
Abstract
Virulent Mycobacterium tuberculosis (Mtb) inhibits apoptosis and triggers necrosis of host macrophages to evade innate delay in the initiation of adaptive immunity. Necrosis is a mechanism used by bacteria to exit macrophage, evade the host defenses, and disseminate while apoptosis is associated with diminished pathogen viability. We have recently demonstrated that eicosanoids regulate cell death program of either human or murine macrophages infected with Mtb. We have defined prostaglandin E2 (PGE2) as a pro-apoptotic host lipid mediator which protects against necrosis. In contrast, lipoxin A4 (LXA4) is a pro-necrotic lipid mediator which suppresses PGE2 synthesis, resulting in mitochondrial damage and inhibition of plasma membrane repair mechanisms; this ultimately leads to the induction of necrosis. Thus, the balance between PGE2 and LXA4 determines whether Mtb-infected macrophages undergo apoptosis or necrosis and this balance determines the outcome of infection.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others


Keywords
- Mycobacterium tuberculosis (Mtb)
- Macrophages
- Necrosis
- Apoptosis
- Extrinsic pathway
- Intrinsic pathway
- Mitochondrial outer membrane permeabilization (MOMP)
- Mitochondrial permeability transition (MPT)
- Cell death program
- B-cell lymphoma 2 (Bcl-2)
- BH3 interacting domain (BID)
- Bcl-2 associated X Protein (BAX)
- Bcl-2 homologous antagonist killer (BAK)
- FLICE-inhibitory protein (FLIPS)
- Lipoxins (LX)
- Prostaglandins (PG)
- Eicosanoids
- Plasma membrane microdisruptions
- Mycobacterial antigens
- BCG vaccine
- T cell response
1 Introduction
Mycobacterium tuberculosis (Mtb) is an extremely successful bacterium that is transmitted person-to-person by the aerosol route. The World Health Organization (WHO) has estimated that more than 2 billion persons are latently infected with Mtb. From this large reservoir of asymptomatic infected people emerges 8–10 million cases of active TB each year resulting in the deaths of nearly 1.7 million people each year [1]. The increased incidence of TB has been attributed to three major factors: the HIV pandemic, the emergence of multidrug-resistant strains of Mtb, and the failure of the major vaccine, BCG, to prevent pulmonary tuberculosis [2–4].
The success of this pathogen is closely linked to its ability to alter the intracellular environment of the alveolar macrophage. When inhaled, Mtb enters the lower respiratory tract and reaches distal alveoli where initially infects alveolar macrophages. Although macrophages excel at phagocytizing and destroying biological particles including dead cells and bacteria, Mtb has adapted to the harsh intracellular environment, which allows it to survive and replicate within these phagocytic cells. By subverting or avoiding critical components of macrophage immunity including phagolysosomal fusion, microbicidal effectors, and as will be discussed in this chapter, cell death pathways, Mtb evades both innate and adaptive immune responses. Therefore, delineating how Mtb and macrophages interact is fundamental to understand immunity to Mtb.
Manipulation of macrophage death pathways is one mechanism that allows Mtb to evade host defenses. Three major outcomes are observed following productive Mtb infection of human or murine macrophages in vitro: (a) necrosis, a form of death characterized by plasma membrane disruption; (b) apoptosis, a form of death in which the plasma membrane integrity is preserved; and (c) survival of the infected macrophages. Characterization of these different phenotypes is challenging because of the asynchronous nature of intracellular infection and heterogeneity among the bacteria and macrophages. Other factors such as the percentage of infected macrophages and variation in the number of bacteria internalized by each macrophage can affect the kinetics of cell death when studied in vitro. Nevertheless, a spectrum of all three phenotypes can be observed following infection of normal macrophages with virulent Mtb. In general, highly virulent Mtb strains predominantly induce necrosis [5]. The concept that virulent Mtb induce necrosis in part by actively inhibiting macrophage apoptosis [6], has gained additional support by the identification of bacterial mutants that induce apoptosis instead of necrosis [7, 8]. The different cellular fates of Mtb infected macrophages are of great interest as the death modality influences the outcome of infection. In particular, apoptotic death reduces the viability of different mycobacterial species [9, 10] including Mtb [11, 12, 13]. Here, we discuss the cellular mechanisms that regulate the death modality of Mtb-infected macrophages and lead to important functional consequences on immunity to Mtb.
2 Macrophage Apoptosis is a Host Defense Mechanism Against Mtb
The discovery that many attenuated strains of mycobacteria induce more apoptosis than their wild-type counterparts supports the hypothesis that virulent mycobacteria inhibit macrophage apoptosis. Indeed, there exists a reciprocal relationship between virulence and apoptosis. As such, Mtb infection predominantly results in necrosis, while attenuated mutant strains including BCG and H37Ra primarily induce apoptosis. Now, investigators are identifying single gene mutations in Mtb that shift the balance from necrosis to apoptosis [7, 8]. Although it is not yet clear whether virulent Mtb block the triggering of apoptosis or inhibit downstream events that give rise to the typical cellular changes associated with apoptosis, it can be argued that by inducing necrosis, Mtb evades host defenses and provides a pathway for its exit from the infected cell and its dissemination. Detailed analysis of “necrosis” reveals to be heterogeneous and certain subtypes have been defined that have unique cellular triggers and molecular mechanisms. For instance, pyroptosis and necroptosis are forms of necrosis that are dependent on caspase 1 and receptor-interacting proteins 1 and 3 (RIP-1/3), respectively [14–16]. Thus, the idea that necrosis is a passive, accidental, and unregulated form of cell death is an old dogma that needs to be revisited. In fact, how Mtb induces necrosis is a question that remains unanswered.
In contrast to necrosis, the past decades have made tremendous progress in unraveling the signaling pathways that lead to initiation of apoptosis. Hallmarks of apoptosis include the segmentation of DNA [17], exposure of phosphatidylserine on the outer leaflet of the plasma membrane, and finally, packaging of cellular components into membrane-bound blebs [18, 19]. During apoptosis the dying cell produces ‘find me’ and ‘eat me’ signals that aid its rapid clearance by phagocytes through the process of efferocytosis [13, 20].
Apoptosis is initiated by two major pathways
-
(1)
The extrinsic pathway: The induction of apoptosis by attenuated Mtb in human monocyte-derived macrophages is mediated by the executioner caspases 3 and 7 and requires two distinct signals: one is a lipid and the other is a protein [21, 22] the protein-dependent signal can be reconstituted by TNF. Indeed, the induction of apoptosis in macrophages by Mtb requires the action of TNF to activate the extrinsic death receptor-dependent pathway (Fig. 1). Although both bacterial strains produce comparable amounts of TNF [23], the avirulent strain H37Ra much more potently induces apoptosis than the virulent H37Rv strain. One potential explanation for this observation is that soluble TNFR-2 is shed by macrophages infected with virulent Mtb, and neutralizes TNF, resulting in a “TNF-poor” microenvironment [24]. This model is also consistent with data that caspase 8 activation, which is an essential and early step in the induction of apoptosis by the extrinsic pathway, is inhibited in H37Rv-infected macrophages (Remold, unpublished observation). Recently, some of the early components of the extrinsic apoptotic pathway activated by Mtb in the murine macrophage cell line RAW 264 have been identified [25]. In Mtb-infected cells, TNF production induces reactive oxygen species (ROS)-dependent activation of apoptosis signal-regulating kinase (ASK1; A002816), a member of the mitogen-activated protein kinase family causing FLIPs phosphorylation. Phosphorylated FLIPs interacts with the E3 ubiquitin ligase c-CYBL facilitating proteasomal FLIPs degradation involving the tyrosine kinase c-Abl. FLIPs degradation then enables activation of caspase 8 leading to caspase 3/7 activation and apoptosis.
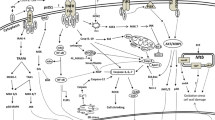
Fig. 1 
Two major pathways leading to macrophage death following M. tuberculosis infection. Some macrophages death programs are triggered by the extrinsic pathway (surface receptor mediated): Ligation of death receptor (e.g., Fas or TNF-α receptors) is followed by activation of caspase 8 which leads to apoptosis. Alternatively, the intrinsic pathway (mitochondria-mediated) is activated: Permeabilization of mitochondria outer membrane potential (MOMP) leads to the release of apoptotic mediators such as cytochrome c from the mitochondrial intermembrane space into the cytosol leading to formation of apoptosome complex and activation of caspases (9, 3, and 7) which in turn induce apoptosis. However, during necrosis mitochondrial permeability transition (MPT) causes mitochondrial inner membrane perturbation (MIMP), collapse of the membrane potential, uncoupling of the respiratory chain, and overproduction of reactive oxygen species (ROS). Cyclophilin D (CypD) is a mitochondrial protein which is involved in MPT and necrosis
-
(2)
The intrinsic pathway: Induction of apoptosis in vertebrate cells most commonly proceeds through the intrinsic apoptotic pathway, which is functionally defined by mitochondrial outer membrane permeabilization (MOMP) [26]. MOMP is a central event that can lead to apoptosis as it results in release of apoptotic mediators, including cytochrome c, Smac-DIABLO, AIF, and other factors from the mitochondrial inter-membrane space and ultimately results in the activation of caspases 9, 3, and 7 (Fig. 1). Although these events usually occur independently of other changes in the mitochondria, they can also be associated with the opening of the mitochondrial inner membrane pore (PT pore), which leads to mitochondrial permeability transition (MPT), loss of the mitochondrial inter-membrane potential (Δψm), and necrosis. We found that Mtb infection, whether virulent or avirulent, induces such changes in the mitochondrial membranes and that these changes are the key events that determine the death modality of infected macrophages.

3 Mitochondrial Damage and Macrophage Death
Considering mitochondria as a key player in regulation of cell death program, the different combinations of MOMP and mitochondrial permeability transition (MPT, the opening of a pore in the inner mitochondrial membrane) in model experimental systems, and their effect on the cellular outcome are reviewed below. Thus the changes in MOMP and MPT induced by either virulent or avirulent Mtb infection will be discussed in the context of these scenarios.
In Scenario I, MPT causes the mitochondria to become leaky to water, which results in swelling, dysfunction, and eventually necrosis [26]. Irreversible MPT can lead to outer mitochondrial membrane damage, which manifests itself as MOMP, in this case a by-product of MPT. This scenario emerges when hepatocytes under oxidative stress or due to other toxic treatment undergo both necrosis and apoptosis [27].
However, in Scenario II, MOMP and apoptosis can occur independently of MPT. This is the case when MOMP is induced by members of the Bcl-2 family of apoptosis-inducing proteins, which do not affect the mitochondrial inner membrane [28]. Specifically, processing of the Bcl-2 protein by BID leads to activation of the pro-apoptotic Bcl-2 family proteins, BAX and BAK, causing MOMP and translocation of pro-apoptotic factors including cytochrome c into the cytosol, activation of caspase 9, and eventually caspase 3. This process neither induces nor requires MPT [29].
In Scenario III, effector molecules capable of damaging the mitochondrial inner membrane gain access to the mitochondrial inter-membrane space if the mitochondrial outer membrane is permeable. This seems to be the mechanism by which Ndufs1, the 30 kDa subunit of mitochondrial complex 1 of the electron transport chain, is damaged by caspase 3 [30]. Caspase 3 is thought to access the mitochondrial inter-membrane space via pores generated in the mitochondrial outer membrane, which allow pro-apoptotic factors including cytochrome c to escape into the cytosol [29]. Damage of Ndufs1 disrupts the electron transport chain in the inner membrane leading to ROS accumulation and necrosis.
Finally, MPT can also occur independently of MOMP (Scenario IV). This is thought to be how granzyme A damages components of the mitochondrial inner membrane [31]. Hsp70 and Hsp90 are candidate molecules that serve as cytosolic chaperones for granzyme A and allow the protease to enter the mitochondrial inter-membrane space without damaging the mitochondrial outer membrane leading to cleavage of Ndufs3 [31].
In macrophages infected with attenuated Mtb, apoptosis is associated with MOMP yet MPT is not induced, as described in Scenario II [29]. Inhibition of MOMP diminishes only apoptosis, but does not affect MPT [5]. Silencing of the gene for the pro-apoptotic Bcl-2 protein BAX, which is required for the release of cytochrome c and AIF from the mitochondrial inter-membrane space, abrogates Mtb-induced apoptosis, but does not affect MPT or necrosis [5]. In contrast, virulent Mtb induce both MOMP and MPT leading to irreversible mitochondrial swelling and necrosis [5]. MPT can be inhibited by cyclosporin A (which selectively blocks the function of cyclophilin D in the mitochondrial inner membrane), has a requirement for mitochondrial Ca++ loading and is independent of Bcl-2 family member-induced apoptosis [32]. Inhibition of MPT vis Cyclophilin D, downregulates only necrosis, but does not affect the degree of MOMP or apoptosis [33, 34]. It is not clear at present whether in Mtb infected macrophages MPT is dependent on opening of pores in the mitochondrial outer membrane (MOMP—Scenario III) or whether toxic molecular species enter the mitochondrial inter membrane space via chaperones (Scenario IV). The different mechanisms induced by virulent and avirulent Mtb indicate that in Mtb-infected macrophages MOMP and MPT are independent phenomena; virulent Mtb are unique in their induction of MPT that leads to the destruction of the mitochondrial outer membrane causing secondary cytochrome c release (see Scenario I) [5]. In summary, induction of apoptosis or necrosis in Mtb-infected macrophages depends on highly specific mechanisms leading to different types of mitochondrial membrane perturbation. Attenuated and virulent Mtb alike cause transient MOMP characterized by cytochrome c release from the mitochondrial inter-membrane space, which requires BAX. In contrast, only virulent H37Rv causes MPT.
While cell death is a tightly regulated process, the host–pathogen interaction adds several layers of complexity. How the death modality of infected cells affects the outcome of infection, particularly during different clinical states in people (e.g., latency versus disease), remains a pertinent question. Here, other investigators working on the genetics of susceptibility to Mtb provide an important perspective. Gene expression profiling finds that several genes related to apoptosis are expressed less in active TB patients than in latently infected people, suggesting that decreased apoptotic activity is associated with the reactivation of latent infection [35]. Using a more targeted approach, Abebe et al. found that patients with active TB in Ethiopia had elevated expression of genes associated with the extrinsic apoptosis pathway including TNF, Fas, FasL, and caspase 8. However, the expression of FLIP, an intrinsic inhibitor of caspase 8 was also significantly elevated [36]. Although the upregulation of TNF, Fas/FasL, and caspase 8 may be the signature of an immune response capable of inducing apoptosis in infected cells, the authors propose a model in which Mtb inhibits the extrinsic apoptosis pathway by upregulating FLIP to evade an apoptotic death. Finally, the eicosanoid biosynthetic pathways, which regulate the death modality of infected human and murine macrophages, have now been identified as important genetic loci that regulate susceptibility to tuberculosis and leprosy in people [37–39]. While the genetic and functional data require greater scrutiny and functional correlation, they independently provide scientific motivation to better understand how death is regulated in Mtb-infected macrophages.
4 Host Lipid Mediators Modulate Mtb-Infected Macrophage Death Modality
As bacterial factors can affect the death modality, host factors also determine whether an infected cell undergoes apoptosis or necrosis. In particular, the eicosanoids appear to be critical regulators of apoptosis following Mtb infection [11, 12, 40]. Mtb induces apoptosis and triggers concomitant antimycobacterial activity of human macrophages based on the activity of cytosolic phospholipase A2-γ (cPLA2-γ), a group IV cytosolic PLA2, which catalyzes the release of arachidonic acid from the sn-2 position of membrane phospholipids [22]. Arachidonic acid and its diverse products regulate death in several cell types [41]. For example, arachidonic acid products are second messengers in TNF-induced apoptosis [42], and oxygen radicals, which are produced during lipoxygenation of arachidonic acid, induce ROS production, which can induce cell death [43]. Arachidonic acid also activates sphingomyelinase leading to ceramide production and apoptosis [44]. Which of these mechanisms are important in vivo is not clear [45].
An interesting area of research focuses on the role of the eicosanoids prostaglandin E2 (PGE2) and lipoxin A4 (LXA4) in regulating programmed cell death of macrophages [11, 12, 40]. The cyclooxygenases COX1 and COX2 convert arachidonic acid into the central intermediate PGH2 [46], which is converted by specific synthases into diverse prostanoids [47]. Interaction of these prostanoid species, which includes the prostaglandins PGD2, PGE2, PGF2α, PGI2 and thromboxane, with an array of specific prostanoid receptors affects many cellular pathways. In the case of PGE2, interaction with one of four receptors, EP1, EP2, EP3, and EP4 triggers intracellular pathways that either promote or inhibit inflammation [48]. Importantly, the functional outcome of PGE2 signaling is largely determined by its interaction with its specific receptors [48]. For example, EP1 mediates the elevation of intracellular Ca++. By contrast, EP2, which is involved in joint inflammation and neutrophil recruitment, and EP4, which induces cell migration in tumor invasion, both lead to an increase in intracellular cAMP levels. EP2 signaling results in PKA activation and triggering EP4 activates adenylate cyclase and phosphatidylinositol 3 kinase. Triggering EP3 decreases cAMP concentrations and is known to mediate fever and angiogenesis [48].
D’Avila et al. find that lipid bodies form at distinct cytoplasmic sites following infection of murine macrophages with the attenuated M. bovis strain BCG. These lipid bodies are the site of COX2 activity and PGE2 generation [49]. Indeed, PGE2 production has been a consistent finding following BCG infection of mice and macrophages [50]. We find that macrophages infected with attenuated Mtb also activate the PGE2 production, which prevents necrosis and leads instead to an apoptotic death (Fig. 2) [12]. In contrast, virulent Mtb strains, such as H37Rv or Erdman, only minimally induce the production of PGE2 by macrophages [12]. This raises the possibility that virulent Mtb actively inhibits PGE2 production. Thus, an important strategy that Mtb exploits to avoid death by apoptosis is the subversion of host eicosanoid biosynthetic pathways [11, 12].

Virulent mycobacteria tip the balance between PGE2 and LXA4 production in macrophages. Infection with virulent Mtb induces LXA4, which inhibits the production of COX-2 dependent PGE2. In the absence of PGE2 mitochondria are damaged and membrane microdisruptions remain unrepaired triggering macrophage necrosis. We hypothesize that bacterial inhibition of prostaglandin production is an immune evasion strategy that allows Mtb to avoid the consequences of apoptosis, which leads to early immune response
Lipoxins are also generated from arachidonic acid but require the action of different enzymes including 5- and 15-lipoxygenases [51]. Lipoxins are anti-inflammatory and modulate chemokine and cytokine expression, monocyte trafficking and efferocytosis (phagocytosis of apoptotic cells) [52]. In contrast to attenuated strains, virulent Mtb induces LXA4 production, which inhibits cyclooxygenase-2 production effectively shutting down PGE2 biosynthesis, and provides an explanation for how Mtb inhibits PGE2 production [11, 12]. In a PGE2-poor microenvironment, the macrophage cannot prevent mitochondrial damage nor enable repair of plasma membrane disruptions effectively [5, 11, 12]. Both processes are required to prevent macrophage necrosis and induce apoptotic cell death [11, 12]. Virulent Mtb in pre-necrotic macrophages continues to replicate and once the cells are lysed, propagate the infection by spreading to uninfected macrophages. Thus, the balance of PGE2 and LXA4 production by the infected macrophage regulates the relative amount of apoptosis and necrosis following Mtb infection and has important functional consequences for innate control of intracellular Mtb infection.
Induction of LXA4 by virulent Mtb inhibits PGE2 production and triggers mitochondrial permeability transition (MPT) leading to irreversible mitochondrial damage [12]. By triggering LXA4 production in the host macrophage virulent Mtb inhibits prostanoid production by blocking COX2 mRNA accumulation. By contrast, attenuated Mtb induce only minimal amounts of LXA4 and cause instead production of substantial amounts of PGE2. We found that when macrophage are infected with attenuated Mtb, PGE2 actively suppresses mitochondrial inner membrane perturbation, which is the outcome in an infection with virulent Mtb [12]. Therefore, infection with virulent H37Rv, a PGE2 non-inducer, causes MPT, which is suppressed by reconstitution with PGE2.
Our model that lipoxin production by Mtb-infected macrophages is associated with increased bacterial replication and greater virulence is strengthened by the recent genetic analysis of zebrafish susceptibility to M. marinum [53]. Multiple mutant classes with different innate susceptibilities to M. marinum were identified by Tobin et al. [53]. A hypersusceptible zebrafish mutant was found to map to the LTA4H locus, which encodes leukotriene A4 hydrolase (LTA4H), an enzyme that is required for the final step of leukotriene B4 (LTB4) synthesis. While LTA4H deficiency results in the loss of LTB4 production, addition of LTB4 did not complement the genetic defect nor increase host resistance. In the absence of LTA4H, its substrate, LTA4, accumulates and can lead to redirected eicosanoid synthesis and increase lipoxin synthesis. Therefore, Tobin et al. hypothesize that the increased susceptibility of the zebrafish LTA4H mutant is due to an increase in lipoxin production. The same study presents human genetic data that polymorphisms in the LTA4H gene are associated with susceptibility to pulmonary and meningeal tuberculosis [39, 53]. Thus, from fish to man, eicosanoids appear to play an unexpected role in susceptibility to tuberculosis.
5 Blocking Plasma Membrane Repair
The ESAT-6 secretion system 1 (ESX-1), a specialized Type VII secretion system, is required for the secretion of certain virulence factors including the immunodominant antigens early secreted antigen 6 kilodaltons (ESAT6) and culture filtrate protein 10 (CFP10). Although known to contribute to bacterial virulence, why ESX-1 is required for bacterial survival in the host is unknown. Some data indicate that ESAT6 damages host cell membranes [54, 55]. We hypothesized that disruption of the plasma membrane by Mtb is one mechanism that induces necrosis of the macrophage. Interaction of mycobacteria with the host macrophage results in plasma membrane microdisruptions. Microdisruptions induced by attenuated Mtb are rapidly resealed by plasma membrane repair mechanisms that include recruitment of lysosomal and Golgi apparatus-derived vesicles to the macrophage surface lesions [11, 56, 57]. Lysosomal or Golgi membrane recruitment to the plasma membrane can be assessed by measuring LAMP1 or mannosidase II translocation to the macrophage surface [58, 59]. Active membrane repair prevents necrosis and is required for induction of apoptosis. By contrast, if resealing of the plasma membrane microdisruptions inflicted by the bacteria is inhibited, as is the case with virulent Mtb infection and necrosis ensues.
Ca++ sensors are of crucial importance for the recruitment of both lysosomal and Golgi vesicles to the membrane lesions. Gene silencing of the lysosomal Ca++ sensor synaptotagmin 7 (SYT7) impairs the recruitment of lysosomal, but not Golgi membranes to the cell surface [11, 60]. The recruitment of Golgi-derived vesicles to the cell surface, which occurs independently of lysosomal vesicle recruitment, requires the expression of neuronal calcium sensor 1 (NCS-1), a Ca++ sensor that is particularly abundant in the Golgi [11, 61]. Silencing NCS-1 gene expression, or the use of brefeldin A, a Golgi-specific transport inhibitor, both inhibit translocation of Golgi membranes. These data show that both lysosomal and Golgi membranes are involved in plasma membrane repair and are recruited independently to plasma membrane lesions of infected macrophage.
Plasma membrane resealing is cAMP dependent [62], and addition of forskolin, an activator of adenylate cyclase, results in greater translocation of lysosomal membranes to the cell surface [11]. The protective effect of PGE2 on mitochondrial stability is mediated through the PGE2 receptor EP2 [12] and binding of PGE2 to either EP2 or EP4 causes increased cAMP accumulation [63]. Consistent with this, PGE2 treatment of human macrophages infected with virulent H37Rv reconstitutes repair mediated by lysosomal membranes. By contrast, PGE2 does not affect Golgi mediated repair [11]. Although the protective effects of PGE2 on mitochondria require the EP2 receptor, PGE2-dependent lysosomal membrane translocation requires PI3 K activation, which indicates that signaling through EP4 is involved [11]. These findings have important functional consequences for control of intracellular mycobacterial replication. First, Alox-5 −/− mice (unable to produce LXA4 and other Alox-5-dependent products) survive longer than wild-type (WT) control mice after low dose aerosol infection with virulent Mtb [64]. Conversely, Ptges -/- (unable to produce PGE2) mice succumb earlier than WT mice (unpublished observation: Divangahi, Behar, and Remold). However, as many cell types produce eicosanoids, these results do not provide information about the role of eicosanoids during innate immunity. In experiments using macrophages from Ptges -/- and Alox-5 -/- mice, we found that Ptges -/- macrophages were unable to control intracellular Mtb infection, while Alox5 -/- macrophages limited Mtb replication better than WT macrophages [11]. This phenotype is replicated in vivo when Mtb infected Ptges -/-, Alox5 -/- and WT macrophages were adoptively transferred into the lungs of V(D)J recombination-activating protein 1-deficient (Rag -/-) recipient mice. Recipient mice that received infected Alox5 -/- macrophages had a substantially lower mycobacterial lung burden than recipients that received infected Ptges -/- or WT macrophages. Since Rag1 -/- mice lack B and T cells, the greater capacity of Rag1 -/- mice to control pulmonary infection following transfer of Mtb infected Alox5 -/- macrophages must be attributed to an intrinsic property of Alox5 -/- macrophages or a unique interaction between Alox5 -/- macrophages and the innate immune system [11].
One conceivable explanation for the role of PGE2 in fostering membrane repair is that PGE2 is required for the generation of SYT7, the lysosomal Ca++ sensor essential for plasma membrane repair. Virulent Mtb stimulate LXA4 production in macrophages, which inhibits PGE2 production by down regulation of COX2 mRNA accumulation [12]. Indeed we find that in contrast to LAMP1 expression, SYT7 transcription is specifically induced by PGE2. Likewise, Alox5 -/- macrophages infected with virulent Mtb express more SYT7 than WT or Ptges -/- macrophages [11]. Although it is not known how PGE2 modulates SYT7 expression, collectively these data indicate that PGE2 is an essential mediator of SYT7 expression and is therefore of critical importance for the prevention of necrosis and induction of apoptosis. Cumulatively, these studies show that the balance of PGE2 and LXA4 production by infected macrophages affects the outcome of infection in the microenvironment of the lung (Fig. 2).
6 The Fate of Mtb-Infected Macrophages Determines Cross-Presentation of Mycobacterial Antigens
As elegantly discussed in Chap. 8 by Dr. Behar, an alternate possibility is that phagocytosis of dying infected macrophages leads to acquisition of bacterial antigens by DC, as has been shown for influenza and Listeria [65, 66]. The relevance of these processes to mycobacterial antigen presentation was first investigated by Schaible et al. [67]. Extracellular vesicles derived from infected DC and macrophages were identified that were free of viable bacteria but contained mycobacterial lipids and proteins. The origin of these vesicles was not entirely clear, but they appear to be apoptotic blebs or possibly exosomes. While infected macrophages were not efficient to directly stimulate CD8+ T cells, their co-culture with uninfected DC led to the transfer of mycobacterial antigens to DC, which became competent to cross-present the antigens to CD8+ T cells. Presentation was TAP-1-dependent and required an intact class I MHC pathway. Thus, the antigenic cargo contained in these vesicles could be cross-presented by DC to CD8+ T cells. As these studies were done with previously activated T cells, the observed T cell activation was not true cross-priming but would be more accurately categorized as cross-presentation. Nevertheless, the uptake of antigen-containing vesicles by DC provides a mechanism by which uninfected DC can acquire Mtb antigens and prime naïve T cells.
Winau et al. used similar vesicles purified from BCG-infected murine macrophages to immunize mice [68]. Again, the purified apoptotic bodies contained bacterial antigens but no bacteria. CD8+ T cell priming was observed and required an intact class I MHC pathway. Successful T cell priming was associated with DC homing to the tissue sites where the purified vesicles were injected. Interestingly, initiation of the endosomal processing pathway abrogated CD8+ T cell priming—a feature that may be unique to cross-presentation of class I MHC-restricted peptides. The generation of CD8+ T cell responses in naïve mice indicates that true cross-priming occurred. Remarkably, not only did a CD8+ T cell response develop, but also vaccination with the vesicles generated immunity that protected mice against challenge with virulent Mtb.
The studies by Schaible and Winau provide the foundation for the “Detour Model” as proposed by Kaufmann [69]. They convincingly show that the mycobacterial antigens contained in purified vesicles are taken up by both human and murine DC and enter the class I MHC pathway. However, these studies fall short of demonstrating whether apoptosis of infected macrophage is required for the transfer of antigens to DC and whether this process occurs in vivo indicating physiological significance. Additionally, the apoptotic vesicles used in the studies by Winau et al. and Schaible et al. were derived from BCG-infected macrophages [68, 69] and it is not clear whether infection of macrophage with wild-type virulent Mtb would lead to apoptosis and enhanced T cell immunity. Finally, while immunization with purified vesicles cross-primes antigen-specific T cells, it is uncertain whether the generation of vesicles from infected macrophages is required for CD8+ T cell priming in vivo. The finding that Mtb infected DC traffic from the lung to the regional LN with kinetics mirroring T cell priming could be consistent with Mtb-infected DC directly priming Mtb-specific T cells and could indicate the existence of a priming pathway independent of the “Detour Pathway” [69]. Thus, the role of apoptosis and cross-priming in the generation of adaptive immunity during virulent Mtb infection remained an important unanswered question. To confirm the existence of these pathways and to begin to elucidate their relevance, a better understanding of the host factors regulating cell death during Mtb infection was required.
7 The Role of Eicosanoids in Apoptosis-Mediated Cross-Presentation
Eicosanoids have been identified as important host lipid mediators that regulate inflammation and susceptibility following mycobacterial infection. One effect of eicosanoids is the regulation of cell death in both human and murine macrophages infected with Mtb [11, 12]. As discussed above, prostanoids such as the host lipid mediator PGE2 induce plasma membrane repair and prevent mitochondrial damage; together these events protect infected macrophages against necrosis and instead promote apoptosis. Importantly, products of 5-lipoxygenase including LXA4 are produced by macrophages after infection with virulent Mtb. LXA4 inhibits COX-2 activity, which shuts down prostaglandin synthesis. As predicted, macrophages from mice that lack 5-lipoxygenase, produce prostaglandins even after infection with virulent Mtb and undergo more apoptosis than necrosis. Interestingly, Alox5 -/- mice are more resistant to Mtb. Studies from Bafica et al. found that a more pronounced Th1 cytokine response is detected in the lungs of infected Alox5 -/- mice compared to WT controls mice [64].
In order to determine whether apoptotic macrophages contribute to adaptive immunity, we established a novel adoptive transfer model in which macrophages from wild-type or knockout mice were infected in vitro with Mtb and then transferred by intra-tracheal instillation into normal recipient mice. This strategy was used to determine whether the macrophage genotype influences the T cell response and control of infection [40]. By using knockout macrophages that are prone to undergo either apoptosis (e.g., Alox5 -/-) or necrosis (e.g., Ptges -/-) following infection, we determined how these two different cellular fates alter the course of infection in vivo. One advantage of this adoptive transfer infection model is that the development of tuberculosis occurs in a developmentally normal host with an intact immune system, which avoids the pitfalls of studying Mtb infection in knockout mice in which the genetic lesion affects multiple cell types and physiological processes.
We used the CD8+ T cell response to TB10.4, a mycobacterial antigen that elicits an immunodominant response following low dose aerosol Mtb infection, to track the CD8+ T cell response following intra-tracheal transfer of Mtb-infected macrophages [40]. An earlier TB10.4-specific CD8+ T cell response was detected both in the draining pulmonary LN and in the lung following transfer of pro-apoptotic macrophages compared to wild-type macrophages. Importantly, the cellular fate of the infected macrophages was crucial; pre-treatment of the pro-apoptotic macrophages with inhibitors of caspase 8 and caspase 9, which prevented apoptosis of the infected macrophages, abrogated the enhancement of the CD8+ T cell response [40].
To determine how the Mtb-infected macrophages enhanced the CD8+ T cell response, the infected macrophage adoptive transfer model was adapted for use with OT-I TCR-transgenic mice (carry a transgenic CD8 T cell receptor (TCR) for the MHC class I-restricted OVA257–264 peptide), so early events in T cell priming could be easily assessed. Similar to the intra-tracheal adoptive transfer of Mtb-infected macrophages, OT-I CD8+ T cell priming was detected earlier after the transfer of Mtb-infected OVA-pulsed Alox5 -/- macrophages compared to wild-type macrophages. Importantly, the infected macrophages did not directly activate CD8+ T cells; instead, T cell priming required endogenous DC, since DC depletion abrogated OT-I CD8+ T cell expansion. Similar to the results of Winau et al. [68], CD8+ T cell priming required TAP-1 and an intact class I MHC pathway. These experiments show that CD8+ T cell priming requires cross-presentation of antigen acquired by DC from apoptotic Mtb-infected macrophage via the detour pathway. In addition, after the transfer of Mtb-infected pro-apoptotic Alox5 -/- macrophages, not only was there an earlier and more robust Mtb-specific CD8+ T cell response, but the CD4+ T cell response to ESAT6 and Ag85B was also enhanced [40]. This may not be too surprising if DC phagocytosis of apoptotic vesicles transfers their cargo of Mtb antigens to the endocytic system, which intersects with the MHC II processing pathway. However, the mechanisms that govern this potential transfer have yet to be elucidated. Thus, while apoptosis has been directly linked to increased CD8+ T cell responses via cross-presentation, it also enhances class II MHC-restricted antigen presentation. This has important implications for the finding that vaccination with apoptosis-inducing bacterial vaccines or apoptotic vesicles induces protection against virulent Mtb: namely, the protective immunity elicited may be due to a combination of Mtb-specific CD4+ and CD8+ T cells.
Moreover, the pro-apoptotic mutants of Mtb prime a greater T cell response and enhance host control of infection [7]. This has generated considerable interest in whether pro-apoptotic mutants of Mtb could be used as a vaccine strategy. For example, vaccination with attenuated BCG or Mtb that induce greater macrophage apoptosis or with purified apoptotic bodies [68] may stimulate an enhanced T cell response. Collectively, these studies have provided important evidence that during pulmonary Mtb infection apoptosis of infected macrophages: (1) leads to innate control of early bacterial growth; and (2) acts as a reservoir of antigen that facilitates initiation of acquired T cell immunity via cross-priming by DC.
8 Conclusions
The finding that macrophages infected with virulent Mtb undergo necrosis while macrophages infected with attenuated mutant strains of Mtb undergo apoptosis, suggests that wild-type Mtb actively inhibits apoptosis. This forms the foundation for the concept that apoptosis is an innate macrophage defense mechanism. Apoptosis is associated with a reduction in the viability of intracellular Mtb and provides an important link to the establishment of T cell immunity. Investigation of the interaction between Mtb and macrophages finds that three distinct mechanisms contribute to macrophages necrosis. First, Mtb inhibits plasma membrane repair. Second, virulent Mtb causes inner mitochondrial membrane damage. Third, Mtb inhibits generation of the apoptotic cellular envelope. These three effects predispose the infected macrophages to necrosis. In part, these events occur because virulent Mtb inhibits the production of PGE2, a prostaglandin that is important for stimulation of membrane repair and protection of the mitochondrion. However, it is also important to note that some investigators have found that PGE2 can impair immunity to other bacterial infections [70, 71] or Influenza viral infection (Divangahi, unpublished observation). Thus, how virulent Mtb subvert eicosanoid biosynthesis to alter the death modality of macrophages to foil both innate and adaptive immunity is an important area for future investigation. Given our capacity to manipulate eicosanoid-pathways, a better understanding of how their regulation is altered by mycobacteria may lead to novel approach to intervene therapeutically as well as to develop immunomodulatory strategies that can enhance vaccine efficacy.
References
WHO (2012) World health organization: global tuberculosis control 2010
Behar SM, Martin CJ, Nunes-Alves C, Divangahi M, Remold HG (2011) Lipids, apoptosis, and cross-presentation: links in the chain of host defense against Mycobacterium tuberculosis. Microbes Infect 13:749–756
Dye C, Williams BG (2010) The population dynamics and control of tuberculosis. Science 328:856–861
Kaufmann SH (2001) How can immunology contribute to the control of tuberculosis? Nat Rev Immunol 1:20–30
Chen M, Gan H, Remold HG (2006) A mechanism of virulence: virulent Mycobacterium tuberculosis strain H37Rv, but not attenuated H37Ra, causes significant mitochondrial inner membrane disruption in macrophages leading to necrosis. J Immunol 176:3707–3716
Keane J, Remold HG, Kornfeld H (2000) Virulent Mycobacterium tuberculosis strains evade apoptosis of infected alveolar macrophages. J Immunol 164:2016–2020
Hinchey J et al (2007) Enhanced priming of adaptive immunity by a proapoptotic mutant of Mycobacterium tuberculosis. J Clin Invest 117:2279–2288
Velmurugan K et al (2007) Mycobacterium tuberculosis nuoG is a virulence gene that inhibits apoptosis of infected host cells. PLoS Pathog 3:e110
Molloy A, Laochumroonvorapong P, Kaplan G (1994) Apoptosis, but not necrosis, of infected monocytes is coupled with killing of intracellular bacillus Calmette-Guerin. J Exp Med 180:1499–1509
Fratazzi C, Arbeit RD, Carini C, Remold HG (1997) Programmed cell death of Mycobacterium avium serovar 4-infected human macrophages prevents the mycobacteria from spreading and induces mycobacterial growth inhibition by freshly added, uninfected macrophages. J Immunol 158:4320–4327
Divangahi M et al (2009) Mycobacterium tuberculosis evades macrophage defenses by inhibiting plasma membrane repair. Nat Immunol 10:899–906
Chen M et al (2008) Lipid mediators in innate immunity against tuberculosis: opposing roles of PGE2 and LXA4 in the induction of macrophage death. J Exp Med 205:2791–2801
Constance et al (2012) Efferocytosis is an innate antibacterial mechanism. Cell Host Microbe 12:289
Bergsbaken T, Fink SL, Cookson BT (2009) Pyroptosis: host cell death and inflammation. Nat Rev Microbiol 7:99–109
Vandenabeele P, Galluzzi L, Vanden BT, Kroemer G (2010) Molecular mechanisms of necroptosis: an ordered cellular explosion. Nat Rev Mol Cell Biol 11:700–714
Duprez L, Wirawan E, Vanden BT, Vandenabeele P (2009) Major cell death pathways at a glance. Microbes Infect 11:1050–1062
Cohen JJ (1993) Apoptosis. Immunol Today 14:126–130
Fadok VA et al (1992) Exposure of phosphatidylserine on the surface of apoptotic lymphocytes triggers specific recognition and removal by macrophages. J Immunol 148:2207–2216
Fadok VA et al (2000) A receptor for phosphatidylserine-specific clearance of apoptotic cells. Nature 405:85–90
Henson PM, Tuder RM (2008) Apoptosis in the lung: induction, clearance and detection. Am J Physiol Lung Cell Mol Physiol 294:L601–L611
Krysko DV, D’Herde K, Vandenabeele P (2006) Clearance of apoptotic and necrotic cells and its immunological consequences. Apoptosis 11:1709–1726
Duan L, Gan H, Arm J, Remold HG (2001) Cytosolic phospholipase A2 participates with TNF-alpha in the induction of apoptosis of human macrophages infected with Mycobacterium tuberculosis H37Ra. J Immunol 166:7469–7476
Christofferson DE, Yuan J (2010) Necroptosis as an alternative form of programmed cell death. Curr Opin Cell Biol 22:263–268
Thornberry NA, Lazebnik Y (1998) Caspases: enemies within. Science 281:1312–1316
Balcewicz-Sablinska MK, Keane J, Kornfeld H, Remold HG (1998) Pathogenic Mycobacterium tuberculosis evades apoptosis of host macrophages by release of TNF-R2, resulting in inactivation of TNF-alpha. J Immunol 161:2636–2641
Green DR, Kroemer G (2004) The pathophysiology of mitochondrial cell death. Science 305:626–629
Kim JS, He L, Lemasters JJ (2003) Mitochondrial permeability transition: a common pathway to necrosis and apoptosis. Biochem Biophys Res Commun 304:463–470
Chipuk JE, Green DR (2008) How do BCL-2 proteins induce mitochondrial outer membrane permeabilization? Trends Cell Biol 18:157–164
Bossy-Wetzel E, Newmeyer DD, Green DR (1998) Mitochondrial cytochrome c release in apoptosis occurs upstream of DEVD-specific caspase activation and independently of mitochondrial transmembrane depolarization. EMBO J 17:37–49
Ricci JE et al (2004) Disruption of mitochondrial function during apoptosis is mediated by caspase cleavage of the p75 subunit of complex I of the electron transport chain. Cell 117:773–786
Martinvalet D, Dykxhoorn DM, Ferrini R, Lieberman J (2008) Granzyme A cleaves a mitochondrial complex I protein to initiate caspase-independent cell death. Cell 133:681–692
Baines CP et al (2005) Loss of cyclophilin D reveals a critical role for mitochondrial permeability transition in cell death. Nature 434:658–662
Gan H et al (2005) Enhancement of antimycobacterial activity of macrophages by stabilization of inner mitochondrial membrane potential. J Infect Dis 191:1292–1300
Connern CP, Halestrap AP (1992) Purification and N-terminal sequencing of peptidyl-prolyl cis-trans isomerase from rat liver mitochondrial matrix reveals the existence of a distinct mitochondrial cyclophilin. Biochem J 284(2):381–385
Maertzdorf J et al (2011) Human gene expression profiles of susceptibility and resistance in tuberculosis. Genes Immun 12:15–22
Abebe M et al (2010) Expression of apoptosis-related genes in an Ethiopian cohort study correlates with tuberculosis clinical status. Eur J Immunol 40:291–301
Herb F et al (2008) ALOX5 variants associated with susceptibility to human pulmonary tuberculosis. Hum Mol Genet 17:1052–1060
Tobin DM et al (2010) The lta4 h locus modulates susceptibility to mycobacterial infection in zebrafish and humans. Cell 140:717–730
Tobin DM et al (2012) Host genotype-specific therapies can optimize the inflammatory response to mycobacterial infections. Cell 148:434–446
Divangahi M, Desjardins D, Nunes-Alves C, Remold HG, Behar SM (2010) Eicosanoid pathways regulate adaptive immunity to Mycobacterium tuberculosis. Nat Immunol 11:751–758
Wolf LA, Laster SM (1999) Characterization of arachidonic acid-induced apoptosis. Cell Biochem Biophys 30:353–368
Chang DJ, Ringold GM, Heller RA (1992) Cell killing and induction of manganous superoxide dismutase by tumor necrosis factor-alpha is mediated by lipoxygenase metabolites of arachidonic acid. Biochem Biophys Res Commun 188:538–546
Peterson DA et al (1988) Polyunsaturated fatty acids stimulate superoxide formation in tumor cells: a mechanism for specific cytotoxicity and a model for tumor necrosis factor? Biochem Biophys Res Commun 155:1033–1037
Jayadev S, Linardic CM, Hannun YA (1994) Identification of arachidonic acid as a mediator of sphingomyelin hydrolysis in response to tumor necrosis factor alpha. J Biol Chem 269:5757–5763
Finstad HS et al (1998) Cell proliferation, apoptosis and accumulation of lipid droplets in U937–1 cells incubated with eicosapentaenoic acid. Biochem J 336(2):451–459
Rocca B, FitzGerald GA (2002) Cyclooxygenases and prostaglandins: shaping up the immune response. Int Immunopharmacol 2:603–630
Murakami M et al (2000) Regulation of prostaglandin E2 biosynthesis by inducible membrane-associated prostaglandin E2 synthase that acts in concert with cyclooxygenase-2. J Biol Chem 275:32783–32792
Sugimoto Y, Narumiya S (2007) Prostaglandin E receptors. J Biol Chem 282:11613–11617
D’Avila H et al (2006) Mycobacterium bovis bacillus Calmette-Guerin induces TLR2-mediated formation of lipid bodies: Intracellular domains for eicosanoid synthesis in vivo. J Immunol 176:3087–3097
Almeida PE et al (2009) Mycobacterium bovis bacillus Calmette-Guerin infection induces TLR2-dependent peroxisome proliferator-activated receptor gamma expression and activation: functions in inflammation, lipid metabolism, and pathogenesis. J Immunol 183:1337–1345
Levy BD, Clish CB, Schmidt B, Gronert K, Serhan CN (2001) Lipid mediator class switching during acute inflammation: signals in resolution. Nat Immunol 2:612–619
Serhan CN, Chiang N, Van Dyke TE (2008) Resolving inflammation: dual anti-inflammatory and pro-resolution lipid mediators. Nat Rev Immunol 8:349–361
Tobin DM et al (2010) The lta4 h locus modulates susceptibility to mycobacterial infection in zebrafish and humans. Cell 140:717–730
Smith J et al (2008) Evidence for pore formation in host cell membranes by ESX-1-secreted ESAT-6 and its role in Mycobacterium marinum escape from the vacuole. Infect Immun 76:5478–5487
de Jonge MI et al (2007) (2007) ESAT-6 from Mycobacterium tuberculosis dissociates from its putative chaperone CFP-10 under acidic conditions and exhibits membrane-lysing activity. J Bacteriol 189:6028–6034
Roy D et al (2004) A process for controlling intracellular bacterial infections induced by membrane injury. Science 304:1515-1518
Togo T, Alderton JM, Bi GQ, Steinhardt RA (1999) The mechanism of facilitated cell membrane resealing. J Cell Sci 112(5):719–731
Granger BL et al (1990) Characterization and cloning of lgp110, a lysosomal membrane glycoprotein from mouse and rat cells. J Biol Chem 265:12036–12043
Novikoff PM, Tulsiani DR, Touster O, Yam A, Novikoff AB (1983) Immunocytochemical localization of alpha-D-mannosidase II in the Golgi apparatus of rat liver. Proc Natl Acad Sci U S A 80:4364–4368
Martinez I et al (2000) Synaptotagmin VII regulates Ca(2 +)-dependent exocytosis of lysosomes in fibroblasts. J Cell Biol 148:1141–1149
Burgoyne RD, O’Callaghan DW, Hasdemir B, Haynes LP, Tepikin AV (2004) Neuronal Ca2 + -sensor proteins: multitalented regulators of neuronal function. Trends Neurosci 27:203–209
Togo T, Alderton JM, Steinhardt RA (2003) Long-term potentiation of exocytosis and cell membrane repair in fibroblasts. Mol Biol Cell 14:93–106
Regan JW (2003) EP2 and EP4 prostanoid receptor signaling. Life Sci 74:143–153
Bafica A et al (2005) Host control of Mycobacterium tuberculosis is regulated by 5-lipoxygenase-dependent lipoxin production. J Clin Invest 115:1601–1606
Albert ML (2004) Death-defying immunity: do apoptotic cells influence antigen processing and presentation? Nat Rev Immunol 4:223–231
Yrlid U, Wick MJ (2000) Salmonella-induced apoptosis of infected macrophages results in presentation of a bacteria-encoded antigen after uptake by bystander dendritic cells. J Exp Med 191:613–624
Schaible UE et al (2003) Apoptosis facilitates antigen presentation to T lymphocytes through MHC-I and CD1 in tuberculosis. Nat Med 9:1039–1046
Winau F et al (2006) Apoptotic vesicles crossprime CD8 T cells and protect against tuberculosis. Immunity 24:105–117
Winau F, Kaufmann SH, Schaible UE (2004) Apoptosis paves the detour path for CD8 T cell activation against intracellular bacteria. Cell Microbiol 6:599–607
Aronoff DM et al (2009) E-prostanoid 3 receptor deletion improves pulmonary host defense and protects mice from death in severe Streptococcus pneumoniae infection. J Immunol 183:2642–2649
Medeiros AI, Serezani CH, Lee SP, Peters-Golden M (2009) Efferocytosis impairs pulmonary macrophage and lung antibacterial function via PGE2/EP2 signaling. J Exp Med 206:61–68
Acknowledgments
M.D. is supported by the Canadian Institute of Health Research-New Investigator Award. Work in his laboratory is supported by the Canadian Institute of Health Research (CIHR) and The Natural Sciences and Engineering Research Council of Canada (NSERC).
Author information
Authors and Affiliations
Corresponding authors
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2013 Springer Science+Business Media New York
About this chapter
Cite this chapter
Divangahi, M., Behar, S.M., Remold, H. (2013). Dying to Live: How the Death Modality of the Infected Macrophage Affects Immunity to Tuberculosis. In: Divangahi, M. (eds) The New Paradigm of Immunity to Tuberculosis. Advances in Experimental Medicine and Biology, vol 783. Springer, New York, NY. https://doi.org/10.1007/978-1-4614-6111-1_6
Download citation
DOI: https://doi.org/10.1007/978-1-4614-6111-1_6
Published:
Publisher Name: Springer, New York, NY
Print ISBN: 978-1-4614-6110-4
Online ISBN: 978-1-4614-6111-1
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)