
Abstract
The nonprotein amino acid taurine has been shown to counteract the negative effects of a high-fructose diet in rats with regard to insulin resistance and dyslipidemia. Here we examined the long-term (26 weeks) effects of oral taurine supplementation (2% in the drinking water) in fructose-fed Wistar rats.
The combination of fructose and taurine caused a significant increase in fasting glucose compared to the control diet without changing hepatic phosphoenol pyruvate carboxykinase mRNA levels. The combination of fructose and taurine also improved glucose tolerance compared to control. Neither a high-fructose diet nor taurine supplementation induced significant changes in body weight, body fat or total calorie intake, fasting insulin levels, HOMA-IR, or insulin-induced Akt phosphorylation in skeletal muscle.
Fructose alone caused a decrease in liver triglyceride content, with taurine supplementation preventing this. There was no effect of long-term fructose diet and/or taurine supplementation on plasma triglycerides, plasma nonesterified fatty acids, as well as plasma HDL, LDL, and total cholesterol.
In conclusion, the study suggests that long-term taurine supplementation improves glucose tolerance and normalize hepatic triglyceride content following long-term fructose feeding. However, as the combination of taurine and fructose also increased fasting glucose levels, the beneficial effect of taurine supplementation towards amelioration of glucose intolerance and insulin resistance may be questionable.
Access provided by Autonomous University of Puebla. Download conference paper PDF
Similar content being viewed by others


Keywords
- Fasting Glucose
- Improve Glucose Tolerance
- Phosphoenol Pyruvate Carboxykinase
- Taurine Supplementation
- Taurine Level
These keywords were added by machine and not by the authors. This process is experimental and the keywords may be updated as the learning algorithm improves.
1 Introduction
Fructose has recently been hypothesized to be one of the main effectors driving the increased incidence of the metabolic syndrome in countries that have adopted the Western diet (Lustig et al. 2012; Stanhope 2012).
Taurine (2-aminoethanesulfonic acid) is a nonprotein semi-essential amino acid found in large amounts in both tissues (5–50 mM) and plasma (50–100 μM) and known to have a number of physiological functions, such as conjugation with bile acids, intracellular osmolyte for volume regulation, and antioxidant properties (Hansen 2001).
In 1935 it was demonstrated by Ackermann et al. that taurine had a beneficial effect upon glucose homeostasis in diabetic patients (Ackermann and Heinsen 1935). Taurine stimulates glycolysis and glycogenesis, thus presenting insulin-like activity (Dokshina et al. 1976). Furthermore, it is evident that plasma and tissue taurine levels change in a variety of metabolic pathophysiological conditions (Hansen 2001) and taurine concentration in plasma has been demonstrated to decrease in type 2 diabetic patients (De Luca et al. 2001). This reduction in plasma taurine levels has also been seen in diabetic animal models (Franconi et al. 1995, 1996). An increase in taurine intake has been shown to decrease plasma glucose in type 1 diabetic patients (Elizarova and Nedosugova 1996). However, in studies examining prediabetic and diabetic complications, differences in fasting plasma glucose and insulin after taurine supplementation were not reported (Franconi et al. 1996; Nishimura et al. 2002). Together these data indicate that taurine may have a protective role in hyperglycemia, something which is corroborated by recent literature reviews showing an interaction between taurine and diabetes (Franconi et al. 2006; Kim et al. 2007).
Several animal studies have shown that taurine prevents fructose-induced insulin resistance, as judged by both oral glucose tolerance tests and a normalization of total insulin-induced kinase activity. Furthermore, a high-fructose diet in rats has been reported to decrease plasma and liver taurine levels (El Mesallamy et al. 2010; Nandhini et al. 2004, 2005; Nandhini and Anuradha 2002). In a rat model of non-insulin-dependent diabetes, the Otsuka Long Evans Tokushima Fatty (OLETF) rat (Kawano et al. 1992) taurine supplementation caused a decrease in serum triglycerides and cholesterol, with no effect on body weight and abdominal fat mass (Harada et al. 2004; Nakaya et al. 2000).
It has been speculated that the effect of taurine on glucose homeostasis could involve a lipid lowering effect (El Mesallamy et al. 2010) and/or enhancement of insulin signaling (Nandhini et al. 2005). However, the mechanism(s) by which taurine affects glucose homeostasis is largely unknown and no long-term studies of this effect have been performed. Thus, we performed a long-term study in fructose-fed Wistar rats examining fasting glucose and lipid parameters as well as glucose tolerance with or without taurine supplementation.
2 Methods
2.1 Animals, Study Design, and Diet
All experimental procedures complied with guidelines laid down by The Danish Animal Experiments Inspectorate and by the local animal facility at the University of Copenhagen, Denmark. Male Wistar Hannover GALAS (HanTac:WH) rats (Taconic, Ejby, Denmark) were fed ad libitum, housed two rats per cage, and kept at a 12-h light/dark cycle. Animals as well as food and water intake were measured biweekly.
Twenty-four 5-week-old rats were fed four different diet regimes for 26 weeks. The rats were divided randomly into four groups of six, ensuring a similar start weight of each group, and fed a control diet or a high-fructose diet with 10% (w/v) fructose (Sigma-Aldrich, St. Louis, MO, USA) in the drinking water either with or without 2% (w/v) taurine supplementation. The carbohydrate control diet contained 68% energy from glucose (corn starch or maltodextrin), 21% casein, and 12% corn oil (Research Diets, New Brunswick, NJ, USA). The fructose-rich diet contained 50% energy from fructose, 18% energy from glucose (corn starch or maltodextrin), 21% casein, and 12% energy from corn oil (Research Diets). As the animals receiving the fructose diet also obtained fructose from the drinking water, approximately 62% of their energy intake was derived from fructose (data not shown).
Fructose and taurine were supplemented in drinking water by dissolving fructose and/or taurine directly in the water used in the animal facility. The taurine used for supplementation was of the chemically synthesized variety (Sigma-Aldrich).
An Echo MRI 4-in-1 for Small Animals (EchoMRI, Houston, TX, USA) was used to monitor total body fat mass of unanesthetized animals. Values are averages of duplicate measurements.
2.2 Oral Glucose Tolerance Test
Animals were fasted for 16–18 h before an oral glucose tolerance test (OGTT) was carried out. The required amount of glucose (2 g per kg) was given as a solution (45% w/v) by gavage. Blood glucose levels were determined before glucose administration (time −30 min) in blood from the tail vein followed by a blood sample of 100 μl for plasma fasting insulin. Blood glucose levels were furthermore determined in blood from tail vein at 0, 30, 60, 90, and 120 min after glucose administration using two different automated Accu-Check Glucometers (Roche, Basal, Switzerland), thus measuring blood glucose at all time points in duplicate. The area under the curve (AUC) of the glucose tolerance test measurements was calculated from between 0 and 120 min (following glucose administration) from baseline (before glucose bolus).
2.3 Insulin Signaling
Overnight fasted rats were sedated with a mixture of Hypnorm (active ingredients fentanyl and fluanisone at a concentration of 0.079 and 2.5 mg/ml, respectively) and Dormicum (active ingredient midazolam at a concentration of 1.25 mg/ml) in water given as 0.3 ml per 100 g of body weight. Quadriceps muscle was dissected from one leg, and the rat was cut open. A liver lobe was removed by disconnecting the blood supply using suture. An 18 G catheter was inserted into vena portae for injection of 4.2 nmol/kg of insulin in a 2 ml volume of Krebs–Henseleit bicarbonate buffer (KHB) (an aqueous solution at pH 7.4 of 4.74 mM KCl, 1.18 mM KH2PO4, 1.18 mM MgSO4, 118.5 mM NaCl, 24.7 mM NaHCO3, 2.5 mM CaCl2) with 0.1% BSA (free from free fatty acids, Sigma) to directly measure insulin signaling in vivo. After 5 min of insulin stimulation, another liver lobe and the other quadriceps muscle were dissected. All tissues were quick frozen in liquid nitrogen and stored at −80°C for further analysis.
2.4 Quantitative Real-Time PCR
RNA was extracted from liver tissue using Qiazol (Qiagen, Valencia, CA, USA) as described by the manufacturer. In short, samples were homogenized in Qiazol with 5 mm steal beads, using a Qiagen Tissuelyzer (Qiagen) three times for 1 min at 30 Hz. Upon lysis, RNA was extracted with chloroform (Sigma) and precipitated using isopropanol (Sigma). Finally, precipitated RNA was washed with 75% ethanol and dissolved in 30 μl diethyl pyrocarbonate-treated water (DEPC water) (Sigma). Quality of RNA was assured using a Bioanalyzer (Agilent, Santa Clara, CA, USA) as described by the manufacturer. RNA concentrations were measured at a Saveen Werner Nanodrop Spectrophometer ND-1000 (Saveen Werner, Limhamn, Sweden).
Total RNA was mixed (at a concentration >0.15 μg/μl for a total of 2 μg RNA in 20 μl volume) with reverse transcriptase, random hexamer primers and nucleotides, and cDNA synthesis performed using the High Capacity cDNA Reverse Transcription Kit with RNase inhibitor (Applied Biosystems, Carlsbad, CA, USA) according to the manufacturer’s instruction employing an Eppendorf Thermo cycler (Applied Biosystems). A non-reverse transcription and a non-template control reaction were included in all cases and the cDNA product was diluted to a volume of 200 μl and stored at −20°C until further analysis.
Amplification mixtures were amplified using a SYBR Green mastermix (Applied Biosystems) according to standard conditions ((95°C 10 min) × 1, (95°C 15 s, 60°C 1 min, 95°C 15 s, 60°C 15 s, 95°C 15 s) × 50 cycles in a total volume of 10 μl with a melting curve from 60 to 100°C) in 384-well plates in triplicate on an ABI PRISM 7900 sequence detector (Applied Biosystems). Target gene mRNA levels were normalized to TATA-binding protein (TBP) mRNA levels.
Primers: PCK1-forward: 5′-TGGAGACCACAGGATGAGGAACCG-3′, PCK1-reverse: 5′-AATGGGACATTGGCTGGCAGGG-3′, TBP-forward: 5′-CCCACCAGCAGTTCAGTA-3′, TBP-reverse: 5′-CAATTCTGGGTTTGATCATTC-3′.
2.5 Western Blot
Muscle biopsies were mixed with a modified RIPA lysis buffer with protease and phosphatase inhibitors (an aqueous solution at pH 7.4 of 50 mM Tris–HCl, 150 mM NaCl, 1 mM EDTA, 1 mM EGTA, 0.25% deoxycholate, 1% Triton X-100, 1 μg/ml pepstatin A, 1 mM Na3VO4, 1 mM NaF, phosphatase inhibitor 1 and 2 (Sigma), and a complete protease inhibitor cocktail (Roche)). Tissue samples were mechanically homogenized using the Tissuelyzer (Qiagen) for 1 min at 30 Hz and 15-min incubation on ice, repeated three times. Samples were rotated end over for 1 h at 4°C before being centrifuged for 30 min at 20,000 × g at 4°C and the supernatant transferred to a new tube. Protein concentration was determined using the Bio-Rad DC kit (Bio-Rad) with BSA used as standard. All determinations were done in triplicate.
Twenty-five micrograms of protein lysate per lane were boiled in NuPAGE LDS Sample Buffer (Invitrogen), separated on 4–12% Bis-Tris NuPAGE gels (Invitrogen), and transferred to PVDF membranes (Hybond-P, GE Healthcare, Little Chalfont, United Kingdom). Membranes were then blocked for 1 h at room temperature in blocking buffer (Tris-buffered saline with 0.1% Tween-20 and 5% Skim milk (Sigma-Aldrich, St. Louis, MO, USA)). The membranes were then incubated overnight at 4°C in 5% BSA in Tris-buffered saline with 0.1% Tween-20 containing a primary antibody against phospho-AKT (#4060, Cell Signaling Technology, Inc., Danvers, MA) at a 1:1,000 dilution. The membranes were then washed three times for 5 min in wash buffer (Tris-buffered saline with 0.1% Tween-20) and incubated for 1 h at room temperature with secondary antibody (Goat Anti-rabbit HRP, P0448, Dako, Glostrup, Denmark) at a 1:10,000 dilution in blocking buffer, followed by three-times 5-min washes in wash buffer. The protein bands were detected using ECL (Pierce ECL substrate, Pierce) and quantified using a CCD image sensor (Syngene G:BOX Cemi XR5, VWR, Copenhagen, Denmark) and ImageJ software (Abramoff et al. 2004) was used to estimate the optical density of the bands. The above method was also used to quantify total AKT(pan) (#4691, Cell Signaling Technology). The amount of p-AKT was expressed as arbitrary units relative to total AKT content.
2.6 Biochemical Analysis
Fasting plasma insulin concentrations were measured using the ultrasensitive rat insulin ELISA kit as described by the manufacturer (Mercodia, Uppsala, Sweden). Tissue insulin sensitivity was evaluated by the previously validated (Bonora et al. 2000) homeostasis model assessment (HOMA) using the HOMA index of insulin resistance (HOMA-IR) = fasting insulin (mU/L) × fasting glucose (mM)/22.5 (Matthews et al. 1985).
Plasma nonesterified fatty acids (NEFA) were measured at 546 nm using NEFA-HR (2) Kit according to instructions from the manufacturer (WAKO, Richmond, VA, USA) at 37°C.
Triglycerides were measured in 10 μl plasma or 50 mg liver tissue, hydrolyzed in 0.5 M KOH/85% ethanol at 60°C for 30 min (Kates 1986). After cooling, MgSO4 was added to 0.1 M and samples were vortexed and centrifuged at 16,000 × g for 20 min at 4°C. Glycerol was measured spectrophotometrically at 340 nm as described (Wieland 1984).
High-density lipoprotein (HDL) cholesterol and low-density lipoprotein (LDL) cholesterol in rat plasma were measured at 450 nm using an ELISA Kit according to instructions from the manufacturer (Novatein Biosciences, Cambridge, MA, USA) at 37°C. Total cholesterol was calculated as HDL + LDL.
2.7 Statistical Analysis
Data are presented as means ± standard error of the mean (SEM). Statistical analyses were carried out using Bonferroni corrected student’s t-tests. The mRNA data were log-transformed before statistical analysis in order to obtain a normal distribution. All statistical analyses were performed using SAS 9.2 (The SAS Institute, Cary, NC, USA). A p value less than 0.05 was considered significant.
3 Results
3.1 Body Weight, Body Fat, Food Intake, and Water Intake
All animal groups demonstrated a steady weight gain rate (data not shown). No significant differences in weight gain or total body fat were observed (Table 5.1). Interestingly, the fructose-fed groups had a decreased food intake, but also an increase in water intake, making the total calorie intake equal due to the 10% fructose in the water (Table 5.1). Due to the difference in water intake, the fructose-fed animals obtained double the amount of taurine throughout the 26-week study period compared to control. No effect of taurine upon food intake, weight gain, or body fat was observed in either study.
3.2 Glucose Homeostasis, Glucose Tolerance, and Insulin Signaling
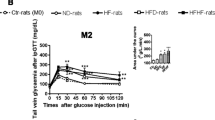
The combination of a high-fructose diet and taurine supplementation caused a significant increase in fasting glucose compared to the control diet alone, whereas taurine or fructose on their own had no significant effect on fasting glucose levels (Table 5.1). Neither a high-fructose diet nor taurine supplementation or the combination induced any change in fasting insulin levels or HOMA-IR (Table 5.1). Taurine supplementation of the fructose-fed animals caused a significant improvement in glucose tolerance, whereas this effect was not significant when animals were fed the control diet. We examined if this was due to an increase in insulin sensitivity in skeletal muscle by measuring insulin-induced Akt phosphorylation in quadriceps, but despite a higher value in the taurine-supplemented fructose group, we did not find a significant difference (Table 5.1). We also examined if the increase in fasting glucose could be due to an increased expression of the rate-limiting enzyme of gluconeogenesis, phosphoenol pyruvate carboxykinase (PCK1), but found no difference in mRNA levels between groups (Table 5.1).
3.3 Lipid Parameters
Fructose caused a significant decrease in hepatic triglyceride content, with taurine supplementation rescuing this effect. No effect of taurine alone on hepatic triglyceride content was observed (Table 5.1). No difference between groups was observed with regard to plasma triglycerides, nonesterified free fatty acids, HDL, LDL, or total cholesterol (Table 5.1).
4 Discussion
The current study set out to examine the effect of long-term taurine supplementation in rats receiving a high-fructose diet. Surprisingly we saw both beneficial and detrimental effects of taurine upon glucose metabolism.
4.1 Taurine and Glucose Metabolism
Taurine has in studies from two different laboratories been shown to counteract the effects of a high-fructose diet on glucose metabolism in Wistar rats, with no observable negative side effects of taurine supplementation (El Mesallamy et al. 2010; Nandhini et al. 2004, 2005; Nandhini and Anuradha 2002).
In the current study, taurine seems to have a beneficial effect on glucose tolerance in fructose-fed rats, but not when given together with a control diet. As taurine has previously been shown to improve insulin signaling (Nakaya et al. 2000; Nandhini et al. 2005) we examined whether or not this could be due to increased insulin-induced skeletal muscle signaling through AKT. However, although we did see an increase in AKT phosphorylation in the group receiving fructose and taurine, the increase was not significant compared to the other groups, ruling out this mechanism of action as an explanation of the improvement in glucose tolerance, although further studies are needed to confirm this. Taurine has been shown to potentiate insulin release (Carneiro et al. 2009), and although some disagree (Kulakowski and Maturo 1984), one might speculate that this could be one possible explanation for the improved glucose tolerance. Unfortunately we did not measure insulin levels during the glucose tolerance tests and although we found no difference in fasting insulin levels, this question remains unanswered.
We examined whether or not the increase in fasting glucose could be due to an increase in hepatic gluconeogenesis by measuring the mRNA level of the rate-determining gluconeogenic enzyme PCK1 (Pilkis and Granner 1992), but found no evidence of PCK1 being increased. Increased glycogenolysis could also explain the increase in fasting glucose, but this is contradicted by previous findings that taurine can attenuate glycogenolysis (Lau-Cam and Patel 2006; Patel and Lau-Cam 2006). Taurine has been shown to potentiate insulin release (Carneiro et al. 2009), but that would, if anything, cause a decrease and not an increase in fasting glucose. As such we are at present unable to explain the mechanism by which taurine might increase fasting glucose levels.
The discrepancy between our results and previous studies, in terms of increased fasting glucose when taurine is given together with fructose, may relate to the duration of the experiments. In reports using the same general strain of rats and the same concentration of taurine in the drinking water, the duration of the experiments was about 30 days (El Mesallamy et al. 2010; Nandhini et al. 2004, 2005; Nandhini and Anuradha 2002) making the duration of this study six times longer than previous studies. Furthermore, substrain differences in the outbred Wistar rat strain might also be a possible explanation as some rat strains seem to respond differently to fructose (Stark et al. 2000). The Wistar substrain used in the current study seems surprisingly tolerant towards fructose and displays only small diabetogenic effects when fed fructose compared to other Wistar substrains that show extreme increases in HOMA-IR and become severely glucose intolerant after being fructose fed (El Mesallamy et al. 2010; Nandhini et al. 2005; Perret et al. 2007).
4.2 Taurine and Lipid Metabolism
Earlier studies have shown an improvement in hepatic and plasma lipid parameters such as plasma triglycerides and free fatty acids as well as hepatic triglyceride content following taurine administration (Nardelli et al. 2011; Yan et al. 1993). In the current study we found no effect of taurine upon plasma lipid parameters in direct contradiction with earlier shorter studies (El Mesallamy et al. 2010; Nardelli et al. 2011). However, taurine did seem to normalize the hepatic triglyceride content following a high-fructose diet indicating an interaction between taurine and liver lipid metabolism or lipid storage. The differences between this and previous studies are most likely due to the Wistar rat substrain used in the current study that displayed no fructose-induced changes in plasma lipids, indicating a large degree of tolerance towards fructose. As such, any normalization by taurine upon plasma lipid parameters would not have been noticed in the current study.
4.3 Taurine and Fructose Perspective
Dietary fructose is thought, at elevated concentrations, to be a contributing factor to the marked increase in insulin resistance, obesity, and type 2 diabetes seen in the last few decades (Lustig et al. 2012; Stanhope 2012; Tappy et al. 2010; Tappy and Lê 2010) and many studies have reported that a high-fructose diet in rodents can cause insulin resistance, dyslipidemia, and type 2 diabetes (Basciano et al. 2005; Samuel 2011; Tappy et al. 2010). However, several human and animal studies have found none or only a small effect of a fructose diet on glucose metabolism (Abdullah et al. 2009; Tappy and Lê 2010). Thus, in some rat strains, long-term high-fructose diets cause an adaptation to the diet, completely abolishing the effect of a high-fructose diet on glucose and lipid homeostasis and glucose tolerance (Stark et al. 2000). Furthermore, fructose effects may depend on the composition of the control diet, i.e., effects of fructose have been reported to be significant only when compared to chow but not if compared to a diet with fructose substituted by starch (Kim et al. 1999).
An increase in fasting blood glucose has been linked to overall mortality and development of metabolic and cardiovascular disease (Seshasai et al. 2011; Singh et al. 2010). The results of the current study are particularly worrying, as taurine is an often used ingredient in energy drinks, and thereby automatically consumed together with a large amount of fructose (from either sucrose or high-fructose corn syrup used as sweeteners). There is a good case from epidemiological studies for fructose ingestion as a precipitator of insulin resistance albeit age, obesity, study duration, and other dietary factors are possible confounding factors (Stanhope 2012). Thus, the exact circumstances in which increased fructose consumption may be detrimental in humans are currently not well established. However, this study points towards a possible link between glucose homeostasis and a combined intake of fructose and taurine. As such further studies are needed to examine this interaction between taurine and fructose.
5 Conclusion
In summary, the current study indicates that long-term taurine supplementation may be beneficial with regard to glucose tolerance when given together with fructose. However, taurine also increases fasting glucose when given together with fructose. Thus, this study did not corroborate earlier findings showing that taurine has a solely beneficial effect on glucose and lipid homeostasis. Despite these negative findings, when taking into account earlier studies and the current study, it seems quite clear that there is a connection between taurine and glucose homeostasis and possibly a connection to dietary fructose, but also that a number of unanswered questions remain.
Abbreviations
- Fru:
-
Fructose
- HDL:
-
High-density lipoprotein cholesterol
- HOMA-IR:
-
Homeostasis model assessment of insulin resistance
- LDL:
-
Low-density lipoprotein cholesterol
- OGTT:
-
Oral glucose tolerance test
- Tau:
-
Taurine
References
Abdullah MM, Riediger NN, Chen Q et al (2009) Effects of long-term consumption of a high-fructose diet on conventional cardiovascular risk factors in Sprague–Dawley rats. Mol Cell Biochem 327:247–256. doi:10.1007/s11010-009-0063-z
Abramoff M, Magalhaes P, Ram S (2004) Image Processing with ImageJ. J Biophotonics Int 11:36–42
Ackermann D, Heinsen H (1935) Über die physiologische Wirkung des Asterubins und anderer, zum Teil neu dargestellter schwelfelhaltiger Guanidinderivate. Hoppe Seyles Z Physiol Chemie 235:115–121
Basciano H, Federico L, Adeli K (2005) Fructose, insulin resistance, and metabolic dyslipidemia. Nutr Metab (Lond) 2:5. doi:doi: 10.1186/1743-7075-2-5
Bonora E, Targher G, Alberiche M et al (2000) Homeostasis model assessment closely mirrors the glucose clamp technique in the assessment of insulin sensitivity: studies in subjects with various degrees of glucose tolerance and insulin sensitivity. Diabetes Care 23:57–63
Carneiro EM, Latorraca MQ, Araujo E et al (2009) Taurine supplementation modulates glucose homeostasis and islet function. J Nutr Biochem 20:503–511. doi:10.1016/j.jnutbio.2008.05.008
Dokshina GA, Silaeva TI, Lartsev EI (1976) Insulin-like effects of taurine. Vopr Med Khim 22:503–507
El Mesallamy HO, El-Demerdash E, Hammad LN, El Magdoub HM (2010) Effect of taurine supplementation on hyperhomocysteinemia and markers of oxidative stress in high fructose diet induced insulin resistance. Diabetol Metab Syndr 2:46. doi:10.1186/1758-5996-2-46
Elizarova EP, Nedosugova LV (1996) First experiments in taurine administration for diabetes mellitus. The effect on erythrocyte membranes Adv Exp Med Biol 403:583–588
Franconi F, Bennardini F, Mattana A et al (1995) Plasma and platelet taurine are reduced in subjects with insulin-dependent diabetes mellitus: effects of taurine supplementation. Am J Clin Nutr 61:1115–1119
Franconi F, Loizzo A, Ghirlanda G, Seghieri G (2006) Taurine supplementation and diabetes mellitus. Curr Opin Clin Nutr Metab Care 9:32–36
Franconi F, Miceli M, Fazzini A et al (1996) Taurine and diabetes. Humans and experimental models Adv Exp Med Biol 403:579–582
Hansen SH (2001) The role of taurine in diabetes and the development of diabetic complications. Diabetes Metab Res Rev 17:330–346
Harada N, Ninomiya C, Osako Y et al (2004) Taurine alters respiratory gas exchange and nutrient metabolism in type 2 diabetic rats. Obes Res 12:1077–1084. doi:10.1038/oby.2004.135
Kates M (1986) Techniques in Lipidology. Elsevier, New York, p 142
Kawano K, Hirashima T, Mori S et al (1992) Spontaneous long-term hyperglycemic rat with diabetic complications. Otsuka Long-Evans Tokushima Fatty (OLETF) strain. Diabetes 41:1422–1428
Kim JY, Nolte LA, Hansen PA et al (1999) Insulin resistance of muscle glucose transport in male and female rats fed a high-sucrose diet. Am J Physiol 276:R665–R672
Kim S-J, Gupta RC, Lee HW (2007) Taurine-diabetes interaction: from involvement to protection. Curr Diabetes Rev 3:165–175
Kulakowski EC, Maturo J (1984) Hypoglycemic properties of taurine: not mediated by enhanced insulin release. Biochem Pharmacol 33:2835–2838
Lau-Cam CA, Patel JP (2006) Comparison of the effects of taurine with those of related sulfur-containing compounds on pyridoxal-induced adrenomedullary catecholamine release and glycogenolysis in the rat. Adv Exp Med Biol 583:203–212
De Luca G, Calpona PR, Caponetti A et al (2001) Preliminary report: amino acid profile in platelets of diabetic patients. Metab Clin Exp 50:739–741. doi:10.1053/meta.2001.24193
Lustig RH, Schmidt LA, Brindis CD (2012) Public health: the toxic truth about sugar. Nature 482:27–29. doi:10.1038/482027a
Matthews DR, Hosker JP, Rudenski AS et al (1985) Homeostasis model assessment: insulin resistance and beta-cell function from fasting plasma glucose and insulin concentrations in man. Diabetologia 28:412–419
Nakaya Y, Minami A, Harada N et al (2000) Taurine improves insulin sensitivity in the Otsuka Long-Evans Tokushima Fatty rat, a model of spontaneous type 2 diabetes. Am J Clin Nutr 71:54–58
Nandhini ATA, Anuradha CV (2002) Taurine modulates kallikrein activity and glucose metabolism in insulin resistant rats. Amino Acids 22:27–38
Nandhini ATA, Thirunavukkarasu V, Anuradha CV (2004) Stimulation of glucose utilization and inhibition of protein glycation and AGE products by taurine. Acta Physiol Scand 181:297–303. doi:10.1111/j.1365-201X.2004.01287.x
Nandhini ATA, Thirunavukkarasu V, Anuradha CV (2005) Taurine modifies insulin signaling enzymes in the fructose-fed insulin resistant rats. Diabetes Metab 31:337–344
Nardelli TR, Ribeiro RA, Balbo SL et al (2011) Taurine prevents fat deposition and ameliorates plasma lipid profile in monosodium glutamate-obese rats. Amino Acids 41:901–908. doi:10.1007/s00726-010-0789-7
Nishimura N, Umeda C, Ona H, Yokogoshi H (2002) The effect of taurine on plasma cholesterol concentration in genetic type 2 diabetic GK rats. J Nutr Sci Vitaminol 48:483–490
Patel JP, Lau-Cam CA (2006) Taurine attenuates pyridoxal-induced adrenomedullary catecholamine release and glycogenolysis in the rat. Adv Exp Med Biol 583:147–156
Perret P, Slimani L, Briat A et al (2007) Assessment of insulin resistance in fructose-fed rats with 125I-6-deoxy-6-iodo-D-glucose, a new tracer of glucose transport. Eur J Nucl Med Mol Imaging 34:734–744. doi:10.1007/s00259-006-0267-3
Pilkis SJ, Granner DK (1992) Molecular physiology of the regulation of hepatic gluconeogenesis and glycolysis. Annu Rev Physiol 54:885–909. doi:10.1146/annurev.ph.54.030192.004321
Samuel VT (2011) Fructose induced lipogenesis: from sugar to fat to insulin resistance. Trends Endocrinol Metab 22:60–65. doi:10.1016/j.tem.2010.10.003
Seshasai SRK, Kaptoge S, Thompson A et al (2011) Diabetes mellitus, fasting glucose, and risk of cause-specific death. N Engl J Med 364:829–841. doi:10.1056/NEJMoa1008862
Singh S, Dhingra S, Ramdath DD et al (2010) Risk factors preceding type 2 diabetes and cardiomyopathy. J Cardiovasc Transl Res 3:580–596. doi:10.1007/s12265-010-9197-3
Stanhope KL (2012) Role of fructose-containing sugars in the epidemics of obesity and metabolic syndrome. Annu Rev Med 63:329–343. doi:10.1146/annurev-med-042010-113026
Stark AH, Timar B, Madar Z (2000) Adaptation of Sprague Dawley rats to long-term feeding of high fat or high fructose diets. Eur J Nutr 39:229–234
Tappy L, Lê K-A (2010) Metabolic effects of fructose and the worldwide increase in obesity. Physiol Rev 90:23–46. doi:10.1152/physrev.00019.2009
Tappy L, Lê KA, Tran C, Paquot N (2010) Fructose and metabolic diseases: new findings, new questions. Nutrition 26:1044–1049. doi:10.1016/j.nut.2010.02.014
Wieland O (1984) Methods of enzymatic analysis vol. VI. Verlag Chemie, Weinheim, pp 504–510
Yan CC, Bravo E, Cantàfora A (1993) Effect of taurine levels on liver lipid metabolism: an in vivo study in the rat. Proc Soc Exp Biol Med 202:88–96
Acknowledgements
This research was supported by The Danish Strategic Research Council grant #09-067124 and #09-059921, Danish Medical Research Council grant #271-07-0732, by Købmand i Odense Johann og Hanne Weimann f. Seedorffs Legat, Gangstedfonden, Ernst Fischers mindelegat, Eva og Hans Carl Adolfs Mindelegat, and Direktør Emil Hertz og Hustru Inger Hertz Fond.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2013 Springer Science+Business Media New York
About this paper
Cite this paper
Larsen, L.H., Ørstrup, L.K.H., Hansen, S.H., Grunnet, N., Quistorff, B., Mortensen, O.H. (2013). The Effect of Long-Term Taurine Supplementation and Fructose Feeding on Glucose and Lipid Homeostasis in Wistar Rats. In: El Idrissi, A., L'Amoreaux, W. (eds) Taurine 8. Advances in Experimental Medicine and Biology, vol 776. Springer, New York, NY. https://doi.org/10.1007/978-1-4614-6093-0_5
Download citation
DOI: https://doi.org/10.1007/978-1-4614-6093-0_5
Published:
Publisher Name: Springer, New York, NY
Print ISBN: 978-1-4614-6092-3
Online ISBN: 978-1-4614-6093-0
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)