. 1963;86:16–21.
Hers HG. Inborn lysosomal diseases. Gastroenterology. 1965;48:625–33.
Engel AG, Gomez MR, Seybold ME, Lambert EH. The spectrum and diagnosis of acid maltase deficiency. Neurology. 1973;23:95–105.
Nascimbeni AC, Fanin M, Tasca E, Angelini C. Molecular pathology and enzyme processing in various phenotypes of acid maltase deficiency. Neurology. 2008;70:617–26.
Hasilik A, Neufeld EF. Biosynthesis of lysosomal enzymes in fibroblasts. Synthesis as precursors of higher molecular weight. J Biol Chem. 1982;255:4937–45.
Hasilik A, Neufeld EF. Biosynthesis of lysosomal enzymes in fibroblasts. Phosphorylation of mannose residues. J Biol Chem. 1982;255:4946–50.
DiMauro S, Stern LZ, Mehler M, et al. Adult-onset acid maltase deficiency: a postmortem study. Muscle Nerve. 1978;1:27–36.
Mehler M, DiMauro S. Residual acid maltase activity in late-onset acid maltase deficiency. Neurology. 1977;27:178–84.
Reuser AJ, Kroos M, Willemsen R, et al. Clinical diversity in glycogenosis type II. Biosynthesis and in situ localization of acid alpha-glucosidase in mutant fibroblasts. J Clin Invest. 1987;79:1689–99.
Settembre C, Fraldi A, Jahreiss L, et al. A block of autophagy in lysosomal storage disorders. Hum Mol Genet. 2008;17:119–29.
Fukuda T, Ewan L, Bauer M, et al. Dysfunction of endocytic and autophagic pathways in a lysosomal storage disease. Ann Neurol. 2006;59:700–8.
Malicdan MC, Noguchi S, Nonaka I, et al. Lysosomal myopathies: an excessive build-up in autophagosomes is too much to handle. Neuromusc Disord. 2008;18:521–9.
Engel AG, Hirschhorn R, Huie MI. Acid maltase deficiency. In: Engel AG, Franzini-Armstrong C, editors. Myology, vol. 2. New York: McGraw-Hill; 2004. p. 1559–86.
Mellies U, Lofaso F. Pompe disease: a neuromuscular disease with respiratory muscle involvement. Respir Med. 2009;103:477–84.
Amato AA, Leep Hunderfund AN, Selcen D, Keegan BM. A 49-year-old woman with progressive shortness of breath. Neurology. 2011;76:830–6.
Kretzchmer HA, Wagner H, Hubner G, et al. Aneurysms and vacuolar degeneration of cerebral arteries in late-onset acid maltase deficiency. J Neurol Sci. 1990;98:169–83.
Carlier R-Y, Laforet P, Wary C, et al. Whole-body muscle MRI in 20 patients suffering from late onset Pompe disease: involvement patterns. Neuromuscul Disord. 2011;21:791–9.
Danon MJ, Oh SJ, DiMauro S, et al. Lysosomal glycogen storage disease with normal acid maltase. Neurology. 1981;31:51–7.
Nishino I, Fu J, Tanji K, et al. Primary LAMP-2 deficiency causes X-linked vacuolar cardiomyopathy and myopathy (Danon disease). Nature. 2000;406:906–10.
Shanske S, DiMauro S. Late-onset acid maltase deficiency. Biochemical studies of leukocytes. J Neurol Sci. 1981;50:57–62.
van der Ploeg AT. Where do we stand in enzyme replacement therapy in Pompe disease? Neuromusc Dis. 2010;20:733–4.
Kishnani PS, Goldenberg PC, DeArmey SL, et al. Cross-reacting immunological material status affects treatment outcomes in Pompe disease infants. Mol Genet Metab. 2010;99:26–33.
van der Ploeg AT, Clemens PR, Corzo D, et al. A randomized study of alglucosidase alfa in late-onset Pompe disease. N Engl J Med. 2010;362:1396–406.
Hagemans MLC, Janssens ACJW, Winkel LPF, et al. Late-onset Pompe disease primarily affects quality of life in physical health domains. Neurology. 2004;63:1688–92.
Laloui K, Wary C, Carlier R-Y, et al. Making diagnosis of Pompe disease at a presymptomatic stage: to treat or not to treat? Neurology. 2011;77:594–5.
Van den Hout JMP, Reuser AJJ, de Klerk JBC, et al. Enzyme therapy for Pompe disease with recombinant human alpha-glucoside from rabbit milk. J Inherit Metab Dis. 2001;24:266–74.
Van den Hout J, Kamphoven JHJ, Winkel LPF, et al. Long-term intravenous treatment of Pompe disease with recombinant human alpha-glucosidase from milk. Pediatrics. 2004;113:e448–57.
Chakrapani A, Vellodi A, Robinson P, et al. Treatment of infantile Pompe disease with aglucosidase alpha: the UK experience. J Inherit Metab Dis. 2010;33:747–50.
Mellies U, Stehling F, Dohna-Schwake C, et al. Respiratory failure in Pompe disease: treatment with noninvasive ventilation. Neurology. 2005;64:1465–7.
Slonim AE, Bulone L, Minikes J, et al. Benign course of glycogen storage disease type IIb in two brothers: nature or nurture? Muscle Nerve. 2006;33:571–4.
Kishnani P, Austin SL, Arn P, et al. Glycogen storage disease type III diagnosis and management guidelines. Genet Med. 2010;12:446–63.
DiMauro S, Hartwig GB, Hays AP, et al. Debrancher deficiency: neuromuscular disorder in five adults. Ann Neurol. 1979;5:422–36.
Wary C, Nadaj-Pakleza A, Laforet P, et al. Investigating glycogenosis type III with multi-parametric functional NMR imaging and spectroscopy. Neuromuscul Disord. 2010;20:548–58.
Shen J, Bao Y, Liu H-M, et al. Mutations in exon 3 of the glycogen debranching enzyme gene are associated with glycogen storage disease type III that is differentially expressed in liver and muscle. J Clin Invest. 1996;98:352–7.
Bruno C, van Diggelen OP, Cassandrini D, et al. Clinical and genetic heterogeneity of branching enzyme deficiency (glycogenosis type IV). Neurology. 2004;63:1053–8.
Tay SKH, Akman HO, Chung WK, et al. Fatal infantile neuromuscular presentation of glycogen storage disease type IV. Neuromuscul Disord. 2004;14:253–60.
Janecke AR, Dertinger S, Ketelsen UP, et al. Neonatal type IV glycogen storage disease associated with “null” mutations in glycogen branching enzyme 1. J Pediatr. 2004;145:705–9.
Konstantinidou AE, Anninos H, Gyftodimou Y, et al. Neonatal neuromuscular variant of glycogen storage disease type IV: histopathological findings leading to the diagnosis. Histopathology. 2006;48:878–80.
Taratuto AL, Akman HO, Saccoliti M, et al. Branching enzyme deficiency/glycogenosis storage disease type IV presenting as a severe congenital hypotonia: muscle biopsy and autopsy findings, biochemical and molecular studies. Neuromuscul Disord. 2010;20:783–90.
Alegria A, Martins E, Dias M, et al. Glycogen storage disease type IV presenting as hydrops fetalis. J Inherit Metab Dis. 1999;22:330–2.
Akman HO, Karadimas CL, Gyftodimou Y, et al. Prenatal diagnosis of glycogen storage disease type IV. Prenat Diagn. 2006;26(10):951–5.
Assereto S, van Diggelen OP, Diogo L, et al. Null mutations and lethal congenital form of glycogen storage disease type IV. Biochem Biophys Res Comm. 2007;361:445–50.
Herrick MK, Twiss JL, Vladutiu GD, et al. Concomitant branching enzyme and phosphorylase deficiencies. An unusual glycogenosis with extensive neuronal polyglucosan storage. J Neuropathol Exp Neurol. 1994;53:239–46.
Mochel F, Schiffmann R, Steenweg M, et al. Adult polyglucosan body disease: natural history and key MRI findings. Ann Neurol. 2012;72(3):433–41.
Akman HO, Sheiko T, Tay SKH, et al. Generation of a novel mouse model that recapitulates early and adult onset glycogenosis type IV. Hum Mol Genet. 2011;20:4430–9.
Tein I, DeVivo DC, Bierman F, et al. Impaired skin fibroblast carnitine uptake in primary systemic carnitine deficiency manifested by childhood carnitine-responsive cardiomyopathy. Pediatr Res. 1990;28:247–55.
Pons R, Carrozzo R, Tein I, et al. Deficient muscle carnitine transport in primary carnitine deficiency. Pediatr Res. 1997;42:583–7.
Nezu J-i, Tamai I, Oku A, et al. Primary systemic carnitine deficiency is caused by mutations in a gene encoding sodium ion-dependent carnitine transporter. Nat Genet. 1999;21:91–4.
Engel AG, Angelini C. Carnitine deficiency of human skeletal muscle with associated lipid storage myopathy: a new syndrome. Science. 1973;179:899–902.
Tein I, DiMauro S, Xie Z-W, De Vivo DC. Valproic acid impairs carnitine uptake in cultured human skin fibroblasts. An in vitro model for pathogenesis of valproic acid-associated carnitine deficiency. Pediatr Res. 1993;34:281–7.
Dalakas M, Leon-Monzon ME, Bernardini I, et al. Zidovudine-induced mitochondrial myopathy is associated with muscle carnitine deficiency and lipid storage. Ann Neurol. 1994;35:482–7.
Lafolla AK, Thompson RJ, Roe CR. Medium-chain acyl-coenzyme A dehydrogenase deficiency: clinical course in 120 affected children. J Pediatr. 1994;124:409–15.
Olsen RK, Olpin S, Andresen BS, et al. ETFDH mutations as major cause of riboflavin-responsive multiple acyl-CoA dehydrogenase deficiency. Brain. 2007;130:2045–54.
Chanarin L, Patel A, Slavin G, et al. Neutral-lipid storage disease: a new disorder of lipid metabolism. Br Med J. 1975;1:553–5.
Bruno C, Bertini E, Di Rocco M, et al. Clinical and genetic characterization of Chanarin-Dorfman syndrome. Biochem Biophys Res Comm. 2008;369:1125–8.
Miranda AF, DiMauro S, Eastwood AB, et al. Lipid storage, ichthyosis, and steatorrhea. Muscle Nerve. 1979;2:1–13.
Hirano K-I, Ikeda Y, Zaima N, et al. Triglyceride deposit cardiomyovasculopathy. N Engl J Med. 2008;359:2396–8.
Campagna F, Nanni L, Quagliarini F, et al. Novel mutations in the adipose triglyceride kipase gene causing neutral lipid storage diseases with myopathy. Biochem Biophys Res Comm. 2008;377:843–6.
Akman HO, Davidzon G, Tanji K, et al. Neutral lipid storage disease with subclinical myopathy due to a retrotransposal insertion in the PNPLA2 gene. Neuromuscul Disord. 2010;20:397–402.
Legros F, Chatzoglou E, Frachon P, et al. Functional characterization of novel mutations in the human cytochrome b gene. Eur J Hum Genet. 2001;9:510–8.
Massie R, Wong L-JC, Milone M. Exercise intolerance due to cytochrome b mutation. Muscle Nerve. 2010;42:136–40.
Andreu AL, Bruno C, Shanske S, et al. Missense mutation in the mtDNA cytochrome b gene in a patient with myopathy. Neurology. 1998;51:1444–7.
Andreu AL, Bruno C, Dunne TC, et al. A nonsense mutation (G15059A) in the cytochrome b gene in a patient with exercise intolerance and myoglobinuria. Ann Neurol. 1999;45:127–30.
Bruno C, Santorelli FM, Assereto S, et al. Progressive exercise intolerance associated with a new muscle-restricted nonsense mutation (G142X) in the mitochondrial cytochrome b gene. Muscle Nerve. 2003;28:508–11.
Lamantea E, Carrara F, Mariotti C, et al. A novel nonsense mutation (Q352X) in the mitochondrial cytochrome b gene associated with a combined defect of complexes I and III. Neuromuscul Disord. 2002;12:49–52.
Mancuso M, Filosto M, Stevens JC, et al. Mitochondrial myopathy and complex III deficiency in a patient with a new stop-codon mutation (G339X) in the cytochrome b gene. J Neurol Sci. 2003;209:61–3.
Dumoulin R, Sagnol I, Ferlin T, et al. A novel gly290asp mitochondrial cytochrome b mutation linked to a complex III deficiency in progressive exercise intolerance. Mol Cell Probes. 1996;10:389–91.
DiMauro S, Haller RG. Metabolic myopathies: substrate use defects. In: Schapira AHV, Griggs RC, editors. Muscle diseases. Boston: Butterworth-Heinemann; 1999. p. 225–49.
Acknowledgements
Some of the work described was supported by a grant from the Muscular Dystrophy Association. Dr. Paradas is supported by the Consejería de Salud, Junta de Andalucía, Spain.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2014 Springer Science+Business Media New York
About this chapter
Cite this chapter
DiMauro, S., Akman, H.O., Paradas, C. (2014). Metabolic Myopathies. In: Katirji, B., Kaminski, H., Ruff, R. (eds) Neuromuscular Disorders in Clinical Practice. Springer, New York, NY. https://doi.org/10.1007/978-1-4614-6567-6_63
Download citation
DOI: https://doi.org/10.1007/978-1-4614-6567-6_63
Published:
Publisher Name: Springer, New York, NY
Print ISBN: 978-1-4614-6566-9
Online ISBN: 978-1-4614-6567-6
eBook Packages: MedicineMedicine (R0)